

Abstract
Abstract
The present study aimed to identify the presence of cystathionine-γ-lyase (CSE) and 3-mercaptopyruvate sulphurtransferase (3-MST), which endogenously produce hydrogen sulphide (H2S), and to functionally examine the mechanisms of H2S-induced vasodilatation in the human cutaneous microcirculation. CSE and 3-MST were quantified in forearm skin samples from 5 healthy adults (24 ± 3 years) using western blot analysis. For functional studies, microdialysis fibres were placed in the forearm skin of 12 healthy adults (25 ± 3 years) for graded infusions (0.01–100 mm) of sodium sulphide (Na2S) and sodium hydrogen sulphide (NaHS). To define the mechanisms mediating H2S-induced vasodilatation, microdialysis fibres were perfused with Ringer solution (control), a ATP-sensitive potassium channel (KATP) inhibitor, an intermediate calcium-dependent potassium channel (KCa) inhibitor, a non-specific KCa channel inhibitor or triple blockade. To determine the interaction of H2S-mediated vasodilatation with nitric oxide (NO) and cyclo-oxygenase (COX) signalling pathways, microdialysis fibres were perfused with Ringer solution (control), a non-specific NO synthase inhibitor, a non-selective COX inhibitor or combined inhibition during perfusion of increasing doses of Na2S. CSE and 3-MST were expressed in all skin samples. Na2S and NaHS elicited dose-dependent vasodilatation. Non-specific KCa channel inhibition and triple blockade blunted Na2S-induced vasodilatation (P < 0.05), whereas KATP and intermediate KCa channel inhibition had no effect (P > 0.05). Separate and combined inhibition of NO and COX attenuated H2S-induced vasodilatation (all P < 0.05). CSE and 3-MST are expressed in the human microvasculature. Exogenous H2S elicits cutaneous vasodilatation mediated by KCa channels and has a functional interaction with both NO and COX vasodilatatory signalling pathways.
Key points
Hydrogen sulphide (H2S) is vasoprotective, attenuates inflammation and modulates blood pressure in animal models; however, its specific mechanistic role in the human vasculature remains unclear.
In the present study, we report the novel finding that the enzymes responsible for endogenous H2S production, cystathionine-γ-lyase and 3-mercaptopyruvate sulphurtransferase, are expressed in the human cutaneous circulation.
Functionally, we show that H2S-induced cutaneous vasodilatation is mediated, in part, by tetraethylammonium-sensitive calcium-dependent potassium channels and not by ATP-sensitive potassium channels. In addition, nitric oxide and cyclo-oxygenase-derived byproducts are required for full expression of exogenous H2S-mediated cutaneous vasodilatation.
Future investigations of the potential role for H2S with respect to modulating vascular function in humans may have important clinical implications for understanding the mechanisms underlying vascular dysfunction characteristic of multiple cardiovascular pathologies.
Introduction
Hydrogen sulphide (H2S), once considered a malodorous toxic gas, is now recognized as a third endogenous gasotransmitter, in addition to nitric oxide (NO) and carbon monoxide. H2S has significant vasoactive properties and plays a role in the modulation of vascular function (Zhao et al. 2001; Yang et al. 2008; Papapetropoulos et al. 2014). H2S is produced endogenously through enzymatic activity, non-enzymatic pathways and intracellular sulphur stores (Li et al. 2011).
In peripheral vascular tissue, H2S is synthesized via the pyridoxal phosphate-dependent enzyme cystathionine-γ-lyase (CSE) and by 3-mercaptopyruvate sulphurtransferase (3-MST), and its release is dependent upon cholinergic stimulation (Mustafa et al, 2011). In the aorta of healthy rats, inhibition of endogenous H2S production blunted acetylcholine-dependent vasodilatation (Paredes et al. 2012). Furthermore, CSE knockout mice exhibit decreased endogenous H2S synthesis, resulting in significant impairments in both endothelium-dependent vasodilatation and hyperpolarization and also in the development of pronounced hypertension (Yang et al. 2008; Mustafa et al. 2011). Collectively, these studies suggest an important functional role for H2S as an endothelium-derived hyperpolarizing factor (EDHF) in the healthy vasculature. The expression of CSE, the primary enzymatic source of H2S in the endothelium (Hosoki et al. 1997; Webb et al. 2008; Yang et al. 2008), has been demonstrated in isolated human mammary arteries (Webb et al. 2008); however, the specific mechanistic role of H2S in the microvasculature of healthy humans remains unclear.
Exogenous administration of H2S elicits vascular relaxation in both in vitro and in vivo animal preparations (Zhao et al. 2001; Sun et al. 2011; Tian et al. 2012; Chitnis et al. 2013). In these models, H2S-induced vascular smooth muscle relaxation occurs predominantly through activation of ATP-sensitive potassium channels (KATP) and subsequent membrane hyperpolarization (Zhao et al. 2001). Recently, small, intermediate, and large conductance calcium-dependent potassium channels (SKCa, IKCa and BKCa, respectively) have emerged as additional signalling pathways by which H2S mediates vasodilatation in resistance vessels (Mustafa et al. 2011; Jackson-Weaver et al. 2013). By contrast to both the NO- and cyclo-oxygenase (COX)-dependent pathways, which result in cGMP-induced vascular smooth muscle relaxation, H2S-induced vasodilatation occurs independent from cGMP, inducing direct hyperpolarization of the vascular smooth muscle (Zhao et al. 2001). In addition to its direct hyperpolarizing effects, murine studies suggest that H2S also interacts with both NO and byproducts of COX metabolism to modulate vascular tone (Hu et al. 2008; Minamishima et al. 2009). However, to date, no studies have examined H2S-induced vasodilatation or potential interactions with the NO and COX signalling pathways in vivo in the healthy human vasculature.
The human cutaneous circulation is an accessible regional vascular bed that allows for the in vivo investigation of the specific molecular mechanisms mediating the regulation of vascular function in healthy, preclinical and diseased adults (Abularrage et al. 2005; Rossi et al. 2006; Holowatz et al. 2008). Deficits in cutaneous vascular function are highly correlated with, and predictive of, vascular dysfunction evident in both the coronary and renal circulations (Khan et al. 2008; Coulon et al. 2012). Therefore, the cutaneous circulation has significant utility for examining the mechanisms underlying microvascular signalling and potential functional alterations with cardiovascular pathology (Abularrage et al. 2005; Rossi et al. 2006).
As a necessary first step to understanding the functional role of H2S signalling in health and disease, the present study aimed to identify the presence of CSE and 3-MST in the microcirculation and to examine the mechanisms of H2S-induced vasodilatation in young healthy humans. We hypothesized that CSE and 3-MST would be expressed in the microvessels of the cutaneous circulation and, functionally, that H2S-donors would elicit dose-dependent increases in cutaneous vasodilatation predominately through KATP channels. We further hypothesized that both NO and byproducts of COX metabolism contribute, in part, to exogenous H2S-induced cutaneous vasodilatation.
Methods
Subjects
All experimental procedures were approved by the Institutional Review Board at The Pennsylvania State University. Verbal and written consent were obtained voluntarily from all subjects prior to participation according to guidelines set forth by the Declaration of Helsinki. Six adults participated in Protocol 1, and subjects for Protocols 2 and 3 were from a pool of 10 adults. All subjects underwent a complete medical screening including a resting 12-lead electrocardiogram, physical examination and 12-h fasting blood chemistry (Quest Diagnostics, Pittsburgh, PA, USA). All subjects were normotensive, normochlesterolemic, non-obese, normally active, without dermatological disease, and were not taking any medications. Women were tested during the early follicular phase of their menstrual cycle or during the placebo phase if taking oral contraceptives.
In vitro microvascular biochemical analysis
On a separate day from the in vivo studies, forearm skin samples were obtained via punch biopsy in five subjects (24 ± 3 years), as described previously (Smith et al. 2011). Skin samples were homogenized in radioimmunoprecipitation assay buffer containing protease and phosphatase inhibitors. Homogenates were centrifuged twice at 15 000 g at 4°C for 20 min. For western blot analysis, 1 μg of total protein were resolved by SDS-PAGE and electrotransferred to nitrocellulose membrane (Hybond-ECL; Amersham Life Sciences, Little Chalfont, UK). Antibodies used were rabbit-polyclonal anti-CSE antibody (dilution 1:1000; Protein Tech, Chicago, IL, USA), rabbit polyclonal anti-3-MST antibody (dilution 1:1000; Atlas Antibodies, Stockholm, Sweden) and mouse monoclonal anti-GAPDH antibody (dilution 1:5000; Novus Biologicals, Littleton, CO, USA). Rat liver homogenate was used as a positive control for CSE expression and rat carotid artery homogenate was used as a positive control for 3-MST. Densitometry analysis was performed using ImageJ software (NIH, Bethesda, MD, USA).
In vivo vascular function analysis
Protocols were performed in a thermoneutral (20–22°C) laboratory with the subject semisupine and the experimental arm at heart level. Integrated laser-Doppler flowmetry was used to measure red cell flux, an index of skin blood flow. Local skin temperature was controlled using a local heater (clamped at 33°C) placed directly above each microdialysis membrane (MoorLAB, Temperature Monitor SH02; Moor Instruments, Devon, UK). Laser-Doppler probes were secured in each local heater and used to continuously measure skin blood flow over each microdialysis fibre. An automated brachial cuff (Cardiocap 5; GE Healthcare) was used to measure arterial blood pressure on the contralateral arm every 5 min throughout the protocol. Procedures for preparation of pharmacological agents were held to a rigorous time schedule to ensure consistency between subjects and among experimental visits.
Protocol 1
Two intradermal microdialysis fibres (10 mm, 20 kDa cut-off membrane, MD 2000; Bioanalytical Systems, West Lafayette, IN, USA) were inserted into the forearm for local delivery of pharmacological agents as described previously (FDA IND 105 572) (Smith et al. 2011). All pharmacological solutions were mixed in lactated Ringer solution immediately before use and filtered using syringe microfilters (Acrodisc; Pall, Ann Arbor, MI, USA). The pH of each solution was 7.0 (Z113425-EA; Sigma-Aldrich, St Louis, MO, USA) and solutions were wrapped in foil to prevent photodegradation. Microdialysis sites were perfused with sodium sulphide (Na2S) or sodium hydrogen sulphide (NaHS), both of which act as fast releasing H2S donors (Papapetropoulos et al. 2014), at a rate of 2 μl min−1 (Bee Hive controller and Baby Bee microinfusion pumps; Bioanalytical Systems). Following resolution of the insertion trauma hyperaemia (∼60–90 min) and a 20 min stable baseline period, increasing concentrations of Na2S or NaHS were progressively perfused through the microdialysis fibre (0.01, 0.1, 1, 10 and 100 mm). Pilot work in our laboratory indicated that each donor elicited maximal H2S-induced vasodilatation at a concentration of ∼60 mm in healthy adults; therefore, a purposefully wide range of doses was used in the present study. After the dose–response protocol, all sites were flushed with Ringer solution before 28 mm sodium nitroprusside was perfused at 4 μl min-1 and temperature of the local heaters was increased to 43°C to elicit maximal cutaneous vascular conductance (CVCmax).
Protocol 2
Five intradermal microdialysis probes were inserted for local delivery of the pharmacological agents: lactated Ringer solution (control), 5 mm glybenclamide (GLY; KATP channel inhibitor), 0.1 mm senicapoc (SENI; IKCa channel inhibitor), 50 mm tetraethylammonium (TEA; non-specific KCa channel inhibitor) or GLY + SENI + TEA (Zhao et al. 2001; Lorenzo & Minson, 2007; Ataga et al. 2008; Bellien et al. 2008). Based on extensive pilot work in our laboratory, as well as the results from Protocol 1, 5 mm Na2S was used to elicit cutaneous vasodilatation, a dosage that approximates the half maximal concentration (EC50). Thus, following a 20 min stable baseline, 5 mm Na2S was co-perfused with the site-specific pharmacological agent until a stable plateau in skin blood flow was obtained and CVCmax was then elicited as described above.
Protocol 3
Three intradermal microdialysis probes were inserted for local delivery of the pharmacological agents: lactated Ringer solution (control), 20 mm l-NAME to inhibit NO production via non-specific NO synthase inhibition, or 10 mm ketorolac (KETO) to inhibit downstream vasodilatator products of COX. In a subset of subjects (n = 6), a fourth microdialysis probe was perfused with 20 mm l-NAME + 10 mm KETO to inhibit both NO and COX vasodilatatory pathways concurrently. Pilot testing in our laboratory indicates that maximal NO synthase and COX inhibition occur with these concentrations of l-NAME and KETO (Holowatz et al. 2005). Following a 20 min stable baseline, Na2S was co-perfused with the site specific pharmacological agent in progressively increasing concentrations (0.01, 0.1 , 1 , 10 and 100 mm), and a stable plateau in skin blood flow was obtained at each dose. Following the dose–response protocol, CVCmax was elicited.
Statistical analysis
Data were collected continuously at 40 Hz and stored for offline analysis (Windaq; DataQ Instruments, Akron, OK, USA). CVC was calculated as laser-Doppler flux divided by mean arterial pressure (MAP). Data were normalized and expressed as a percentage of maximal CVC (%CVCmax). CVC was averaged during 5 min of baseline, during the plateau (∼3 min) of each NaHS/Na2S dose (Protocols 1 and 3), and during 5 min of Na2S-induced vasodilatation (Protocol 2).
For Protocol 1, NaHS/Na2S doses were transformed to logarithmic concentrations and normalized, with the lowest value of the data set at 0% and the highest value of the data set at 100%. In addition, constraints were set for the top (100) and bottom (0) to best fit parameters of the model, which allows for the comparison of the dose–response curves on a similar scale and is useful when comparing curve position among the same subjects (Cook & Bielkiewicz, 1984). For Protocol 3, Na2S doses were transformed to logarithmic concentrations but, because of differences in baseline CVC between pharmacological agents, no constraints were imposed. Sigmoidal dose–response curves with variable slope were generated using a four-parameter non-linear regression modelling (Prism; GraphPad, San Diego, CA, USA) (Wenner et al. 2011; Greaney et al. 2014). NaHS/Na2S-induced cutaneous vasodilatation was compared between microdialysis sites in each protocol by the effective concentration resulting in 50% of the maximal response (logEC50), as described previously (Wenner et al. 2011; Greaney et al. 2014). The differences between treatments were analysed using an F test for repeated measures comparisons (Prism, version 5.0), which takes into account all points over the entire curve as opposed to each specific dose (Wenner et al. 2011). A one-way repeated-measure ANOVA (SigmaPlot, version 12.5; Systat Software Inc., Chicago, IL, USA) was used to determine differences in the increase in cutaneous vasodilatation from baseline for each drug treatment (Protocol 2). Results are reported as the mean ± SEM, and the α level was set at P < 0.05.
Results
Sample western blots for CSE and 3-MST expression in the human microvasculature are presented in Fig.1. All cutaneous samples tested (four men, one woman; 24 ± 3 years; resting MAP 88 ± 3 mmHg; body mass index 26 ± 6 kg m−2) expressed both CSE and 3-MST, confirming their presence in the human cutaneous microvasculature.
Figure 1.
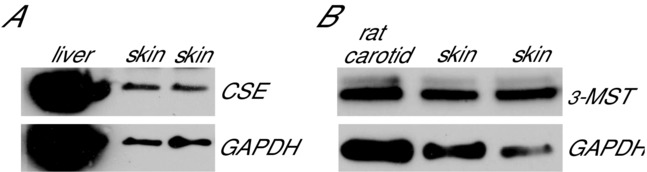
Western blots depicting CSE and 3-MST
Representative western blots depicting CSE (A) and 3-MST (B) protein expression in human cutaneous biopsy samples. All human whole skin homogenates expressed CSE and 3-MST.
Subject characteristics for the in vivo functional protocols are presented in Table1. NaHS and Na2S dose–response curves are illustrated in Fig.2. Both H2S donors elicited dose-dependent vasodilatation (Fig.2). There were no differences in cutaneous vascular responsiveness, assessed as the logEC50, between the H2S donors (NaHS: 0.83 ± 0.12 vs. Na2S: 0.55 ± 0.17; P = 0.19). Because there were no differences in the cutaneous vasodilatatory response to either H2S donor, Na2S was utilized in subsequent protocols.
Table 1.
Subject characteristics
| Protocol 1 | Protocols 2 and 3 | |
|---|---|---|
| Sex | 4 men, 2 women | 5 men, 5 women |
| Age (years) | 25 ± 3 | 24 ± 3 |
| BMI (kg m–2) | 25 ± 1 | 25 ± 1 |
| MAP (mmHg) | 86 ± 4 | 86 ± 8 |
| LDL (mg dL−1) | 68 ± 19 | 67 ± 16 |
| HDL (mg dL−1) | 55 ± 13 | 56 ± 12 |
| Total CHO (mg dL−1) | 140 ± 31 | 143 ± 8 |
| HbA1C (%) | 5 ± 0.2 | 5 ± 0.2 |
Data are expressed as the mean ± SEM. BMI, body mass index; MAP, mean arterial pressure; LDL, low-density lipoprotein; HDL, high-density lipoprotein; Total CHO, total cholesterol; HbA1C, glycated haemoglobin.
Figure 2.
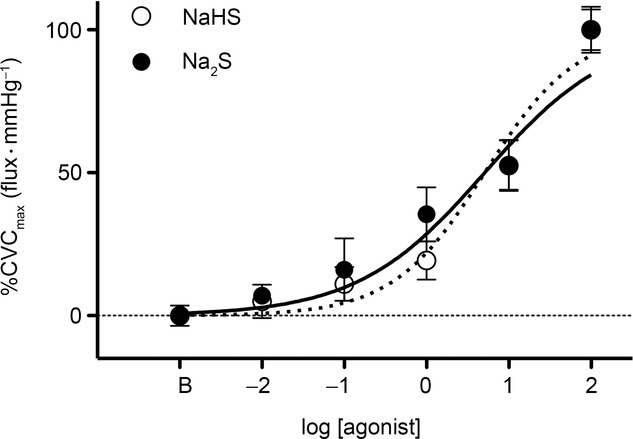
Exogenous H2S dose–response curves
Cutaneous vasodilatation during perfusion of the H2S donors NaHS and Na2S.
As expected, Na2S induced cutaneous vasodilatation above baseline. TEA and GLY + SENI + TEA significantly reduced Na2S-induced cutaneous vasodilatation, whereas GLY or SENI alone had no effect (Fig.3).
Figure 3.
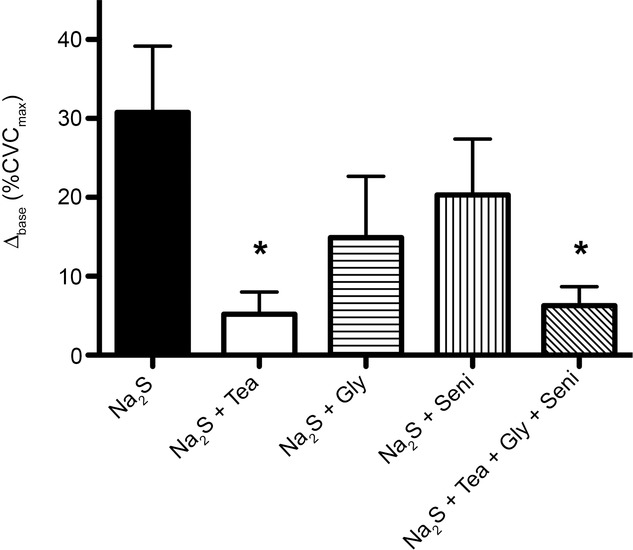
Exogenous H2S with K+ channel inhibition
Summary data, expressed as the difference between baseline skin blood flow and the cutaneous vasodilatation elicited by perfusion of 5 mm Na2S (Δbase) for each pharmacological treatment site. TEA and GLY + SENI + TEA resulted in blunted vasodilatation in response to Na2S. *P < 0.05 vs. Na2S.
Na2S dose–response curves with concomitant pharma-cological inhibition of NO and COX are depicted in Fig.4. There was a difference in baseline CVC between treatments (Ringer solution: 7.9 ± 1.1; KETO: 16.5 ± 3.1; l-NAME: 6.1 ± 0.8; KETO + l-NAME: 10.8 ± 2.2%CVCmax; P < 0.01). Perfusion of KETO or l-NAME alone blunted cutaneous H2S-induced vasodilatation, as indicated by the rightward shift in the dose–response curve (Fig.4A and B) and increased logEC50 (Ringer solution: 0.90 ± 0.18; KETO: 1.45 ± 0.12; l-NAME: 1.50 ± 0.07; P < 0.01 for both). Combined inhibition further increased the logEC50 from control (l-NAME + KETO: 1.72 ± 0.10; P < 0.01); however, the attenuation in cutaneous vasodilatation during combined inhibition was not statistically different from that elicited by either treatment alone (Fig.4C).
Figure 4.
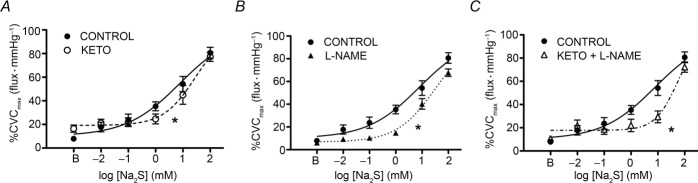
H2S interactions with NO and COX
Summary data for exogenous H2S-induced cutaneous vasodilatation during concurrent KETO (A), l-NAME (B) and KETO + l-NAME (C) administration. NO and COX inhibition, separately and combined, blunted exogenous H2S-induced cutaneous vasodilatation. *P < 0.05 vs. control.
Discussion
The principal new findings of the present study were: (1) CSE and 3-MST are expressed in the human cutaneous microvasculature; (2) H2S donors elicited dose-dependent vasodilatation in the cutaneous circulation of healthy young adults; (3) exogenous H2S-induced cutaneous vasodilatation is mediated, in part, by KCa channels; and (4) NO and COX inhibition attenuated exogenous H2S-induced cutaneous vasodilatation. Taken together, these in vitro molecular findings suggest that the enzymes producing H2S are expressed in the microvasculature and, coupled with the in vivo functional findings, further suggest that H2S, via KCa channels, may participate as a vasoactive molecule in the human cutaneous vasculature. These data confirm several lines of evidence in rodent models with respect to H2S-induced vasodilatation and suggest a potential functional interaction between H2S and both NO and byproducts of COX metabolism in the control of microvascular function in healthy humans.
These findings demonstrate that CSE and 3-MST, enzymes responsible for endogenous endothelial-derived H2S production, are expressed in the cutaneous circulation in humans (Hosoki et al. 1997; Shibuya et al. 2009). The localization of CSE and 3-MST to the human cutaneous microvasculature further validates the use of intradermal microdialysis as an in vivo experimental technique to pharmaco-dissect the role of H2S in contributing to vascular homeostasis in healthy humans. The skin is an accessible and representative vascular bed for the study of the vascular signalling mechanisms (Abularrage et al. 2005; Rossi et al. 2006; Holowatz et al. 2008). CSE has been detected in isolated human mammary arteries (Webb et al. 2008) and, when considered together with the results of the present investigation, provides further support for the ubiquitous expression of CSE, as well as the subsequent endogenous production of H2S, in the human vasculature. A secondary enzymatic source of H2S, 3-MST, was also detected. 3-MST has been reported to localize to both the vascular endothelium and smooth muscle (Shibuya et al. 2009; Yadav et al. 2013), and probably represents an additional source of H2S in the cutaneous circulation.
The H2S donor molecules used in the present study, NaHS and Na2S, have been widely used in rodent models to examine H2S-mediated vascular function (Siebert et al. 2008; Wang et al. 2010; Liu et al. 2012; Chen et al. 2013). Both donors are inorganic salts that quickly dissolve in solution, resulting in the immediate formation of H2S in a pH-dependent manner, and are therefore considered ‘fast H2S generators’ (Papapetropoulos et al. 2014). The use of these ‘fast H2S generators’ in the present study resulted in significant dose-dependent vasodilatation in the cutaneous circulation. Furthermore, there was no difference in cutaneous vascular responsiveness between donors. To date, only one other study has examined the functional in vivo effects of exoge-nous H2S in humans; that study utilized a bolus intravenous infusion of the H2S donor IK-1001 in healthy adults (Toombs et al. 2010). IK-1001 did not elicit alterations in systemic blood pressure; however, no direct assessment of conduit or microvessel H2S-mediated vasoreactivity was performed (Toombs et al. 2010). Future studies measuring H2S production and utilizing direct pharmacological inhibition of endogenous H2S are needed to confirm the physiological function of H2S in the human vasculature.
Rodent studies suggests that H2S elicits vasodilatation predominantly through activation of KATP channels and subsequent membrane hyperpolarization (Zhao et al. 2001; Cheng et al. 2004; Webb et al. 2008; Tay et al. 2010; Kimura, 2011; Szabo et al. 2011). More recently, SKCa, IKCa and BKCa have emerged as additional pathways by which H2S mediates vasodilatation in resistance vessels (Mustafa et al. 2011; Jackson-Weaver et al. 2013). In the present study, KATP channel inhibition with GLY did not significantly affect the vasodilatatory response to exogenous Na2S. By contrast to our initial hypothesis, the data suggest that KATP channels do not appear to mediate exogenous H2S-induced cutaneous vasodilatation. Instead, H2S-induced cutaneous vasodilatation is mediated, at least in part, by TEA-sensitive KCa channels. The differences between the findings of the present investigation regarding the specific channels mediating H2S-induced vasodilatation and those reported using murine models may be a result of the use of in vivo vs. in vitro assessment of vascular signalling mechanisms, as well as the concentrations of H2S delivered to the tissue. In addition, very few selective pharmacological SKCa, IKCa, or BKCa channel inhibitors are currently approved for use in humans (Ataga et al. 2008; Brunt & Minson, 2012), making it challenging to precisely examine these mechanisms in an in vivo experimental paradigm. However, SENI, a specific antagonist for IKCa3.1 channels (Ataga et al. 2008), did not affect the cutaneous vasodilatation in response to exogenous H2S, suggesting that this mechanism does not contribute to the cutaneous vascular responses to H2S in humans.
In the present study, we also investigated potential cross-talk that may exist in vivo between H2S signalling and the endothelial-derived signalling molecules NO and downstream byproducts of COX metabolism. Our data suggest that both NO and COX-derived metabolic byproducts are required for the full expression of exogenous H2S-induced cutaneous vasodilatation. The literature regarding the interaction between H2S and NO is equivocal (Li et al. 2006; Cai et al. 2007; Kubo et al. 2007; Wang et al. 2010; Kondo et al. 2013). For example, mice treated orally with H2S have an upregulation of endothelial NO synthase and increased NO bioavailability (Kondo et al. 2013). By contrast, H2S has also been reported to inhibit endothelial NO synthase activity via alterations in the co-factor tetrahydrobiopterin (Kubo et al. 2007). The present findings suggest a synergistic interaction between exogenous H2S and NO in mediating cutaneous vasodilatation in young adults, as indicated by attenuated H2S-induced cutaneous vasodilatation during NO synthase inhibition with l-NAME. Limited data exist regarding the signalling interaction between H2S and COX-derived byproducts. H2S upregulates the COX-2/prostacyclin pathway in isolated human pulmonary artery smooth muscle cells, creating a pro-vasodilatatory environment (Li et al. 2014). We found a significant interaction, albeit weaker, between H2S and COX byproducts, as indicated by the reduced vascular sensitivity to exogenous H2S during concurrent COX inhibition. By contrast to the blunted vasodilatatory response during COX inhibition in the present study, the use of the COX inhibitor flurbiprofen during H2S-induced vasodilatation with a slow-release donor resulted in augmented vasodilatation in pre-constricted bovine conduit vessels (Chitnis et al. 2013). The reasons for these differing conclusions are unclear, although they may be related to the choice of COX inhibitor, the type of H2S donor, the vascular bed examined or the difference between in vitro and in vivo investigation of signalling mechanisms. Surprisingly, concurrent inhibition of both NO and COX signalling pathways did not further attenuate cutaneous vasodilatation in response to exogenous H2S. However, when considered collectively, the findings of the present study indicate that exogenous H2S interacts with both NO and COX-derived metabolic byproducts to mediate cutaneous vasodilatation in healthy humans.
As a necessary first step, the present study utilized exogenous sources of H2S to characterize cutaneous vascular end-organ responsiveness and the mechanisms by which these responses occur. We did not specifically test the functional role of H2S in the control of cutaneous blood flow during physiological stimuli (i.e. local skin and reflex heating). However, given the extensive work that has been carroed out aiming to determine the mechanisms underlying the control of skin blood flow during these physiological stimuli, we speculate that H2S may contribute as a putative EDHF during local heating-induced and reflex-mediated cutaneous vasodilatation. Local heating-induced increases in cutaneous blood flow are primarily mediated by endothelial NO synthase- (Bruning et al. 2012) and epoxyeicossatrienioc acid-dependent mechanisms (Brunt & Minson, 2012); however, as an EDHF, H2S may also contributing to local thermal hyperaemia. During reflex cutaneous vasodilatation, both NO and COX-derived vasodilatator products, along with EDHFs, contribute to the full expression of this response (McCord et al. 2006). Given the role of cholinergic nerves (Kellogg et al. 1995) and the downstream interactions of these pathways in reflex cutaneous vasodilatation (McCord et al. 2006), it is plausible that H2S may also play a role as a putative EDHF during reflex vasodilatation.
These studies served to characterize the mechanisms mediating the functional vasodilatatory role of H2S in the healthy human vasculature. We demonstrated the presence of the enzymes responsible for endogenous H2S production in the cutaneous microvasculature and suggest a role for H2S in mediating cutaneous vascular relaxation. These findings have potential implications for an important functional role for H2S in vivo. Future studies designed to delineate the physiological role of H2S in modulating vascular function in humans are warranted, particularly studies aimed at measuring H2S production and utilizing direct pharmacological inhibition of endogenous H2S. There is substantial evidence from rodent studies suggesting that dysregulation of H2S probably plays an important role in the pathogenesis of hypertension-associated vascular dysfunction via its effects on blood pressure regulation, vessel function and remodelling, and inflammatory processes (Yan et al. 2004; Jiang et al. 2005; Liu et al. 2010; Taniguchi et al. 2011). Future investigation of the potential role for H2S in the modulation of vascular regulation in humans may therefore have important clinical implications for understanding the mechanisms underlying the vascular dysfunction characteristic of multiple cardiovascular pathologies.
Acknowledgments
The present study was conducted in the Department of Kinesiology at The Pennsylvania State University, University Park, PA, USA. Both the time and effort expended by all the volunteer subjects is greatly appreciated. The authors are grateful for the assistance of Susan Slimak RN, Anna Stanhewicz PhD, Jane Pierzga MS and Yehudit Bergman MS.
Glossary
- 3-MST
mercaptopyruvate sulphurtransferase
- BKCa
large conductance calcium-dependent potassium channels
- CSE
cystathionine-Υ;-lyase
- COX
cyclo-oxygenase
- CVC
cutaneous vascular conductance
- CVCmax
maximal cutaneous vascular conductance
- EDHF
endothelium-derived hyperpolarizing factor
- GLY
glybenclamide
- H2S
hydrogen sulphide
- IKCa
intermediate conductance calcium-dependent potassium channels
- KCa
calcium-dependent potassium channel
- KATP
ATP-sensitive potassium channel
- KETO
ketorolac
- MAP
mean arterial pressure
- Na2S
sodium sulphide
- NaHS
sodium hydrogen sulphide
- NO
nitric oxide
- SENI
senicapoc
- SKCa
small conductance calcium-dependent potassium channels
- TEA
tetraethylammonium
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
J.L.K., J.L.G., L.S. and L.M.A. are responsible for the conception and design of the research. J.L.K. performed experiments. J.L.K. and J.L.G analysed data. J.L.K., J.L.G., L.S. and L.M.A. interpreted the results of the experiments. J.L.K. drafted the manuscript. J.L.K., J.L.G., L.S. and L.M.A. edited and revised the manuscript. J.L.K., J.L.G., L.S. and L.M.A. approved the final version of the manuscript submitted for publication.
Funding
This research was supported by HL120471-01 (J.L.G.) and HL093-238-04 (L.M.A.).
References
- Abularrage CJ, Sidawy AN, Aidinian G, Singh N, Weiswasser JM. Arora S. Evaluation of the microcirculation in vascular disease. J Vasc Surg. 2005;42:574–581. doi: 10.1016/j.jvs.2005.05.019. [DOI] [PubMed] [Google Scholar]
- Ataga KI, Smith WR, De Castro LM, Swerdlow P, Saunthararajah Y, Castro O, Vichinsky E, Kutlar A, Orringer EP, Rigdon GC. Stocker JW. Efficacy and safety of the Gardos channel blocker, senicapoc (ICA-17043), in patients with sickle cell anemia. Blood. 2008;111:3991–3997. doi: 10.1182/blood-2007-08-110098. [DOI] [PubMed] [Google Scholar]
- Bellien J, Thuillez C. Joannides R. Contribution of endothelium-derived hyperpolarizing factors to the regulation of vascular tone in humans. Fundam Clin Pharmacol. 2008;22:363–377. doi: 10.1111/j.1472-8206.2008.00610.x. [DOI] [PubMed] [Google Scholar]
- Bruning RS, Santhanam L, Stanhewicz AE, Smith CJ, Berkowitz DE, Kenney WL. Holowatz LA. Endothelial nitric oxide synthase mediates cutaneous vasodilation during local heating and is attenuated in middle-aged human skin. J Appl Physiol. 2012;112:2019–2026. doi: 10.1152/japplphysiol.01354.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunt VE. Minson CT. KCa channels and epoxyeicosatrienoic acids: major contributors to thermal hyperaemia in human skin. J Physiol. 2012;590:3523–3534. doi: 10.1113/jphysiol.2012.236398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai WJ, Wang MJ, Moore PK, Jin HM, Yao T. Zhu YC. The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc Res. 2007;76:29–40. doi: 10.1016/j.cardiores.2007.05.026. [DOI] [PubMed] [Google Scholar]
- Chen W, Liu N, Zhang Y, Qi Y, Yang J, Deng Z, Li X. Xie X. [Exogenous hydrogen sulfide protects against myocardial injury after skeletal muscle ischemia/reperfusion by inhibiting inflammatory cytokines and oxidative stress in rats] Nan Fang Yi Ke Da Xue Xue Bao. 2013;33:554–558. [PubMed] [Google Scholar]
- Cheng Y, Ndisang JF, Tang G, Cao K. Wang R. Hydrogen sulfide-induced relaxation of resistance mesenteric artery beds of rats. Am J Physiol Heart Circ Physiol. 2004;287:H2316–H2323. doi: 10.1152/ajpheart.00331.2004. [DOI] [PubMed] [Google Scholar]
- Chitnis MK, Njie-Mbye YF, Opere CA, Wood ME, Whiteman M. Ohia SE. Pharmacological actions of the slow release hydrogen sulfide donor GYY4137 on phenylephrine-induced tone in isolated bovine ciliary artery. Exp Eye Res. 2013;116:350–354. doi: 10.1016/j.exer.2013.10.004. [DOI] [PubMed] [Google Scholar]
- Cook DA. Bielkiewicz B. A computer-assisted technique for analysis and comparison of dose-response curves. J Pharmacol Methods. 1984;11:77–89. doi: 10.1016/0160-5402(84)90017-2. [DOI] [PubMed] [Google Scholar]
- Coulon P, Constans J. Gosse P. Impairment of skin blood flow during post-occlusive reactive hyperhemy assessed by laser Doppler flowmetry correlates with renal resistive index. J Hum Hypertens. 2012;26:56–63. doi: 10.1038/jhh.2010.117. [DOI] [PubMed] [Google Scholar]
- Greaney JL, Stanhewicz AE, Kenney WL. Alexander LM. Lack of limb or sex differences in the cutaneous vascular responses to exogenous norepinephrine. J Appl Physiol (1985) 2014 doi: 10.1152/japplphysiol.00575.2014. jap 00575 02014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holowatz LA, Thompson-Torgerson CS. Kenney WL. The human cutaneous circulation as a model of generalized microvascular function. J Appl Physiol (1985) 2008;105:370–372. doi: 10.1152/japplphysiol.00858.2007. [DOI] [PubMed] [Google Scholar]
- Holowatz LA, Thompson CS, Minson CT. Kenney WL. Mechanisms of acetylcholine-mediated vasodilatation in young and aged human skin. J Physiol. 2005;563:965–973. doi: 10.1113/jphysiol.2004.080952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoki R, Matsuki N. Kimura H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem Biophys Res Commun. 1997;237:527–531. doi: 10.1006/bbrc.1997.6878. [DOI] [PubMed] [Google Scholar]
- Hu LF, Pan TT, Neo KL, Yong QC. Bian JS. Cyclooxygenase-2 mediates the delayed cardioprotection induced by hydrogen sulfide preconditioning in isolated rat cardiomyocytes. Pflugers Arch. 2008;455:971–978. doi: 10.1007/s00424-007-0346-8. [DOI] [PubMed] [Google Scholar]
- Jackson-Weaver O, Osmond JM, Riddle MA, Naik JS, Gonzalez Bosc LV, Walker BR. Kanagy NL. Hydrogen sulfide dilates rat mesenteric arteries by activating endothelial large-conductance Ca(2)(+)-activated K(+) channels and smooth muscle Ca(2)(+) sparks. Am J Physiol Heart Circ Physiol. 2013;304:H1446–H1454. doi: 10.1152/ajpheart.00506.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang HL, Wu HC, Li ZL, Geng B. Tang CS. [Changes of the new gaseous transmitter H2S in patients with coronary heart disease] Di Yi Jun Yi Da Xue Xue Bao. 2005;25:951–954. [PubMed] [Google Scholar]
- Kellogg DL, Jr, Pergola PE, Piest KL, Kosiba WA, Crandall CG, Grossmann M. Johnson JM. Cutaneous active vasodilation in humans is mediated by cholinergic nerve cotransmission. Circ Res. 1995;77:1222–1228. doi: 10.1161/01.res.77.6.1222. [DOI] [PubMed] [Google Scholar]
- Khan F, Patterson D, Belch JJ, Hirata K. Lang CC. Relationship between peripheral and coronary function using laser Doppler imaging and transthoracic echocardiography. Clin Sci (Lond) 2008;115:295–300. doi: 10.1042/CS20070431. [DOI] [PubMed] [Google Scholar]
- Kimura H. Hydrogen sulfide: its production and functions. Exp Physiol. 2011;96:833–835. doi: 10.1113/expphysiol.2011.057455. [DOI] [PubMed] [Google Scholar]
- Kondo K, Bhushan S, King AL, Prabhu SD, Hamid T, Koenig S, Murohara T, Predmore BL, Gojon G, Sr, Gojon G, Jr, Wang R, Karusula N, Nicholson CK, Calvert JW. Lefer DJ. H(2)S protects against pressure overload-induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation. 2013;127:1116–1127. doi: 10.1161/CIRCULATIONAHA.112.000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo S, Kurokawa Y, Doe I, Masuko T, Sekiguchi F. Kawabata A. Hydrogen sulfide inhibits activity of three isoforms of recombinant nitric oxide synthase. Toxicology. 2007;241:92–97. doi: 10.1016/j.tox.2007.08.087. [DOI] [PubMed] [Google Scholar]
- Li XH, Du JB, Bu DF, Tang XY. Tang CS. Sodium hydrosulfide alleviated pulmonary vascular structural remodeling induced by high pulmonary blood flow in rats. Zhongguo Yao Li Xue Bao. 2006;27:971–980. doi: 10.1111/j.1745-7254.2006.00353.x. [DOI] [PubMed] [Google Scholar]
- Li Y, Liu G, Cai D, Pan B, Lin Y, Li X, Li S, Zhu L, Liao X. Wang H. H2S inhibition of chemical hypoxia-induced proliferation of HPASMCs is mediated by the upregulation of COX-2/PGI2. Int J Mol Med. 2014;33:359–366. doi: 10.3892/ijmm.2013.1579. [DOI] [PubMed] [Google Scholar]
- Li L, Rose P. Moore PK. Hydrogen sulfide and cell signaling. Annu Rev Pharmacol Toxicol. 2011;51:169–187. doi: 10.1146/annurev-pharmtox-010510-100505. [DOI] [PubMed] [Google Scholar]
- Liu C, Gu X. Zhu YZ. Synthesis and biological evaluation of novel leonurine-SPRC conjugate as cardioprotective agents. Bioorg Med Chem Lett. 2010;20:6942–6946. doi: 10.1016/j.bmcl.2010.09.135. [DOI] [PubMed] [Google Scholar]
- Liu L, Liu H, Sun D, Qiao W, Qi Y, Sun H. Yan C. Effects of H(2)S on myogenic responses in rat cerebral arterioles. Circ J. 2012;76:1012–1019. doi: 10.1253/circj.cj-11-0890. [DOI] [PubMed] [Google Scholar]
- Lorenzo S. Minson CT. Human cutaneous reactive hyperaemia: role of BKCa channels and sensory nerves. J Physiol. 2007;585:295–303. doi: 10.1113/jphysiol.2007.143867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCord GR, Cracowski JL. Minson CT. Prostanoids contribute to cutaneous active vasodilation in humans. Am J Physiol Regul Integr Comp Physiol. 2006;291:R596–R602. doi: 10.1152/ajpregu.00710.2005. [DOI] [PubMed] [Google Scholar]
- Minamishima S, Bougaki M, Sips PY, Yu JD, Minamishima YA, Elrod JW, Lefer DJ, Bloch KD. Ichinose F. Hydrogen sulfide improves survival after cardiac arrest and cardiopulmonary resuscitation via a nitric oxide synthase 3-dependent mechanism in mice. Circulation. 2009;120:888–896. doi: 10.1161/CIRCULATIONAHA.108.833491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa AK, Sikka G, Gazi SK, Steppan J, Jung SM, Bhunia AK, Barodka VM, Gazi FK, Barrow RK, Wang R, Amzel LM, Berkowitz DE. Snyder SH. Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ Res. 2011;109:1259–1268. doi: 10.1161/CIRCRESAHA.111.240242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapetropoulos A, Whiteman M. Giuseppe C. Pharmacological tools for hydrogen sulfide research: a brief, introductory guide for beginners. Br J Pharmacol. 2014 doi: 10.1111/bph.12806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paredes DA, Jackson-Weaver O, DeLeon X. Kanagy N. Endogenous H2S opposes vasoconstriction in conjunction with NO in rat aorta. Faseb J. 2012;26 1129.16. [Google Scholar]
- Rossi M, Carpi A, Galetta F, Franzoni F. Santoro G. The investigation of skin blood flowmotion: a new approach to study the microcirculatory impairment in vascular diseases? Biomed Pharmacother. 2006;60:437–442. doi: 10.1016/j.biopha.2006.07.012. [DOI] [PubMed] [Google Scholar]
- Shibuya N, Mikami Y, Kimura Y, Nagahara N. Kimura H. Vascular endothelium expresses 3-mercaptopyruvate sulfurtransferase and produces hydrogen sulfide. J Biochem. 2009;146:623–626. doi: 10.1093/jb/mvp111. [DOI] [PubMed] [Google Scholar]
- Siebert N, Cantre D, Eipel C. Vollmar B. H2S contributes to the hepatic arterial buffer response and mediates vasorelaxation of the hepatic artery via activation of K(ATP) channels. Am J Physiol Gastrointest Liver Physiol. 2008;295:G1266–G1273. doi: 10.1152/ajpgi.90484.2008. [DOI] [PubMed] [Google Scholar]
- Smith CJ, Santhanam L, Bruning RS, Stanhewicz A, Berkowitz DE. Holowatz LA. Upregulation of inducible nitric oxide synthase contributes to attenuated cutaneous vasodilation in essential hypertensive humans. Hypertension. 2011;58:935–942. doi: 10.1161/HYPERTENSIONAHA.111.178129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Tang Q, Du JP. Jin HF. Hydrogen sulfide and vascular relaxation. Chin Med J. 2011;124:3816–3819. [PubMed] [Google Scholar]
- Szabo G, Veres G, Radovits T, Gero D, Modis K, Miesel-Groschel C, Horkay F, Karck M. Szabo C. Cardioprotective effects of hydrogen sulfide. Nitric oxide: Biol Chem /Official J Nitric Oxide Soc. 2011;25:201–210. doi: 10.1016/j.niox.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tain XY, Wong WT, Sayed N, Luo J, Tsang SY, Bain ZX, Lu Y, Cheang WS, Yao X, Chen ZY. Huang Y. NaHS relaxes rat cerebral artery in vivo via inhibition of I-type voltage sensitive Ca2+ channels. Pharmacol Res. 2012;65:239–246. doi: 10.1016/j.phrs.2011.11.006. [DOI] [PubMed] [Google Scholar]
- Taniguchi S, Kang L, Kimura T. Niki I. Hydrogen sulphide protects mouse pancreatic beta-cells from cell death induced by oxidative stress, but not by endoplasmic reticulum stress. Br J Pharmacol. 2011;162:1171–1178. doi: 10.1111/j.1476-5381.2010.01119.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay AS, Hu LF, Lu M, Wong PT. Bian JS. Hydrogen sulfide protects neurons against hypoxic injury via stimulation of ATP-sensitive potassium channel/protein kinase C/extracellular signal-regulated kinase/heat shock protein 90 pathway. Neuroscience. 2010;167:277–286. doi: 10.1016/j.neuroscience.2010.02.006. [DOI] [PubMed] [Google Scholar]
- Toombs CF, Insko MA, Wintner EA, Deckwerth TL, Usansky H, Jamil K, Goldstein B, Cooreman M. Szabo C. Detection of exhaled hydrogen sulphide gas in healthy human volunteers during intravenous administration of sodium sulphide. Br J Clin Pharmacol. 2010;69:626–636. doi: 10.1111/j.1365-2125.2010.03636.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MJ, Cai WJ, Li N, Ding YJ, Chen Y. Zhu YC. The hydrogen sulfide donor NaHS promotes angiogenesis in a rat model of hind limb ischemia. Antioxid Redox Signal. 2010;12:1065–1077. doi: 10.1089/ars.2009.2945. [DOI] [PubMed] [Google Scholar]
- Webb GD, Lim LH, Oh VM, Yeo SB, Cheong YP, Ali MY, El Oakley R, Lee CN, Wong PS, Caleb MG, Salto-Tellez M, Bhatia M, Chan ES, Taylor EA. Moore PK. Contractile and vasorelaxant effects of hydrogen sulfide and its biosynthesis in the human internal mammary artery. J Pharmacol Exp Ther. 2008;324:876–882. doi: 10.1124/jpet.107.133538. [DOI] [PubMed] [Google Scholar]
- Wenner MM, Wilson TE, Davis SL. Stachenfeld NS. Pharmacological curve fitting to analyze cutaneous adrenergic responses. J Appl Physiol (1985) 2011;111:1703–1709. doi: 10.1152/japplphysiol.00780.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav PK, Yamada K, Chiku T, Koutmos M. Banerjee R. Structure and kinetic analysis of H2S production by human mercaptopyruvate sulfurtransferase. J Biol Chem. 2013;288:20002–20013. doi: 10.1074/jbc.M113.466177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Du J. Tang C. The possible role of hydrogen sulfide on the pathogenesis of spontaneous hypertension in rats. Biochem Biophys Res Commun. 2004;313:22–27. doi: 10.1016/j.bbrc.2003.11.081. [DOI] [PubMed] [Google Scholar]
- Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, Snyder SH. Wang R. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322:587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Zhang J, Lu Y. Wang R. The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J. 2001;20:6008–6016. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]