

Abstract
The majority of antibiotic-induced diarrhea is caused by Clostridium difficile (C. difficile). Hospitalizations for C. difficile infection (CDI) have tripled in the last decade, emphasizing the need to better understand how the organism colonizes the intestine and maintain infection. The mucus provides an interface for bacterial-host interactions and changes in intestinal mucus have been linked host health. To assess mucus production and composition in healthy and CDI patients, the main mucins MUC1 and MUC2 and mucus oligosaccharides were examined. Compared with healthy subjects, CDI patients demonstrated decreased MUC2 with no changes in surface MUC1. Although MUC1 did not change at the level of the epithelia, MUC1 was the primary constituent of secreted mucus in CDI patients. CDI mucus also exhibited decreased N-acetylgalactosamine (GalNAc), increased N-acetylglucosamine (GlcNAc), and increased terminal galactose residues. Increased galactose in CDI specimens is of particular interest since terminal galactose sugars are known as C. difficile toxin A receptor in animals. In vitro, C. difficile is capable of metabolizing fucose, mannose, galactose, GlcNAc, and GalNAc for growth under healthy stool conditions (low Na+ concentration, pH 6.0). Injection of C. difficile into human intestinal organoids (HIOs) demonstrated that C. difficile alone is sufficient to reduce MUC2 production but is not capable of altering host mucus oligosaccharide composition. We also demonstrate that C. difficile binds preferentially to mucus extracted from CDI patients compared with healthy subjects. Our results provide insight into a mechanism of C. difficile colonization and may provide novel target(s) for the development of alternative therapeutic agents.
Keywords: C. difficile, MUC1, MUC2, intestinal organoid, mucus oligosaccharides
clostridium difficile infection (CDI) represents an increasing public health problem as it is a primary cause of antibiotic-induced diarrhea and colitis. Since the cause and current treatment of C. difficile is antibiotics, CDI will likely persist as a major health concern. Multiple studies have focused on the effects of C. difficile toxin production on the intestinal epithelium; however, the human C. difficile mucus interaction and the process of colonization remains unknown. Gastrointestinal (GI) mucus consists of a firmly attached inner mucus layer, devoid of bacteria under normal conditions, and a loose outer mucus layer, which is colonized by bacteria (54, 55). The intestinal mucus is composed primarily of MUC2, and secreted MUC2 forms a gel-like structure that provides a physical barrier to protect the intestinal epithelium from the gut microbiota (6, 8, 53–55, 59, 107). The colon harbors the densest bacterial population and contains the thickest intestinal mucus as a result of MUC2 secretion (96). Secreted mucus and adherent mucus are continuously renewed and can be rapidly secreted by the host to limit host-bacterial interaction (65, 71). In addition to host-modulated mucus changes, several bacterial species are also capable of altering host mucus (27, 31, 63, 65, 70, 71, 96). Secretion of MUC2 mucus has been proposed to be one mechanism used by commensal bacteria to improve barrier function and exclude pathogens (67, 70, 77). If bacteria are able to penetrate the secreted MUC2 mucus layer, they are able to interact with the glycocalyx consisting of primarily cell-surface mucins, such as MUC1 (96).
To reach the epithelium, GI pathogens must develop specific virulence strategies to address the presence of mucus (71). The mucus barrier has been shown to provide partial protection against several enteric bacterial pathogens, including Yersinia enterocolitica, Shigella flexneri, Citrobacter rodentium, and Salmonella enterica Serovar Typhimurium (8, 69, 76, 107). Although multiple GI pathogens have been shown to evade or alter the host mucus barrier (8, 69, 76, 107), no studies have examined how C. difficile infection affects host mucus production or composition. It has been well documented that antibiotic disruption of the gut microbiota provides an open niche that favors C. difficile colonization and toxin production (3, 5, 9–12, 14). C. difficile has been shown to bind to mucus in cell lines and animal models (16, 32, 45, 56, 60, 98, 103, 104), but data on C. difficile mucus binding and colonization in humans are scarce. Adherence of GI pathogens to the intestinal mucus has been hypothesized to allow for optimal delivery of toxins to the host. C. difficile toxin A has been shown to bind to α-Gal-(1,3)-β-Gal-(1,4)-β-GlcNAc in animal models (19, 22, 28, 40, 41, 47, 60, 82, 83, 98), but the receptors in humans have not been identified. Furthermore, it is unknown whether C. difficile is able to manipulate the mucus oligosaccharide composition thereby exposing or stimulating production of binding and toxin receptors. A better understanding of C. difficile pathogenesis, especially during the colonization phase, is critical for developing new therapeutics for treatment of prevention of CDI.
In this study, we demonstrate that patients with CDI secrete acidic mucus that consists primarily of MUC1. Patients with CDI have decreased MUC2 expression indicating a mucus barrier defect. Injection of human intestinal organoids (HIOs) with C. difficile demonstrates that C. difficile alone is sufficient to decrease MUC2 production. In addition, we will show that patients with CDI exhibit an altered mucus oligosaccharide composition and in vitro C. difficile is able to use free oligosaccharides in media for proliferation. C. difficile injection into HIOs reveals that C. difficile alone is not sufficient to alter the mucus oligosaccharide composition. However, HIOs injected with CDI stool supernatant are sufficient to elicit the changes observed in biopsy specimens and surgical resections from patients with CDI. Finally, we demonstrate that C. difficile binds better to mucus from CDI patients compared with control patient mucus.
METHODS
Patient information.
Colonic biopsy specimens or surgical sections were obtained from healthy volunteers or patients with recurrent CDI at the University of Cincinnati Medical Center Hospital, Cincinnati, OH. All subjects provided informed consent approved by the University of Cincinnati Institutional Review Board. Patients with recurrent CDI were defined as >1 C. difficile-positive laboratory test [as determined by ELISA or lysosome-associated membrane protein (LAMP) test] and completion of at least one round of antibiotic treatment. Biopsy specimens were collected from five healthy volunteers, fixed in neutral buffered formalin, and embedded in paraffin. Healthy subjects were defined as presenting without previous or current GI symptoms and history of chronic intestinal disease or cancer. Healthy subject demographics were as follows: average age: 52, age range: 45–63, three females, and two males. Formalin-fixed paraffin sections were obtained from five de-identified patients with current CDI diagnosis (C. difficile-positive toxin test). Demographics for the CDI patients were as follows: average patient age: 44, age range: 28–65, two females, and three males. Selected patients and volunteers did not have history of cancer, diverticulosis, colostomy, inflammatory bowel disease, or small bowel obstruction.
Stool mucus was prepared from feces collected from healthy and recurrent CDI patients. For mucus extraction, stool was collected from seven healthy subjects (average age: 41; age range: 28–64; female: 5; male: 3) and seven recurrent CDI patients (average age: 47; age range: 34–71; female: 3; male: 4). Total stool DNA was prepared from feces collected from nine healthy (average age: 43; age range: 28–57; females: 5; males: 4) and nine recurrent CDI patients (average age: 51; age range 32–64; females: 6; males: 3). The same criteria for patient inclusion for biopsy specimen collection were used for stool samples.
Histology.
Transverse colon biopsy specimens, which had been previously formalin fixed and embedded in paraffin, were obtained for CDI patients. For consistency, transverse colon biopsy specimens from healthy subjects were collected, fixed overnight at 4°C in neutral-buffered formalin, and embedded in paraffin. Serial 6- to 7-μm-thick sections were applied to glass slides. Intestine architecture was examined by hematoxylin and eosin stain. Mucus composition was examined by periodic acid-Schiff-Alcian Blue stain. Briefly, deparaffinized slides were exposed to 3% acetic acid, followed by staining with Alcian Blue, 1% periodic acid-Shiff reagent, and sodium metabisulfite (Fisher Scientific, Walthman, MA). Expression of mucus oligosaccharides was examined using FITC-conjugated lectins: Ulex europaeus agglutinin 1 (UEA-1; fucose), concanavalin A (ConA; mannose), Dolichos biflorus agglutinin [DBA; N-acetylgalactosamine (GalNAc)], peanut agglutinin (PNA; galactose), and wheat germ agglutinin [WGA; N-acteylglucosamine (GlcNAc); Vector Laboratories, Burlingame, CA] were used as previously described (29, 49, 68). Briefly, sections were deparaffinized, blocked with PBS containing 10% BSA, and stained with FITC-labeled lectin (10 μg/ml) for 1 h at room temperature. Sections were then washed three times in PBS, counterstained with Hoechst (Fisher Scientific), and analyzed by confocal laser scanning microscopy (Zeiss LSM Confocal 710; Carl Zeiss, Germany).
Expression of MUC1 was examined with an anti-human MUC1 antibody (dilution 1:100, RB-9222; Thermo Fisher Scientific), MUC2 was examined with an anti-human MUC2 antibody (dilution 1:100, MS-1037; Thermo Fisher Scientific), and C. difficile binding was examined with rabbit anti-C. difficile cell surface protein antibody (dilution 1:100, ab93728; Abcam, Cambridge, MA). Briefly, sections were stripped of paraffin and incubated for 40 min at 97°C with Tris-EDTA-SDS buffer as previously described (99). Sections were then blocked with PBS containing 10% serum and stained with primary antibody overnight at 4°C. Sections were then washed three times in PBS, incubated with goat-anti-rabbit IgG Alexa Fluor 488 secondary antibody (1:100 dilution; Life Technologies, Grand Island, NY) for 1 h at room temperature, and counterstained with Hoechst (Fisher Scientific). For MUC1 and MUC2 stains, MUC1 was stained with goat-anti-rabbit IgG Alexa Fluor 488 (1:100 dilution) and MUC2 was stained with donkey anti-mouse IgG Alexa Fluor 633 (1:100 dilution) and counterstained with Hoechst (Fisher Scientific). Sections were analyzed by confocal laser scanning microscopy (Zeiss LSM Confocal 710). Digital images of slides were evaluated by tabulating mean pixel intensity of the respective color channel on each image using ImageJ software [National Institutes of Health (NIH)] and reported as relative fluorescence. Five regions of interests per image, four images per slide, and n = 5 healthy and CDI patients were used for semiquantitation of stain intensity.
Stool mucus extraction.
Crude mucus was extracted from human feces as previously described (57, 79, 88). Briefly, stool was diluted in 4 vol of ice-cold PBS (PBS; 10 mM phosphate, pH 7.2) containing 0.5 μg/μl sodium azide (prevents bacterial growth), 1 mM phenylmethylsulfonyl fluoride, iodoacetamide, and 10 mM EDTA to inhibit proteases. The suspension was shaken for 1 h at 43°C and centrifuged for 20 min at 4°C at 14,000 g to pellet stool solids. From the clear supernatant, the mucus was precipitated twice with ice-cold 60% ethanol and resuspended in ultrapure water. Mucus was then lyophilized and reconstituted based on weight. Mucus was stored at −80°C until used. Crude mucus protein concentration was determined by Bio-Rad protein assay performed according to the instructions of the manufacturer (Bio-Rad Laboratories, Richmond, CA). The original nonlyophilized crude mucus contained different amounts of proteins, with healthy mucus containing 0.4 μg/μl and CDI mucus 2.2 μg/μl. Pooled lyophilized crude mucin preparation was reconstituted to 7 μg/μl protein. C. difficile mucus binding was examined in two ways: microtiter plate and slide. For the microtiter plate assay, 100 μl of mucus were immobilized in polystyrene microtiter plate wells (Maxisorp; Nunc) by overnight incubation at 4°C as previously described (79), which covers the well with sufficient mucus. The wells were washed twice with PBS to remove excess mucus. The microtiter plate was then placed in an anaerobic hood for 2 h to create an anaerobic environment. Once anaerobic, 100 μl of C. difficile ATTC-1870 culture [106 colony-forming units (CFU)] were added to each well and incubated for 3 h at 37°C within the Coy anaerobic chamber. After incubation, the wells were washed twice with PBS to remove unattached bacteria. Mucus was then scrapped off the microtiter plate and grown on C. difficile-selective agar plates (Fisher Scientific). For the slide binding assay, 50 μl of mucus (385 μg protein) were added to glass slides and spread by an inoculating loop. Slides were allowed to air dry and then heat fixed using a Bunsen burner. Slides were blocked with PBS containing 10% serum. An ImmEdge pen (Fisher Scientific) was used to select areas for C. difficile binding. Slides were placed in an anaerobic chamber for 2 h, and then 100 μl of C. difficile (106 CFU) were added to each well and incubated for 3 h at 37°C within the anaerobic chamber. Slides were then washed three times in PBS to remove unattached bacteria, and slides were incubated with an anti-C. difficile antibody (dilution 1:100, ab93728; Abcam) for 1 h at room temperature. Slides were then washed three times in PBS, incubated with goat-anti-rabbit IgG Alexa Fluor 488 secondary antibody (1:100 dilution) (Life Technologies), coverslipped, and analyzed by confocal laser scanning microscopy (Zeiss LSM Confocal 710) and ImageJ software (NIH).
HIOs and microinjection.
Organoids resembling human proximal colon, hereafter referred to as HIOs, were generated by the Cincinnati Children's Hospital Medical Center (CCHMC) Pluripotent Stem Cell Facility. HIOs were generated by directed differentiation of human pluripotent stem cells (hPSCs) by the facility and obtained in matrigel. hPSCs were differentiated by culturing with ActivinA, fibroblast growth factor 4 (FGF4), and Wnt3a. Addition of epidermal growth factor (EGF), R-spondin, and Noggin to HIOs in matrigel generated three-dimensional growth (105). HIOs resemble the proximal colon and contain differentiated cell types (105). HIOs were injected with broth (control), C. difficile culture (grown in tryptone yeast TY broth; Ref. 33), and healthy or CDI patient stool supernatant using a Nanoject microinjector (Drummon Scientific, Broomall, PA). They were then incubated overnight as previously described (29). To generate stool supernatant, in an anaerobic hood 0.5 g of healthy or CDI stool were added to 4.5 ml tryptic soy broth (TSB; Fisher Scientific) in an anaerobic hood. Samples were vortexed and centrifuged at 150 for 10 min to pellet solid materials, and liquid supernatants containing bacteria were used for injection. After overnight incubation, injected HIOs were processed for immunostaining or RNA. RNA was extracting from HIOs using TRIzol reagent according to the manufacturer's instructions (Invitrogen, Life Technologies). To process HIOs for immunostaining, HIOs were fixed in Carnoy's fixative (60% ethanol, 30% chloroform, and 10% glacial acetic acid) for 30 min at room temperature, washed with PBS, transferred to sucrose (30% in PBS), and incubated overnight at 4°C. HIOs were placed in optimal cutting temperature (OCT) medium, frozen at −80°C, and cut to 7-μm sections on a cryostat. Slides were stained with anti-human MUC1 antibody (dilution 1:100, RB-9222; Thermo Fisher Scientific) or anti-human MUC2 antibody (dilution 1:100, MS-1037; Thermo Fisher Scientific) overnight at 4°C. Sections were countered stained with Alexa Fluor 488 or 633 secondary antibodies for 1 h at room temperature and also counterstained with Hoechst (0.1 μg/ml; Fisher Scientific) for 10 min at room temperature. Expression of mucus oligosaccharides in HIOs was examined with 10 μg/ml FITC-conjugated lectins: ConA (mannose), DBA (GalNAc), PNA (galactose), and WGA (GlcNAc) (Vector Laboratories, Burlingame, CA). Sections were stained for 1 h at room temperature followed by Hoechst staining (Fisher Scientific) for 10 min at room temperature. All slides were analyzed by confocal laser scanning microscopy (Zeiss LSM Confocal 710; Carl Zeiss), and the relative fluorescence intensity was quantified using ImageJ software (NIH) and reported as relative fluorescence. Five regions of interests per image, four images per slide, and n = 3 were used for semiquantitation of stain intensity.
Quantitative real time-PCR amplification of 16S sequences.
Quantitative (q)RT-PCR standard curves were generated from pure cultures of Micrococcus luteus, Staphylococcus aureus, Escherichia coli, Burkholderia cepacia, Bacteroidetes thetaiotaomicron ATCC 29741 (Thermo Fisher Scientific, Waltham, MA), and Rhizobium leguminosarum (Carolina Biological Supply, Burlington, NC) as previously described (29). Total DNA was isolated from stool using a QIAamp DNA Stool kit (Qiagen, Valencia, CA) with the addition of a 95°C incubation and lysozyme (10 mg/ml, 37°C for 30 min) incubation as previously described (20, 29, 36, 75, 92, 93). qPCR was performed using a Step One Real Time PCR machine [Applied Biosystems (ABI) Life Technologies] with SYBR Green PCR master mix (ABI) and bacteria-specific primers (Table 1). Standard curves were used to correlate cycle of threshold values (CT) with calculated bacteria number as previously described to access the abundance of specific intestinal bacterial phyla (4, 29, 78, 92). Percent bacterial phyla were determined with the calculated CFU values for each bacterial phylum represented as a percentage of the total bacteria.
Table 1.
Quantitative PCR primer sequences bacterial phyla
| Type | Bacteria | Forward | Reverse | Reference |
|---|---|---|---|---|
| Total | Universal (total bacteria) | ACTCCTACGGGAGGCAGCAG | ATTACCGCGGCTGCTGG | (4, 34, 42) |
| Phyla | Bacteriodetes | GGCGACCGGCGCACGGG | GRCCTTCCTCTCAGAACCC | (42) |
| Phyla | Firmicutes | GGAGYATGTGGTTTAATTCGAAGCA | AGCTGACGACAACCATGCAC | (34) |
| Phyla | Actinobacteria | CGCGGCCTATCAGCTTGTTG | ATTACCGCGGCTGCTGG | (34) |
| Phyla | α-Proteobacteria | ACTCCTACGGGAGGCAGCAG | TCTACGRATTTCACCYCTAC | (34) |
| Phyla | β-Proteobacteria | CCGCACAGTTGGCGAGATGA | CGACAGTTATGACGCCCTCC | (34) |
| Phyla | y-Proteobacteria | GAGTTTGATCATGGCTCA | GTATTACCGCGGCTGCTG | (61) |
qRT-PCR of mRNA.
To examine MUC1 and MUC2 message level, total RNA was extracted from HIOs with TRIzol reagent (Invitrogen) according to the manufacturer's instructions and as previously described. qRT-PCR was performed using SYBR Green PCR master mix (ABI) on a Step One Real Time PCR Machine (ABI) with the following gene-specific qRT-PCR primers that were derived from previous literature: Muc1 forward 5′- CCAGACCCCTGCACTCTGAT-3′; Muc1 reverse 5′-CGCTTGACAAAGGGCATGA-3′; Muc2 forward 5′-TGCCCACCTCCTCAAAGAC-3′; Muc2 reverse 5′-TAGTTTCCGTTGGAACAGTGAA-3′ (38); human GAPDH forward 5′-TGCACCACCAACTGCTTAGC-3′; and GAPDH reverse 5′-GGCATGGACTGTGGTCATGAG-3′ (86). Data were reported as the ΔΔCT using GAPDH as the standard. Differences in mRNA expression were determined by qRT-PCR and expressed as the ΔΔCT relative fold difference.
Bacterial strains and culture conditions.
C. difficile ATTC BAA-1870 was grown in media with 8 mM Na+, pH 6.0 mimicking healthy conditions and 75 mM Na+, pH 7.0 mimicking CDI conditions (see Ref. 29a, Fig. 2). To determine the ability of mucus oligosaccharides to promote C. difficile growth, fucose, GlcNAc, GalNAc (Thermo Fisher Scientific), mannose, or galactose (Sigma-Aldrich) was added to tryptone-yeast extract-glucose (TYG) media at a final concentration of 1 mM, and their presence was then analyzed for an influence on bacterial growth. Bacteria were grown under anaerobic conditions at 37°C to early stationary phase in normal TYG media [12 h, optical density at 560 nm (OD560nm) ∼1] and used to inoculate media containing varying the Na+ concentration ([Na+]). To assess restoration of growth, C. difficile was grown first in high [Na+], pH 7.0 overnight and then used to inoculate low [Na+] pH 6.0 cultures with or without oligosaccharides. Growth from all experiments was measured as the optical density (OD560nm) with an Amersham Biosciences Ultospec 3100 Spectrophotometer (GE Healthcare Life Sciences, Pittsburgh, PA) after 24 h. C. difficile titers were determined by bacterial cell counts using a Petroff-Hauser chamber (Hausser Scientific; Horsham, PA) and also by CFU (17, 18).
Fig. 2.
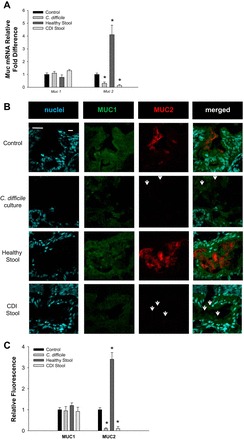
Human intestinal organoids (HIOs) injected with C. difficile and CDI stool supernatant have decreased MUC2 expression. A: Muc1 and Muc2 mRNA levels indicate decreased expression of Muc2in HIOs injected with CDI stool and C. difficile. B: confocal images MUC1 (green) and MUC2 expression (red) counterstained with Hoechst (blue) in HIOs. HIOs have similar surface MUC1 expression (green) but decreased MUC 2(red). Arrows indicate significant differences verses respective controls. Representative micrographs of observations from n = 5 healthy and CDI patient biopsy specimens. Scale bar = 50 μM. C: semiquantitative analysis of MUC1 and MUC2 stain for control (broth injected), C. difficile culture, healthy stool, and CDI stool. *P < 0.005, two-way ANOVA, Holm-Sidak.
Statistics.
Data are presented as means ± SE. Comparisons between groups were made with either one or two-way ANOVA, and the Holm-Sidak post hoc test was used to determine significance between pair wise comparisons using SigmaPlot (Systat Software, San Jose, CA). A P < 0.05 value was considered significant while n is the number of experiments performed.
RESULTS
Mucus provides both an interface and barrier between luminal bacteria and the host (85). Studies have demonstrated that some bacteria are capable of adhering to intestinal mucus and/or epithelium (84) and changing the rate of mucus secretion (26, 62, 77). Although it has been demonstrated that several GI pathogens alter mucus production, little data exist on C. difficile-mucus alteration in the patient setting. Hematoxylin and eosin stains of healthy and CDI colon reveal the presence of mucus and the formation of pseudomembranes in patients with CDI compared with healthy (Fig. 1A, black arrows). Periodic acid-Schiff-Alcian Blue stains of CDI specimens depict the secretion of acidic mucins (Fig. 1A, dark blue). To determine the type of mucin present in CDI mucus, intestinal mucins MUC1 (cell-surface) and MUC2 (secreted) were examined in human specimens by immunofluorescence (Fig. 1, B and C). Although MUC1 secretion was observed, no changes were observed in MUC1 levels at the epithelium. Interestingly, MUC2 levels were significantly decreased in CDI patients compared with healthy subjects (P < 0.001). To assess whether MUC1 was the predominant mucus in CDI stool, mucus was extracted from healthy and CDI patient stool and MUC1 and MUC2 were analyzed by immunofluorescence. Healthy subject stool was composed primarily of secreted MUC2. In contrast, CDI stool was found to be composed almost entirely of MUC1 (Fig. 1, D and E). These data indicate that CDI patients have decreased secreted MUC2 and the secretion of the normally adherent MUC1. To determine if C. difficile is capable of altering host mucus production, C. difficile, CDI, and healthy stool supernatants were injected into HIOs (Fig. 2, A–C). Injection of C. difficile and CDI stool supernatant both resulted in decreased MUC2 expression at the level of mRNA (Fig. 2A) and protein (Fig. 2, B and C). This indicates that C. difficile alone is sufficient to inhibit MUC2 expression.
Fig. 1.
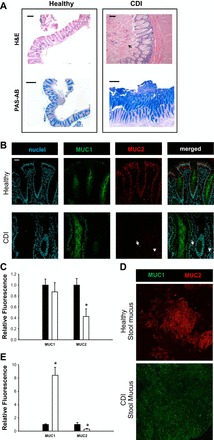
Clostridium difficile infection (CDI) patient mucus production. A: hematoxylin and eosin stains (H&E) of healthy and CDI patient biopsy specimens and surgical resections demonstrate that CDI patients have regions of pseudomembranous colitis (necrotic epithelium and mucus; black arrows) and altered morphology. Periodic acid-Schiff (PAS)-Alcian Blue (AB) stains demonstrate that CDI patients secrete acidic mucus (dark blue). Scale bar = 500 μm. B: confocal images MUC1 (green) and MUC2 expression (red) counterstained with Hoechst (blue) of healthy subjects and recurrent CDI patients. CDI patient have similar surface MUC1 expression (green) but decreased MUC 2 (red). Representative micrographs of observations from healthy subjects and CDI patient biopsy specimens (n = 5). Scale bar = 50 μM. C: semiquantitative analysis of MUC1 and MUC2 stain in healthy (black bar) and CDI (white bar) biopsy specimen. D: confocal images MUC1 (green) and MUC2 expression (red) in stool extracted mucus. CDI stool mucus consists primarily of MUC1 (green). Representative micrographs of observations healthy and CDI patient stool (n = 6). Scale bar = 50 μM. E: semiquantitative analysis of MUC1 and MUC2 stain in healthy (black bar) and CDI (white bar) stool mucus. *P < 0.005, two-way ANOVA, Holm-Sidak.
In addition to serving as a barrier, the mucus serves as an interface between the bacteria and the host: the mucus provides binding sites for both commensal and pathogenic bacteria (64, 87) and bacterial energy/food sources (2, 6, 26, 107). Gut mucus oligosaccharides GlcNAc, galactose (Gal), GalNAc, fucose (Fuc), N-acetylneuraminic acid (NeuNAc), and mannose (53–55) are attached to mucin glycoproteins and can be used by bacteria to maintain their relative niches. To address whether or not CDI patients exhibit an altered stool microbiota composition compared with healthy subjects, stool DNA was examined. Bacterial analysis of stool DNA revealed that patients with CDI have an altered gut microbiota with increased Bacteroidetes (P < 0.001) and Proteobacteria (P = 0.02) members compared with healthy subjects (Fig. 3A). These data indicate that patients with recurrent CDI have an altered gut microbiota compared with healthy subjects.
Fig. 3.
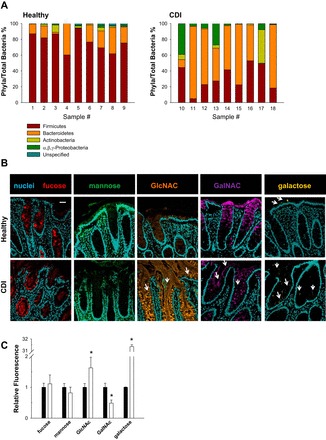
CDI patients have an altered gut microbiota and altered mucus oligosaccharide composition. A: relative phyla percentage was calculated as the percentage of bacterial phylum compared with total bacteria for healthy subjects and recurrent CDI patient stool by quantitative PCR. Each patient (1–18; n = 9 per group) is presented as a single bar to highlight interperson variation. CDI stool microbiota are significantly different from healthy stool microbiota (P < 0.001, two-way ANOVA, Holm-Sidak). B: confocal images of healthy subjects and CDI patient specimens stained with a panel of lectins to identify mucus oligosaccharides fucose (red), mannose (green), N-acetylglucosamine (GlcNAc, orange), N-acetylgalactosamine (GalNAc, purple), and galactose (yellow) counterstained with Hoechst (blue). Arrows indicate significant differences in CDI compared with control. Representative micrographs of observations from healthy subjects and CDI patient specimens (n = 5). Scale bar = 50 μM. C: semiquantitative analysis of lectin stain where fluorescence of oligosaccharide was calculated relative to unstained tissue for healthy (black bar) and CDI (white bar) biopsy and surgical resections. *P < 0.00, two-way ANOVA, Holm-Sidak; n = 5.
Although C. difficile has been shown to be devoid of oligosaccharide-cleaving enzymes (106), we hypothesized that an altered gut microbiota in CDI patients may modify the mucus oligosaccharide composition to create a favorable environment for C. difficile. To determine if CDI patients have altered intestinal mucus composition, fucose, mannose, galactose, GalNAc, and GlcNAc levels were examined by FITC-lectin (Fig. 3, B and C). CDI patients exhibited increased GlcNAc (P = 0.003) and decreased levels of GalNAc (P < 0.001). CDI patients also had increased galactose (P < 0.001) residues, a known receptor for C. difficile toxin A in mice, hamsters, rabbits, and pigs (40, 41, 47, 81). No changes in intensity were observed in fucose or mannose levels between healthy subjects and CDI patients.
To determine if C. difficile is capable of altering the host mucus oligosaccharide composition or mucus production, broth (control), C. difficile, CDI, and healthy stool supernatants were injected into HIOs (Fig. 4, A and B). Injection of C. difficile alone did not change HIO oligosaccharide composition. In contrast, injection of whole CDI stool supernatant resulted in changes similar to those observed in the mucus oligosaccharide composition of patients with CDI. These data point to another factor, likely another bacterial group within the altered gut microbiota in CDI patients, which is responsible for exposure or production of altered mucus oligosaccharide composition.
Fig. 4.
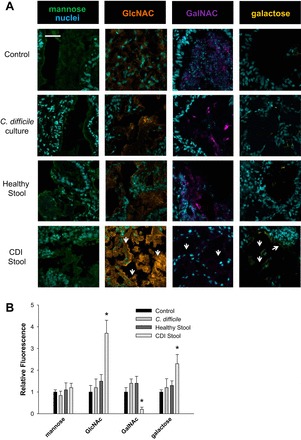
HIOs injected with CDI stool supernatant have altered mucus oligosaccharide composition. A: confocal images of HIOs stained with a panel of lectins to identify mucus oligosaccharides mannose (green), GlcNAc (orange), GalNAc (purple), and galactose (yellow) counterstained with Hoechst (blue). Arrows indicate significant difference in CDI stool vs. healthy stool. Representative micrographs of observations from n = 3. Scale bar = 50 μM. B: semiquantitative analysis of mannose, GlcNAc, GalNAc, and galactose stain for control (broth injected), C. difficile culture, healthy stool, and CDI stool. *P < 0.005, two-way ANOVA, Holm-Sidak; n = 6–9 organoids.
To better determine if C. difficile could use cleaved mucus oligosaccharides for growth, C. difficile ATTC BAA-1870 was grown in vitro in conditions mimicking healthy subjects (8 mM Na+, pH 6.0; data not shown) and CDI patients (75 mM Na+, pH 7.0; data not shown) with the addition of oligosaccharides found in the mucus (Fig. 5). Under conditions which mimic a healthy colon (8 mM Na+, pH 6.0), the additions of mannose, fucose, GlcNAc, GalNAc, and galactose were all found to enhance C. difficile growth at low [Na+] (P = 0.004; Fig. 5A). At pH 7.0, no differences were observed between high [Na+] and addition of oligosaccharides (Fig. 5B). To determine if use of oligosaccharides was reversible, C. difficile was grown overnight in high [Na+] pH 7.0 then transferred to low [Na+] pH 6.0 with or without oligosaccharides (Fig. 5C). Under these conditions (“high [Na+] to low [Na+]”), C. difficile had highest growth with galactose (P < 0.001) compared with no oligosaccharides and all other oligosaccharides examined. Differences were found between growth within no oligosaccharides between all three conditions (P < 0.001). In addition, differences were found between low [Na+] pH 6.0 and “high [Na+] to low [Na+]” with mannose (P = 0.001) and GalNAc (P = 0.007) and galactose (P = 0.015). These data indicate that C. difficile is able to use mucus oligosaccharides to drive proliferation at lower [Na+] and pH, which mimic healthy subjects, yet oligosaccharides do not influence C. difficile growth at higher [Na+] and pH levels; this use of oligosaccharides is not restored to the same degree when C. difficile grown at higher [Na+] and pH are placed in low [Na+] and pH.
Fig. 5.
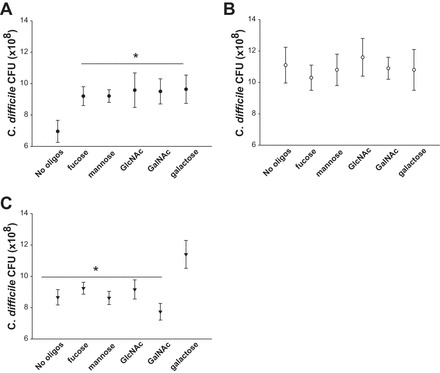
In vitro growth of C. difficile ATTC BAA-1870 in the presence of oligosaccharides. A: growth of C. difficile in tryptone-yeast extract-glucose (TYG) broth at 8 mM Na+ at pH 6.0 mimicking healthy stool with the addition of fucose, mannose, galactose, GlcNAc, and GalNAc at 24-h time point. There is a significant increase in growth between 8 mM Na+ without oligosaccharides and the addition of 1 mM fucose, mannose, galactose, GlcNAc, and GalNAc (P < 0.001). CFU, colony-forming units. B: growth of C. difficile in TYG broth at 75 mM Na+ at pH 7.0 mimicking CDI stool conditions with the addition of fucose, mannose, galactose, GlcNAc, and GalNAc at 24-h time point. Differences were observed between healthy (at 8 mM Na+ at pH 6.0) and CDI (75 mM Na+ at pH 7.0) conditions (P < 0.001); however, there was no further effect of oligosaccharide addition on growth (P = 0.194). C: growth of C. difficile when transferred from overnight culture of TYG broth at 75 mM Na+ at pH 7.0 to TYG broth at 8 mM Na+ at pH 6.0 with or without oligosaccharides at the 24-h time point. Addition of galactose (P < 0.001) yielded the highest growth compared with no oligosaccharides and all other oligosaccharides examined. Differences were found between growth within no oligosaccharides between all 3 conditions (P < 0.001). *P < 0.05, two-way ANOVA, Holm-Sidak; n = 3 experiments; n = 4.
C. difficile has been shown to be capable of binding to intestinal mucus in cell lines (32, 103), but limited data exist on the C. difficile mucus binding location in humans. Immunofluorescence revealed that C. difficile appeared to localize near the epithelium in CDI patients (Fig. 6A). To address if there is a binding site exposed in the mucus from patients with CDI, C. difficile binding was examined in mucus extracted from stool of healthy and CDI individuals. C. difficile binding was found to be substantially higher in CDI mucus compared with healthy subjects when compared by CFU (2.4-fold) and immunofluorescence (Fig. 6, A and B). C. difficile binding to stool and tissue mucus, which is composed primarily of MUC1, indicates that cell-surface mucus MUC1 may be a key binding site. The data presented herein demonstrate a number of new details about CDI infection and point to the intestinal environment and mucus as potential targets for the treatment of CDI.
Fig. 6.
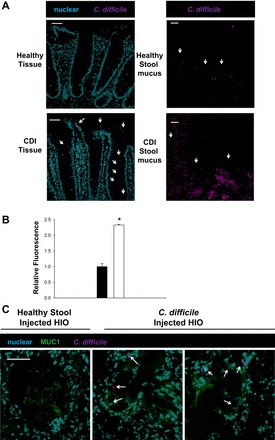
C. difficile mucus binding. A: CDI patients have C. difficile bound to tissue specimens. Confocal images from CDI patients depicting C. difficile (purple) and nuclei (blue). Representative micrographs of observations from n = 5 CDI patient specimens and n = 6 stool mucus. Healthy subjects showed no C. difficile binding while CDI patients have C. difficile localized near the epithelium. In addition, C. difficile have enhanced binding to stool mucus from patients with CDI. Scale bar = 50 μM. B: semiquantitative analysis of C. difficile stain fluorescence relative to healthy (black bar) and CDI (white bar) stool mucus. *P < 0.005, one-way ANOVA, Holm-Sidak; n = 5. C: confocal images of HIOs injected with healthy stool, C. difficile, and CDI stool supernatant depicting C. difficile (purple), MUC1 (green), and nuclei (blue). White arrows indicate C. difficile near MUC1 stains.
DISCUSSION
Little is known about the mechanism of C. difficile colonization, host binding, and mucus-C. difficile interaction. In this study, we demonstrate the following: 1) patients with CDI secrete acidic mucins composed of MUC1 (as detected in tissue and stool). At the level of the tissue, CDI patients have decreased MUC2 levels and C. difficile alone is sufficient to decrease MUC2 expression as demonstrated by HIOs injected with C. difficile. 2) Patients with CDI exhibit an altered gut microbiota and altered mucus oligosaccharide composition with increased levels of GlcNAc and galactose, decreased levels of GalNAc, and no changes in fucose or mannose levels. In vitro C. difficile growth can use fucose, mannose, galactose, GlcNAc, and GalNAc under healthy conditions (8 mM Na+, pH 6.0) to drive proliferation, but these residues do not contribute in elevated growth at high [Na+] and pH 7.0. HIOs injected with C. difficile or stool supernatant confirm that C. difficile alone is not sufficient to alter mucus oligosaccharide composition. 3) C. difficile binds better to CDI stool mucus, which is composed of secreted MUC1. Figure 7 highlights our current working model of C. difficile colonization based on the data presented herein. Normally, Firmicutes is the dominant member of the human colon at the level of the luminal and mucosa-associated bacterial populations (21). The introduction of antibiotics has been shown to alter the gut microbiota by decreasing the resident Firmicutes and increasing Proteobacteria (46, 80). In this antibiotic depleted environment, C. difficile is able to use oligosaccharides cleaved by antibiotic-resistant bacteria to proliferate. C. difficile also decreases MUC2 production to gain closer access to host. C. difficile then binds to cell adhesive MUC1 and delivers its toxin load. Toxin production alters the intestinal environment (data not shown), which further shapes the reemerging gut microbiota resulting in increased Bacteroidetes members and maintaining decreased Firmicutes members. We predict that the host attempts to prevent bacterial-host interaction by the secretion of normally adherent MUC1 mucus but this is limited in effectiveness as C. difficile is able localize near some host crypts. Exposure and/or production of terminal galactose mucus toxin receptors may also perpetuate the C. difficile infection by introducing toxin load to more host cells, continuing the altered environment and C. difficile proliferation. C. difficile mucus oligosaccharide binding may also allow the bacteria to penetrate deeper into the intestinal mucus and interact with the host in a manner similar to other pathogens (25, 26, 56, 71, 102).
Fig. 7.
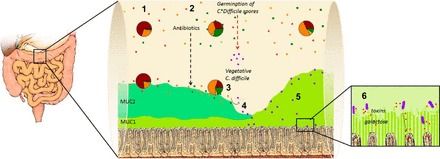
Current working model of C. difficile colonization. 1: Firmicutes is the dominant member of the human colon at the level of the luminal and mucosa-associated bacterial populations (21). 2: Antibiotic use, which decreases Firmicutes and increases Proteobacteria (46, 80), opens a niche for C. difficile. 3: After germination, C. difficile uses oligosaccharides cleaved by antibiotic-resistant bacteria to proliferate and decreases MUC2 production. 4: C. difficile then bind to cell adhesive MUC1 and deliver its toxin load, altering the intestinal environment, which shapes the gut microbiota to be increased in Bacteroidetes and decreased in Firmicutes. It remains unknown what changes are occurring in the mucosa-associated bacteria under these conditions. 5: MUC1 mucus is secreted by the host to limit bacterial-host interaction but is relatively ineffective. 6: Exposure and/or production of terminal galactose mucus toxin receptors by an alternate bacteria group likely continues C. difficile infection by introducing toxin to new host cells, continuing the altered environment and C. difficile proliferation.
Intestinal mucus is highly hydrated and requires specific fixation to preserve secreted mucins. Formalin fixation followed by paraffin embedding is suboptimal for mucus since secreted mucins are extracted during the paraffin-embedding clearing step, which typically requires prolonged incubation with organic solvents (23). Fixation with Carnoy's fixative has been demonstrated to preserve the secreted mucus layer. Due to the nature of CDI only previously processed formalin-fixed sections could be obtained. For consistency we choose to process healthy subject biopsy specimens in the same manner. As a result, the secreted mucus architecture is likely not fully retained (54) and the mucus observed in healthy and CDI biopsy specimens likely represents compressed secreted and adhered mucus composition. To account for discrepancies in secreted mucus in the biopsy specimens, stool mucus was examined and confirmed decreased MUC2 in CDI patients. In addition, in subsequent HIO experiments the HIOs were fixed in Carnoy's fixative to maintain normal mucus architecture.
Another potential caveat lies in the immature nature of HIOs. HIOs that have not been exposed to bacterial species have low level MUC2 secretion. In the future, we would like to examine HIOs first stimulated to secrete MUC2 or mature enteroids with C. difficile injection to fully confirm that C. difficile is able to decreased MUC2 production in the presence of an actively mucus secreting epithelium.
Under normal conditions, secreted Muc2 is the predominant mucus component (53–55) and forms a gel-like and viscous layer that protects the epithelium from both commensal bacteria and pathogens (8, 59, 107). Several pathogens have been shown to alter mucus production as a mechanism of infection (8, 69, 76, 107). However, before this study, limited studies have addressed the interaction of C. difficile and host mucus. We show that CDI patients have decreased secreted MUC2 (tissue and stool). However, the possibility remains that loss of MUC2 may be due to a combination of C. difficile-specific MUC2 inhibition and the loss of other MUC2-stimulating bacterial members. CDI patients do exhibit an altered stool microbiota, and alterations in the gut microbiota may not properly stimulate MUC2 production as it would under normal conditions. In addition, several GI pathogens, such as C. rodentium, have been demonstrated to cause dramatic goblet cell depletion (7, 65). It is possible that C. difficile either directly or indirectly through toxin production alters goblet cell differentiation or causes goblet cells depletion. C. difficile toxin A has been shown to exert a rapid inhibition of compound mucin exocytosis when added to HT29-Cl.16E cells (13). This may represent one mechanism C. difficile uses to minimize mucus production before it inhibits MUC2. Additionally, intestinal inflammation, a common hallmark in CDI, has also been associated with goblet cell depletion (97). Regardless of the mechanism, the loss of MUC2 has consequences for both pathogens and commensal bacteria. MUC2−/− mice have been shown to have enhanced interaction commensal bacteria with the epithelium (8). Loss of MUC2 in CDI patients may also result in increased interaction of both C. difficile and commensal bacteria with the epithelium in patients, which may exacerbate intestinal inflammation.
Decreased expression of MUC2 in patients with CDI likely allows for increased C. difficile binding to cell-surface mucins such as MUC1. Adherence of GI pathogens to the intestinal mucus likely allows toxin delivery (35). C. difficile mucus binding may likewise serve for introduction of toxin A and B to the epithelium. Toxin A has been shown to bind to the linear B type α-Gal-(1,3)-β-Gal-(1,4)-β-GlcNAc in animal models (19, 22, 28, 40, 41, 47, 60, 82, 83, 98). Although anti-Gal IgGs are present in humans (39, 43, 82), it has been hypothesized that C. difficile has an alternate receptor in humans since α-galactosyl epitopes are minimal in normal human intestine. Our data provide evidence that mucus galactose residues are exposed in CDI patients and may thus be the toxin receptor in humans as well as in the aforementioned animal models. This work is consistent with that of Smith et al. (98), who demonstrated that toxin A can bind human colonic biopsy specimens, but pretreatment of cells with α- or β-galactosidases, which cleave terminal galactose residues, reduced toxin binding. Increased galactose residues in patients with CDI may indicate increased toxin A receptor, but this has yet to be shown. Interestingly, mice overexpressing β-1,4-galactosyltransferase I (βGalT1), an enzyme involved in adding galactose to mucus glycoproteins, are resistant to TNF-induced systemic inflammatory response syndrome (SIRS) and dextran sodium sulfate (DSS)-induced colitis (101). Vanhoorren et al. (101) hypothesized that the addition of galactose was a host response to select for Firmicutes members (vs. Bacteroidetes), which may protect against inflammation. Since CDI patients have intestinal inflammation (73), increased galactose may represent a host response designed to protect against inflammation and/or alter the gut microbiota back towards high Firmicutes and lower Bacteroidetes. Alternatively, since CDI patients have an altered gut microbiota that are capable of altering the mucus oligosaccharide composition, increased galactose may also represent increased exposure as a result of degradation by other bacterial members in CDI patients. Regardless of the mechanism, our data suggest that CDI patients have an altered gut microbiota, which may indirectly result in a pathological galactose residue increase.
Our data demonstrate that C. difficile uses oligosaccharides under healthy patient conditions (low [Na+], pH 6.0) but do not use oligosaccharides to the same level under altered conditions (high [Na+], pH 7.0). This shift from using oligosaccharides at low [Na+] and a more acidic pH to not using oligosaccharides at high [Na+] with a more alkaline pH may represent the shift in C. difficile gene expression from colonization to virulence as previously described (52). Transformation from a colonization phase (use of oligosaccharides) to virulence phase (toxin production) may alter expression of bacterial oligosaccharide transporters thereby minimizing the need for oligosaccharides for growth under CDI conditions (high [Na+] and pH 7.0). A lag in the reexpression of oligosaccharide transporter may provide clues for why C. difficile did not use oligosaccharides when switched from high [Na+], pH 7.0 to low [Na+], pH 6.0, although this has yet to be determined.
Intestinal mucus is involved in protection of the enterocytes from bacteria and also provides a binding site, nutrient source, and matrix for bacterial growth (57, 101). Studies have demonstrated that several bacterial groups are capable of altering and using the mucus oligosaccharide (15, 26, 29, 30, 37, 48, 49). Release of oligosaccharides and mucin degradation is a multistep process involving a variety of microbial proteases, which degrade mucin oligosaccharides based on the size, branching, linkage, and the presence or absence of terminal sialic acid or sulfate groups (26, 72). Interestingly, C. difficile does not appear to synthesize oligosaccharide hydrolytic enzymes but does require oligosaccharides to prevent being out-competed by other bacteria (106). The lack of enzymatic activity in C. difficile and work with HIOs injected with C. difficile suggests that C. difficile uses oligosaccharides cleaved by other bacterial group(s). Only certain bacteria groups are capable of secreting oligosaccharide degrading enzymes (29, 30, 51, 53, 90, 94) including the genera Ruminococcus (Firmicutes), Bacteroides (Bacteroidetes), Bifidobacterium (Actinobacteria), Lactobacillus (Firmicutes), and Clostridium (Firmicutes) (15, 24, 26, 48, 66, 89–91). Released oligosaccharides can then be used by non-mucin degrading bacteria, including C. difficile. It is unclear which bacterial group(s) are responsible for the altered oligosaccharide composition; however, C. difficile has been shown in a mouse model to use mucus oligosaccharides released by Bacteroides thetaiotaomicron (74). In the NHE3−/− mouse terminal ileum, increased B. thetaiotaomicron resulted in increased mucus fucose concentrations (29). However, B. thetaiotaomicron is not increased in the distal colon of NHE3−/− mice or in the stool of CDI patients (data not shown). It may be possible that B. thetaiotaomicron is increased in the ileum of patients with CDI, similar to NHE3−/− mice, but further studies will be required to test this hypothesis. Although Bacteroidetes is increased in recurrent CDI patients, the genus Bacteroides is decreased in CDI patients stool (50, 100), which is consistent with data showing specific Bacteroides members can inhibit C. difficile growth (100). This suggests that a bacterial group other than Bacteroides is responsible for releasing or exposing C. difficile toxin epitopes and oligosaccharides. CDI patients have increased mouse intestinal Bacteroidetes (MIB) (data not shown), which is closely related to Bacteroides and found in both mice and humans (58, 92). It is possible that MIB may contribute to some of the changes observed in the CDI patient mucus, but this remains to be established. In mice, antibiotic treatment alters the gut microbiota (with increased Proteobacteria) (1, 44, 95) and increases free sialic acid oligosaccharides, which can be used by C. difficile (74). This suggests that members of the antibiotic-induced disrupted gut microbiota may also play a role in C. difficile colonization. These studies implicate an altered gut microbiota and subsequent alteration in mucus oligosaccharide availability in C. difficile colonization and infection. Taken together, the findings presented herein shed light on potential mechanisms of C. difficile pathogenesis and colonization. This work also demonstrates the production and exposure of potential CDI toxin receptors. Increased knowledge of these binding and/or toxin oligosaccharides could be used to improve current CDI therapeutic regimens.
GRANTS
This work was supported in part by National Institute of Diabetes and Digestive and Kidney Diseases Grant DK-079979 (to R. T. Worrell).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: M.A.E., M.B.Y., B.D., B.R.Y., and R.T.W. conception and design of research; M.A.E., M.B.Y., K.A.E., J.W., and B.R.Y. performed experiments; M.A.E., M.B.Y., K.A.E., J.W., and R.T.W. analyzed data; M.A.E., M.B.Y., B.D., B.R.Y., and R.T.W. interpreted results of experiments; M.A.E. and R.T.W. prepared figures; M.A.E. and R.T.W. drafted manuscript; M.A.E., M.B.Y., K.A.E., J.W., B.D., D.J.H., B.R.Y., and R.T.W. edited and revised manuscript; R.T.W. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Lindsey Ferreira, Stacy Huang, and Amy Engevik for technical assistance with this project. This work was in partial fulfillment of the Ph.D. degree for M. A. Engevik.
REFERENCES
- 1.Antonopoulos DA, Huse SM, Morrison HG, Schmidt TM, Sogin ML, Young VB. Reproducible community dynamics of the gastrointestinal microbiota following antibiotic perturbation. Infect Immun 77: 2367–2375, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aristoteli LP, Willcox MD. Mucin degradation mechanisms by distinct Pseudomonas aeruginosa isolates in vitro. Infect Immun 71: 5565–5575, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Badger VO, Ledeboer NA, Graham MB, Edmiston CE Jr. Clostridium difficile: epidemiology, pathogenesis, management, and prevention of a recalcitrant healthcare-associated pathogen. JPEN J Parenter Enteral Nutr 36: 645–662, 2012. [DOI] [PubMed] [Google Scholar]
- 4.Barman M, Unold D, Shifley K, Amir E, Hung K, Bos N, Salzman N. Enteric salmonellosis disrupts the microbial ecology of the murine gastrointestinal tract. Infect Immun 76: 907–915, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bauer MP, van Dissel JT. Alternative strategies for Clostridium difficile infection. Int J Antimicrob Agents 33, Suppl 1: S51–56, 2009. [DOI] [PubMed] [Google Scholar]
- 6.Berg RD. The indigenous gastrointestinal microflora. Trends Microbiol 4: 430–435, 1996. [DOI] [PubMed] [Google Scholar]
- 7.Bergstrom KS, Guttman JA, Rumi M, Ma C, Bouzari S, Khan MA, Gibson DL, Vogl AW, Vallance BA. Modulation of intestinal goblet cell function during infection by an attaching and effacing bacterial pathogen. Infect Immun 76: 796–811, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bergstrom KS, Kissoon-Singh V, Gibson DL, Ma C, Montero M, Sham HP, Ryz N, Huang T, Velcich A, Finlay BB, Chadee K, Vallance BA. Muc2 protects against lethal infectious colitis by disassociating pathogenic and commensal bacteria from the colonic mucosa. PLoS Pathog 6: e1000902, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borriello SP, Barclay FE. An in-vitro model of colonisation resistance to Clostridium difficile infection. J Med Microbiol 21: 299–309, 1986. [DOI] [PubMed] [Google Scholar]
- 10.Borriello SP, Heaton S, Drasar BS. Antibiotic susceptibility testing of anaerobes: comparison of three methods, including an automated micro-method, and evaluation of the effect of different media. J Appl Bacteriol 46: 57–63, 1979. [DOI] [PubMed] [Google Scholar]
- 11.Borriello SP, Ketley JM, Mitchell TJ, Barclay FE, Welch AR, Price AB, Stephen J. Clostridium difficile–a spectrum of virulence and analysis of putative virulence determinants in the hamster model of antibiotic-associated colitis. J Med Microbiol 24: 53–64, 1987. [DOI] [PubMed] [Google Scholar]
- 12.Borriello SP, Larson HE. Antibiotic and pseudomembranous colitis. J Antimicrob Chemother 7, Suppl A: 53–65, 1981. [DOI] [PubMed] [Google Scholar]
- 13.Branka JE, Vallette G, Jarry A, Bou-Hanna C, Lemarre P, Van PN, Laboisse CL. Early functional effects of Clostridium difficile toxin A on human colonocytes. Gastroenterology 112: 1887–1894, 1997. [DOI] [PubMed] [Google Scholar]
- 14.Britton RA, Young VB. Interaction between the intestinal microbiota and host in Clostridium difficile colonization resistance. Trends Microbiol 20: 313–319, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bry L, Falk PG, Midtvedt T, Gordon JI. A model of host-microbial interactions in an open mammalian ecosystem. Science 273: 1380–1383, 1996. [DOI] [PubMed] [Google Scholar]
- 16.Calabi E, Calabi F, Phillips AD, Fairweather NF. Binding of Clostridium difficile surface layer proteins to gastrointestinal tissues. Infect Immun 70: 5770–5778, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caldwell DR, Arcand C. Inorganic and metal-organic growth requirements of the genus Bacteroides. J Bacteriol 120: 322–333, 1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caldwell DR, Keeney M, Barton JS, Kelley JF. Sodium and other inorganic growth requirements of bacteroides amylophilus. J Bacteriol 114: 782–789, 1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Castagliuolo I, LaMont JT, Qiu B, Nikulasson ST, Pothoulakis C. A receptor decoy inhibits the enterotoxic effects of Clostridium difficile toxin A in rat ileum. Gastroenterology 111: 433–438, 1996. [DOI] [PubMed] [Google Scholar]
- 20.Castillo M, Martin-Orue SM, Manzanilla EG, Badiola I, Martin M, Gasa J. Quantification of total bacteria, enterobacteria and lactobacilli populations in pig digesta by real-time PCR. Vet Microbiol 114: 165–170, 2006. [DOI] [PubMed] [Google Scholar]
- 21.Chen W, Liu F, Ling Z, Tong X, Xiang C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS One 7: e39743, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clark GF, Krivan HC, Wilkins TD, Smith DF. Toxin A from Clostridium difficile binds to rabbit erythrocyte glycolipids with terminal Gal alpha 1–3Gal beta 1–4GlcNAc sequences. Arch Biochem Biophys 257: 217–229, 1987. [DOI] [PubMed] [Google Scholar]
- 23.Cohen M, Varki NM, Jankowski MD, Gagneux P. Using unfixed, frozen tissues to study natural mucin distribution. J Vis Exp 67: e3928, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Corfield AP, Wagner SA, Clamp JR, Kriaris MS, Hoskins LC. Mucin degradation in the human colon: production of sialidase, sialate O-acetylesterase, N-acetylneuraminate lyase, arylesterase, and glycosulfatase activities by strains of fecal bacteria. Infect Immun 60: 3971–3978, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Repentigny L, Aumont F, Bernard K, Belhumeur P. Characterization of binding of Candida albicans to small intestinal mucin and its role in adherence to mucosal epithelial cells. Infect Immun 68: 3172–3179, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deplancke B, Gaskins HR. Microbial modulation of innate defense: goblet cells and the intestinal mucus layer. Am J Clin Nutr 73: 1131S–1141S, 2001. [DOI] [PubMed] [Google Scholar]
- 27.Dohrman A, Miyata S, Gallup M, Li JD, Chapelin C, Coste A, Escudier E, Nadel J, Basbaum C. Mucin gene (MUC 2 and MUC 5AC) upregulation by Gram-positive and Gram-negative bacteria. Biochim Biophys Acta 1406: 251–259, 1998. [DOI] [PubMed] [Google Scholar]
- 28.Eglow R, Pothoulakis C, Itzkowitz S, Israel EJ, O'Keane CJ, Gong D, Gao N, Xu YL, Walker WA, LaMont JT. Diminished Clostridium difficile toxin A sensitivity in newborn rabbit ileum is associated with decreased toxin A receptor. J Clin Invest 90: 822–829, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Engevik MA, Aihara E, Montrose MH, Shull GE, Hassett DJ, Worrell RT. Loss of NHE3 alters gut microbiota composition and influences Bacteroides thetaiotaomicron growth. Am J Physiol Gastrointest Liver Physiol 305: G697–G711, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29a.Engevik MA, Engevik KA, Yacyshyn MB, Wang J, Hassett DJ, Darien B, Yacyshyn BR, Worrell RT. Human Clostridium difficile infection: inhibition of NHE3 and microbiota profile. Am J Physiol Gastrointest Liver Physiol. First published December 31, 2014; 10.1152/ajpgi.00090.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Engevik MA, Hickerson A, Shull GE, Worrell RT. Acidic conditions in the NHE2−/− intestine result in an altered mucosa-associated bacterial population with changes in mucus oligosaccharides. Cell Physiol Biochem 32: 111–128, 2013. [DOI] [PubMed] [Google Scholar]
- 31.Epple HJ, Kreusel KM, Hanski C, Schulzke JD, Riecken EO, Fromm M. Differential stimulation of intestinal mucin secretion by cholera toxin and carbachol. Pflügers Arch 433: 638–647, 1997. [DOI] [PubMed] [Google Scholar]
- 32.Eveillard M, Fourel V, Barc MC, Kerneis S, Coconnier MH, Karjalainen T, Bourlioux P, Servin AL. Identification and characterization of adhesive factors of Clostridium difficile involved in adhesion to human colonic enterocyte-like Caco-2 and mucus-secreting HT29 cells in culture. Mol Microbiol 7: 371–381, 1993. [DOI] [PubMed] [Google Scholar]
- 33.Fang A, Gerson DF, Demain AL. Production of Clostridium difficile toxin in a medium totally free of both animal and dairy proteins or digests. Proc Natl Acad Sci USA 106: 13225–13229, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fierer N, Jackson JA, Vilgalys R, Jackson RB. Assessment of soil microbial community structure by use of taxon-specific quantitative PCR assays. Appl Environ Microbiol 71: 4117–4120, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Finlay BB, Falkow S. Common themes in microbial pathogenicity. Microbiol Rev 53: 210–230, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fite A, Macfarlane GT, Cummings JH, Hopkins MJ, Kong SC, Furrie E, Macfarlane S. Identification and quantitation of mucosal and faecal desulfovibrios using real time polymerase chain reaction. Gut 53: 523–529, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Freitas M, Axelsson LG, Cayuela C, Midtvedt T, Trugnan G. Microbial-host interactions specifically control the glycosylation pattern in intestinal mouse mucosa. Histochem Cell Biol 118: 149–161, 2002. [DOI] [PubMed] [Google Scholar]
- 38.Fu J, Wei B, Wen T, Johansson ME, Liu X, Bradford E, Thomsson KA, McGee S, Mansour L, Tong M, McDaniel JM, Sferra TJ, Turner JR, Chen H, Hansson GC, Braun J, Xia L. Loss of intestinal core 1-derived O-glycans causes spontaneous colitis in mice. J Clin Invest 121: 1657–1666, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Galili U, Mandrell RE, Hamadeh RM, Shohet SB, Griffiss JM. Interaction between human natural anti-alpha-galactosyl immunoglobulin G and bacteria of the human flora. Infect Immun 56: 1730–1737, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Genth H, Dreger SC, Huelsenbeck J, Just I. Clostridium difficile toxins: more than mere inhibitors of Rho proteins. Int J Biochem Cell Biol 40: 592–597, 2008. [DOI] [PubMed] [Google Scholar]
- 41.Greco A, Ho JG, Lin SJ, Palcic MM, Rupnik M, Ng KK. Carbohydrate recognition by Clostridium difficile toxin A. Nat Struct Mol Biol 13: 460–461, 2006. [DOI] [PubMed] [Google Scholar]
- 42.Guo X, Xia X, Tang R, Zhou J, Zhao H, Wang K. Development of a real-time PCR method for Firmicutes and Bacteroidetes in faeces and its application to quantify intestinal population of obese and lean pigs. Lett Appl Microbiol 47: 367–373, 2008. [DOI] [PubMed] [Google Scholar]
- 43.Hamadeh RM, Jarvis GA, Galili U, Mandrell RE, Zhou P, Griffiss JM. Human natural anti-Gal IgG regulates alternative complement pathway activation on bacterial surfaces. J Clin Invest 89: 1223–1235, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hazenberg MP, Van de Boom M, Bakker M, Van de Merwe JP. Effect of antibiotics on the human intestinal flora in mice. Antonie Van Leeuwenhoek 49: 97–109, 1983. [DOI] [PubMed] [Google Scholar]
- 45.Hennequin C, Janoir C, Barc MC, Collignon A, Karjalainen T. Identification and characterization of a fibronectin-binding protein from Clostridium difficile. Microbiology 149: 2779–2787, 2003. [DOI] [PubMed] [Google Scholar]
- 46.Hill DA, Hoffmann C, Abt MC, Du Y, Kobuley D, Kirn TJ, Bushman FD, Artis D. Metagenomic analyses reveal antibiotic-induced temporal and spatial changes in intestinal microbiota with associated alterations in immune cell homeostasis. Mucosal Immunol 3: 148–158, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ho JG, Greco A, Rupnik M, Ng KK. Crystal structure of receptor-binding C-terminal repeats from Clostridium difficile toxin A. Proc Natl Acad Sci USA 102: 18373–18378, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hooper LV, Midtvedt T, Gordon JI. How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu Rev Nutr 22: 283–307, 2002. [DOI] [PubMed] [Google Scholar]
- 49.Hooper LV, Xu J, Falk PG, Midtvedt T, Gordon JI. A molecular sensor that allows a gut commensal to control its nutrient foundation in a competitive ecosystem. Proc Natl Acad Sci USA 96: 9833–9838, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hopkins MJ, Macfarlane GT. Changes in predominant bacterial populations in human faeces with age and with Clostridium difficile infection. J Med Microbiol 51: 448–454, 2002. [DOI] [PubMed] [Google Scholar]
- 51.Hoskins LC, Boulding ET. Mucin degradation in human colon ecosystems. Evidence for the existence and role of bacterial subpopulations producing glycosidases as extracellular enzymes. J Clin Invest 67: 163–172, 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Janoir C, Deneve C, Bouttier S, Barbut F, Hoys S, Caleechum L, Chapeton-Montes D, Pereira FC, Henriques AO, Collignon A, Monot M, Dupuy B. Adaptive strategies and pathogenesis of Clostridium difficile from in vivo transcriptomics. Infect Immun 81: 3757–3769, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Johansson ME, Ambort D, Pelaseyed T, Schutte A, Gustafsson JK, Ermund A, Subramani DB, Holmen-Larsson JM, Thomsson KA, Bergstrom JH, van der Post S, Rodriguez-Pineiro AM, Sjovall H, Backstrom M, Hansson GC. Composition and functional role of the mucus layers in the intestine. Cell Mol Life Sci 68: 3635–3641, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Johansson ME, Larsson JM, Hansson GC. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc Natl Acad Sci USA 108, Suppl 1: 4659–4665, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci USA 105: 15064–15069, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Juge N. Microbial adhesins to gastrointestinal mucus. Trends Microbiol 20: 30–39, 2012. [DOI] [PubMed] [Google Scholar]
- 57.Juntunen M, Kirjavainen PV, Ouwehand AC, Salminen SJ, Isolauri E. Adherence of probiotic bacteria to human intestinal mucus in healthy infants and during rotavirus infection. Clin Diagn Lab Immunol 8: 293–296, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kibe R, Sakamoto M, Yokota H, Benno Y. Characterization of the inhabitancy of mouse intestinal bacteria (MIB) in rodents and humans by real-time PCR with group-specific primers. Microbiol Immunol 51: 349–357, 2007. [DOI] [PubMed] [Google Scholar]
- 59.Kim YS, Ho SB. Intestinal goblet cells and mucins in health and disease: recent insights and progress. Curr Gastroenterol Rep 12: 319–330, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krivan HC, Clark GF, Smith DF, Wilkins TD. Cell surface binding site for Clostridium difficile enterotoxin: evidence for a glycoconjugate containing the sequence Gal alpha 1–3Gal beta 1–4GlcNAc. Infect Immun 53: 573–581, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee J, Kim S, Jung J, Oh BS, Kim IS, Hong SK. Analysis of total bacteria, enteric members of γ-proteobacteria and microbial communities in seawater as indirect indicators for quantifying biofouling. Environ Eng Res 14: 19–25, 2009. [Google Scholar]
- 62.Leitch GJ. Cholera enterotoxin-induced mucus secretion and increase in the mucus blanket of the rabbit ileum in vivo. Infect Immun 56: 2871–2875, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lencer WI, Reinhart FD, Neutra MR. Interaction of cholera toxin with cloned human goblet cells in monolayer culture. Am J Physiol Gastrointest Liver Physiol 258: G96–G102, 1990. [DOI] [PubMed] [Google Scholar]
- 64.Lillehoj EP, Hyun SW, Kim BT, Zhang XG, Lee DI, Rowland S, Kim KC. Muc1 mucins on the cell surface are adhesion sites for Pseudomonas aeruginosa. Am J Physiol Lung Cell Mol Physiol 280: L181–L187, 2001. [DOI] [PubMed] [Google Scholar]
- 65.Linden SK, Sutton P, Karlsson NG, Korolik V, McGuckin MA. Mucins in the mucosal barrier to infection. Mucosal Immunol 1: 183–197, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Macfarlane S, Hopkins MJ, Macfarlane GT. Toxin synthesis and mucin breakdown are related to swarming phenomenon in Clostridium septicum. Infect Immun 69: 1120–1126, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mack DR, Ahrne S, Hyde L, Wei S, Hollingsworth MA. Extracellular MUC3 mucin secretion follows adherence of Lactobacillus strains to intestinal epithelial cells in vitro. Gut 52: 827–833, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Magalhaes A, Gomes J, Ismail MN, Haslam SM, Mendes N, Osorio H, David L, Le Pendu J, Haas R, Dell A, Boren T, Reis CA. Fut2-null mice display an altered glycosylation profile and impaired BabA-mediated Helicobacter pylori adhesion to gastric mucosa. Glycobiology 19: 1525–1536, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mantle M, Basaraba L, Peacock SC, Gall DG. Binding of Yersinia enterocolitica to rabbit intestinal brush border membranes, mucus, and mucin. Infect Immun 57: 3292–3299, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mattar AF, Teitelbaum DH, Drongowski RA, Yongyi F, Harmon CM, Coran AG. Probiotics up-regulate MUC-2 mucin gene expression in a Caco-2 cell-culture model. Pediatr Surg Int 18: 586–590, 2002. [DOI] [PubMed] [Google Scholar]
- 71.McGuckin MA, Linden SK, Sutton P, Florin TH. Mucin dynamics and enteric pathogens. Nat Rev Microbiol 9: 265–278, 2011. [DOI] [PubMed] [Google Scholar]
- 72.Miner-Williams WM, Moughan PJ, Fuller MF. Analysis of an ethanol precipitate from ileal digesta: evaluation of a method to determine mucin. Sci Rep 3: 3145, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ng J, Hirota SA, Gross O, Li Y, Ulke-Lemee A, Potentier MS, Schenck LP, Vilaysane A, Seamone ME, Feng H, Armstrong GD, Tschopp J, Macdonald JA, Muruve DA, Beck PL. Clostridium difficile toxin-induced inflammation and intestinal injury are mediated by the inflammasome. Gastroenterology 139: 542–552, 552.e541–543, 2010. [DOI] [PubMed] [Google Scholar]
- 74.Ng KM, Ferreyra JA, Higginbottom SK, Lynch JB, Kashyap PC, Gopinath S, Naidu N, Choudhury B, Weimer BC, Monack DM, Sonnenburg JL. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature 502: 96–99, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Norkina O, Burnett TG, De Lisle RC. Bacterial overgrowth in the cystic fibrosis transmembrane conductance regulator null mouse small intestine. Infect Immun 72: 6040–6049, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nutten S, Sansonetti P, Huet G, Bourdon-Bisiaux C, Meresse B, Colombel JF, Desreumaux P. Epithelial inflammation response induced by Shigella flexneri depends on mucin gene expression. Microbes Infect 4: 1121–1124, 2002. [DOI] [PubMed] [Google Scholar]
- 77.Ohland CL, Macnaughton WK. Probiotic bacteria and intestinal epithelial barrier function. Am J Physiol Gastrointest Liver Physiol 298: G807–G819, 2010. [DOI] [PubMed] [Google Scholar]
- 78.Ott SJ, Musfeldt M, Ullmann U, Hampe J, Schreiber S. Quantification of intestinal bacterial populations by real-time PCR with a universal primer set and minor groove binder probes: a global approach to the enteric flora. J Clin Microbiol 42: 2566–2572, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ouwehand AC, Isolauri E, Kirjavainen PV, Salminen SJ. Adhesion of four Bifidobacterium strains to human intestinal mucus from subjects in different age groups. FEMS Microbiol Lett 172: 61–64, 1999. [DOI] [PubMed] [Google Scholar]
- 80.Perez-Cobas AE, Gosalbes MJ, Friedrichs A, Knecht H, Artacho A, Eismann K, Otto W, Rojo D, Bargiela R, von Bergen M, Neulinger SC, Daumer C, Heinsen FA, Latorre A, Barbas C, Seifert J, dos Santos VM, Ott SJ, Ferrer M, Moya A. Gut microbiota disturbance during antibiotic therapy: a multi-omic approach. Gut 62: 1591–1601, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pothoulakis C. Pathogenesis of Clostridium difficile-associated diarrhoea. Eur J Gastroenterol Hepatol 8: 1041–1047, 1996. [DOI] [PubMed] [Google Scholar]
- 82.Pothoulakis C, Galili U, Castagliuolo I, Kelly CP, Nikulasson S, Dudeja PK, Brasitus TA, LaMont JT. A human antibody binds to alpha-galactose receptors and mimics the effects of Clostridium difficile toxin A in rat colon. Gastroenterology 110: 1704–1712, 1996. [DOI] [PubMed] [Google Scholar]
- 83.Pothoulakis C, Gilbert RJ, Cladaras C, Castagliuolo I, Semenza G, Hitti Y, Montcrief JS, Linevsky J, Kelly CP, Nikulasson S, Desai HP, Wilkins TD, LaMont JT. Rabbit sucrase-isomaltase contains a functional intestinal receptor for Clostridium difficile toxin A. J Clin Invest 98: 641–649, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Probert HM, Gibson GR. Bacterial biofilms in the human gastrointestinal tract. Curr Issues Intest Microbiol 3: 23–27, 2002. [PubMed] [Google Scholar]
- 85.Pullan RD, Thomas GA, Rhodes M, Newcombe RG, Williams GT, Allen A, Rhodes J. Thickness of adherent mucus gel on colonic mucosa in humans and its relevance to colitis. Gut 35: 353–359, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Quinkler M, Bujalska IJ, Kaur K, Onyimba CU, Buhner S, Allolio B, Hughes SV, Hewison M, Stewart PM. Androgen receptor-mediated regulation of the alpha-subunit of the epithelial sodium channel in human kidney. Hypertension 46: 787–798, 2005. [DOI] [PubMed] [Google Scholar]
- 87.Rajkumar R, Devaraj H, Niranjali S. Binding of Shigella to rat and human intestinal mucin. Mol Cell Biochem 178: 261–268, 1998. [DOI] [PubMed] [Google Scholar]
- 88.Roos S, Karner F, Axelsson L, Jonsson H. Lactobacillus mucosae sp. nov, a new species with in vitro mucus-binding activity isolated from pig intestine. Int J Syst Evol Microbiol 501: 251–258, 2000. [DOI] [PubMed] [Google Scholar]
- 89.Ruas-Madiedo P, Gueimonde M, Fernandez-Garcia M, de los Reyes-Gavilan CG, Margolles A. Mucin degradation by Bifidobacterium strains isolated from the human intestinal microbiota. Appl Environ Microbiol 74: 1936–1940, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Salyers AA. Energy sources of major intestinal fermentative anaerobes. Am J Clin Nutr 32: 158–163, 1979. [DOI] [PubMed] [Google Scholar]
- 91.Salyers AA, Vercellotti JR, West SE, Wilkins TD. Fermentation of mucin and plant polysaccharides by strains of Bacteroides from the human colon. Appl Environ Microbiol 33: 319–322, 1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Salzman NH, de Jong H, Paterson Y, Harmsen HJ, Welling GW, Bos NA. Analysis of 16S libraries of mouse gastrointestinal microflora reveals a large new group of mouse intestinal bacteria. Microbiology 148: 3651–3660, 2002. [DOI] [PubMed] [Google Scholar]
- 93.Salzman NH, Hung K, Haribhai D, Chu H, Karlsson-Sjoberg J, Amir E, Teggatz P, Barman M, Hayward M, Eastwood D, Stoel M, Zhou Y, Sodergren E, Weinstock GM, Bevins CL, Williams CB, Bos NA. Enteric defensins are essential regulators of intestinal microbial ecology. Nat Immunol 11: 76–83, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schwab C, Ganzle M. Lactic acid bacteria fermentation of human milk oligosaccharide components, human milk oligosaccharides and galactooligosaccharides. FEMS Microbiol Lett 315: 141–148, 2011. [DOI] [PubMed] [Google Scholar]
- 95.Sekirov I, Tam NM, Jogova M, Robertson ML, Li Y, Lupp C, Finlay BB. Antibiotic-induced perturbations of the intestinal microbiota alter host susceptibility to enteric infection. Infect Immun 76: 4726–4736, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sheng YH, Hasnain SZ, Florin TH, McGuckin MA. Mucins in inflammatory bowel diseases and colorectal cancer. J Gastroenterol Hepatol 27: 28–38, 2012. [DOI] [PubMed] [Google Scholar]
- 97.Shirazi T, Longman RJ, Corfield AP, Probert CS. Mucins and inflammatory bowel disease. Postgrad Med J 76: 473–478, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Smith JA, Cooke DL, Hyde S, Borriello SP, Long RG. Clostridium difficile toxin A binding to human intestinal epithelial cells. J Med Microbiol 46: 953–958, 1997. [DOI] [PubMed] [Google Scholar]
- 99.Syrbu SI, Cohen MB. An enhanced antigen-retrieval protocol for immunohistochemical staining of formalin-fixed, paraffin-embedded tissues. Methods Mol Biol 717: 101–110, 2011. [DOI] [PubMed] [Google Scholar]
- 100.Tvede M, Rask-Madsen J. Bacteriotherapy for chronic relapsing Clostridium difficile diarrhoea in six patients. Lancet 1: 1156–1160, 1989. [DOI] [PubMed] [Google Scholar]
- 101.Vanhooren V, Vandenbroucke RE, Dewaele S, Van Hamme E, Haigh JJ, Hochepied T, Libert C. Mice overexpressing beta-1,4-galactosyltransferase I are resistant to TNF-induced inflammation and DSS-induced colitis. PLoS One 8: e79883, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Vimal DB, Khullar M, Gupta S, Ganguly NK. Intestinal mucins: the binding sites for Salmonella typhimurium. Mol Cell Biochem 204: 107–117, 2000. [DOI] [PubMed] [Google Scholar]
- 103.Waligora AJ, Barc MC, Bourlioux P, Collignon A, Karjalainen T. Clostridium difficile cell attachment is modified by environmental factors. Appl Environ Microbiol 65: 4234–4238, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Waligora AJ, Hennequin C, Mullany P, Bourlioux P, Collignon A, Karjalainen T. Characterization of a cell surface protein of Clostridium difficile with adhesive properties. Infect Immun 69: 2144–2153, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wells JM, Brugman S. Building additional complexity to in vitro-derived intestinal tissues. Stem Cell Res Ther 4: S1, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wilson KH, Perini F. Role of competition for nutrients in suppression of Clostridium difficile by the colonic microflora. Infect Immun 56: 2610–2614, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zarepour M, Bhullar K, Montero M, Ma C, Huang T, Velcich A, Xia L, Vallance BA. The mucin Muc2 limits pathogen burdens and epithelial barrier dysfunction during Salmonella enterica serovar Typhimurium colitis. Infect Immun 81: 3672–3683, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]