

Abstract
Translocation of bacteria and their products across the intestinal barrier is common in patients with liver disease, and there is evidence that experimental liver fibrosis depends on bacterial translocation. The purpose of our study was to investigate liver fibrosis in conventional and germ-free (GF) C57BL/6 mice. Chronic liver injury was induced by administration of thioacetamide (TAA) in the drinking water for 21 wk or by repeated intraperitoneal injections of carbon tetrachloride (CCl4). Increased liver fibrosis was observed in GF mice compared with conventional mice. Hepatocytes showed more toxin-induced oxidative stress and cell death. This was accompanied by increased activation of hepatic stellate cells, but hepatic mediators of inflammation were not significantly different. Similarly, a genetic model using Myd88/Trif-deficient mice, which lack downstream innate immunity signaling, had more severe fibrosis than wild-type mice. Isolated Myd88/Trif-deficient hepatocytes were more susceptible to toxin-induced cell death in culture. In conclusion, the commensal microbiota prevents fibrosis upon chronic liver injury in mice. This is the first study describing a beneficial role of the commensal microbiota in maintaining liver homeostasis and preventing liver fibrosis.—Mazagova, M., Wang, L., Anfora, A. T., Wissmueller, M., Lesley, S. A., Miyamoto, Y., Eckmann, L., Dhungana, S., Pathmasiri, W., Sumner, S., Westwater, C., Brenner, D. A., Schnabl, B. Commensal microbiota is hepatoprotective and prevents liver fibrosis in mice.
Keywords: bacterial translocation, innate immune system, microbiome
Acute liver injury of any origin results in apoptosis and cell death of hepatocytes. Dead hepatocytes are cleared by the immune system and are replaced by regenerating hepatic parenchymal cells. Ongoing and chronic liver injury exhausts the proliferative capacity of the liver, and dead hepatocytes are no longer replaced by new hepatocytes. Fibrogenic cells in the liver, mainly hepatic stellate cells, are activated into myofibroblasts, produce extracellular matrix proteins, and deposit a fibrous scar in the extracellular space of the liver parenchyma. The process of fibrosis may result in end-stage liver disease or cirrhosis, which eventually disrupts the metabolic functions of the liver. Other than removing the causative agent, there is currently no specific therapy for patients with chronic liver disease, and liver transplantation is often indicated (1).
Patients with chronic liver disease show an increase in intestinal permeability, which facilitates bacteria and bacterial products to cross the intestinal barrier to extraintestinal organs including the systemic circulation (2–5). Translocated bacteria or their products are important mediators of experimental liver fibrosis by binding to receptors of the innate immune system in the liver (6). Several studies have identified a role for LPS signaling in experimental liver fibrosis. Mutant mice with a disruption of the LPS-TLR4 signaling pathway are resistant to liver injury and fibrosis (7–9). TLR4 has also been shown in patients as an important modulator of liver fibrosis (10). TLR9 is a pattern recognition receptor that is activated by CpG motifs present specifically in bacterial DNA, and TLR9-deficient mice are resistant to experimental liver fibrosis (11). We have recently demonstrated that intestinal inflammation and bacterial translocation contribute to liver fibrosis via TLR2 signaling on monocytes in the intestinal lamina propria and tumor necrosis factor receptor I signaling on intestinal epithelial cells in mice (12). However, the TLR3 ligand poly (I:C) inhibits liver fibrosis via activation of NK cell killing of hepatic stellate cells and production of IFN-β and IFN-γ (13–15). Furthermore, TLR7-mediated type I IFN signaling prevents experimental liver fibrosis (16). Together, bacterial products have different effects on the activation of the liver innate immune system and on progression of hepatic fibrosis.
In the present study, we examined the absence of the commensal microbiota on the development of liver fibrosis following toxic liver injury induced by administration of TAA or repeated injections of CCl4.
MATERIALS AND METHODS
Animal models of liver fibrosis
TAA-induced liver fibrosis in GF mice
GF male C57BL/6 mice were bred at and obtained from The National Gnotobiotic Rodent Resource Center at the University of North Carolina (Chapel Hill, NC, USA), and maintained in GF isolators at the Genomics Institute of the Novartis Research Foundation (San Diego, CA, USA) (17). Conventional male C57BL/6 mice were purchased from Charles River (Wilmington, MA, USA) and kept in a specific pathogen-free vivarium at the University of California, San Diego. TAA (Sigma-Aldrich, St. Louis, MO, USA) was dissolved in the drinking water at a concentration of 0.3 g/L, sterilized by filtration, and administered for a total of 21 wk. The drinking water was exchanged every 1–2 wk. The same lot of TAA was used for GF and conventional mice. During the 21 wk of treatment with TAA, 3 conventional mice and 1 GF mouse died. The results shown in this study are based on the following number of animals: nontreated (vehicle) conventional (n = 5) and GF mice (n = 3), TAA-treated conventional (n = 16) and GF (n = 12) mice.
CCl4-induced liver fibrosis in GF mice
For CCl4 experiments, GF male C57BL/6 mice were bred at and obtained from GF & Gnotobiotic Mouse Facilities at the University of Michigan (Ann Arbor, MI, USA), and maintained in GF isolators at the Medical University of South Carolina Gnotobiotic Animal Core (Charleston, SC, USA) (18). C57BL/6 mice were treated with CCl4 (4 µl/g body weight; 1:8 dilution with corn oil), or corn oil as control (4 µl/g body weight) by intraperitoneal injections 3 times per week. Injections were repeated for a total of 12 times. Due to the moribund appearance of some GF mice, mice were recovered for an additional 2 d following the second injection of CCl4, and the third injection was given at a dilution of 1:32. Conventional C57BL/6 mice were purchased from Charles River, kept in a specific pathogen-free vivarium at the University of California, San Diego, and treated in parallel with the same dose using the same lot of CCl4. Mice were harvested 3 d following the last injection. During the treatment with CCl4, 1 conventional mouse and 2 GF mice died. The results shown in this study are based on the following number of animals: oil-injected conventional (n = 4) and GF mice (n = 3), CCl4-injected conventional (n = 11) and GF (n = 7) mice.
CCl4-induced liver fibrosis in Myd88 and Trif-deficient mice
Mice deficient in Myd88 and Trif (Myd88−/−/TrifLPS2/LPS2) mice have been described and are on a C57BL/6 genetic background (19). As control, male C57BL/6 mice were bred in a specific pathogen-free vivarium at the University of California, San Diego, and treated on the same animal rack in parallel with Myd88−/−/TrifLPS2/LPS2 mice. Mice were treated with CCl4 (2 µl/g body weight; first and second injection with 1:100 and 1:50 dilutions with corn oil, respectively, followed by 1:4 dilutions), or corn oil as control (2 µl/g body weight) by intraperitoneal injections every third day. Injections were repeated for a total of 12 times. Mice were harvested 2 d following the last injection. During the treatment with CCl4, 2 wild-type mice and 1 Myd88−/−/TrifLPS2/LPS2 mouse died. The results shown in this study are based on the following number of animals: oil-injected wild-type (n = 5) and Myd88−/−/TrifLPS2/LPS2 mice (n = 4), CCl4-injected wild-type (n = 8–13) and Myd88−/−/TrifLPS2/LPS2 mice (n = 16) mice.
To induce intestinal dysbiosis, male C57BL/6 mice were gavaged with a mixture of vancomycin 100 mg/kg body weight, neomycin 200 mg/kg body weight, metronidazol 200 mg/kg body weight, and ampicillin 200 mg/kg body weight once daily for 1 wk. Following eradication of the commensal microbiota, mice were gavaged with 1010 colony-forming units of Escherichia coli (or PBS as control) and injected intraperitoneally with CCl4 (2 µl/g body weight; 1:4 dilution with corn oil; n = 10 in each group) or corn oil as control (2 µl/g body weight; n = 5 in each group) 3 times per week for a total of 12 times. Mice were harvested 2 d following the last injection. We used E. coli serotype O74:K:H39 isolated from a patient with peritonitis (ATCC #23535). E. coli was grown freshly in nutrient agar/broth for each gavage.
Mice were age-matched within each individual experiment and had access to water and food ad libitum. For Kupffer cell isolation, GF and conventional C57BL/6 mice were bred and maintained at the University of California, San Diego. All animals received humane care in compliance with guidelines from each individual institution and are in accordance with those set by the National Institutes of Health (Bethesda, MD, USA).
Isolation and culture of hepatocytes, Kupffer cells, and hepatic stellate cells
Isolation of liver cell fractions from normal liver using magnetic cell sorting has been described elsewhere (20, 21). Hepatic stellate cells were cultured on plastic and harvested on culture day 5. Kupffer cells were cultured overnight before stimulation with LPS (100 ng/ml) or poly (I:C) (10 μg/ml) for 3 h. Kupffer cells isolated from GF and conventional C57BL/6 mice were stimulated for 6 h. To induce cell death, hepatocytes were cultured in Medium 199 (without phenol red) containing 0% fetal calf serum in the presence of CCl4 (0.5–1 mM) for 24 h. Cell death was assessed by measuring alanine aminotransferase (ALT) in the supernatant using Infinity kit (Thermo Fisher Scientific, Waltham, MA, USA), or by cell death determination using a Cytotoxicity Detection Kit (Roche, Indianapolis, IN, USA).
Staining procedures and ALT measurement
Formalin-fixed liver samples were embedded in paraffin and stained with anti–4-hydroxynonenal (4-HNE) (Alpha Diagnostic, San Antonio, TX, USA) antibody using standard staining protocols as described (22). To detect dead cells in the liver, sections were analyzed by staining using an In Situ Cell Death Detection kit (Roche). Forty high-power fields per sample were counted for positively stained cells. Sections were also stained with Sirius red to assess fibrosis as described (21). ALT levels were measured using a kit from Infinity (Thermo Fisher Scientific).
Western blot analysis
Western blot analysis was performed as described (23) using primary antibodies for α-tubulin, Growth-arrest and DNA-damage-inducible (Gadd)-153 (CHOP; all Santa Cruz Biotechnology, Santa Cruz, CA, USA), smooth muscle α-actin (SMA; Dako, Carpinteria, CA, USA), and cytochrome P450 Cyp2e1 (Millipore, Billerica, MA, USA). Bands were quantitated using NIH ImageJ analysis.
Real-time PCR analysis
RNA extraction and real-time quantitative PCR were performed as described elsewhere (24) using specific primer sets obtained from the NIH qPrimerDepot. All data are normalized to 18S gene expression. Values are presented relative to vehicle control mice. DNA from mouse feces was extracted as previously described (22, 25). To quantify fecal bacteria, the following published bacterial primer sequences were used: 16S rRNA gene (26), Clostridium cluster I (27), Firmicutes (28), and Bacteroidetes (29).
Liquid chromatography–mass spectrometry analysis of indole-3-propionic acid
Indole-3-propionic acid (IPA) Stock solution (MP Biochemicals, Santa Ana, CA, USA) was prepared in methanol. A 10 ng/ml solution of IPA in 80% methanol was used to the tune the mass spectrometer. A multiple reaction monitoring (MRM) transition of m/z 187.9/59.1 was used to monitor the IPA signal. All mass spectrometry/mass spectrometry data were acquired on an API 4000 Q-trap mass (AB SCIEX, Framingham, MA, USA) equipped with Waters’ Acquity Ultra Performance Liquid Chromatography (Waters Corporation, Milford, MA, USA) operating in the ElectroSpray Ionization (ESI) negative ion mode. The ESI conditions were as follows: capillary voltage −4.5 kV; declustering potential −25 V; entrance potential −10 V; collision energy −24 V; collision cell exit potential −7 V; curtain gas 20; ion source gas 1 60; ion source gas 2 60; temperature 450°C. Nitrogen was used as nebulizer and auxiliary gas. The mass spectrometer was operated in MRM mode. IPA was separated from the matrix on an Agilent Eclipse XDB-C18 (4.6 × 50 mm, 5 µm) column operating at 30°C using 95:5 water:acetonitrile with 0.15% acetic acid as Mobile Phase A and 95:5 acetonitrile:water with 0.15% acetic acid as Mobile Phase B (MPB). IPA was eluted off the column using a gradient flow starting at 100% MPA and ending at 100% MPB over 2 min at a flow rate of 0.6 ml/min. A solution standard curve was constructed in water with a linear range of 10–1000 ng/ml. A 25 µl aliquot of a reconstituted plasma extract (which was previously used for an NMR analysis, not presented), was diluted using 225 µl of HPLC grade water. IPA extracts from plasma were subjected to net 30× dilution during sample preparation. A front and back standard curve was used to bracket the sample analysis. Analyst 1.42 was used to process the concentration response data by using the area under the MRM transition peak (m/z 187.9/59.1).
Statistical analysis
Mann–Whitney U rank-sum test, two-tailed Student’s test, or ANOVA with Newman-Keuls multiple comparison test was used for statistical analysis (Prism, GraphPad Software, Incorporated, La Jolla, CA, USA). Data are presented as mean ± sem. P ≤ 0.05 was selected as the level of significance.
RESULTS
TAA-induced liver fibrosis is increased in GF mice
To determine the role of commensal microbiota in the pathogenesis of liver fibrosis, conventional and GF mice were administered TAA via the drinking water for 21 wk. Liver fibrosis was initially measured assessing hepatic collagen α1(I) mRNA levels. Expression levels of the collagen α1(I) gene were significantly higher in livers of GF mice compared with conventional mice treated with TAA (P = 0.01; Fig. 1A). To assess fibrosis as a consequence of chronic liver injury, deposition of collagen was stained with Sirius red. Deposition of collagen was not observed in GF or conventional mice receiving normal drinking water. Bridging fibrosis occurred in GF mice after 21 wk of TAA in the drinking water, and conventional mice exposed to TAA for the same period of time showed portal fibrosis with some septa extending into the liver lobules (Fig. 1B). Quantification of the Sirius red-positive area confirmed a significantly increased deposition of collagen in GF mice compared with conventional mice following TAA exposure (P = 0.007; Fig. 1C). Activated hepatic stellate are the major matrix producing cell type in liver fibrosis. SMA is a marker for activated hepatic stellate cells/myofibroblasts and was more highly expressed in livers of GF mice than in conventional mice after 21 wk of TAA (Fig. 1D). Although 1 vehicle-treated GF mouse showed strong SMA reactivity on Western blot, this particular mouse did not show other features of hepatic stellate cell activation including increased hepatic collagen mRNA expression or protein deposition. In addition, 4 other nontreated GF mice did not show features of increased stellate cell activation at baseline (not shown). Because Cyp2e1 is required to bioactivate TAA to its electrophilic derivative (30), hepatic expression of Cyp2e1 was determined. Cyp2e1 protein was similarly expressed in GF mice compared with conventional mice following TAA administration (Fig. 1E). To investigate whether the absence of the intestinal microbiota results in differences in hepatic inflammation that would explain the increase in liver fibrosis of GF mice, cytokine and chemokine expression in the liver was examined. GF mice subjected to TAA showed similar hepatic gene expression levels of the inflammatory cytokines IL-1β, IL-6, and TNF-α (P = 0.01 for IL-1β conventional vs. GF following TAA; Fig. 1F), and chemokines Ccl2 (also known as monocyte chemotactic protein-1), Ccl3 (macrophage inflammatory protein-1α) and Ccl5 (RANTES) compared with conventional mice treated with TAA (Fig. 1G). Taken together, GF mice show greater liver fibrosis following chronic liver injury induced by TAA treatment for 21 wk.
Figure 1.

Chronic liver injury results in enhanced liver fibrosis in GF mice. GF and conventional mice (Conv) were treated with TAA in the drinking water for 21 wk. As control, GF and conventional mice received drinking water as vehicle (veh) alone. A) Hepatic collagen α1(I) (Col1a1) mRNA. B, C) Collagen deposition was evaluated by Sirius red staining and quantitated by image analysis. Representative sections stained with Sirius red are shown. D, E) Western blots for hepatic SMA and Cyp2e1 proteins. F, G) Hepatic gene expression of cytokines and chemokines. *P < 0.05; **P < 0.01.
Enhanced hepatic reactive oxygen species accumulation and increased liver cell death in GF mice after treatment with TAA
We next assessed reactive oxygen species (ROS) by staining for 4-HNE, which is a highly reactive aldehyde generated by the exposure of polyunsaturated fatty acids to peroxides and ROS and is used as a marker for oxidative stress (31). TAA-induced ROS was found to be higher in the livers of GF mice than conventional mice. Nontreated mice did not show any staining for 4-HNE in the liver (Fig. 2A). Gadd-153 belongs to the CCAAT/enhancer binding protein family of transcription regulatory proteins and is induced by ROS at post-transcriptional levels (32). Livers of TAA-treated GF mice had significantly higher levels of Gadd153 protein than livers of conventional mice (P = 0.03; Fig. 2B). Gadd153 activates a cell death response downstream of ROS (33, 34). Consistent with increased levels of Gadd153, GF mice showed a significantly higher rate of liver cell death than conventional mice after TAA treatment (P = 0.02; Fig. 2C, D). However, there was no significant increase in plasma ALT levels in TAA-treated conventional and GF mice compared with their nontreated counterparts. TAA is administered continuously via the drinking water and causes continuous but mild hepatocyte death. Taken together, GF mice experience enhanced hepatic ROS accumulation following TAA treatment, which is associated with more cell death of liver cells.
Figure 2.

Increased hepatic reactive oxidative stress and liver cell death in GF mice following TAA administration. A) Liver sections from conventional (Conv) and GF, vehicle, and TAA-treated mice were stained immunohistochemically for 4-HNE. Representative sections are shown. B) Liver lysates were analyzed by immunoblotting with antibodies for Gadd153 (CHOP) and tubulin. The intensity of the bands was quantified using ImageJ, and the level of Gadd153 protein was normalized to the level of tubulin protein as loading control. C) Liver sections were stained to detect dead liver cells using an In Situ Cell Death Detection kit (Roche). Representative sections are shown. D) Forty high-power fields (HPF) per single slide were counted for positively stained cells. *P < 0.05.
GF mice are more susceptible to CCl4-induced liver injury and fibrosis
To rule out the possibility that the enteric microbiota metabolizes orally administered TAA prior to absorption, a second model of liver fibrosis was investigated in GF mice. Repeated intraperitoneal injections with CCl4 are an experimental animal model of hepatic fibrosis that bypasses the gastrointestinal tract. We additionally switched the source of GF mice and the location of the GF facility to control for mouse genetic and environmental confounding variables. Similar to TAA-induced liver fibrosis, hepatic collagen α1 (I) mRNA expression (Fig. 3A), collagen deposition (P = 0.05; Fig. 3B, C) and activation of hepatic stellate cells were increased in GF compared with conventional mice following CCl4 injections (Fig. 3D). Hepatic Cyp2e1 protein expression was similar between CCl4-treated GF and conventional mice (Fig. 3E). Conventional mice subjected to CCl4 injections showed similar hepatic levels of the inflammatory cytokines IL-1β, IL-6, and TNF-α compared with GF mice (Fig. 3F). Although the chemokine Ccl2 showed no difference, hepatic gene expression of Ccl3 and Ccl5 were higher in GF mice compared with conventional mice (P = 0.04 for Ccl5 conventional vs. GF mice following CCl4; Fig. 3G). TAA was administered continuously via the drinking water in a low dose causing mild hepatocyte death; CCl4 was injected every third day and caused periodic hepatotoxicity after each injection. CCl4-induced liver injury was dramatically higher in GF mice compared with conventional mice as evidenced by plasma ALT levels (P = 0.002; Fig. 3H). These results indicate that GF mice show exacerbated toxin-induced liver injury and fibrosis.
Figure 3.

Enhanced hepatic fibrosis in GF mice subjected to CCl4 injections. GF and conventional (Conv) mice were treated with CCl4 for a total of 12 times. As control, GF and conventional mice received oil (vehicle) injections. A) Hepatic collagen α1(I) (Col1a1) mRNA. B, C) Collagen deposition was evaluated by Sirius red staining and quantitated by image analysis. Representative sections stained with Sirius red are shown. D, E) Western blots for hepatic SMA and Cyp2e1 proteins. F, G) Hepatic gene expression of cytokines and chemokines. (H) Plasma ALT levels. *P ≤ 0.05; **P < 0.01.
Mice deficient in Myd88 and Trif show increased liver injury and fibrosis
Upon recognition of pathogen-associated molecular patterns (PAMPs) by TLRs, intracellular domain of TLRs recruits 2 specific adaptor molecules, Myd88 and/or Trif, to orchestrate downstream signals (35, 36). Mice deficient in both Myd88 and TRIF (Myd88−/−/TrifLPS2/LPS2) (19) are unresponsive to TLR ligands. To determine whether lack of innate immune recognition receptor signaling affects hepatic fibrosis, toxic liver injury was induced by repeated intraperitoneal injections of CCl4. Increased hepatic collagen mRNA expression was found in Myd88−/−/TrifLPS2/LPS2 mice compared with wild-type mice (Fig. 4A). Hepatic collagen deposition and activation of hepatic stellate cells were significantly higher in Myd88−/−/TrifLPS2/LPS2 mice following CCl4 injections (P = 0.04; Fig. 4B–D). Hepatic Cyp2e1 protein expression was reduced similarly in wild-type and Myd88−/−/TrifLPS2/LPS2 mice following CCl4 treatment (Fig. 4E). Hepatic expression of inflammatory molecules was similar to the results in GF mice following CCl4 administration. Hepatic TNF-α, Ccl2, Ccl3, and Ccl5 gene expression was higher in Myd88−/−/TrifLPS2/LPS2 mice compared with wild-type mice following CCl4, but this did not reach statistical significance. No difference was found for IL-1β and IL-6 (Fig. 4F and G). CCl4-induced liver injury was significantly higher in Myd88−/−/TrifLPS2/LPS2 mice compared with wild-type mice as determined by plasma ALT levels (P = 0.03; Fig. 4H). We confirmed previous reports that the impact of Myd88 or TLR deficiency on the composition of the intestinal microbiota is minimal under homeostatic conditions (37). The abundance of the two dominant bacterial phyla Bacteroidetes and Firmicutes is similar between wild-type and Myd88−/−/TrifLPS2/LPS2 mice (Supplemental Fig. 1). These and the preceding results indicate that lack of innate immune signaling enhances toxin-induced liver injury and fibrosis in mice.
Figure 4.

Myd88/Trif-deficient mice are more susceptible to toxin-induced liver fibrosis. C57BL/6 and Myd88−/−/TrifLPS2/LPS2 mice were injected with oil as control or CCl4 for a total of 12 times. A) Hepatic collagen α1(I) (Col1a1) mRNA. B, C) Collagen deposition was evaluated by Sirius red staining and quantitated by image analysis. Representative sections stained with Sirius red are shown. D, E) Western blots for hepatic SMA and Cyp2e1 proteins. F, G) Hepatic gene expression of cytokines and chemokines. H) Plasma ALT levels. WT, wild-type. *P < 0.05.
Myd88/Trif-deficient hepatocytes are more susceptible to toxin-induced cell death
Liver fibrosis is a process that is modulated by several factors including hepatocyte damage, inflammation, and extracellular matrix deposition. To explain the beneficial effect of the microbiota in liver fibrosis, we took advantage of our genetic mouse model lacking innate immune signaling and we isolated parenchymal and nonparenchymal cells from Myd88−/−/TrifLPS2/LPS2 mice. Hepatic stellate cells isolated from Myd88−/−/TrifLPS2/LPS2 mice did not show significant differences in the expression of fibrogenic genes SMA, collagen α1(I), and TGF-β1, proinflammatory gene Ccl2, or the proliferation marker proliferating cell nuclear antigen during culture activation compared with cells isolated from wild-type mice (Fig. 5A).
Figure 5.
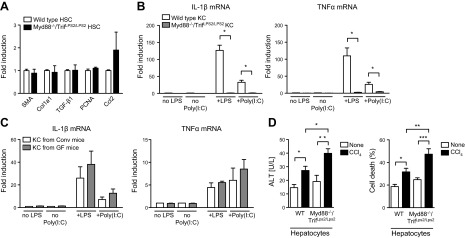
Toxin-induced cell death is higher in cultured Myd88/Trif-deficient hepatocytes. Nonparenchymal and parenchymal liver cells were isolated from C57BL/6 and Myd88−/−/TrifLPS2/LPS2 mice. A) Primary hepatic stellate cells (HSC) were cultured for 5 d. Expression of fibrotic genes (SMA, Col1a1, and TGF-β1), proliferation marker (PCNA), and Ccl2 was analyzed by quantitative PCR. The mean of 4 different hepatic stellate cell isolations is shown. B) Primary Kupffer cells (KC) were cultured for 1 d before stimulation with LPS (100 ng/ml) or poly (I:C) (10 μg/ml) for 3 h. LPS- and poly (I:C)-induced expression of IL-1β and TNF-α was analyzed by quantitative PCR. The mean of 3 different Kupffer cell isolations is shown. C) Primary Kupffer cells isolated from GF or conventional C57BL/6 mice (Conv) were stimulated with LPS (100 ng/ml) or poly (I:C) (10 μg/ml) for 6 h. IL-1β and TNF-α was analyzed by quantitative PCR. The mean of 3 different Kupffer cell isolations is shown. D) Primary mouse hepatocytes were treated with CCl4 (0.5 mM) for 24 h. CCl4-induced cell death was assessed by measuring ALT in the supernatant and by determining cell death rate. The mean of 6 different hepatocyte isolations is shown. *P < 0.05; **P < 0.01; ***P < 0.001.
Kupffer cells are resident macrophages in the liver and a major source of inflammatory mediators in liver disease. Experimental depletion of Kupffer cells inhibits liver fibrosis (38). Kupffer cells are activated by translocated bacterial products in liver fibrosis but exhibit tolerance to bacterial products such as LPS upon repeat challenge (39). To investigate whether changes in Kupffer cell biology contribute to the observed fibrosis phenotype in GF and Myd88−/−/TrifLPS2/LPS2 mice, primary Kupffer cells were isolated from wild-type and Myd88−/−/TrifLPS2/LPS2 mice and stimulated with LPS or poly (I:C). LPS binds to TLR4 and propagates its intracellular signaling via Myd88 and Trif; poly (I:C) is a TLR3 agonist and activates Trif downstream of the TLR3 receptor. Wild-type Kupffer cells showed significantly higher LPS- and poly (I:C)-induced expression of IL-1β and TNF-α compared with Myd88/Trif-deficient Kupffer cells (P = 0.03 for LPS-induced IL-1β and TNF-α mRNA in wild-type vs. Myd88/Trif-deficient Kupffer cells, P = 0.047 for poly(I:C)-induced IL-1β and TNF-α mRNA in wild-type vs. Myd88/Trif-deficient Kupffer cells; Fig. 5B). Kupffer cells were also isolated from conventional and GF C57BL/6 mice and treated with LPS or poly (I:C) in culture. Baseline, LPS- or poly (I:C)-induced TNF-α and IL-1β expression was not significantly different between Kupffer cells isolated from GF and conventional mice (Fig. 5C). Thus, Kupffer cells are unlikely mediating an inflammatory and hence fibrotic response in the absence of microbiota.
We then focused on hepatocytes as the liver cell population that potentially could be more susceptible to toxins in GF mice. As evidenced by increased ALT in the supernatant and more cell death (released LDH), CCl4 caused cell death in cultured primary hepatocytes isolated from wild-type C57BL/6 mice (P = 0.02 for ALT; P = 0.04 for LDH) or isolated from Myd88/Trif deficient mice (P = 0.002 for ALT; P = 0.0001 for LDH). Hepatocytes isolated from Myd88−/−/TrifLPS2/LPS2 mice showed significantly higher cytotoxicity than wild-type hepatocytes in response to CCl4 (P = 0.02 for ALT; P = 0.002 for LDH; Fig. 5D). Taken together, increased susceptibility of hepatocytes to toxin-induced cell death provides a possible explanation for exacerbated liver injury and fibrosis in GF mice.
Hepatocytes show less Cyp26a1 expression in Myd88−/−/TrifLPS2/LPS2 and GF mice
To provide a possible explanation between the absence of microbiota and increased toxin-induced liver injury, we performed microarrays using pooled liver mRNA from conventional and GF mice that have either been treated or not treated with liver toxins. Several of the P450 genes were found to have lower expression in livers of GF mice. Among them, we confirmed the reduced expression of Cyp26a1. Cyp26a1 mRNA expression was lower in the liver of GF mice compared with conventional mice following TAA treatment (P = 0.009; Fig. 6A) or CCl4 injections (P = 0.007; Fig. 6B). Cyp26a1 was induced in conventional mice following TAA (P = 0.03; Fig. 6A) or CCl4 treatment (P = 0.02; Fig. 6B). Likewise, Myd88/Trif-deficient hepatocytes showed decreased Cyp26a1 gene expression at baseline and after CCl4 treatment in culture (P = 0.043; Fig. 6C). Because alterations in P450 enzyme expression can result in an accumulation of toxic products, this might contribute to increased toxin-induced hepatotoxicity in GF mice.
Figure 6.

Cyp26a1 gene expression in GF and Myd88/Trif-deficient hepatocytes. A) GF and conventional mice (Conv) were treated with TAA in the drinking water for 21 wk. As control, GF and conventional mice received drinking water as vehicle alone. Hepatic Cyp26a1 mRNA expression. B) GF and conventional (Conv) mice were treated with CCl4 for a total of 12 times. As control, GF and conventional mice received oil (vehicle) injections. Hepatic Cyp26a1 mRNA expression. C) Hepatocytes were isolated from C57BL/6 and Myd88−/−/TrifLPS2/LPS2 mice and treated with CCl4 (1 mM) for 24 h. Cyp26a1 mRNA expression. The mean of 6 different hepatocyte isolations is shown. D) Liquid chromatography–mass spectrometry/mass spectrometry was used to quantitate the levels of IPA in plasma of the GF and conventional mice administered vehicle or TAA. E) Fecal levels of Clostridium Cluster I as determined by quantitative PCR. *P < 0.05; **P < 0.01.
To further delineate a mechanism resulting in enhanced liver fibrosis in GF mice upon chronic liver injury, we subjected plasma of untreated and TAA-treated conventional and GF mice to liquid chromatography–mass spectrometry/mass spectrometry analysis for IPA. We found that plasma of GF mice is devoid of IPA compared with conventional mice (Fig. 6D), confirming previously published results reported by us (17). IPA is a deamination product of dietary tryptophan, and it is a powerful antioxidant (40). Production of IPA is completely dependent on the presence of the gut microflora in particular Clostridium sporogenes (17). We confirmed that C. sporogenes is present in the feces of our conventional mice (Fig. 6E). IPA protects primary neurons and hepatocytes from oxidative stress-induced cell death (40, 41). As we observed increased hepatic oxidative stress in GF compared with conventional mice following TAA treatment (Fig. 2A), the absence of IPA in GF mice might render hepatocytes more susceptible to cell death.
Liver fibrosis in mice with dysbiosis induced by E. coli supplementation
Dysbiosis has been linked to progression from liver steatosis to nonalcoholic steatohepatitis (42) and to the progression of cholestatic liver fibrosis (43). To investigate whether changes in the intestinal microbial composition is linked to progression of toxin-induced liver injury and fibrosis, dysbiosis was induced by repeated oral gavage of nonpathogenic E. coli. Following 12 injections of CCl4, mice supplemented with E. coli showed a similar degree of liver fibrosis compared with control gavaged mice (Supplemental Fig. 2A–C). Liver injury as measured by systemic ALT levels was slightly higher in dysbiotic mice compared with control mice (P = 0.045; Supplemental Fig. 2D) indicating that dysbiosis induced by gram-negative E. coli alone is a minor contributor to CCl4-induced liver fibrosis.
DISCUSSION
There is an evolving concept that gut-derived bacterial products as a result of increased intestinal permeability and bacterial translocation play a critical role in fibrosis progression during chronic liver disease (44). First, patients with liver disease have increased systemic levels of bacterial products such as LPS and bacterial DNA, and the severity of liver disease correlates with plasma levels of bacteria-derived LPS (4). Second, reducing the intestinal bacterial burden using nonabsorbable antibiotics or stabilizing the intestinal barrier ameliorates experimental liver fibrosis (8, 12, 45–48). Third, mice with a single genetic deletion in innate immune signaling that are unable to sense bacterial products or propagate signaling are resistant to liver fibrosis (7–9, 11). Together, these clinical and experimental studies emphasize the importance of the commensal microbiota as a proinflammatory signal in chronic liver disease. On the other hand, TLR3 and TLR7 inhibit experimental liver fibrosis (13, 16). We therefore wanted to investigate the role of translocated bacteria and their products for liver fibrosis using GF mice. We made the novel observation that the microbiota is required for liver homeostasis in chronic liver injury. Both GF and Myd88/Trif-deficient mice have increased liver fibrosis upon chronic liver injury induced by administration of hepatotoxins.
Our findings fit into a broader picture of mutual symbiosis between the host and the commensal microbiota. GF mice have a severe phenotype due to the lack of bacteria, and this phenotype includes modulation of metabolic reactions, vitamin deficiencies, and dysregulation of the immune system (17, 49). The intestinal microbiota can be considered as an important part of the human body that provides additional benefits for the host such as extraction of calories from otherwise indigestible carbohydrates, bile acid metabolism, and other metabolic functions (50). A similar concept has been established for other diseases such as colitis and inflammatory bowel disease. Activation of the immune system against the commensal microbiota contributes to chronicity of disease (51). Reducing the intestinal microbiota with antibiotics protects conventional mice from dextran sodium sulfate-induced colitis (52, 53), but GF mice have exacerbated experimental colitis (54, 55). The microbiota and microbial products are required for the development of regulatory T cells with secretion of IL-10 and hence for homeostasis of the colon in disease (55–57).
The question arises as to how the bacterial microbiota protects the liver and prevents hepatic fibrosis. Using Myd88/Trif-deficient mice that are lacking downstream TLR signaling, hepatocytes were identified to be more susceptible to toxin-induced cell death. This is consistent with elevated ALT levels following CCl4 injections in GF or Myd88/Trif-deficient mice in vivo. We have confirmed findings from two previous studies demonstrating the requirement of the microbiota for the hepatic expression of P450 enzymes (58, 59). We also observed that P450 enzymes such as Cyp26a1 are down-regulated in the liver of GF mice following chronic liver injury. It is feasible that a lower expression of these phase I xenobiotic metabolizing enzymes results in an accumulation of toxic products and increased oxidative stress and hepatotoxicity, such as we have observed in injured livers of GF or Myd88/Trif-deficient mice. A low baseline level of bacterial products might be sufficient for hepatic protection from toxic stimuli. Higher systemic levels of microbial products likely do not confer additional resistance, but rather activate hepatic stellate cells and Kupffer cells/recruited macrophages to increase liver damage. This overwhelms the finely tuned balance between parenchymal cell death and regeneration and results in a deposition of scarring tissue as hepatocytes lose their capacity to regenerate. Liver fibrosis results from ongoing hepatocyte death and a diminished ability of hepatocytes to regenerate (60, 61). Hepatic stellate cells then activate and deposit matrix proteins in the extracellular space of the liver, as seen in our study. However, additional mechanisms may be involved in hepatic protection and maintenance of liver homeostasis in conventional mice. It is certainly conceivable that following chronic liver injury, products from the microbiota directly target parenchymal cells in the liver. And indeed, microbial-derived IPA might provide hepatic protection from oxidative stress. Though these possibilities deserve future scrutiny, our work described here suggests such an unappreciated, protective aspect of the gut-liver axis. This microbially mediated protection leads to increased resistance of hepatocytes to cell damage and increased self-defense.
A second established concept contributing to liver disease is intestinal dysbiosis. Disruption of the gut microbiota can result in a variety of diseases including liver diseases such as nonalcoholic steatohepatitis (42, 62). Dysbiosis following the onset of toxic liver disease was characterized by an increased prevalence of the phylum Firmicutes, namely members in the genus Lactobacillus, Dorea, and Lachnospiraceae incertae sedis. There was also a significant increase in the number of operational taxonomic units in the phylum Actinobacteria, particularly members in the genus Coriobacteriaceae in the CCl4-treated group. Toxic liver injury also resulted in a reduction in cecal microbial diversity (25). In this study, we demonstrate that experimental dysbiosis induced by repeated gavage of E. coli results in a slight increase in liver injury but not an exacerbation of liver fibrosis. This is consistent with a recent study demonstrating that high-fat diet-induced dysbiosis exacerbates cholestatic but not toxic liver fibrosis (43). Dysbiosis might cause intestinal inflammation, disruption of the gut barrier, and bacterial translocation (6, 63, 64). Subsequently, translocated bacterial products induce hepatic inflammation and liver damage (6). However, translocation of bacterial products depends on the degree of gut leakiness. We have previously reported that intestinal leakiness is lower following toxin-induced liver disease compared with other types of liver damage such as cholestasis (25). Thus, intestinal dysbiosis might not contribute as much to the progression of toxic liver fibrosis, as it does for other types of chronic liver diseases.
Taken together, this is the first study demonstrating a beneficial role of the normal microbiota in chronic liver disease. Identification of protective bacterial species and products by which they reduce liver injury and fibrosis will facilitate rational attempts to manipulate the intestinal microbiome toward a protective state and eubiosis. This will ultimately improve patient outcome in chronic liver disease.
Supplementary Material
Acknowledgments
The authors thank Drs. Richard Glynne, Alan Hofmann, Reiner Wiest, Balfour Sartor, Dimitrios Moskophidis, and Eric Johnson for helpful discussion. The study was supported in part by the U.S. National Institutes of Health (NIH) Grants K08-DK081830, R01-AA020703 (to B.S.), U01-AA021856 (to B.S. and D.A.B.), and 2P42-ES010337 (to D.A.B.). The project described was supported by Award Number 1I01BX002213 from the Biomedical Laboratory Research & Development Service of the VA Office of Research and Development (to B.S.). The authors acknowledge The National Gnotobiotic Rodent Resource Center Grants P40RR018603 and P30-DK34987 at the University of North Carolina in Chapel Hill for providing GF mice. The MUSC Gnotobiotic Animal Core is supported by the NIH Grant P30 GM103331. The IPA analysis was conducted under RTI Internal Research and Development Funds and in part by the NIH Eastern Regional Comprehensive Metabolomics Resource Core (Grant 1U24DK097193). None of the authors has a financial, personal or professional conflict of interest to disclose.
Glossary
- ALT
alanine aminotransferase
- CCl4
carbon tetrachloride
- GF
germ-free
- Gadd
growth-arrest and DNA-damage-inducible
- 4-HNE
4-hydroxynonenal
- IPA
indole-3-propionic acid
- P450
cytochrome P450
- PAMPs
pathogen-associated molecular patterns
- ROS
reactive oxygen species
- TAA
thioacetamide
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Schnabl B., Scholten D., Brenner D. A. (2008) What is the potential role of antifibrotic agents for the treatment of liver disease? Nat. Clin. Pract. Gastroenterol. Hepatol. 5, 496–497 [DOI] [PubMed] [Google Scholar]
- 2.Pascual S., Such J., Esteban A., Zapater P., Casellas J. A., Aparicio J. R., Girona E., Gutiérrez A., Carnices F., Palazón J. M., Sola-Vera J., Perez-Mateo M. (2003) Intestinal permeability is increased in patients with advanced cirrhosis. Hepatogastroenterology 50, 1482–1486 [PubMed] [Google Scholar]
- 3.Francés R., Zapater P., González-Navajas J. M., Muñoz C., Caño R., Moreu R., Pascual S., Bellot P., Pérez-Mateo M., Such J. (2008) Bacterial DNA in patients with cirrhosis and noninfected ascites mimics the soluble immune response established in patients with spontaneous bacterial peritonitis. Hepatology 47, 978–985 [DOI] [PubMed] [Google Scholar]
- 4.Lin R. S., Lee F. Y., Lee S. D., Tsai Y. T., Lin H. C., Lu R. H., Hsu W. C., Huang C. C., Wang S. S., Lo K. J. (1995) Endotoxemia in patients with chronic liver diseases: relationship to severity of liver diseases, presence of esophageal varices, and hyperdynamic circulation. J. Hepatol. 22, 165–172 [DOI] [PubMed] [Google Scholar]
- 5.Wigg A. J., Roberts-Thomson I. C., Dymock R. B., McCarthy P. J., Grose R. H., Cummins A. G. (2001) The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut 48, 206–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schnabl B., Brenner D. A. (2014) Interactions between the intestinal microbiome and liver diseases. Gastroenterology 146, 1513–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Isayama F., Hines I. N., Kremer M., Milton R. J., Byrd C. L., Perry A. W., McKim S. E., Parsons C., Rippe R. A., Wheeler M. D. (2006) LPS signaling enhances hepatic fibrogenesis caused by experimental cholestasis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G1318–G1328 [DOI] [PubMed] [Google Scholar]
- 8.Seki E., De Minicis S., Osterreicher C. H., Kluwe J., Osawa Y., Brenner D. A., Schwabe R. F. (2007) TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 13, 1324–1332 [DOI] [PubMed] [Google Scholar]
- 9.Hritz I., Mandrekar P., Velayudham A., Catalano D., Dolganiuc A., Kodys K., Kurt-Jones E., Szabo G. (2008) The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology 48, 1224–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang H., Shiffman M. L., Friedman S., Venkatesh R., Bzowej N., Abar O. T., Rowland C. M., Catanese J. J., Leong D. U., Sninsky J. J., Layden T. J., Wright T. L., White T., Cheung R. C. (2007) A 7 gene signature identifies the risk of developing cirrhosis in patients with chronic hepatitis C. Hepatology 46, 297–306 [DOI] [PubMed] [Google Scholar]
- 11.Gäbele E., Mühlbauer M., Dorn C., Weiss T. S., Froh M., Schnabl B., Wiest R., Schölmerich J., Obermeier F., Hellerbrand C. (2008) Role of TLR9 in hepatic stellate cells and experimental liver fibrosis. Biochem. Biophys. Res. Commun. 376, 271–276 [DOI] [PubMed] [Google Scholar]
- 12.Hartmann P., Haimerl M., Mazagova M., Brenner D. A., Schnabl B. (2012) Toll-like receptor 2-mediated intestinal injury and enteric tumor necrosis factor receptor I contribute to liver fibrosis in mice. Gastroenterology 143, 1330–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Radaeva S., Sun R., Jaruga B., Nguyen V. T., Tian Z., Gao B. (2006) Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology 130, 435–452 [DOI] [PubMed] [Google Scholar]
- 14.Jeong W. I., Park O., Radaeva S., Gao B. (2006) STAT1 inhibits liver fibrosis in mice by inhibiting stellate cell proliferation and stimulating NK cell cytotoxicity. Hepatology 44, 1441–1451 [DOI] [PubMed] [Google Scholar]
- 15.Yin S., Gao B. (2010) Toll-like receptor 3 in liver diseases. Gastroenterol. Res. Pract. 2010, 750904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roh Y. S., Park S., Kim J. W., Lim C. W., Seki E., Kim B. (2014) Toll-like receptor 7-mediated type I interferon signaling prevents cholestasis- and hepatotoxin-induced liver fibrosis. Hepatology 60, 237–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wikoff W. R., Anfora A. T., Liu J., Schultz P. G., Lesley S. A., Peters E. C., Siuzdak G. (2009) Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 106, 3698–3703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Westwater C., Schofield D. A., Nicholas P. J., Paulling E. E., Balish E. (2007) Candida glabrata and Candida albicans; dissimilar tissue tropism and infectivity in a gnotobiotic model of mucosal candidiasis. FEMS Immunol. Med. Microbiol. 51, 134–139 [DOI] [PubMed] [Google Scholar]
- 19.Brandl K., Sun L., Neppl C., Siggs O. M., Le Gall S. M., Tomisato W., Li X., Du X., Maennel D. N., Blobel C. P., Beutler B. (2010) MyD88 signaling in nonhematopoietic cells protects mice against induced colitis by regulating specific EGF receptor ligands. Proc. Natl. Acad. Sci. USA 107, 19967–19972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang L., Hartmann P., Haimerl M., Bathena S. P., Sjöwall C., Almer S., Alnouti Y., Hofmann A. F., Schnabl B. (2014) Nod2 deficiency protects mice from cholestatic liver disease by increasing renal excretion of bile acids. J. Hepatol. 60, 1259–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iwaisako K., Haimerl M., Paik Y. H., Taura K., Kodama Y., Sirlin C., Yu E., Yu R. T., Downes M., Evans R. M., Brenner D. A., Schnabl B. (2012) Protection from liver fibrosis by a peroxisome proliferator-activated receptor δ agonist. Proc. Natl. Acad. Sci. USA 109, E1369–E1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yan A. W., Fouts D. E., Brandl J., Stärkel P., Torralba M., Schott E., Tsukamoto H., Nelson K. E., Brenner D. A., Schnabl B. (2011) Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology 53, 96–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schnabl B., Kweon Y. O., Frederick J. P., Wang X. F., Rippe R. A., Brenner D. A. (2001) The role of Smad3 in mediating mouse hepatic stellate cell activation. Hepatology 34, 89–100 [DOI] [PubMed] [Google Scholar]
- 24.Hartmann P., Chen P., Wang H. J., Wang L., McCole D. F., Brandl K., Stärkel P., Belzer C., Hellerbrand C., Tsukamoto H., Ho S. B., Schnabl B. (2013) Deficiency of intestinal mucin-2 ameliorates experimental alcoholic liver disease in mice. Hepatology 58, 108–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fouts D. E., Torralba M., Nelson K. E., Brenner D. A., Schnabl B. (2012) Bacterial translocation and changes in the intestinal microbiome in mouse models of liver disease. J. Hepatol. 56, 1283–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maeda H., Fujimoto C., Haruki Y., Maeda T., Kokeguchi S., Petelin M., Arai H., Tanimoto I., Nishimura F., Takashiba S. (2003) Quantitative real-time PCR using TaqMan and SYBR Green for Actinobacillus actinomycetemcomitans, Porphyromonas gingivalis, Prevotella intermedia, tetQ gene and total bacteria. FEMS Immunol. Med. Microbiol. 39, 81–86 [DOI] [PubMed] [Google Scholar]
- 27.Wise M. G., Siragusa G. R. (2007) Quantitative analysis of the intestinal bacterial community in one- to three-week-old commercially reared broiler chickens fed conventional or antibiotic-free vegetable-based diets. J. Appl. Microbiol. 102, 1138–1149 [DOI] [PubMed] [Google Scholar]
- 28.Guo X., Xia X., Tang R., Zhou J., Zhao H., Wang K. (2008) Development of a real-time PCR method for Firmicutes and Bacteroidetes in faeces and its application to quantify intestinal population of obese and lean pigs. Lett. Appl. Microbiol. 47, 367–373 [DOI] [PubMed] [Google Scholar]
- 29.Nakanishi Y., Murashima K., Ohara H., Suzuki T., Hayashi H., Sakamoto M., Fukasawa T., Kubota H., Hosono A., Kono T., Kaminogawa S., Benno Y. (2006) Increase in terminal restriction fragments of Bacteroidetes-derived 16S rRNA genes after administration of short-chain fructooligosaccharides. Appl. Environ. Microbiol. 72, 6271–6276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kang J. S., Wanibuchi H., Morimura K., Wongpoomchai R., Chusiri Y., Gonzalez F. J., Fukushima S. (2008) Role of CYP2E1 in thioacetamide-induced mouse hepatotoxicity. Toxicol. Appl. Pharmacol. 228, 295–300 [DOI] [PubMed] [Google Scholar]
- 31.Paik Y. H., Iwaisako K., Seki E., Inokuchi S., Schnabl B., Osterreicher C. H., Kisseleva T., Brenner D. A. (2011) The nicotinamide adenine dinucleotide phosphate oxidase (NOX) homologues NOX1 and NOX2/gp91(phox) mediate hepatic fibrosis in mice. Hepatology 53, 1730–1741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lai W. L., Wong N. S. (2005) ROS mediates 4HPR-induced posttranscriptional expression of the Gadd153 gene. Free Radic. Biol. Med. 38, 1585–1593 [DOI] [PubMed] [Google Scholar]
- 33.Gotoh T., Terada K., Oyadomari S., Mori M. (2004) hsp70-DnaJ chaperone pair prevents nitric oxide- and CHOP-induced apoptosis by inhibiting translocation of Bax to mitochondria. Cell Death Differ. 11, 390–402 [DOI] [PubMed] [Google Scholar]
- 34.Kim D. G., You K. R., Liu M. J., Choi Y. K., Won Y. S. (2002) GADD153-mediated anticancer effects of N-(4-hydroxyphenyl)retinamide on human hepatoma cells. J. Biol. Chem. 277, 38930–38938 [DOI] [PubMed] [Google Scholar]
- 35.Seki E., Schnabl B. (2012) Role of innate immunity and the microbiota in liver fibrosis: crosstalk between the liver and gut. J. Physiol. 590, 447–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Akira S., Uematsu S., Takeuchi O. (2006) Pathogen recognition and innate immunity. Cell 124, 783–801 [DOI] [PubMed] [Google Scholar]
- 37.Ubeda C., Lipuma L., Gobourne A., Viale A., Leiner I., Equinda M., Khanin R., Pamer E. G. (2012) Familial transmission rather than defective innate immunity shapes the distinct intestinal microbiota of TLR-deficient mice. J. Exp. Med. 209, 1445–1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rivera C. A., Bradford B. U., Hunt K. J., Adachi Y., Schrum L. W., Koop D. R., Burchardt E. R., Rippe R. A., Thurman R. G. (2001) Attenuation of CCl(4)-induced hepatic fibrosis by GdCl(3) treatment or dietary glycine. Am. J. Physiol. Gastrointest. Liver Physiol. 281, G200–G207 [DOI] [PubMed] [Google Scholar]
- 39.Enomoto N., Ikejima K., Bradford B., Rivera C., Kono H., Brenner D. A., Thurman R. G. (1998) Alcohol causes both tolerance and sensitization of rat Kupffer cells via mechanisms dependent on endotoxin. Gastroenterology 115, 443–451 [DOI] [PubMed] [Google Scholar]
- 40.Karbownik M., Reiter R. J., Garcia J. J., Cabrera J., Burkhardt S., Osuna C., Lewiński A. (2001) Indole-3-propionic acid, a melatonin-related molecule, protects hepatic microsomal membranes from iron-induced oxidative damage: relevance to cancer reduction. J. Cell. Biochem. 81, 507–513 [PubMed] [Google Scholar]
- 41.Chyan Y. J., Poeggeler B., Omar R. A., Chain D. G., Frangione B., Ghiso J., Pappolla M. A. (1999) Potent neuroprotective properties against the Alzheimer beta-amyloid by an endogenous melatonin-related indole structure, indole-3-propionic acid. J. Biol. Chem. 274, 21937–21942 [DOI] [PubMed] [Google Scholar]
- 42.Henao-Mejia J., Elinav E., Jin C., Hao L., Mehal W. Z., Strowig T., Thaiss C. A., Kau A. L., Eisenbarth S. C., Jurczak M. J., Camporez J. P., Shulman G. I., Gordon J. I., Hoffman H. M., Flavell R. A. (2012) Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482, 179–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.De Minicis S., Rychlicki C., Agostinelli L., Saccomanno S., Candelaresi C., Trozzi L., Mingarelli E., Facinelli B., Magi G., Palmieri C., Marzioni M., Benedetti A., Svegliati-Baroni G (2014) Dysbiosis contributes to fibrogenesis in the course of chronic liver injury in mice. Hepatology 59, 1738–1749 [DOI] [PubMed] [Google Scholar]
- 44.Cesaro C., Tiso A., Del Prete A., Cariello R., Tuccillo C., Cotticelli G., Del Vecchio Blanco C., Loguercio C. (2011) Gut microbiota and probiotics in chronic liver diseases. Dig. Liver Dis. 43, 431–438 [DOI] [PubMed] [Google Scholar]
- 45.Adachi Y., Moore L. E., Bradford B. U., Gao W., Thurman R. G. (1995) Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology 108, 218–224 [DOI] [PubMed] [Google Scholar]
- 46.Zhang M., Song G., Minuk G. Y. (1996) Effects of hepatic stimulator substance, herbal medicine, selenium/vitamin E, and ciprofloxacin on cirrhosis in the rat. Gastroenterology 110, 1150–1155 [DOI] [PubMed] [Google Scholar]
- 47.Chen P., Torralba M., Tan J., Embree M., Zengler K., Stärkel P., Pijkeren J. P., DePew J., Loomba R., Ho S. B., Bajaj J. S., Mutlu E. A., Keshavarzian A., Tsukamoto H., Nelson K. E., Fouts D. E., Schnabl B (2014) Supplementation of saturated long-chain fatty acids maintains intestinal eubiosis and reduces ethanol-induced liver injury in mice [Epub ahead of print]. Gastroenterology 10.1053/j.gastro.2014.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang W., Gu Y., Chen Y., Deng H., Chen L., Chen S., Zhang G., Gao Z. (2010) Intestinal flora imbalance results in altered bacterial translocation and liver function in rats with experimental cirrhosis. Eur. J. Gastroenterol. Hepatol. 22, 1481–1486 [DOI] [PubMed] [Google Scholar]
- 49.Nicholson J. K., Holmes E., Kinross J., Burcelin R., Gibson G., Jia W., Pettersson S. (2012) Host-gut microbiota metabolic interactions. Science 336, 1262–1267 [DOI] [PubMed] [Google Scholar]
- 50.Bäckhed F., Ding H., Wang T., Hooper L. V., Koh G. Y., Nagy A., Semenkovich C. F., Gordon J. I. (2004) The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 101, 15718–15723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim S. C., Tonkonogy S. L., Albright C. A., Tsang J., Balish E. J., Braun J., Huycke M. M., Sartor R. B. (2005) Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology 128, 891–906 [DOI] [PubMed] [Google Scholar]
- 52.Rath H. C., Schultz M., Freitag R., Dieleman L. A., Li F., Linde H. J., Schölmerich J., Sartor R. B. (2001) Different subsets of enteric bacteria induce and perpetuate experimental colitis in rats and mice. Infect. Immun. 69, 2277–2285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brandl K., Rutschmann S., Li X., Du X., Xiao N., Schnabl B., Brenner D. A., Beutler B. (2009) Enhanced sensitivity to DSS colitis caused by a hypomorphic Mbtps1 mutation disrupting the ATF6-driven unfolded protein response. Proc. Natl. Acad. Sci. USA 106, 3300–3305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kitajima S., Morimoto M., Sagara E., Shimizu C., Ikeda Y. (2001) Dextran sodium sulfate-induced colitis in germ-free IQI/Jic mice. Exp. Anim. 50, 387–395 [DOI] [PubMed] [Google Scholar]
- 55.Pils M. C., Bleich A., Prinz I., Fasnacht N., Bollati-Fogolin M., Schippers A., Rozell B., Müller W. (2011) Commensal gut flora reduces susceptibility to experimentally induced colitis via T-cell-derived interleukin-10. Inflamm. Bowel Dis. 17, 2038–2046 [DOI] [PubMed] [Google Scholar]
- 56.Ostman S., Rask C., Wold A. E., Hultkrantz S., Telemo E. (2006) Impaired regulatory T cell function in germ-free mice. Eur. J. Immunol. 36, 2336–2346 [DOI] [PubMed] [Google Scholar]
- 57.Strauch U. G., Obermeier F., Grunwald N., Gürster S., Dunger N., Schultz M., Griese D. P., Mähler M., Schölmerich J., Rath H. C. (2005) Influence of intestinal bacteria on induction of regulatory T cells: lessons from a transfer model of colitis. Gut 54, 1546–1552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Björkholm B., Bok C. M., Lundin A., Rafter J., Hibberd M. L., Pettersson S. (2009) Intestinal microbiota regulate xenobiotic metabolism in the liver. PLoS ONE 4, e6958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rabot S., Membrez M., Bruneau A., Gerard P., Harach T., Moser M., Raymond F., Mansourian R., Chou C. J. (2010) Germ-free C57BL/6J mice are resistant to high-fat-diet-induced insulin resistance and have altered cholesterol metabolism. FASEB J. 24, 4948–4959 [DOI] [PubMed] [Google Scholar]
- 60.Rudolph K. L., Chang S., Millard M., Schreiber-Agus N., DePinho R. A. (2000) Inhibition of experimental liver cirrhosis in mice by telomerase gene delivery. Science 287, 1253–1258 [DOI] [PubMed] [Google Scholar]
- 61.Ueki T., Kaneda Y., Tsutsui H., Nakanishi K., Sawa Y., Morishita R., Matsumoto K., Nakamura T., Takahashi H., Okamoto E., Fujimoto J. (1999) Hepatocyte growth factor gene therapy of liver cirrhosis in rats. Nat. Med. 5, 226–230 [DOI] [PubMed] [Google Scholar]
- 62.Henao-Mejia J., Elinav E., Thaiss C. A., Licona-Limon P., Flavell R. A. (2013) Role of the intestinal microbiome in liver disease. J. Autoimmun. 46, 66–73 [DOI] [PubMed] [Google Scholar]
- 63.Abu-Shanab A., Quigley E. M. (2010) The role of the gut microbiota in nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 7, 691–701 [DOI] [PubMed] [Google Scholar]
- 64.Chen P., Stärkel P., Turner J. R., Ho S. B., Schnabl B. (2014) Dysbiosis-induced intestinal inflammation activates TNFRI and mediates alcoholic liver disease in mice [Epub ahead of print]. Hepatology 10.1002/hep.27489 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.