

Abstract
Abstract
Endothelial cell Ca2+ signalling is integral to blood flow control in the resistance vasculature yet little is known of how its regulation may be affected by advancing age. We tested the hypothesis that advanced age protects microvascular endothelium by attenuating aberrant Ca2+ signalling during oxidative stress. Intact endothelial tubes (width, ∼60 μm; length, ∼1000 μm) were isolated from superior epigastric arteries of Young (3–4 months) and Old (24–26 months) male C57BL/6 mice and loaded with Fura-2 dye to monitor [Ca2+]i. At rest there was no difference in [Ca2+]i between age groups. Compared to Young, the [Ca2+]i response to maximal stimulation with acetylcholine (3 μm, 2 min) was ∼25% greater in Old, confirming signalling integrity with advanced age. Basal H2O2 availability was ∼33% greater in Old while vascular catalase activity was reduced by half. Transient exposure to elevated H2O2 (200 μm, 20 min) progressively increased [Ca2+]i to ∼4-fold greater levels in endothelium of Young versus Old. With no difference between age groups at rest, Mn2+ quench of Fura-2 fluorescence revealed 2-fold greater Ca2+ influx in Young during elevated H2O2; this effect was attenuated by ∼75% using ruthenium red (5 μm) as a broad-spectrum inhibitor of transient receptor potential channels. Prolonged exposure to H2O2 (200 μm, 60 min) induced ∼7-fold greater cell death in endothelium of Young versus Old. Thus, microvascular endothelium can adapt to advanced age by reducing Ca2+ influx during elevated oxidative stress. Protection from cell death during oxidative stress will sustain endothelial integrity during ageing.
Key points
Calcium signalling in endothelial cells of resistance arteries is integral to blood flow regulation. Oxidative stress and endothelial dysfunction can prevail during advanced age and we questioned how calcium signalling may be affected.
Intact endothelium was freshly isolated from superior epigastric arteries of Young (∼4 months) and Old (∼24 months) male C57BL/6 mice. Under resting conditions, with no difference in intracellular calcium levels, hydrogen peroxide (H2O2) availability was ∼1/3 greater in endothelium of Old mice while vascular catalase activity was reduced by nearly half.
Compared to Old, imposing oxidative stress (200 μm H2O2) for 20 min increased intracellular calcium to 4-fold greater levels in endothelium of Young in conjunction with twice the calcium influx. Prolonged (60 min) exposure to H2O2 induced 7-fold greater cell death in endothelium of Young.
Microvascular adaptation to advanced age may protect endothelial cells during elevated oxidative stress to preserve functional viability of the intima.
Introduction
In the resistance vasculature, the role of the endothelium in governing tissue perfusion is driven by dynamic regulation of intracellular Ca2+ concentration [Ca2+]i. For example, elevations in endothelial cell (EC) [Ca2+]i activate small- and intermediate-conductance KCa channels (SKCa and IKCa, respectively) along with production of nitric oxide (NO) and prostacyclin (Crutchley et al. 1983; Kruse et al. 1994; Campbell et al. 1996; Tran & Watanabe, 2006; Ledoux et al. 2008). These complementary signalling events serve to relax the surrounding smooth muscle cells (SMCs), with vasodilatation increasing tissue blood flow. With advanced age, blood flow regulation by the endothelium is characterized by greater levels of reactive oxygen species (ROS) and diminished bioavailability of NO (Taddei et al. 2001; Seals et al. 2011; Muller-Delp et al. 2012). In turn, these changes may impair EC function and compromise tissue perfusion (Feletou & Vanhoutte, 2006; Seals et al. 2011). These adverse effects of advanced age often coincide with a sedentary lifestyle and impaired functional work capacity (Dinenno et al. 1999, 2001; Donato et al. 2006). Of even greater concern, compromising the physiological integrity of the endothelium presages the advance of cardiovascular disease (Yeboah et al. 2007; Lind et al. 2011).
Superoxide generation occurs through mitochondrial respiration, substrate conversion via cytosolic and membrane-associated oxidases, and the uncoupling of nitric oxide synthase (NOS) (Drouin et al. 2007; Widlansky & Gutterman, 2011; Muller-Delp et al. 2012; Bachschmid et al. 2013). While able to react with NO and generate peroxynitrite (Fleming & Busse, 1999; van der Loo et al. 2000; Donato et al. 2007; Trott et al. 2009; Sindler et al. 2013), superoxide anions can be converted rapidly to the stable (and membrane-permeant) ROS hydrogen peroxide (H2O2) either spontaneously or via mitochondrial, cytosolic and membrane-associated superoxide dismutases (Widlansky & Gutterman, 2011; Muller-Delp et al. 2012; Bachschmid et al. 2013). While H2O2 can contribute to endothelial-dependent dilatation (Miura et al. 2003; Widlansky & Gutterman, 2011; Muller-Delp et al. 2012), high levels of H2O2 can elevate EC [Ca2+]i, impair normal function and initiate cell death (Volk et al. 1997; Hecquet et al. 2008; Sun et al. 2012). However, it is unknown how advanced age affects Ca2+ responses to H2O2 in endothelium of the resistance vasculature, or whether there may be associated changes in EC viability.
In the present study, we tested the hypothesis that advanced age protects microvascular endothelium by attenuating aberrant Ca2+ signalling during exposure to elevated H2O2. Endothelial tubes freshly isolated from skeletal muscle resistance arteries of Young (3–4 months) and Old (24–26 months) mice were exposed to H2O2 at a concentration (200 μm) shown to hyperpolarize ECs to their K+ equilibrium potential within ∼20 min through activating KCa (Behringer et al. 2013). The present findings reveal that, with no difference in [Ca2+]i under basal conditions, resting levels of H2O2 were higher in Old. Exposure to elevated H2O2 (200 μm) increased [Ca2+]i to ∼4-fold higher levels and evoked cell death to a ∼7-fold greater extent in endothelium of Young compared to Old, suggesting that microvascular ECs adapt during advanced age to attenuate the rise in [Ca2+]i and minimize cell death when subjected to an abrupt increase in oxidative stress.
Methods
Animal care and use
All procedures were approved by the Institutional Animal Care and Use Committee of the University of Missouri– Columbia. Experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals of the National Research Council (8th edn, 2011). Young (∼4 months) and Old (∼24 months) male C57BL/6 mice were obtained concurrently from the National Institute on Aging colonies (Charles River Laboratories, Wilmington, MA, USA). Prior to experiments, mice were acclimatized in the same room of the animal care facility at the University of Missouri School of Medicine for at least 1 week on a 12/12 h light–dark cycle at ∼23°C with fresh tap water and standard rodent chow available ad libitum. Young and Old mice were studied in random order under the same conditions within each experimental protocol. On the morning of an experiment, a mouse was anaesthetized with an intraperitoneal injection of pentobarbital sodium (60 mg kg−1) and abdominal fur was removed by shaving. The origin of each superior epigastric artery (SEA) was exposed near the sternum and ligated with 6-0 silk suture. The entire abdominal musculature was then excised and placed in 4°C dissection buffer (pH 7.4, 285–290 mOsm) composed of (in mm): 137 NaCl, 5 KCl, 1 MgCl2, 10 Hepes, 10 glucose, 0.01 sodium nitroprusside (SNP) and 0.1% bovine serum albumin (BSA) (US Biochemical, Cleveland, OH, USA). Following tissue removal, the anaesthetized mouse was killed via cardiac excision or an overdose of pentobarbital via cardiac injection.
Isolation of endothelial tubes
Intact endothelial tubes were isolated from the SEA as described (Socha et al. 2011, 2012; Socha & Segal, 2013). Briefly, the abdominal musculature was transferred to a dissection chamber at 4°C and pinned onto a transparent silicone rubber block (Sylgard 184; Dow-Corning, Midland, MI, USA). While viewing through a stereomicroscope, each SEA was dissected from the site of ligation to the first major branch point (isolated segment length, 3–3.5 mm) and the lumen was flushed with chilled dissection buffer to remove residual blood. Each SEA was cut into 2–3 smaller segments (length, ∼1–2 mm) and transferred into 12 × 75 mm culture tubes on ice containing 4 ml of dissection buffer. The culture tube was warmed to room temperature (∼24°C) and vessel segments were transferred to a 12 × 75 mm culture tube containing 1 ml of preheated (37°C) dissociation buffer composed of (in mm): 137 NaCl, 5 KCl, 1 MgCl2, 10 Hepes, 10 glucose, 2 CaCl2, 0.1% BSA, 0.62 mg ml−1 papain, 1.5 mg ml−1 collagenase and 1.0 mg ml−1 dithioerythritol, pH 7.4. Vessel segments were incubated for 30 min at 37°C then transferred to enzyme-free dissociation buffer at room temperature. A vessel segment was transferred to a recording chamber (RC-27 N; Warner Instruments, Hamden, CT, USA). During visual inspection at 40× magnification, surrounding SMCs and adventitia were dissociated from the endothelium using gentle trituration through the heat-polished tip (internal diameter, ∼120 μm) of a micropipette pulled (P-97; Sutter Instruments, Novato, CA, USA) from borosilicate glass capillary tubes (#1B100-4; World Precision Instruments, Sarasota, FL, USA). The freshly isolated endothelial tube was positioned at the centre of the chamber and aligned along the chamber axis in the direction of fluid flow. Each end was pressed gently against the chamber bottom (a 24 × 50 mm coverslip) using a blunt-tipped pinning pipette (heat-polished borosilicate glass; external diameter, ∼100 μm) secured in three-axis micromanipulators (DT3-100; Siskiyou Design, Grants Pass, OR, USA). The endothelial tube was extended to approximate in situ length by slowly retracting each pinning pipette and superfused (3 ml min−1) with physiological salt solution [PSS, 290–295 mOsm; pH 7.4, composed of (in mm): 137 NaCl, 5 KCl, 1 MgCl2, 10 Hepes, 10 glucose and 2 CaCl2].
Calcium photometry
The endothelial tube was equilibrated for ∼15 min during superfusion with PSS at room temperature (∼24°C) then incubated for 30 min without flow in Fura-2 AM dye (# F14185; Invitrogen, Carlsbad, CA, USA). The dye was dissolved in DMSO and diluted to 5 μm in PSS with final DMSO < 0.5% (Socha et al. 2011). Superfusion with PSS was resumed to wash out excess dye and allow dye within cells to de-esterify while temperature was raised to 32°C during 30 min of equilibration. Calcium photometry was performed with an IonOptix (Milford, MA, USA) system (Socha et al. 2011). Using a 20× objective (Nikon Fluor20; NA 0.75; Nikon Corporation, Tokyo, Japan), Fura-2 was excited alternately (250 Hz) at 340 and 380 nm with a 75 W Xe lamp while fluorescence emission was collected at 510 nm and expressed as the ratio F340/F380. Auto-fluorescence of the endothelium was recorded prior to dye loading and subtracted from respective recordings. The imaging window for collecting fluorescence emission was 320 μm long and adjusted to accommodate the width of each tube (range, 50–75 μm) while observing it on a video monitor using transmitted light from a halogen lamp (600 nm long pass filter) directed to a charge coupled device camera (LCL-902C; Watec, Orangeburg, NY, USA). To determine minimum and maximum F340/F380 values in Young and Old mice, tubes were superfused with PSS containing 0 mm Ca2+ and 5 mm EGTA for 1 h followed by PSS containing 3 μm ionomycin and 10 mm Ca2+, respectively (Socha et al. 2011). Following Fura-2 dye loading and equilibration, an index of basal [Ca2+]i was determined by averaging the F340/F380 ratio over 30 s under resting conditions. To evaluate [Ca2+]i responses to G-protein coupled receptor (GPCR) activation, 3 μm ACh, which evokes peak responses for [Ca2+]i and hyperpolarization (Behringer et al. 2012, 2013), was added to the superfusion solution for 2 min while the F340/F380 ratio was recorded. Data collected during the final 30 s of the [Ca2+]i response to ACh were averaged for analysis.
Dichlorofluorescein fluorescence
Endothelial tubes were secured in the recording chamber (see Calcium photometry above). To determine whether basal H2O2 availability in endothelium increases with advanced age, cells were loaded with the H2O2-sensitive fluorescent probe dichlorofluorescein (DCFH; 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate; #C-400; Molecular Probes, Eugene, OR, USA). For dye loading, DCFH was dissolved in DMSO and then diluted to 15 μm in PSS (pH 7.4; DMSO < 0.5%). The tube was immersed at room temperature (24°C) for 30 min without flow, then superfusion with PSS (3 ml min−1) was restored to remove excess dye, the signal was allowed to stabilize (1–2 min) and initial baseline fluorescence (F0) was determined. These experiments were maintained at room temperature because intracellular DCFH levels remained stable at 24°C for at least 30 min. In contrast, DCFH extrusion was pronounced within several min at 32°C (time controls not shown). Utilizing a 40× objective (NA = 0.9, Nikon) and recording from a 140 × 50 μm window, DCFH was excited at 460–500 nm while emission was recorded continuously at 510–560 (505 nm dichroic mirror; Chroma Technology Corp., Bellows Falls, VT, USA) for at least 30 min. Linear regression was performed on the first 10 min, which corresponded to the steepest rise in fluorescence.
Control experiments were performed to confirm the sensitivity of intracellular DCFH to H2O2. To inhibit the accumulation of DCFH fluorescence resulting from H2O2 produced endogenously, endothelial tubes were pre-incubated in PSS (pH 7.4) containing the superoxide dismutase mimetic TEMPOL (4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl, 1 mm) + PEG-catalase (500 U ml−1) to decompose intracellular H2O2 (Behringer et al. 2013). To estimate the [H2O2] required to increase DCFH fluorescence above its basal rate of accumulation, H2O2 was added to the superfusion solution (pH 7.4) at defined concentrations. Data were acquired using Ion Wizard software (IonOptix) and were analysed after subtraction of background fluorescence.
Catalase activity
To determine whether scavenging of H2O2 within the vessel wall is reduced with advanced age, catalase activity was measured using a colorimetric assay. Preliminary experiments determined the amount of vessel required. Thus, for each mouse, first- and second-order superior and inferior epigastric arteries were dissected bilaterally, flushed of blood, then pooled and cut into segments ∼100 μm long. Each pooled sample (one per mouse) was placed in 100 μl lysis buffer consisting of: 50 mm Hepes, 150 mm NaCl, 1 mm MgCl2, 1% Triton X-100, 2% glycerol and protease inhibitor cocktail (modified from Nystoriak et al. 2011). Samples were vortexed for 1 min, sonicated for 20 min, vortexed again for 1 min, sonicated for another 10 min, then centrifuged at 7000 g for 10 min at 4°C. The lysate supernatant was retained and used for assay. Protein concentrations for each sample were measured using the Micro BCA Assay (#23235; ThermoFisher, Waltham, MA, USA). A standard curve (0–200 μg μl−1 BSA) was prepared and protein concentrations were measured on a NanoDrop 2000c (ThermoFisher). The assay for catalase activity (#CAT100; Sigma-Aldrich, St Louis, MO, USA) was run at room temperature according to the manufacturer's instructions. Standard curves for H2O2 concentration were prepared fresh daily and each assay included positive and negative controls in addition to samples from Young and Old mice. Each reaction included 75 μl of lysate supernatant (containing 10–15 μg protein) and lasted 5 min. Assays were read at 520 nm in a quartz cuvette on the NanoDrop 2000c. Specific catalase activity is reported as μmol H2O2 consumed min−1 ml−1 (μg protein)−1.
Hydrogen peroxide exposure
To determine whether advanced age impairs Ca2+ signalling in microvascular endothelium during acute oxidative stress, endothelial tubes were loaded with Fura-2 dye as described above to evaluate Ca2+ responses to H2O2. After warming to 32°C, the protocol consisted of an initial baseline recording, addition of ACh (3 μm) to the superfusion solution for 2 min, then washout and recovery to resting baseline (10 min) during superfusion with control PSS. In accordance with the protocol developed for earlier studies (Behringer et al. 2013), H2O2 (200 μm) was included in the superfusion solution (pH 7.4) for 20 min followed by 30 min washout and recovery with control PSS. Fura-2 emission was recorded for the final 10 s of each 5 min period during H2O2 exposure and throughout recovery with F340/F380 averaged for each recording. In separate experiments to determine whether Ca2+ signals during H2O2 exposure were derived from intra- or extracellular sources, the 20 min exposure to H2O2 was performed using PSS that was nominally Ca2+-free (i.e. prepared without CaCl2). Superfusion with control PSS (2 mm Ca2+) was restored during the 30 min recovery period with Fura-2 emission sampled as above. To determine whether advanced age affected EC viability during oxidative stress, the duration of exposure to H2O2 (200 μm) was extended to 60 min.
Manganese quench
Quenching of Fura-2 fluorescence by Mn2+ correlates with the rate of divalent cation flux across the plasma membrane, rendering this method an accurate assessment of Ca2+ influx (Potocnik & Hill, 2001; Tas et al. 2008). An endothelial tube was loaded with Fura-2 dye as described above and studied at 32°C. To obtain an index of Ca2+ influx at rest, the tube was superfused with PSS in which the CaCl2 was replaced with MnCl2 for 5 min, during which the peak rate of decrease in the Fura-2 signal was recorded at its isosbestic wavelength (360 nm). To determine how oxidative stress influenced Ca2+ influx, the tube was superfused with 200 μm H2O2 in PSS containing 2 mm Ca2+ for 10 min, then with 200 μm H2O2 in nominally Ca2+-free PSS for 10 min. As determined in preliminary experiments, this Ca2+-free interval prior to Mn2+ addition prevented Ca2+ influx, thereby avoiding cytosolic Ca2+ accumulation and its binding to Fura-2 (which would limit Mn2+ binding sites for the quench assay). Ca2+-free PSS containing 2 mm MnCl2 was then added for 5 min during which the Fura-2 signal was recorded. This protocol resulted in measurements of H2O2-induced cation permeability coinciding with 20 min exposure to H2O2. Linear regression was performed on a 30 s interval during the steepest portion of the Fura-2 response during MnCl2 exposure (i.e. during maximal Mn2+ influx) to determine the maximal rate of fluorescence quenching, thereby providing an index of the peak rate of cation influx across the plasma membrane.
Transient receptor potential (TRP) channel inhibition
To determine whether TRP channels were involved in [Ca2+]i responses, the broad-spectrum TRP channel inhibitor ruthenium red (RuR; Ramsey et al. 2006) was dissolved directly into PSS (5 μm; pH 7.4) and included during superfusion with H2O2. Having determined that [Ca2+]i responses to exogenous H2O2 in the endothelium of Young were far greater than in Old mice (see Results), endothelial tubes from Young mice were loaded with Fura-2 as described above. After recording baseline F340/F380 values and control responses to 3 μm ACh, the endothelium was superfused for 20 min with PSS containing 200 μm H2O2 + 5 μm RuR followed by control PSS for 30 min of recovery. Fura-2 fluorescence emission was recorded for 10 s at the end of each 5 min period and averaged.
Cell viability assay
To determine whether EC death was responsible for the Fura-2 signal during the 20 min exposure to H2O2, a Live/Dead Viability/Cytotoxicity assay for mammalian cells (#L-3224; Life Technologies,; Carlsbad, CA, USA) was performed. Briefly, following isolation, tubes were superfused with 200 μm H2O2 for 20 min at 32°C as done for evaluating [Ca2+]i responses. Following a 2 min wash with control PSS while cooling back to 24°C, the Live/Dead assay was performed at room temperature according to the manufacturer's recommendations. Thus, individual tubes were incubated for 30 min in PSS containing 2 μm calcein-AM (hydrolysed and retained in viable cells) and 4 μm ethidium bromide (EtBr; stains nuclei of dead and dying cells). The tube was washed again for 2 min in control PSS. As there was negligible EtBr staining following 20 min of H2O2 for either age group (see Results), additional experiments were performed in which the duration of H2O2 exposure was extended to 60 min prior to performing the Live/Dead assay. Images of each tube were acquired using a 40× oil immersion objective (NA = 1.25) on a Leica SP5 laser scanning confocal microscope equipped with LAS Software (Leica Microsystems, Buffalo Grove, IL, USA). Both dye molecules were excited with the 488 nm line of an argon laser with emission acquired at 500–535 nm (calcein) and 595–700 nm (EtBr). Transmitted light images were acquired concurrently. To quantify cell damage in each tube, the number of ECs stained with EtBr was expressed as a percentage of the total number of fluorescently labelled ECs [i.e. (EtBr-positive cells)/(EtBr-positive cells + calcein-positive cells)].
Data analysis and statistics
One EC tube was studied from each SEA. Unpaired Student's t-tests were used to evaluate differences between age groups in F340/F380 values at rest, in response to ACh and during Mn+2 quench, during DCFH fluorescence and for catalase activity. Changes in F340/F380 over time during H2O2 exposure were analysed using two-way ANOVA with Bonferroni post-tests. Statistical analyses were performed using GraphPad Prism (GraphPad Software, La Jolla, CA, USA). Summary data are presented as means ± SEM. Differences were accepted as statistically significant at P < 0.05.
Results
Calcium photometry
Time controls confirmed that baseline F340/F380 did not change significantly for at least 2 h in either Young or Old mice (n = 4 each). Thus, consistent with earlier findings (Socha et al. 2011), resting [Ca2+]i remained stable at 32°C. To substantiate comparisons of F340/F380 between Young and Old, minimum and maximum F340/F380 values for the Ca2+ signal were determined as described previously for endothelial tubes (Socha et al. 2011). Thus, minimum F340/F380 values were 0.61 ± 0.02 for Young (n = 3) and 0.64 ± 0.02 for Old (n = 4) with respective maximum F340/F380 values of 5.13 ± 0.76 and 5.92 ± 1.35; neither value was significantly different between age groups (P = 0.44 and 0.61, respectively). With F340/F380 as an index of [Ca2+]i, resting values were not different between endothelium of Young versus Old mice (Fig.1A). In response to stimulation with ACh (3 μm), the mean steady-state [Ca2+]i response was 25% higher in Old versus Young (P < 0.05) (Fig.1C), confirming the integrity of signalling through muscarinic GPCRs in advanced age.
Figure 1.
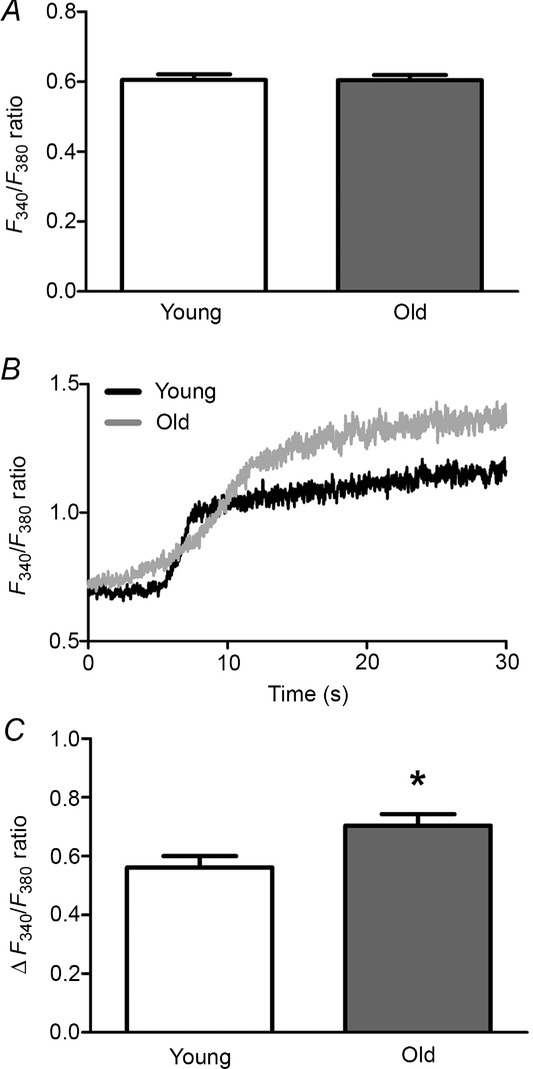
Enhanced [Ca2+]i response to ACh in microvascular endothelium with advanced age
A, resting F340/F380 ratios were not different between endothelium of Young versus Old mice indicating similar [Ca2+]i at rest. B, representative F340/F380 recordings from endothelium of Young and Old in response to 3 μm ACh (added at time 0). C, maximal change in [Ca2+]i (ΔF340/F380) of endothelium from Young and Old mice during sustained response to 3 μm ACh. Summary data are means ± SEM; n = 33 for Young, n = 31 for Old. *P < 0.05, Old versus Young.
Hydrogen peroxide availability under basal conditions
Baseline raw fluorescence (F0) units for DCFH at the onset of recordings were 474 ± 32 for Young and 402 ± 34 for Old (means ± SEM, n = 10 per age group) and not significantly different between age groups (P = 0.15). To provide an internal control for variability in dye loading and other factors that may influence F0, these data are presented relative to F0 (i.e. F/F0). The rate of DCFH fluorescence accumulation was 33% greater in endothelium of Old versus Young (P < 0.05; Fig.2A–C). In the presence of TEMPOL (1 mm) + PEG-catalase (500 U ml−1) DCFH fluorescence did not change over time (Fig.2A–C), confirming the applicability of this probe for detecting H2O2 in our experiments. In lysates of epigastric arteries, endogenous catalase activity was lower by nearly half in Old compared to Young (P < 0.05, Fig.2D). Thus, microvascular endothelium of Old mice exhibits greater H2O2 availability under basal conditions in conjunction with diminished capacity of the vessel wall to remove H2O2 when compared to Young. To provide an index of H2O2 availability under basal conditions, H2O2 was added to the superfusion solution. In endothelial tubes of Young (n = 3), the slope of fluorescence accumulation was unaffected by 1 μm H2O2 but began to increase following addition of 5 μm H2O2. For reference, 200 μm H2O2 promptly increased DCFH fluorescence and saturated the signal within 2–3 min (data not shown).
Figure 2.

Enhanced H2O2 availability with diminished H2O2 decomposition during advanced age
A, representative recordings of DCFH in endothelium of Young mice illustrating progressive increase in fluorescence over 10 min during superfusion with PSS containing 2 mm Ca2+ that was prevented by treatment with TEMPOL (1 mm, superoxide dismutase mimetic) + PEG-catalase (500 U ml−1, H2O2 scavenger). B, as in A for Old mice; note greater rate of fluorescence accumulation versus Young. C, summary data for rates of fluorescence accumulation in endothelium of Young and Old mice determined from recordings as represented by those in A and B. Rates of fluorescence accumulation were depressed by TEMPOL + PEG-catalase to values not different between Young and Old (n = 10 per age group with PSS; n = 4 per group for TEMPOL + PEG-catalase). *P < 0.05, Old versus Young, #P < 0.05, TEMPOL + PEG-catalase versus PSS for Young; ΨP < 0.05, TEMPOL + PEG-catalase versus PSS for Old. D, catalase activity measured via H2O2 decomposition by lysates of epigastric arteries from Young and Old mice (n = 5 per age group). *P < 0.05, Old versus Young. Summary data in C and D are means ± SEM.
Effect of H2O2 on endothelial Ca2+ and cellular integrity
Exposing the endothelium of Young mice to 200 μm H2O2 for 20 min progressively increased [Ca2+]i. After a delay of several min, the increase was significant by 20 min (Fig.3C) and was nearly twice that observed in response to maximal stimulation with 3 μm ACh (Fig.1C). During the ensuing washout of H2O2, this aberrant elevation in [Ca2+]i persisted for the initial 5 min before recovering to control levels within 20–30 min (Fig.3A). In endothelium of Old mice, exposure to 200 μm H2O2 also led to a rise in [Ca2+]i with a time course similar to that for Young (Fig.3B), although the [Ca2+]i response was far less (∼25%; P < 0.05) than observed for Young. In separate experiments, omitting Ca2+ from the superfusion solution abolished the response to H2O2 in both Young (Fig.3A) and Old (Fig.3B) when compared to responses in the presence of 2 mm [Ca2+]o. Within 5 min of reintroducing 2 mm Ca2+ during H2O2 washout, [Ca2+]i increased in endothelium of Young (Fig.3A) but not in Old (Fig.3B), further indicating a persistent effect of H2O2 on Ca2+ entry.
Figure 3.
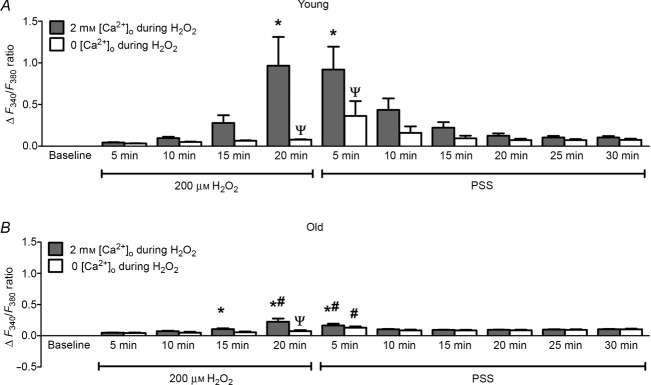
Reduced [Ca2+]i response of microvascular endothelium to H2O2 with advanced age
A, exposing endothelium from Young mice to 200 μm H2O2 for 20 min progressively increased [Ca2+]i (ΔF340/F380). [Ca2+]i recovered during 30 min washout of H2O2 with PSS containing 2 mm Ca2+. Removing extracellular Ca2+ (0 [Ca2+]o) during H2O2 exposure prevented the rise in [Ca2+]i in endothelial tubes of Young mice yet [Ca2+]i increased within 5 min of H2O2 washout with PSS containing 2 mm Ca2+. B, as in A for Old mice; note diminished [Ca2+]i response compared to Young in the presence of 2 mm [Ca2+]o and negligible [Ca2+]i response in 0 [Ca2+]o. Summary data are means ± SEM. For both age groups n = 9 for 2 mm [Ca2+]o during H2O2, and n = 7 for 0 [Ca2+]o during H2O2. *P < 0.05, versus Control. #P < 0.05, Old versus Young for given time and condition, ΨP < 0.05 for 0 [Ca2+]o versus 2 mm [Ca2+]o exposed to 200 μm H2O2.
The above data implicate a key role for extracellular Ca2+ in the [Ca2+]i response during elevated H2O2. Thus, Ca2+ influx across the plasma membrane was evaluated through measuring Fura-2 quench by Mn+2 (Fig.4). Consistent with the similarity in resting [Ca2+]i, there was no difference in the quench rate of Fura-2 fluorescence at rest between endothelium of Young versus Old mice (Fig.4A and B). During exposure to 200 μm H2O2, the quench rate increased for endothelium of Young and Old; however, the rate was ∼2-fold greater (P < 0.05) in Young (Fig.4C and D). Given that TRP channels encompass redox-sensitive cation channels in the plasma membrane of ECs (Poteser et al. 2006; Xu et al. 2008; Sun et al. 2012) and are integral to Ca2+ influx (Ramsey et al. 2006), we tested whether H2O2-induced Ca2+ influx was mediated by TRP channels. In endothelium of Young, inclusion of 5 μm RuR [a broad-spectrum blocker of TRP channels (Ramsey et al. 2006)] during exposure to 200 μm H2O2 abrogated the [Ca2+]i response by ∼70% (Fig.5).
Figure 4.
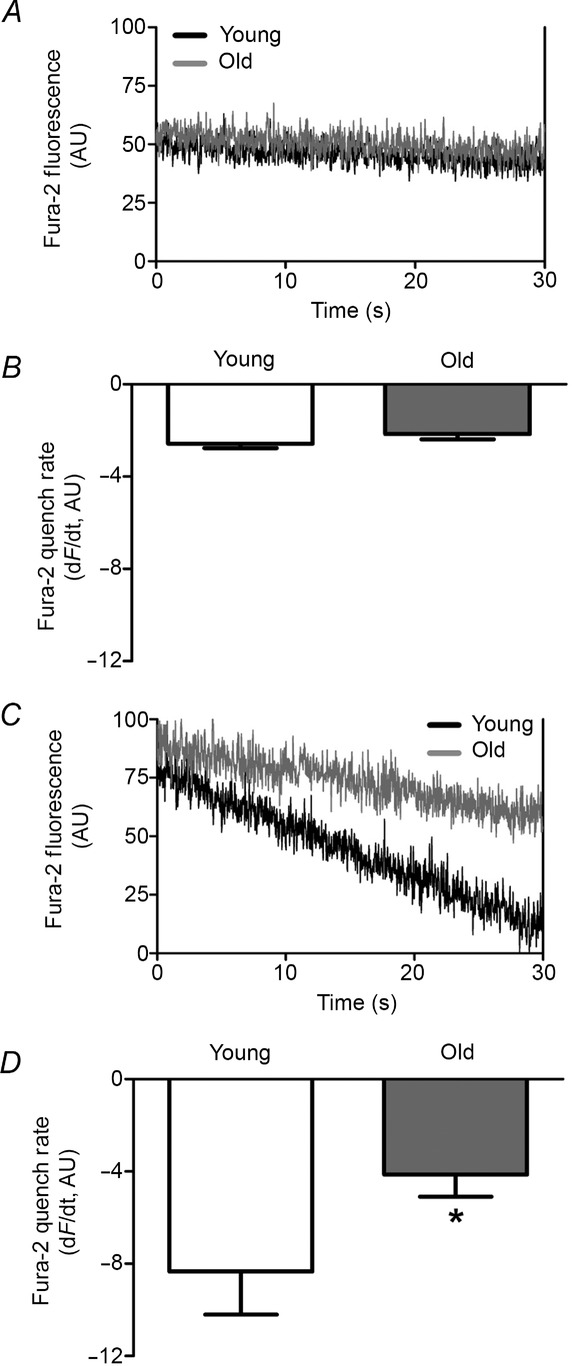
Enhanced Mn2+ quenching of Fura-2 fluorescence during H2O2 is reduced with advanced age
A, representative Fura-2 recording at isosbestic wavelength (360 nm) during superfusion with 2 mm MnCl2 (replacing CaCl2 in PSS) of endothelium from Young and Old mice at rest. Note similar rates of Fura-2 quench. B, summary data (means ± SEM) indicate no difference in Mn2+ influx between endothelium of Young (n = 6) and Old (n = 8) mice. C, as in A during exposure to 200 μm H2O2. Note increased rate of Fura-2 quench in both age groups compared to A with greater rate in Young versus Old. D, summary data (means ± SEM) indicate greater rate of Mn2+ influx in endothelium of Young (n = 6) versus Old (n = 6) during exposure to 200 μm H2O2. *P < 0.05, Old versus Young.
Figure 5.
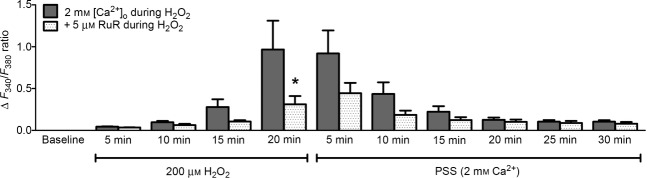
TRP channels contribute to Ca2+ influx induced by H2O2
In endothelium of Young mice, inclusion of 5 μm RuR to inhibit TRP channels reduced influx of Ca2+ during exposure to 200 μm H2O2 by ∼75%. This effect was reversed within 5 min of washout of H2O2 with PSS. Summary data are means ± SEM; n = 9 for 2 mm [Ca2+]o during H2O2 and n = 5 for 5 μm RuR during H2O2. *P < 0.05, Old versus Young.
Elevated levels of H2O2 can damage ECs (Volk et al. 1997; Sun et al. 2012). To determine whether H2O2 exposure was associated with differences in cellular integrity between age groups, Live/Dead cell assays were performed. Following the 20 min exposure to 200 μm H2O2, cells remained viable in the endothelium of both age groups, as confirmed by their uptake and retention of calcein-AM and absence of staining with EtBr (n = 3 per age group; data not shown). Increasing the duration of H2O2 exposure to 60 min resulted in dead and dying cells within endothelial tubes of both age groups (Fig.6B and C). Remarkably, there was a pronounced protective effect of advanced age. Thus, with 566 ± 139 cells counted per endothelial tube in Young (n = 5) and 616 ± 49 cells counted per endothelial tube in Old (n = 5), the percentage of ECs labelled with EtBr was ∼7-fold greater (P < 0.05) in Young compared to Old (Fig.6E). Consistent with the stability of Ca2+ and electrical responses in our previous studies (Socha et al. 2011; Behringer et al. 2012, 2013), time controls performed here confirmed that, in the absence of H2O2, there were no dead or dying cells in endothelial tubes of Young following 60 min under identical conditions (n = 2).
Figure 6.
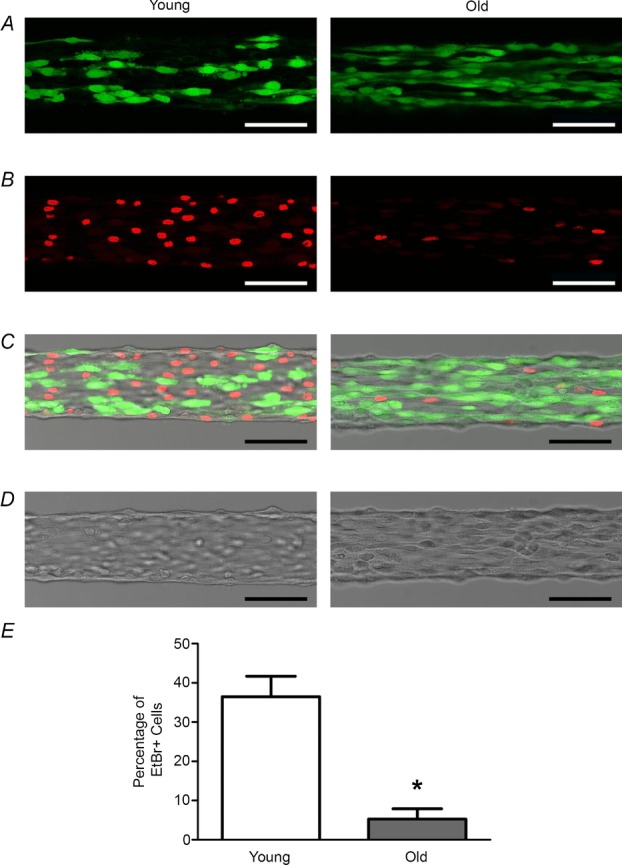
Advanced age abrogates cell death induced by prolonged exposure to H2O2
Following 60 min exposure to 200 μm H2O2, a Live/Dead cell assay was performed on endothelial tubes from Young (left) and Old (right) mice. A, living endothelial cells sequestered and retained calcein-AM (green), confirming their viability. B, dead and dying cells (which have nuclei stained red with EtBr) appear more frequently for endothelium of Young versus Old. C, overlay of images in A and B. D, brightfield images of endothelial tubes in A–C. E, the percentage of EtBr-positive cells is ∼7-fold greater in endothelium of Young versus Old. Summary data are means ± SEM; n = 5 per age group. *P < 0.05, Old versus Young. Scale bars = 50 μm.
Discussion
Calcium signalling is integral to endothelial function and vasomotor control. However, there is a paucity of information regarding the effects of advanced age on endothelial Ca2+ signalling during oxidative stress, particularly in the microcirculation. To address this gap in understanding, we evaluated [Ca2+]i at rest, during muscarinic GPCR activation and in response to acute oxidative stress with H2O2 (200 μm) in endothelium freshly isolated from skeletal muscle resistance arteries of Young and Old mice. Resting [Ca2+]i was not different between age groups, yet the peak steady-state [Ca2+]i response to ACh (3 μm) was greater in endothelium of Old compared to Young. Furthermore, when compared to Young, the constitutive release of H2O2 under basal conditions was greater for endothelium of Old and was accompanied by lower vascular catalase activity. During oxidative stress imposed by exposure to H2O2 for 20 min, the endothelium of Young mice responded with an aberrant increase in [Ca2+]i whereas the endothelium of Old mice did not. As indicated by its dependence on extracellular Ca2+, this difference between age groups may reflect adaptation of microvascular endothelium during advanced age that attenuates influx of Ca2+ across the plasma membrane. Moreover, prolonged (60 min) exposure to H2O2 resulted in far greater cell death in endothelium of Young than in Old. These findings demonstrate a protective effect of advanced age on Ca2+ influx and cell viability in response to acute oxidative stress in native endothelium of resistance microvessels.
Endothelial dysfunction (e.g. impaired endothelial-dependent dilatation, EDD) accompanies advancing age in humans (Taddei et al. 2001; Seals et al. 2011) and is a risk factor for cardiovascular disease (Yeboah et al. 2007; Lind et al. 2011). In the peripheral circulation, the efficacy of endothelium-dependent vasodilators (e.g. ACh and methacholine; which act through muscarinic GPCRs to elevate [Ca2+]i) is diminished in elderly humans (e.g. > 60 years) compared to their younger (e.g. < 30 years) counterparts (Taddei et al. 2001). At the same time, endothelium-independent vasodilatation (e.g. to the NO donor SNP) was maintained (Taddei et al. 2001). Remarkably, little is known of how ageing may affect endothelial [Ca2+]i responses to GPCR agonists or to acute oxidative stress. Therefore, we tested the hypothesis that advanced age impairs endothelial Ca2+ signalling. Under resting conditions, [Ca2+]i was not different between endothelial tubes from Young versus Old mice (Fig.1A). Through binding to muscarinic GPCRs, ACh initiates intracellular Ca2+ release from the endoplasmic reticulum with an ensuing prolonged phase of Ca2+ entry across the plasma membrane from the extracellular fluid (Tran & Watanabe, 2006; Socha et al. 2011). Our use of 3 μm ACh as a maximal criterion stimulus is based upon earlier studies of endothelial tubes, where ACh elevated [Ca2+]i and evoked hyperpolarization through KCa activation in a concentration-dependent manner. Both [Ca2+]i and hyperpolarization plateaued at ≥ 1 μm ACh (Behringer et al. 2012), with no difference in the magnitude of hyperpolarization between Young and Old (Behringer et al. 2013). During the prolonged phase studied here, elevations in [Ca2+]i were ∼33% greater in endothelium of Old versus Young (Fig.1C). This finding confirms the integrity of this signalling pathway while suggesting augmented Ca2+ entry with advanced age. Greater elevation of [Ca2+]i during sustained muscarinic GPCR activation may help to compensate for age-related reductions in NO bioavailability (Muller-Delp, 2006; Seals et al. 2011). Thus, elevated [Ca2+]i would promote the activation of endothelial NOS. However, elevated [Ca2+]i can enter mitochondria, thereby increasing mitochondrial generation of ROS (Camello-Almaraz et al. 2006) and EC oxidative stress.
Augmented superoxide production and greater levels of oxidative stress have been demonstrated in ECs with ageing (Dhalla et al. 2000; Muller-Delp et al. 2012; Bachschmid et al. 2013). Elevation of ROS occurs as a consequence of greater synthesis, diminished removal or a combination thereof. Sources of superoxide (i.e. the precursor to H2O2) in ECs include NADPH oxidase, xanthine oxidase, mitochondria (i.e. respiratory enzymes) and uncoupled NOS (Lambeth, 2004; Forstermann, 2008; Touyz et al. 2011). In addition to constitutive ROS production, ECs are exposed to ROS released by surrounding cells (e.g. SMCs) and by cells carried in the bloodstream (Li & Shah, 2004; Feletou & Vanhoutte, 2006). Once generated, superoxide anions form H2O2 via the superoxide dismutase family of enzymes. Catalase then decomposes H2O2 by converting it to H2O and O2 to protect cells from oxidative damage. Levels of oxidative stress, as well as subsequent cellular responses, can vary between vascular beds (Grinnell et al. 2012). To determine whether oxidative stress was manifest under basal conditions within microvascular ECs during advanced age, we evaluated rates of H2O2 production using the H2O2-sensitive dye DCFH (Carter et al. 1994) after confirming its sensitivity to H2O2 under the conditions of our experiments. Our data illustrate that the availability of H2O2 was significantly greater in the endothelium of Old mice under resting conditions (Fig.2C). This finding helps to explain the enhanced activation of SKCa and IKCa observed for endothelial tubes of Old mice (Behringer et al. 2013). With no difference in [Ca2+]i at rest (Fig.1A), direct activation and/or enhanced sensitivity to Ca2+ of KCa is implied, as channel expression was not different between age groups (Behringer et al. 2013). Complementary experiments revealed that catalase activity was reduced by nearly half in epigastric arteries of Old mice (Fig.2D), supporting higher levels of constitutive oxidative stress in microvascular ECs during advanced age.
Acute elevation of oxidative stress was imposed by superfusion of endothelial tubes with 200 μm H2O2 for 20 min. This concentration of H2O2 and duration of exposure were based upon finding that they effectively activated SKCa and IKCa in the same preparations studied here (Behringer et al. 2013). Moreover, the time course observed for the progression of hyperpolarization is consistent with that found here for the rise in [Ca2+]i; i.e. an initial delay upon exposure followed by a progressive increase during the 20 min exposure (Fig.3). Observing that the effect of H2O2 on [Ca2+]i persisted during the early period of washout suggests that, despite being membrane-permeant, the actions of H2O2 are not instantaneous and that they exert a residual effect following H2O2 removal. Compared to the gradual accumulation of DCFH fluorescence under basal conditions, the fluorescence signal saturated within 2–3 min upon exposure to 200 μm H2O2. Thus 200 μm H2O2 is far greater than levels present under basal conditions. As exposure to 5 μm H2O2 was found to increase DCFH fluorescence above basal accumulation, we suggest that this far lower concentration more closely approximates constitutive levels of H2O2 availability.
Regulation of EC [Ca2+]i occurs though multiple signalling pathways which include stimulation of GPCRs with physiological agonists (Tran & Watanabe, 2006). Thus, ACh binding to muscarinic (M3) receptors generates inositol 1,4,5 trisphosphate, thereby stimulating releasing of Ca2+ from the endoplasmic reticulum followed by sustained influx through the plasma membrane (Tran & Watanabe, 2006). Free Ca2+ is then removed from the cytoplasm by ATPases in respective membranes. Indeed, the maintenance of [Ca2+]i homeostasis is essential for EC function and survival, with aberrant levels of [Ca2+]i leading to dysfunction and cell death (Orrenius et al. 2003; Berridge, 2005). Exposure to 200 μm H2O2 resulted in an ∼4-fold greater increase in [Ca2+]i for endothelium of Young (Fig.3A) compared to Old (Fig.3B). To evaluate the role of Ca2+ entry, extracellular Ca2+ was removed during H2O2 treatment, which effectively prevented the increase in [Ca2+]i. That H2O2 acted via augmenting Ca2+ entry was tested further by using Mn2+ influx to quench Fura-2 (Potocnik & Hill, 2001; Tas et al. 2008). Consistent with resting [Ca2+]i, we found no difference in the rate of Fura-2 fluorescence quenching between endothelium of Young versus Old at rest (Fig.4A and B). However, exposure to 200 μm H2O2 evoked ∼2-fold greater peak rates of Fura-2 fluorescence quenching for endothelium of Young versus Old (Fig.4C and D). These findings indicate that elevated [Ca2+]i in Young during H2O2 exposure can be explained by enhanced Ca2+ influx. In light of greater Ca2+ influx during ACh exposure in endothelium of Old versus Young mice, the corresponding reduction in Ca2+ influx during 200 μm H2O2 exposure in Old mice implies an adaptation that occurs in association with significantly higher basal levels of oxidative stress during advanced age.
TRP channels are Ca2+-permeant, expressed in ECs and sensitive to oxidative stress (Poteser et al. 2006; Yoshida et al. 2006; Xu et al. 2008; Beech, 2013). In accordance with the far greater Ca2+ responses for endothelium of Young versus Old mice, we investigated whether TRP channels were involved in the H2O2-induced Ca2+ influx in endothelium of Young mice. Remarkably, inclusion of the broad-spectrum TRP channel antagonist RuR (Ramsey et al. 2006) attenuated the H2O2-induced [Ca2+]i rise (Fig.5) to levels similar to those observed in the absence of [Ca2+]o (Fig.3). Multiple isoforms of TRP channels are redox sensitive through activation by oxidative or reductive agents. Thus, TRPC5 can be activated directly via sulfhydryl modification by NO and other oxidative agents (Yoshida et al. 2006) or by reducing agents such as thioredoxin (Xu et al. 2008). Furthermore, TRPV1 and TRPA1 can be activated by oxidizing agents through cysteine modification (Yoshida et al. 2006; Macpherson et al. 2007). As recently shown for endothelium of rat cerebral arteries, generation of H2O2 leads to lipid peroxidation products which activate ‘TRPA1 sparklets’ of Ca2+ influx (Sullivan et al. 2015). In addition to these homomeric channels, TRP channel protein isoforms can form redox-sensitive heteromeric channels. For example, association of TRPC3 and TRPC4 subunits have been found to form such channels in ECs (Poteser et al. 2006). The TRPM2 channel, which is robustly expressed in ECs, is activated by H2O2 and the resulting Ca2+ influx can lead to apoptosis (Hecquet et al. 2008; Sun et al. 2012). Owing to the possibility that the H2O2-induced Ca2+ influx could lead to cell death, we performed Live/Dead cell assays and found no evidence thereof following 20 min exposure to 200 μm H2O2. This outcome is in agreement with our earlier studies demonstrating that endothelial tubes remained viable (e.g. membrane potential was maintained) for this period under the same experimental conditions (Behringer et al. 2013). However, when the duration of exposure to 200 μm H2O2 was increased to 60 min, cell death was apparent in both Young and Old (Fig.6). Remarkably, the prevalence of dead and dying cells (EtBr positive) observed in Young was ∼7-fold greater (∼36%) than that in Old (∼5%). In turn, we suggest that the profound reduction in Ca2+ influx observed for endothelium of Old versus Young reflects a protective adaptation during advanced age that is associated with constitutively greater levels of H2O2 under basal conditions (Fig. 2).
The production of ROS within ECs can disrupt signalling pathways and lead to cytotoxicity (Dhalla et al. 2000). For example, superoxide can react with NO to form peroxynitrite and thereby reduce the bioavailability of NO while uncoupling NOS (Landmesser et al. 2003), contributing further to superoxide production. In diabetes, endothelial dysfunction induced by oxidative stress results from the activation of protein kinase C and an increase in glycation end-product formation (Brownlee, 2001). Additionally, NADPH oxidase-induced oxidative stress is a key step in the progression of hypertension (Laursen et al. 1997; Schnackenberg et al. 1998) while mounting evidence implicates ROS in the pathogenesis of atherosclerosis (Madamanchi & Runge, 2007). Furthermore, constitutively elevated H2O2 during advanced age impairs electrical signal transmission along the endothelium via activation of KCa channels, with ‘leaky’ membranes dissipating charge transfer from cell to cell (Behringer et al. 2013).
Scavenging ROS has been found to ameliorate endothelial dysfunction and promote blood flow to skeletal muscle in both humans (Kirby et al. 2009) and mice (Fleenor et al. 2012) of advanced age. Nevertheless, endothelial-derived ROS have increasingly been found to play a key role in cellular signalling pathways under physiological conditions (Ungvari et al. 2006; Thomas et al. 2008; Suvorava & Kojda, 2009; Wolin, 2009; Muller-Delp et al. 2012). Specifically, H2O2 has been identified as an ideal second messenger amongst ROS due to its relatively long half-life compared to superoxide or peroxynitrite. Furthermore, unlike the latter anions, which are membrane-impermeant and react in their vicinity of production, its lack of charge enables H2O2 to readily permeate cell membranes and thereby serve as an autacoid. Indeed, H2O2 has been shown to function as an endothelial-derived vasodilator. Thus, in mesenteric arteries of both humans and mice, endothelial-derived H2O2 can hyperpolarize and thereby relax SMCs (Matoba et al. 2000, 2002) to produce vasodilatation. In human coronary arterioles, H2O2 generated from mitochondria can regulate flow-mediated (i.e. endothelium-dependent) dilatation through activating large-conductance KCa channels of SMCs (Liu et al. 2003). Additionally, recent studies of first-order arterioles from rat soleus muscle indicate that with advanced age there is a shift in EDD from reliance on NO to a greater role for H2O2 (Sindler et al. 2013). Such a shift during ageing would imply an adaptation in the endothelium to reduce the cellular damage otherwise caused by H2O2, which is consistent with the present observations.
Summary and perspective
The present study has investigated Ca2+ signalling in endothelial tubes freshly isolated from resistance arteries of mouse skeletal muscle with respect to the effects of advanced age and acute oxidative stress induced by acute exposure to elevated H2O2. Utilization of this model enables studies of native intact microvascular endothelial function in the absence of confounding interactions with circulating blood, surrounding smooth muscle and adventitia. Our findings indicate that microvascular endothelium of Old mice has higher levels of H2O2 availability under basal conditions. With greater activation of KCa (Behringer et al. 2013) and no difference in [Ca2+]i, at rest in Old versus Young, the present data suggest that constitutive production of H2O2 activates KCa of native microvascular endothelium, consistent with the effects of H2O2 in human endothelial cells (Bychkov et al. 1999; Miura et al. 2003). Exposure to 200 μm H2O2 for 20 min evoked far greater [Ca2+]i increases in the endothelium of Young versus Old mice with a time course similar to that observed for hyperpolarization (Behringer et al. 2013). This rise in [Ca2+]i can be explained by greater Ca2+ influx across the plasma membrane and inhibiting this effect by blocking TRP channels further implicates the activation of membrane ion channels by H2O2. Remarkably, cell injury and death following prolonged (60 min) exposure to H2O2 were far greater in endothelium of Young mice compared to Old. Thus, constitutively elevated basal levels of oxidative stress during advanced age may lead to an adaptive response within the endothelium of resistance microvessels that limits aberrant Ca2+ influx and mitigates the cytotoxic effects of oxidative stress (Dhalla et al. 2000) in association with greater utilization of H2O2 as a mediator of EDD (Widlansky & Gutterman, 2011; Muller-Delp et al. 2012). Indeed, such protective adaptations to advanced age could explain why antioxidant therapy in humans has little beneficial effect and may instead act to increase patient morbidity and mortality (Yoshihara et al. 2010). Our findings collectively indicate a delicate balance between maintaining physiological levels of ROS during normal vascular function (e.g. ‘healthy ageing’) and pathological oxidative stress with associated endothelial dysfunction.
Glossary
- DCFH
6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate
- EDD
endothelial-dependent dilatation
- EC
endothelial cell
- EtBr
ethidium bromide
- GPCR
G-protein coupled receptor
- IKCa
intermediate conductance calcium-activated potassium channel
- KCa
calcium-activated potassium channel
- NO
nitric oxide
- NOS
nitric oxide synthase
- PSS
physiological salt solution
- ROS
reactive oxygen species
- RuR
ruthenium red
- SKCa
small conductance calcium-activated potassium channel
- SMC
smooth muscle cell
- SNP
sodium nitroprusside
- SEA
superior epigastric artery
- TEMPOL
4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl
- TRP
transient receptor potential
Additional information
Competing interests
The authors declare no competing interests.
Author contributions
M.J.S. designed and performed the experiments using Fura-2 dye, analysed and interpreted the data, drafted the manuscript and prepared the figures. E.M.B. evaluated catalase activity, contributed to experimental design, data interpretation and edited the manuscript. R.L.S. evaluated H2O2 levels utilizing DCFH and edited the manuscript. E.J.B., T.L.D. and S.S.S contributed to experimental design, analysis, interpretation and presentation of data, and edited the manuscript. All authors reviewed and approved the final version of the manuscript for publication.
Funding
This research was supported by NIH grants R37-HL041026 and R01-HL086483 to S.S.S., F32-HL107050 to M.J.S., F32-HL118836 to E.M.B., K99-AG047198 to E.J.B. and K01-AG041208 to T.L.D. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author's present address
M. J. Socha: Biology Department, Loyola Science Center, 204 Monroe Avenue, The University of Scranton, Scranton, PA 18510, USA.
References
- Bachschmid MM, Schildknecht S, Matsui R, Zee R, Haeussler D, Cohen RA, Pimental D. Loo B. Vascular aging: chronic oxidative stress and impairment of redox signaling – consequences for vascular homeostasis and disease. Ann Med. 2013;45:17–36. doi: 10.3109/07853890.2011.645498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beech DJ. Characteristics of transient receptor potential canonical calcium-permeable channels and their relevance to vascular physiology and disease. Circ J. 2013;77:570–579. doi: 10.1253/circj.cj-13-0154. [DOI] [PubMed] [Google Scholar]
- Behringer EJ, Shaw RL, Westcott EB, Socha MJ. Segal SS. Aging impairs electrical conduction along endothelium of resistance arteries through enhanced Ca2+-activated K+ channel activation. Arterioscler Thromb Vasc Biol. 2013;33:1892–1901. doi: 10.1161/ATVBAHA.113.301514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behringer EJ, Socha MJ, Polo-Parada L. Segal SS. Electrical conduction along endothelial cell tubes from mouse feed arteries: confounding actions of glycyrrhetinic acid derivatives. Br J Pharmacol. 2012;166:774–787. doi: 10.1111/j.1476-5381.2011.01814.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. Unlocking the secrets of cell signaling. Annu Rev Physiol. 2005;67:1–21. doi: 10.1146/annurev.physiol.67.040103.152647. [DOI] [PubMed] [Google Scholar]
- Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- Bychkov R, Pieper K, Ried C, Milosheva M, Bychkov E, Luft FC. Haller H. Hydrogen peroxide, potassium currents, and membrane potential in human endothelial cells. Circulation. 1999;99:1719–1725. doi: 10.1161/01.cir.99.13.1719. [DOI] [PubMed] [Google Scholar]
- Camello-Almaraz C, Gomez-Pinilla PJ, Pozo MJ. Camello PJ. Mitochondrial reactive oxygen species and Ca2+ signaling. Am J Physiol Cell Physiol. 2006;291:C1082–1088. doi: 10.1152/ajpcell.00217.2006. [DOI] [PubMed] [Google Scholar]
- Campbell WB, Gebremedhin D, Pratt PF. Harder DR. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ Res. 1996;78:415–423. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- Carter WO, Narayanan PK. Robinson JP. Intracellular hydrogen peroxide and superoxide anion detection in endothelial cells. J Leukoc Biol. 1994;55:253–258. doi: 10.1002/jlb.55.2.253. [DOI] [PubMed] [Google Scholar]
- Crutchley DJ, Ryan JW, Ryan US. Fisher GH. Bradykinin-induced release of prostacyclin and thromboxanes from bovine pulmonary artery endothelial cells. Studies with lower homologs and calcium antagonists. Biochim Biophys Acta. 1983;751:99–107. doi: 10.1016/0005-2760(83)90261-8. [DOI] [PubMed] [Google Scholar]
- Dhalla NS, Temsah RM. Netticadan T. Role of oxidative stress in cardiovascular diseases. J Hypertens. 2000;18:655–673. doi: 10.1097/00004872-200018060-00002. [DOI] [PubMed] [Google Scholar]
- Dinenno FA, Jones PP, Seals DR. Tanaka H. Limb blood flow and vascular conductance are reduced with age in healthy humans: relation to elevations in sympathetic nerve activity and declines in oxygen demand. Circulation. 1999;100:164–170. doi: 10.1161/01.cir.100.2.164. [DOI] [PubMed] [Google Scholar]
- Dinenno FA, Seals DR, DeSouza CA. Tanaka H. Age-related decreases in basal limb blood flow in humans: time course, determinants and habitual exercise effects. J Physiol. 2001;531:573–579. doi: 10.1111/j.1469-7793.2001.0573i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato AJ, Eskurza I, Silver AE, Levy AS, Pierce GL, Gates PE. Seals DR. Direct evidence of endothelial oxidative stress with aging in humans: relation to impaired endothelium-dependent dilation and upregulation of nuclear factor-κB. Circ Res. 2007;100:1659–1666. doi: 10.1161/01.RES.0000269183.13937.e8. [DOI] [PubMed] [Google Scholar]
- Donato AJ, Uberoi A, Wray DW, Nishiyama S, Lawrenson L. Richardson RS. Differential effects of aging on limb blood flow in humans. Am J Physiol Heart Circ Physiol. 2006;290:H272–278. doi: 10.1152/ajpheart.00405.2005. [DOI] [PubMed] [Google Scholar]
- Drouin A, Thorin-Trescases N, Hamel E, Falck JR. Thorin E. Endothelial nitric oxide synthase activation leads to dilatory H2O2 production in mouse cerebral arteries. Cardiovasc Res. 2007;73:73–81. doi: 10.1016/j.cardiores.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Feletou M. Vanhoutte PM. Endothelial dysfunction: a multifaceted disorder (The Wiggers Award Lecture) Am J Physiol Heart Circ Physiol. 2006;291:H985–1002. doi: 10.1152/ajpheart.00292.2006. [DOI] [PubMed] [Google Scholar]
- Fleenor BS, Seals DR, Zigler ML. Sindler AL. Superoxide-lowering therapy with TEMPOL reverses arterial dysfunction with aging in mice. Aging Cell. 2012;11:269–276. doi: 10.1111/j.1474-9726.2011.00783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming I. Busse R. NO: the primary EDRF. J Mol Cell Cardiol. 1999;31:5–14. doi: 10.1006/jmcc.1998.0839. [DOI] [PubMed] [Google Scholar]
- Forstermann U. Oxidative stress in vascular disease: causes, defense mechanisms and potential therapies. Nat Clin Pract Cardiovasc Med. 2008;5:338–349. doi: 10.1038/ncpcardio1211. [DOI] [PubMed] [Google Scholar]
- Grinnell K, Duong H, Newton J, Rounds S, Choudhary G. Harrington EO. Heterogeneity in apoptotic responses of microvascular endothelial cells to oxidative stress. J Cell Physiol. 2012;227:1899–1910. doi: 10.1002/jcp.22918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecquet CM, Ahmmed GU, Vogel SM. Malik AB. Role of TRPM2 channel in mediating H2O2-induced Ca2+ entry and endothelial hyperpermeability. Circ Res. 2008;102:347–355. doi: 10.1161/CIRCRESAHA.107.160176. [DOI] [PubMed] [Google Scholar]
- Kirby BS, Voyles WF, Simpson CB, Carlson RE, Schrage WG. Dinenno FA. Endothelium-dependent vasodilatation and exercise hyperaemia in ageing humans: impact of acute ascorbic acid administration. J Physiol. 2009;587:1989–2003. doi: 10.1113/jphysiol.2008.167320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse HJ, Grunberg B, Siess W. Weber PC. Formation of biologically active autacoids is regulated by calcium influx in endothelial cells. Arterioscler Thromb. 1994;14:1821–1828. doi: 10.1161/01.atv.14.11.1821. [DOI] [PubMed] [Google Scholar]
- Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE. Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003;111:1201–1209. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laursen JB, Rajagopalan S, Galis Z, Tarpey M, Freeman BA. Harrison DG. Role of superoxide in angiotensin II-induced but not catecholamine-induced hypertension. Circulation. 1997;95:588–593. doi: 10.1161/01.cir.95.3.588. [DOI] [PubMed] [Google Scholar]
- Ledoux J, Bonev AD. Nelson MT. Ca2+-activated K+ channels in murine endothelial cells: block by intracellular calcium and magnesium. J Gen Physiol. 2008;131:125–135. doi: 10.1085/jgp.200709875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JM. Shah AM. Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1014–1030. doi: 10.1152/ajpregu.00124.2004. [DOI] [PubMed] [Google Scholar]
- Lind L, Berglund L, Larsson A. Sundstrom J. Endothelial function in resistance and conduit arteries and 5-year risk of cardiovascular disease. Circulation. 2011;123:1545–1551. doi: 10.1161/CIRCULATIONAHA.110.984047. [DOI] [PubMed] [Google Scholar]
- Liu Y, Zhao H, Li H, Kalyanaraman B, Nicolosi AC. Gutterman DD. Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circ Res. 2003;93:573–580. doi: 10.1161/01.RES.0000091261.19387.AE. [DOI] [PubMed] [Google Scholar]
- Macpherson LJ, Dubin AE, Evans MJ, Marr F, Schultz PG, Cravatt BF. Patapoutian A. Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature. 2007;445:541–545. doi: 10.1038/nature05544. [DOI] [PubMed] [Google Scholar]
- Madamanchi NR. Runge MS. Mitochondrial dysfunction in atherosclerosis. Circ Res. 2007;100:460–473. doi: 10.1161/01.RES.0000258450.44413.96. [DOI] [PubMed] [Google Scholar]
- Matoba T, Shimokawa H, Kubota H, Morikawa K, Fujiki T, Kunihiro I, Mukai Y, Hirakawa Y. Takeshita A. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in human mesenteric arteries. Biochem Biophys Res Commun. 2002;290:909–913. doi: 10.1006/bbrc.2001.6278. [DOI] [PubMed] [Google Scholar]
- Matoba T, Shimokawa H, Nakashima M, Hirakawa Y, Mukai Y, Hirano K, Kanaide H. Takeshita A. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. J Clin Invest. 2000;106:1521–1530. doi: 10.1172/JCI10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura H, Bosnjak JJ, Ning G, Saito T, Miura M. Gutterman DD. Role for hydrogen peroxide in flow-induced dilation of human coronary arterioles. Circ Res. 2003;92:e31–40. doi: 10.1161/01.res.0000054200.44505.ab. [DOI] [PubMed] [Google Scholar]
- Muller-Delp JM. Aging-induced adaptations of microvascular reactivity. Microcirculation. 2006;13:301–314. doi: 10.1080/10739680600619023. [DOI] [PubMed] [Google Scholar]
- Muller-Delp JM, Gurovich AN, Christou DD. Leeuwenburgh C. Redox balance in the aging microcirculation: new friends, new foes, and new clinical directions. Microcirculation. 2012;19:19–28. doi: 10.1111/j.1549-8719.2011.00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nystoriak MA, O'Connor KP, Sonkusare SK, Brayden JE, Nelson MT. Wellman GC. Fundamental increase in pressure-dependent constriction of brain parenchymal arterioles from subarachnoid hemorrhage model rats due to membrane depolarization. Am J Physiol Heart Circ Physiol. 2011;300:H803–812. doi: 10.1152/ajpheart.00760.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orrenius S, Zhivotovsky B. Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- Poteser M, Graziani A, Rosker C, Eder P, Derler I, Kahr H, Zhu MX, Romanin C. Groschner K. TRPC3 and TRPC4 associate to form a redox-sensitive cation channel. Evidence for expression of native TRPC3-TRPC4 heteromeric channels in endothelial cells. J Biol Chem. 2006;281:13588–13595. doi: 10.1074/jbc.M512205200. [DOI] [PubMed] [Google Scholar]
- Potocnik SJ. Hill MA. Pharmacological evidence for capacitative Ca2+ entry in cannulated and pressurized skeletal muscle arterioles. Br J Pharmacol. 2001;134:247–256. doi: 10.1038/sj.bjp.0704270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey IS, Delling M. Clapham DE. An introduction to TRP channels. Annu Rev Physiol. 2006;68:619–647. doi: 10.1146/annurev.physiol.68.040204.100431. [DOI] [PubMed] [Google Scholar]
- Schnackenberg CG, Welch WJ. Wilcox CS. Normalization of blood pressure and renal vascular resistance in SHR with a membrane-permeable superoxide dismutase mimetic: role of nitric oxide. Hypertension. 1998;32:59–64. doi: 10.1161/01.hyp.32.1.59. [DOI] [PubMed] [Google Scholar]
- Seals DR, Jablonski KL. Donato AJ. Aging and vascular endothelial function in humans. Clin Sci. 2011;120:357–375. doi: 10.1042/CS20100476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindler AL, Reyes R, Chen B, Ghosh P, Gurovich AN, Kang LS, Cardounel AJ, Delp MD. Muller-Delp JM. Age and exercise training alter signaling through reactive oxygen species in the endothelium of skeletal muscle arterioles. J Appl Physiol. 2013;114:681–693. doi: 10.1152/japplphysiol.00341.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Socha MJ, Domeier TL, Behringer EJ. Segal SS. Coordination of intercellular Ca2+ signaling in endothelial cell tubes of mouse resistance arteries. Microcirculation. 2012;19:757–770. doi: 10.1111/micc.12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Socha MJ, Hakim CH, Jackson WF. Segal SS. Temperature effects on morphological integrity and Ca2+ signaling in freshly isolated murine feed artery endothelial cell tubes. Am J Physiol Heart Circ Physiol. 2011;301:H773–783. doi: 10.1152/ajpheart.00214.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Socha MJ. Segal SS. Isolation of microvascular endothelial tubes from mouse resistance arteries. J Vis Exp. 2013;81:e50759. doi: 10.3791/50759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MN, Gonzales AL, Pires PW, Bruhl A, Leo MD, Li W, Oulidi A, Boop FA, Feng Y, Jaggar JH, Welsh DG. Earley S. Localized TRPA1 channel Ca2+ signals stimulated by reactive oxygen species promote cerebral artery dilation. Sci Signal. 2015;8:ra2. doi: 10.1126/scisignal.2005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Yau HY, Wong WY, Li RA, Huang Y. Yao X. Role of TRPM2 in H2O2-induced cell apoptosis in endothelial cells. PLoS One. 2012;7:e43186. doi: 10.1371/journal.pone.0043186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suvorava T. Kojda G. Reactive oxygen species as cardiovascular mediators: lessons from endothelial-specific protein overexpression mouse models. Biochim Biophys Acta. 2009;1787:802–810. doi: 10.1016/j.bbabio.2009.04.005. [DOI] [PubMed] [Google Scholar]
- Taddei S, Virdis A, Ghiadoni L, Salvetti G, Bernini G, Magagna A. Salvetti A. Age-related reduction of NO availability and oxidative stress in humans. Hypertension. 2001;38:274–279. doi: 10.1161/01.hyp.38.2.274. [DOI] [PubMed] [Google Scholar]
- Tas PW, Stossel C. Roewer N. Inhibition of the histamine-induced Ca2+ influx in primary human endothelial cells (HUVEC) by volatile anaesthetics. Eur J Anaesthesiol. 2008;25:976–985. doi: 10.1017/S0265021508004778. [DOI] [PubMed] [Google Scholar]
- Thomas SR, Witting PK. Drummond GR. Redox control of endothelial function and dysfunction: molecular mechanisms and therapeutic opportunities. Antioxid Redox Signal. 2008;10:1713–1765. doi: 10.1089/ars.2008.2027. [DOI] [PubMed] [Google Scholar]
- Touyz RM, Briones AM, Sedeek M, Burger D. Montezano AC. NOX isoforms and reactive oxygen species in vascular health. Mol Interv. 2011;11:27–35. doi: 10.1124/mi.11.1.5. [DOI] [PubMed] [Google Scholar]
- Tran QK. Watanabe H. Calcium signalling in the endothelium. Handb Exp Pharmacol. 2006;176:145–187. doi: 10.1007/3-540-32967-6_5. [DOI] [PubMed] [Google Scholar]
- Trott DW, Gunduz F, Laughlin MH. Woodman CR. Exercise training reverses age-related decrements in endothelium-dependent dilation in skeletal muscle feed arteries. J Appl Physiol. 2009;106:1925–1934. doi: 10.1152/japplphysiol.91232.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungvari Z, Wolin MS. Csiszar A. Mechanosensitive production of reactive oxygen species in endothelial and smooth muscle cells: role in microvascular remodeling? Antioxid Redox Signal. 2006;8:1121–1129. doi: 10.1089/ars.2006.8.1121. [DOI] [PubMed] [Google Scholar]
- van der Loo B, Labugger R, Skepper JN, Bachschmid M, Kilo J, Powell JM, Palacios-Callender M, Erusalimsky JD, Quaschning T, Malinski T, Gygi D, Ullrich V. Luscher TF. Enhanced peroxynitrite formation is associated with vascular aging. J Exp Med. 2000;192:1731–1744. doi: 10.1084/jem.192.12.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk T, Hensel M. Kox WJ. Transient Ca2+ changes in endothelial cells induced by low doses of reactive oxygen species: role of hydrogen peroxide. Mol Cell Biochem. 1997;171:11–21. doi: 10.1023/a:1006886215193. [DOI] [PubMed] [Google Scholar]
- Widlansky ME. Gutterman DD. Regulation of endothelial function by mitochondrial reactive oxygen species. Antioxidants Redox Signal. 2011;15:1517–1530. doi: 10.1089/ars.2010.3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolin MS. Reactive oxygen species and the control of vascular function. Am J Physiol Heart Circ Physiol. 2009;296:H539–549. doi: 10.1152/ajpheart.01167.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu SZ, Sukumar P, Zeng F, Li J, Jairaman A, English A, Naylor J, Ciurtin C, Majeed Y, Milligan CJ, Bahnasi YM, Al-Shawaf E, Porter KE, Jiang LH, Emery P, Sivaprasadarao A. Beech DJ. TRPC channel activation by extracellular thioredoxin. Nature. 2008;451:69–72. doi: 10.1038/nature06414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeboah J, Crouse JR, Hsu FC, Burke GL. Herrington DM. Brachial flow-mediated dilation predicts incident cardiovascular events in older adults: the Cardiovascular Health Study. Circulation. 2007;115:2390–2397. doi: 10.1161/CIRCULATIONAHA.106.678276. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Inoue R, Morii T, Takahashi N, Yamamoto S, Hara Y, Tominaga M, Shimizu S, Sato Y. Mori Y. Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat Chem Biol. 2006;2:596–607. doi: 10.1038/nchembio821. [DOI] [PubMed] [Google Scholar]
- Yoshihara D, Fujiwara N. Suzuki K. Antioxidants: benefits and risks for long-term health. Maturitas. 2010;67:103–107. doi: 10.1016/j.maturitas.2010.05.001. [DOI] [PubMed] [Google Scholar]