

Abstract
Tenascin-X is the largest member of the tenascin (TN) family of evolutionary conserved extracellular matrix glycoproteins, which also comprises TN-C, TN-R and TN-W. Among this family, TN-X is the only member described so far to exert a crucial architectural function as evidenced by a connective tissue disorder (a recessive form of Ehlers-Danlos syndrome) resulting from a loss-of-function of this glycoprotein in humans and mice. However, TN-X is more than an architectural protein, as it displays features of a matricellular protein by modulating cell adhesion. However, the cellular functions associated with the anti-adhesive properties of TN-X have not yet been revealed. Recent findings indicate that TN-X is also an extracellular regulator of signaling pathways. Indeed, TN-X has been shown to regulate the bioavailability of the Transforming Growth Factor (TGF)-β and to modulate epithelial cell plasticity. The next challenges will be to unravel whether the signaling functions of TN-X are functionally linked to its matricellular properties.
Keywords: cell signaling, Ehlers-Danlos syndrome (EDS), epithelial-to-mesenchymal transition (EMT), integrin α11β1, matricellular protein, tenascin-X, TGF-β activation
Abbreviations
- ECM
extracellular matrix
- EDS
Ehlers-Danlos syndrome
- EGF
epidermal growth factor
- EMT
epithelial-to-mesenchymal transition
- FAK
focal adhesion kinase
- FBG
fibrinogen-like domain
- FNIII
fibronectin type III module
- LAP
latency associated peptide
- MMP
matrix metalloproteinase
- SLC
small latent complex
- TGF-β
transforming growth factor-β
- TN
tenascin
- TSP-1
thrombospondin-1
- VEGF
vascular endothelial growth factor
Introduction
The Tenascin-X-encoding gene was serendipitously cloned as an unknown (“X”) gene located within the class III region of the HLA locus on human chromosome 6p21, whose 3′ part overlapped with the steroid 21-hydroxylase-encoding (CYP21B) gene.1 While searching for a CYP21B mutant responsible for congenital adrenal hyperplasia (CAH), a life-threatening disease, Morel et al.1 cloned a novel transcript overlapping the 3′ untranslated region of CYP21B mRNA, but transcribed from the complementary DNA strand.1 Intensive chromosome walking experiments and sequencing efforts of the “X” gene led to the prediction of a 450 kDa glycoprotein consisting of 5 distinct domains: a signal peptide, a hydrophobic domain consisting in 4 heptad repeats, a series of 18.5 repeats of epidermal growth factor (EGF)-like motif, a high number of fibronectin type III (FNIII) module, and a fibrinogen (FBG)-like globular domain at its C-terminus2-4 (Fig. 1A). The predicted protein exhibited a similar modular structure to the prototypic Tenascin-C (TN-C) glycoprotein (formerly called Tenascin/Hexabrachion/Cytotactin)5-7 and to the Tenascin-R (TN-R) protein (Restrictin/J1-160/180/Janusin),8-10 the latter being discovered at the same time as the “X” protein. This newly identified protein was thus further termed Tenascin-X (TN-X) and the corresponding gene TNXB4. TN-X is the largest member of the growing Tenascin family, while the TN-R, together with the latest identified member, Tenascin-W11 (TN-W; also called Tenascin-N12), are the smallest ones.13 Indeed, human TN-W monomer has an apparent molecular weight of 160 kDa,14 whereas human TN-R appears in 2 major protein forms of 160 and 180 kDa,15 and human TN-C has monomers ranging from 190 to 300 kDa.16 In parallel to the TNXB gene cloning and characterization, TN-X was also independently identified as a flexible glycoprotein associated with collagen fibrils, and was initially termed flexilin.17
Figure 1.

TN-X is a tribrachion. (A) Schematic representation of human, mouse and bovine TN-X monomers. Each molecule is composed of the N-terminal oligomerization domain (tenascin assembly domain), followed by 18.5 epidermal growth factor (EGF)-like repeats, between 30 to 32 fibronectin type III (FNIII) repeats and with a fibrinogen (FBG)-like domain at the C-terminus. The FNIII repeats are potentially interrupted by a serine/proline-rich (SPX) region that was identified in the TN-Y molecule, the avian TN-X ortholog.24 For each TN-X ortholog, FNIII repeats (Hu1-32, M1-31 and b0-29) are numbered according to the nomenclature used in the original publications.4,22,23 The alternatively spliced FNIII repeats found in the mouse TN-X are in yellow (M3, and M15-M22).23 Cell-surface receptors of the bovine TN-X are depicted under the corresponding monomer. The heparin-binding site is underlined. Note that the human and bovine TN-X glycoproteins, but not the mouse ortholog, have a RGD sequence in a conserved FNIII module (Hu11 and b10, respectively). (B) Electron Micrographs of rotary shadowed purified recombinant bovine TN-X molecules (upper panels) and their respective putative schematic representation (lower panels). Bars, 50 nm.
In this review, we present all the major findings since the initial identification of TN-X. More precisely, we summarize the structural specificities as well as the histological and ultrastructural distributions of TN-X. We emphasize the architectural role of this glycoprotein that was revealed by an inherited and engineered loss-of-function, respectively, in humans and mice. Finally, we discuss the putative matricellular role of TN-X and highlight the recent findings showing that TN-X is also an extracellular regulator of signaling pathways.
TN-X is a Trimeric Protein with a Peculiar Proline-Rich Motif
The hydrophobic residue-rich heptad repeats located at the N-terminus of this glycoprotein are capable of forming α helices that serve as a structural basis for the trimerization of TN-X monomers. This region is flanked by 7 cysteine residues that are predicted to stabilize the trimer by forming disulfide bonds. However, TN-X lacks the amino-terminal cysteine found in TN-C and TN-W which allows 2 trimers to assemble into a hexamer structure.18,19 Transmission electron microscopy observations of rotary shadowed replicas of recombinant bovine TN-X (Fig. 1B) confirmed that this glycoprotein forms a trimer,20 as does TN-R.21 Note that the flexible appearance of the FNIII repeats endowed this glycoprotein with the name ‘flexilin’17 (Fig. 1B).
The number of FNIII repeats encoded by TNXB genes varies among mammalian species, with 30, 31 and 32 repeats in bovine,22 mouse23 and human,13 respectively (Fig. 1A). Moreover, as for other TNs, several alternatively spliced TN-X mRNA isoforms have been described in mouse, in which some exons encoding FNIIII repeats were absent23 (Fig. 1A). Whether this variability leads to the exposure of distinct functional domains in different tissues is still an open question for TN-X. Alternative splicing events might also explain why a putative exon located between the sequences coding for the second and third FNIII repeats of mammalian TN-X have never been detected by cDNA analysis. The fact that (i) these sequences encode a fragment evocative of the SPX (serine/proline-rich) and Y1/2 regions of the chicken TN-Y, the avian ortholog of mammalian TN-X,13,24 and (ii) that they are in the right open reading frame and do not include stop codons, highly suggests that this exon is probably present in some TN-X mRNA isoforms. This putative TN-X SPX region (Fig. 1A), of about 240 amino acids, is composed of a proline-rich sequence followed by a FNIII-like domain. This SPX region has not been identified in other TNs and may exhibit a peculiar intrinsic function.
TN-X Tissue Distribution is Frequently Reciprocal to that of TN-C during Development, Tissue Homeostasis and Physio-Pathological Conditions
TN-X is ubiquitously expressed during late stages of embryonic development, but not in early embryonic events, indicating that this glycoprotein is mainly involved in organogenesis.4,25 TN-X mRNA is particularly highly detected in developing heart, skeletal muscles and limbs, in a spatially and temporally controlled manner, suggesting that this matrix protein not only supports cell migration, but may also have a role in cell fate determination or cell differentiation.25 In adult, TN-X mRNA is also observed at different levels in a variety of organs and tissues, such as lung, kidney, mammary and adrenal glands, blood vessels, testis, ovaries, with a higher level in the digestive tract (pancreas, stomach, jejunum, ileum, and colon), and the highest expression in the heart, the skin, skeletal muscles, ligaments and tendons.4,26,27 TN-X mRNA is also present in peripheral nerves in adult pigs.27 A closer examination of TN-X protein distribution indicated that this glycoprotein is constitutively localized in connective tissues, such as the peritendineum, epimysium, perimysium, skin dermis, muscularis mucosae, kidney glomeruli and around blood vessels22,26 (Fig. 2A). Positive staining for TN-X is also detectable in peripheral nerves (Fig. 2A).
Figure 2.
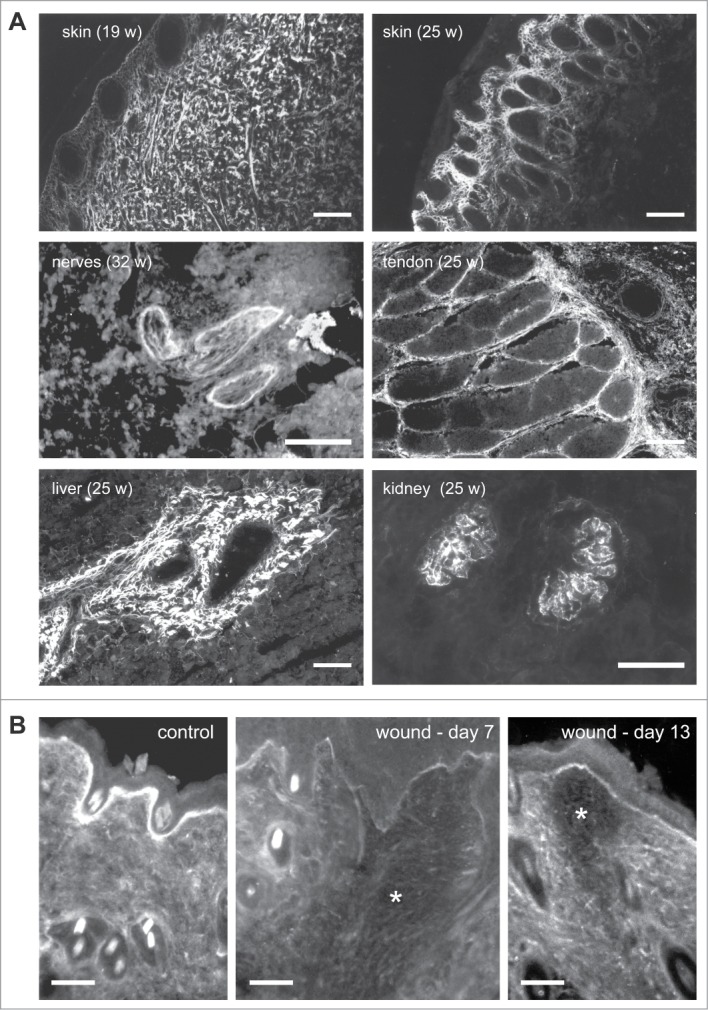
Tissue distribution of TN-X (A) in fetal bovine tissues and (B) during wound healing in mice. (A) Indirect immunofluorescence of TN-X performed on cryostat sections of fetal bovine tissues (at the indicated weeks of gestation) using a monoclonal antibody (8F2) recognizing the FNIII b10 domain of the bovine glycoprotein. (B) Indirect immunofluorescence of TN-X performed on cryostat sections of mouse skin after incisional wound (*). TN-X was detected using polyclonal antibodies directed against the bovine TN-X FNIII b9-b10 repeats. Note that TN-X is hardly detected in the wound (*) even after the completion of the re-epithelialization process (day 7). TN-X staining is re-observed in the deep layers of the wound 13 days after incision. Bars, 50 μm.
Interestingly, both in embryonic and adult tissues, TN-X displays a pattern of expression that is mostly opposed to that of TN-C. For instance, TN-C mRNA is highly detected in spinal cord, spleen and cerebrum, whereas TN-X mRNA is not present or weakly detected in these tissues.26,27 Moreover, when both TN-C and TN-X proteins were detected in the same tissues, such as the digestive tract (esophagus, gut, stomach) and skin dermis, the 2 proteins displayed mostly separated, although adjacent, staining patterns.26 TN-W has a restricted expression pattern in adult tissues (kidney tubules, aortic valve, corneal limbus and periosteum).11 Although TN-W localization has never been compared in detail with that of TN-X, these 2 proteins appear to display mostly distinct expression patterns in adults. Finally, TN-R is strictly restricted to the central nervous system,10,28 where TN-X is virtually absent.
As illustrated below, the opposite localization of both TN-X and TN-C proteins is also observed during physiological processes, such as wound healing. In normal skin, TN-X is widely distributed within the dermis and is also strongly associated with the basement membrane (Fig. 2B). In wounds, TN-X is not detected or only weakly present in the connective tissue, as well as in basement membranes, in the early stages of healing (Fig. 2B). TN-X then becomes re-expressed later during healing, at stages coinciding with matrix assembly and maturation29 (Fig. 2B). Conversely, TN-C is sparsely detected in intact skin dermis, but its expression is induced at the edge of wound, underneath the epidermal-dermal junction accompanying migrating keratinocytes, as well as in the granulation tissue.30 These observations suggest that TN-X might mainly be involved in skin tissue homeostasis, most likely by restricting keratinocyte or fibroblast cell proliferation and migration, whereas TN-C plays the opposite role, by promoting cell division and motility.
In pathological conditions, such as tumorigenesis, TN-X expression is also frequently opposed to that of TN-C. In a swine model of malignant melanoma formation and progression, the level of TN-X protein has been found to be dramatically decreased in the tumor stroma compared to the normal dermis. In contrast, TN-C protein secretion was induced by both tumor cells and surrounding stromal cells.31 Similarly, another study revealed that TN-X, which was expressed in mouse glioma and renal carcinoma cells in vitro, was highly down-regulated in vivo after transplantation of these cells into immune-compromised mice. Indeed, TN-X expression was decreased both in tumor cells and host-mouse stroma in favor of TN-C, whose expression was induced by both tumor and stromal cells.32 Finally, TN-X expression has also been found to be up-regulated in low-grade astrocytomas compared to normal brain. However, the positive TN-X staining decreased significantly with the degree of histological malignancy of human astrocytomas. In contrast, TN-C staining positively correlated with the grade of astrocytomas, being highly expressed in high-grade astrocytomas and glioblastomas.33 These observations further indicate a distinct, possibly antagonistic, role of TN-X and TN-C during pathological disorders. Considering that TN-C has mainly pro-oncogenic actions in tumorigenesis,34 their reciprocal distribution might suggest that TN-X most likely exerts tumor suppressive activities.
An Architectural Function Illustrated by the Ehlers-Danlos Syndrome Associated with TN-X Deficiency
TN-X has an important function in extracellular matrix (ECM) architecture and tissue integrity, as supported by several in vivo and in vitro observations. TN-X was initially biochemically co-purified with types XII and XIV collagens17 and has been further shown to localize between or at the surface of collagen fibrils in tendon, skin dermis and the mesangium of kidney glomeruli.17,35 These in vivo co-localizations are accompanied by multiple interactions between TN-X and several components of the extracellular matrix in vitro, such as types I, III and V fibrillar collagens, types XII and XIV fibril-associated collagens,20,36-38 and the small proteoglycan decorin.39 The regions with which TN-X interacts with fibrillar collagens are not clearly identified as discrepancies have been obtained between independent groups, some arguing that the FNIII region is the main interaction site,37,38 others showing that both the fibrinogen globe and EGF-like repeats are indispensable.20 In contrast to TN-C,40 TN-X does not bind to fibronectin,20,38 further indicating that these 2 TNs exert distinct functions in vivo.
This architectural function is also compatible with an alteration of biomechanical properties of connective tissues observed in TN-X deficient patients suffering from Ehlers-Danlos syndrome (EDS). EDS constitutes a group of diseases characterized by a fragility of soft connective tissues and associated with variable clinical manifestations affecting primarily the skin, ligaments, blood vessels and internal organs.41 As might be expected from TN-X distribution, complete deficiency of this glycoprotein causes an autosomal recessive form of EDS.42,43 Indeed, patients present with joint hypermobility, skin hyperextensibility and easy bruising,43 but they also suffer from generalized muscle weakness and distal contractures.44 This recessive form is phenotypically distinct from classic EDS, as atrophic scarring is not observed and patients do not have poor wound healing.43,45 Truncating mutations and large deletions in both alleles of the TNXB gene are responsible for the complete TN-X deficiency.42,43 Interestingly, TN-X haploinsufficiency leads to a hypermobility form of EDS.46 However, the exact role played by TN-X in these 2 types of EDS is not clearly understood.
TN-X-deficient mice, generated by 2 independent groups,47,48 also display several features mimicking a recessive form of EDS, such as hyperextensible skin47 and muscle weakness.49 Interestingly, the genetic background markedly influences EDS manifestations, as the phenotype was less severe on a homogenous (C57BL/6) compared to a mixed (129/SvJ, C57BL/6 and FVB) genetic background.29,47,49 One possible explanation is that the qualitative and/or quantitative composition of ECM in different tissues may be variable according to mouse strains.
A closer examination of TN-X-deficient mice or patients has suggested several putative architectural functions for this glycoprotein. Firstly, an in-depth analysis of TN-X-deficient mice indicated a role for TN-X as a regulator of collagen expression. Indeed, fibroblasts from TN-X-/- mice displayed a decreased expression of types I and VI collagens, as well as of several collagen fibril-associated molecules (types XII and XIV collagens, lumican and fibromodulin).50,51 However, whether the down-regulation of those matrix protein-encoding genes reflects an adaptive process due to TN-X-deficiency or an active regulatory role exerted by TN-X remains an open question. While the overexpression of full-length TN-X led to an increase of type VI collagen expression in immortalized BALB/3T3 fibroblasts, suggesting that the regulation of this gene by TN-X is a direct process,50 some discrepancies have been observed between mouse and human tissues deficient in TN-X in terms of matrix gene regulation, instead implying an adaptive process. This fact has been illustrated for the type XII collagen whose level is markedly reduced in TN-X-/- mice muscle samples (compared to wild-type mice), but remains unchanged in human biopsies deficient in TN-X (compared to healthy donors).51 Secondly, the analysis of TN-X-deficient mice indicated a role for TN-X as a regulator of collagen deposition into the ECM. Indeed, dermal fibroblasts from TN-X-/- mice deposited fewer type I collagen molecules into the ECM compared to wild-type fibroblasts in vitro.47 This hypothesis is also consistent with the observations showing that dermal tissues from TN-X-deficient mice and patients have a lower amount of type I collagen compared to wild-type littermates or healthy patients.47,52 While several authors have suggested a role for TN-X during type I collagen fibrillogenesis,37,38 showing that TN-X accelerates the rate of fibrillogenesis in vitro, our laboratory demonstrated that recombinant full-length TN-X did not affect purified collagen fibrillogenesis in a cell-free system.53 However, it cannot be ruled out that TN-X might also participate in a cell-mediated fibrillogenesis process.
An ultrastructural analysis of skin from TN-X-deficient patients and mice revealed that the morphology of collagen fibrils was regular,47,54 in contrast to the irregular shapes of collagen fibrils observed in the classic and hypermobility forms of EDS.55 The most striking feature was an increased distance between fibrils.35,47 From those observations and the in vitro interaction studies emerged a model in which the architectural function of TN-X is proposed: TN-X regulates the collagenous network by bridging collagen fibrils through direct interaction with fibrillar collagens and/or, indirectly, via with the cooperation of ECM components such as types XII and XIV collagens and/or decorin35,56 (Fig. 3). Consistent with this model, our laboratory demonstrated that recombinant full-length TN-X increased the stiffness of 3-dimensional collagen gels in vitro.53 In this cell-free system, we hypothesized that TN-X increased collagen gel stiffness by re-enforcing molecular interactions between fibrils. Although the stiffness has never been assessed in biopsies from healthy or TN-X-deficient patients or mice, TN-X absence or haploinsufficiency markedly alters the biomechanical properties of dermis in mice.47 This observation could also be related to the fact that TN-X owns intrinsic elastic properties, due to the remarkable ability of its constitutive FNIII modules to unfold under mechanical stretching and to refold after physical constraint removal.57
Figure 3.
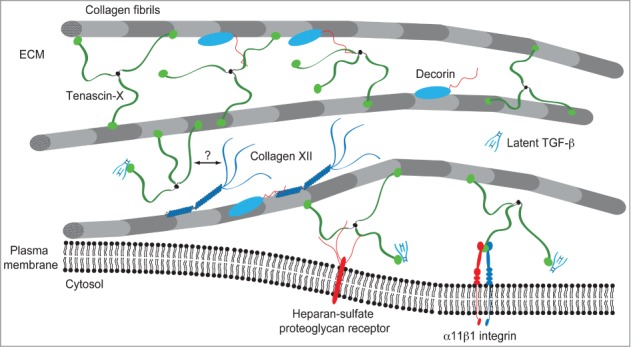
Model of TN-X integration within the collagenous network. TN-X regulates the spacing and cohesiveness between collagen fibrils through direct interaction with collagen molecules and/or through molecular associations with several ECM components interacting with collagen fibrils. Type XII collagen interacts with TN-X through its non-collagenous N-terminal NC3 domain.36 The binding site of this NC3 domain has not been identified in the full-length TN-X. The dermatan sulfate chains of decorin ensure the binding to the heparin-binding site included within the FNIII b10 and b11 domains of bovine TN-X.39 TN-X might also anchor collagen fibrils at the vicinity of cell surface through its interaction with (not yet identified) heparin-sulfate proteoglycan receptor(s) and/or the α11β1 integrin.
Alterations in collagen expression, deposition and inter-fibril spacing may not account for all the observed biomechanical defects due to TN-X deficiency. Indeed, histological examination of skin dermis from TN-X-deficient patients displayed a marked fragmentation of elastic fibers, and abnormal elastic fibers were also observed at the ultrastructural level. Notably, their dermis shows elastic fibers that are mostly immature, devoid of microfibrils and irregularly shaped.52 TN-X has also been shown to localize to elastic fibers in vivo58 and to interact with tropoelastin in vitro through its C-terminal region including the last FNIII domains and the fibrinogen globe.37 It is also conceivable that TN-X might be involved in elastic fibers formation and maturation.29,58
In summary, TN-X appears to be a general regulator of the biomechanical properties of connective tissues due to its intrinsic elastic properties and to its ability to regulate, directly or indirectly, the expression of matrix molecule and/or their correct assembly into supramolecular structures, such as collagen fibrils and elastic fibers.
TN-X is a Matricellular Protein with as yet Unidentified Cell Functions
‘Matricellular’ proteins have been defined as a group of matrix extracellular proteins that do not directly contribute to the formation of structural elements but serve to modulate cell function, through the modulation of cell–matrix interactions.59 This definition implies that matricellular proteins interact with other matrix proteins as well as with cell-surface receptors. TN-R, TN-C and TN-W glycoproteins have been shown to belong to this group of extracellular modulators of cell functions as they exhibit anti-adhesive properties and promote the detachment of cells from other ECM molecules.8,40,60 Is TN-X also a matricellular protein? TN-X shares similar properties with the other TN molecules as it causes in vitro cell rounding22,61 and detachment.62 Anti-adhesive properties of TN-X are characterized by a decreased formation of focal adhesions and are correlated with an impaired phosphorylation of the Focal Adhesion Kinase (FAK) on tyrosine residue at position 397.62 However, the mechanisms by which TN-X modulates cell adhesion are currently not known and require further investigation. Some matricellular proteins are able to interact directly with cell-surface receptors or other membrane-associated signaling molecules, thereby regulating intracellular pathways leading to alteration of cell adhesion. For instance, the amino-terminal heparin-binding (HepI) domain of the matricellular thrombospondin-1 (TSP-1) protein inhibits cell adhesion by interacting with cell-surface calreticulin, resulting in the disassembly of focal adhesions.63 Alternatively, matricellular proteins have been shown to act indirectly, by binding to other ECM molecules and competing with their binding sites for cell-surface receptors. TN-C has been shown to disrupt the adhesion of cells to fibronectin by interacting with the HepII site of fibronectin and preventing it from binding to the signaling cell-surface receptor, syndecan 4.64 It is important to assess whether TN-X has an intrinsic anti-adhesive property by interacting with inhibitory signaling cell-surface receptors, or through the competitive binding to other ECM molecules for cell-surface receptors. Bovine TN-X has been shown to interact with cells using several adhesion sites (Figs. 1A and 3): (i) a conformational heparin-binding site comprising 2 FNIII modules (FNIII b10 and b11 domains) being a natural ligand for heparan-sulfate proteoglycan receptors65; (ii) a cryptic RGD-containing cell adhesion site located between the contiguous FNIII b9 and b10 domains being a ligand for the αvβ3 integrin61; and (iii) the C-terminal FBG-like domain, which is the major cell adhesion site of the whole molecule and involves the α11β1 integrin receptor.61,66 The next challenge will be to determine the involvement of those receptors, and their corresponding binding sites, in the anti-adhesive properties of TN-X.
What are the cell functions regulated by the TN-X-mediated modulation of cell adhesion? To our knowledge, no cell function has been reported to be directly associated with TN-X. Matsumoto et al. (2001) demonstrated that tumor invasion and metastasis of grafted melanoma cells was promoted in host TN-X-/- mice compared to receiving wild-type mice.48 Those observations are in agreement with our hypothesis that TN-X might act as a tumor suppressor protein (related to its decreased expression during tumor progression). Compared to wild-type mice, the level of matrix metalloproteinases (MMP)-2 and MMP-9 has been shown to be increased in TN-X-deficient mice in vivo and this up-regulation is believed to facilitate melanoma cell invasion and dissemination.48 Although Mmp2 gene expression is also increased in fibroblasts isolated from TN-X-/- mice and overexpression of TN-X negatively regulates the level of MMP-2 in a fibroblast cell line,67 it is not clear whether the transcriptional regulation of the MMP-2-encoding gene relies on cell interaction with TN-X, and hence, whether the anti-tumor activity of TN-X relies on the matricellular activity of the glycoprotein. It is also tempting to speculate that the tumor-suppressive activity of TN-X may be linked to the architectural function of this glycoprotein. By modifying the global composition and/or organization of the ECM, TN-X deficiency might promote cell invasiveness and motility.
Independently of its cell adhesion-modulating properties, TN-X has an indirect involvement in cell signaling, enabling it to induce various responses. For instance, TN-X has been shown to interact with vascular endothelial growth factor B (VEGF-B) and to enhance cell endothelial proliferation in combination with this growth factor.68 The interacting site has been mapped to the FNIII M13-M25 repeats of the mouse TN-X. VEGF-B could simultaneously bind to both TN-X and VEGF receptor-1 (VEGF-R1) and this interaction would facilitate VEGFR-1 enzymatic activity.68 Even though the mechanism by which TN-X enhances the ability of VEGF-B to trigger VEGF-R1 signaling activity has not been elucidated, one can speculate that the interaction with TN-X modifies the conformation of VEGF-B and efficiently increases its ability to bind to VEGF-R1.
TN-X regulates the bioavailability of Transforming Growth Factor-β (TGF-β)
TGF-β is a pleiotropic cytokine having a fundamental role in embryonic development (histogenesis and organogenesis) and tissue homeostasis (wound healing, ECM synthesis and remodeling).69 TGF-β is also implicated in several pathological processes, such as fibrosis and tumor progression.69,70 TGF-β is a dimeric polypeptide secreted as a latent complex in which the mature entity remains non-covalently bound to its pro-domain called Latency-Associated Peptide (LAP) (Fig. 4). In some physiological or pathological conditions, mature (also called bioactive) TGF-β has to be liberated from the latent complex, a process called latent TGF-β activation. This activation process is a crucial step in the initiation of a TGF-β signaling and has to be tightly controlled to ensure proper signaling whenever required by cells. Despite the crucial importance of this regulation, very few mechanisms have been described for the conversion of latent TGF-β into its biologically active form.71 Several proteases have demonstrated to be involved in the proteolytic cleavage of the LAP pro-domain enabling the liberation of the TGF-β.72-77 A conformational change of the LAP·TGF-β complex has also been proposed to allow the exposure of the TGF-β entity to cell surface receptors. Different cell surface receptors, such as RGD-dependent integrins,78 mannose 6-phosphate receptor79 and neuropilin-180 have been shown to facilitate latent TGF-β activation at the vicinity of the plasma membrane by inducing a conformational change of the LAP·TGF-β complex. Similarly, the matricellular TSP-1 protein has been shown to bind to, and to activate, the latent TGF-β complex through a conformational unmasking.81,82 Once activated, mature TGF-β initiates a signal via 2 transmembrane serine/threonine-kinase receptors, the types I and II TGF-β receptors (TβRI and TβRII, respectively). Upon interaction with the ligand, the TGF-β receptor complex then phosphorylates and activates the “canonical” Smad pathway or alternative signaling pathways (MAPK, RhoA, PI3K/AKT) in order to regulate different sets of TGF-β-responsive genes.83
Figure 4.

Model of TGF-β activation by the FBG-like domain of TN-X. The small latent TGF-β complex (SLC) interacts with the FBG-like domain of TN-X. In this full-length glycoprotein, the LAP·TGF-β thus constitutes a reservoir of signaling molecule within the ECM. During physiological or pathological events of ECM remodeling, we hypothesized that some proteases would cleave the FBG-like domain of TN-X from the full-length molecule. At the vicinity of cell membrane, the FBG-like domain interacts with the α11β1 integrin receptor, allowing a conformational change of the latent complex, facilitating the presentation of mature TGF-β to TβRII and TβRI receptors. Latent TGF-β activation by the FBG-like domain results in the induction of Smad signaling and subsequent TGF-β response, such as EMT.
We recently demonstrated that full-length TN-X interacted with the latent LAP·TGF-β complex in vitro and in vivo.66 This interaction resides in the C-terminal FBG-like domain of TN-X. More importantly, we found that the FBG-like domain of TN-X was able to activate the latent TGF-β in different cell types in order to induce a TGF-β/Smad intracellular signal.66 As the FBG-mediated activation of latent TGF-β is direct (no protein synthesis required) and does not involve any proteolytic enzymatic activity, we hypothesized that the FBG-like domain could induce a conformational change of the latent complex to unmask the mature entity. We also confirmed that this activation process was not mediated by the RGD-dependent integrins and the matricellular TSP-1 protein, suggesting a novel mechanism of latent TGF-β regulation. Interestingly, cell adhesion to the FBG-like domain was required for the latent TGF-β activation. Indeed, no TGF-β activation was seen in a TGF-β-responsive cell line that is unable to adhere to or interact with the FBG-like domain of TN-X. We identified that the TN-X/FBG cell-surface receptor, i.e. the α11β1 integrin, was essential for latent TGF-β activation. Indeed, the silencing of the α11 integrin subunit fully abrogated the activation process mediated by the FBG-like domain. Conversely, the ectopic expression of α11β1 integrin in a cell line unable to adhere to the FBG-like domain restored its ability to activate latent TGF-β mediated by the C-terminal domain of TN-X.66
We thus defined a new mechanism of latent TGF-β activation that is induced by a matricellular protein other than TSP-1. In our model (Fig. 4), we speculate that under physiological (wound healing) or pathological (fibrosis or tumor progression) conditions, in which the ECM is ultimately remodeled, some proteases would cleave the full-length TN-X molecule to release a C-terminal fragment containing at least the FBG-like domain. In this fragment, the FBG-like domain might sequester the latent TGF-β complex or might interact with free cell-secreted latent TGF-β upon cleavage. When the FBG·LAP·TGF-β complex is recruited to the vicinity of the cell surface and binds to the α11β1 integrin, the FBG-like domain would induce a conformational change of the latent complex, unmasking the mature TGF-β entity and exposing it to the cell-surface receptors. The exact role played by α11β1 integrin in latent TGF-β activation is not clear. This cell-surface receptor might serve as a docking site for the FBG·LAP·TGF-β complex at the membrane, or might have a more active role in the activation process by “helping” the FBG-like domain to trigger the conformational change of the latent complex. The next step will be to elucidate the precise molecular mechanisms by which the FBG-like domain and the α11β1 integrin complex activate the latent TGF-β. It will be of particular importance to identify (i) the specific sequences mediating the interaction between the FBG-like domain and the LAP·TGF-β complex and (ii) and the ‘active’ sites which bind the FBG-like domain to the α11β1 integrin. More importantly, the exact role played by the α11β1 integrin in the activation process remains to be deciphered.
TN-X, a novel regulator of epithelial cell plasticity?
Activated TGF-β is a potent inducer of the epithelial-to-mesenchymal transition (EMT),84 a fundamental cell process governing the conversion of polarized epithelial cells into motile mesenchymal ones.85 Although the full-length TN-X and the FBG-like domain were able to activate latent TGF-β and to induce subsequent Smad intracellular signaling with similar efficiencies, full-length TN-X caused a mild EMT response in contrast to the FBG-like domain that induced a full process.66 Indeed, the full-length TN-X induced a partial delocalization of E-cadherin and actin from adherens junctions (Fig. 5A). Moreover, TN-X downregulated the expression of selected epithelial marker genes, but failed to trigger the expression of mesenchymal genes. The partial EMT observed in the presence of TN-X is peculiar as, to our knowledge, there are no example of such molecules triggering a decrease of epithelial markers without induction of mesenchymal features. For instance, TGF-β efficiently regulates both sets of genes in an opposite manner.84 It would be interesting to determine the molecular mechanisms preventing the expression of mesenchymal genes, while allowing the repression of genes encoding the epithelial markers. We identified that the FNIII-containing region of TN-X was able to antagonize the EMT response induced by the FBG-like domain, indicating that the FNIII repeats and the FBG domain regulate cell plasticity through distinct signaling pathways in the intact molecule66 (Fig. 5B). The specific pathway triggered by the central region of the glycoprotein remains to be determined.
Figure 5.

The FNIII region of TN-X antagonizes the induction of EMT by the FBG-like domain. (A) Actin direct fluorescence in normal murine mammary epithelial (NMuMG) cells seeded onto non-coated dishes or dishes containing equimolar quantities of immobilized recombinant full-length TN-X, FBG-like domain, FNIII repeats (TN-XΔEΔF) or both FNIII repeats and FBG-like domain. Note that compared to the uncoated condition, where almost all cells exhibited a cortical actin staining, the full-length TN-X induced a mild EMT, visualized by a partial delocalization of actin cytoskeleton from cell junctions (*). In contrast, the FBG-like domain caused a full EMT, as illustrated by the acquisition of elongated cell morphology and the organization of actin cytoskeleton into stress fibers. Recombinant human TGF-β1, used here as a positive control, gave similar results to those obtained with the FBG-like domain. However, the FNIII region of TN-X fully inhibited the EMT triggered by the FBG-like domain. Bars, 15 μm. (B) Model representing the regulation of epithelial cell plasticity by TN-X. When separated from the intact TN-X molecule, the FBG-like domain is able to induce a robust EMT response, through its ability to activate latent TGF-β. This response relies on the presence of TβRII/I receptors and α11β1 integrin at the cell surface. In the intact TN-X molecule, the FNIII region antagonizes the EMT induced by the FBG-like domain, most likely via distinct intracellular cues that are initiated by the interaction of certain FNIII repeats to a yet unidentified receptor.
These findings suggest that TN-X is a novel regulator of epithelial cell plasticity, depending on the region that is exposed to the cells. It is tempting to speculate that the FBG-like domain induces an EMT process, through latent TGF-β activation, whereas the FNIII repeats maintain the epithelial cells phenotype or even promote the conversion of mesenchymal-like cells into epithelial ones, a process called mesenchymal-to-epithelial transition.85 If this hypothesis is true, this implies that both regions, FNIII repeats and the FBG-like domain, have to be separated from each other to exert their distinct cell responses.
Conclusion and Perspectives
TN-X is widely expressed in the organism and serves as an architectural support for structural matrix macromolecular complexes, such as collagen fibrils and elastic fibers, providing connective tissues with appropriate biomechanical properties. Besides this architectural function, TN-X behaves as a matricellular protein by regulating cell adhesion-de-adhesion processes. However, in contrast to the other TN molecules, the cellular functions of TN-X have not yet been determined. The regulation of cell plasticity by the central region of the glycoprotein (FNIII repeats) might provide the first clue of a cell function controlled through a cell adhesion-modulating activity for TN-X. Additional experiments will be necessary to identify the exact ‘active’ sequences within the FNIII region, as well as the corresponding cell surface receptor, and to unravel the molecular mechanisms by which the central region of the TN-X inhibits EMT. Our laboratory has also generated evidence that the C-terminal FBG-like domain of TN-X is able to activate latent TGF-β and to induce an EMT in vitro. Whether TN-X activates latent TGF-β in vivo is still an open issue. However, it has been shown that patients suffering from congenital adrenal hyperplasia associated with TN-X deficiency (CAH-X) display some manifestations, such as bifid uvula and cardiac valvular abnormalities, that are overlapping with connective tissue dysplasia phenotypes, which are associated with the dysregulation in the TGF-β signaling pathway and frequently observed in Marfan, Loey's Dietz and Shprintzen-Golberg syndromes.86 Interestingly, the TGF-β family pathway has been demonstrated to be abnormal in patients suffering from CAH-X,87 indicating a putative functional link between TN-X and the TGF-β pathway in vivo. A deeper analysis of TN-X-deficient mice in physiological (wound healing) or pathological (fibrosis or tumor progression) conditions requiring activation of latent TGF-β will provide a functional link between the TN-X and the regulation of mature TGF-β bioavailability in vivo.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the Institut National de la Santé Et de la Recherche Médicale and the Ligue Nationale Contre le Cancer–Comité du Rhône. L.B. Alcaraz was supported by a fellowship from the Ligue Nationale Contre le Cancer.
References
- 1. Morel Y, Bristow J, Gitelman SE, Miller WL. Transcript encoded on the opposite strand of the human steroid 21-hydroxylase/complement component C4 gene locus. Proc Natl Acad Sci U S A 1989; 86:6582-6; PMID:2475872; http://dx.doi.org/ 10.1073/pnas.86.17.6582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Matsumoto K, Arai M, Ishihara N, Ando A, Inoko H, Ikemura T. Cluster of fibronectin type III repeats found in the human major histocompatibility complex class III region shows the highest homology with the repeats in an extracellular matrix protein, tenascin. Genomics 1992; 12:485-91; PMID:1373119; http://dx.doi.org/ 10.1016/0888-7543(92)90438-X [DOI] [PubMed] [Google Scholar]
- 3. Matsumoto K, Ishihara N, Ando A, Inoko H, Ikemura T. Extracellular matrix protein tenascin-like gene found in human MHC class III region. Immunogenetics 1992; 36:400-3; PMID:1382042; http://dx.doi.org/ 10.1007/BF00218048 [DOI] [PubMed] [Google Scholar]
- 4. Bristow J, Tee MK, Gitelman SE, Mellon SH, Miller WL. Tenascin-X: a novel extracellular matrix protein encoded by the human XB gene overlapping P450c21B. J Cell Biol 1993; 122:265-78; PMID:7686164; http://dx.doi.org/ 10.1083/jcb.122.1.265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chiquet-Ehrismann R, Mackie EJ, Pearson CA, Sakakura T. Tenascin: an extracellular matrix protein involved in tissue interactions during fetal development and oncogenesis. Cell 1986; 47:131-9; PMID:2428505; http://dx.doi.org/ 10.1016/0092-8674(86)90374-0 [DOI] [PubMed] [Google Scholar]
- 6. Erickson HP, Inglesias JL. A six-armed oligomer isolated from cell surface fibronectin preparations. Nature 1984; 311:267-9; PMID:6482952; http://dx.doi.org/ 10.1038/311267a0 [DOI] [PubMed] [Google Scholar]
- 7. Grumet M, Hoffman S, Crossin KL, Edelman GM. Cytotactin, an extracellular matrix protein of neural and non-neural tissues that mediates glia-neuron interaction. Proc Natl Acad Sci U S A 1985; 82:8075-9; PMID:2415980; http://dx.doi.org/ 10.1073/pnas.82.23.8075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pesheva P, Spiess E, Schachner M. J1-160 and J1-180 are oligodendrocyte-secreted nonpermissive substrates for cell adhesion. J Cell Biol 1989; 109:1765-78; PMID:2477380; http://dx.doi.org/ 10.1083/jcb.109.4.1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schachner M, Taylor J, Bartsch U, Pesheva P. The perplexing multifunctionality of janusin, a tenascin-related molecule. Perspect Dev Neurobiol 1994; 2:33-41; PMID:7530142 [PubMed] [Google Scholar]
- 10. Rathjen FG, Wolff JM, Chiquet-Ehrismann R. Restrictin: a chick neural extracellular matrix protein involved in cell attachment co-purifies with the cell recognition molecule F11. Dev Camb Engl 1991; 113:151-64 [DOI] [PubMed] [Google Scholar]
- 11. Scherberich A, Tucker RP, Samandari E, Brown-Luedi M, Martin D, Chiquet-Ehrismann R. Murine tenascin-W: a novel mammalian tenascin expressed in kidney and at sites of bone and smooth muscle development. J Cell Sci 2004; 117:571-81; PMID:14709716; http://dx.doi.org/ 10.1242/jcs.00867 [DOI] [PubMed] [Google Scholar]
- 12. Neidhardt J, Fehr S, Kutsche M, Löhler J, Schachner M. Tenascin-N: characterization of a novel member of the tenascin family that mediates neurite repulsion from hippocampal explants. Mol Cell Neurosci 2003; 23:193-209; PMID:12812753; http://dx.doi.org/ 10.1016/S1044-7431(03)00012-5 [DOI] [PubMed] [Google Scholar]
- 13. Tucker RP, Drabikowski K, Hess JF, Ferralli J, Chiquet-Ehrismann R, Adams JC. Phylogenetic analysis of the tenascin gene family: evidence of origin early in the chordate lineage. BMC Evol Biol 2006; 6:60; PMID:16893461; http://dx.doi.org/ 10.1186/1471-2148-6-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Degen M, Brellier F, Kain R, Ruiz C, Terracciano L, Orend G, Chiquet-Ehrismann R. Tenascin-W is a novel marker for activated tumor stroma in low-grade human breast cancer and influences cell behavior. Cancer Res 2007; 67:9169-79; PMID:17909022; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-0666 [DOI] [PubMed] [Google Scholar]
- 15. Pesheva P, Probstmeier R, Lang DM, McBride R, Hsu N-J, Gennarini G, Spiess E, Peshev Z. Early coevolution of adhesive but not antiadhesive tenascin-R ligand-receptor pairs in vertebrates: a phylogenetic study. Mol Cell Neurosci 2006; 32:366-86; PMID:16831557; http://dx.doi.org/ 10.1016/j.mcn.2006.05.008 [DOI] [PubMed] [Google Scholar]
- 16. Jones FS, Jones PL. The tenascin family of ECM glycoproteins: structure, function, and regulation during embryonic development and tissue remodeling. Dev Dyn Off Publ Am Assoc Anat 2000; 218:235-59. [DOI] [PubMed] [Google Scholar]
- 17. Lethias C, Descollonges Y, Boutillon MM, Garrone R. Flexilin: a new extracellular matrix glycoprotein localized on collagen fibrils. Matrix Biol J Int Soc Matrix Biol 1996; 15:11-9; http://dx.doi.org/ 10.1016/S0945-053X(96)90122-5 [DOI] [PubMed] [Google Scholar]
- 18. Kammerer RA, Schulthess T, Landwehr R, Lustig A, Fischer D, Engel J. Tenascin-C hexabrachion assembly is a sequential two-step process initiated by coiled-coil alpha-helices. J Biol Chem 1998; 273:10602-8; PMID:9553121; http://dx.doi.org/ 10.1074/jbc.273.17.10602 [DOI] [PubMed] [Google Scholar]
- 19. Luczak JA, Redick SD, Schwarzbauer JE. A single cysteine, Cys-64, is essential for assembly of tenascin-C hexabrachions. J Biol Chem 1998; 273:2073-7; PMID:9442046; http://dx.doi.org/ 10.1074/jbc.273.4.2073 [DOI] [PubMed] [Google Scholar]
- 20. Lethias C, Carisey A, Comte J, Cluzel C, Exposito J-Y. A model of tenascin-X integration within the collagenous network. FEBS Lett 2006; 580:6281-5; PMID:17078949; http://dx.doi.org/ 10.1016/j.febslet.2006.10.037 [DOI] [PubMed] [Google Scholar]
- 21. Nörenberg U, Wille H, Wolff JM, Frank R, Rathjen FG. The chicken neural extracellular matrix molecule restrictin: similarity with EGF-, fibronectin type III-, and fibrinogen-like motifs. Neuron 1992; 8:849-63; PMID:1375037; http://dx.doi.org/ 10.1016/0896-6273(92)90199-N [DOI] [PubMed] [Google Scholar]
- 22. Elefteriou F, Exposito JY, Garrone R, Lethias C. Characterization of the bovine tenascin-X. J Biol Chem 1997; 272:22866-74; PMID:9278449; http://dx.doi.org/ 10.1074/jbc.272.36.22866 [DOI] [PubMed] [Google Scholar]
- 23. Ikuta T, Sogawa N, Ariga H, Ikemura T, Matsumoto K. Structural analysis of mouse tenascin-X: evolutionary aspects of reduplication of FNIII repeats in the tenascin gene family. Gene 1998; 217:1-13; PMID:9795100; http://dx.doi.org/ 10.1016/S0378-1119(98)00355-2 [DOI] [PubMed] [Google Scholar]
- 24. Hagios C, Koch M, Spring J, Chiquet M, Chiquet-Ehrismann R. Tenascin-Y: a protein of novel domain structure is secreted by differentiated fibroblasts of muscle connective tissue. J Cell Biol 1996; 134:1499-512; PMID:8830777; http://dx.doi.org/ 10.1083/jcb.134.6.1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Burch GH, Bedolli MA, McDonough S, Rosenthal SM, Bristow J. Embryonic expression of tenascin-X suggests a role in limb, muscle, and heart development. Dev Dyn Off Publ Am Assoc Anat 1995; 203:491-504. [DOI] [PubMed] [Google Scholar]
- 26. Matsumoto K, Saga Y, Ikemura T, Sakakura T, Chiquet-Ehrismann R. The distribution of tenascin-X is distinct and often reciprocal to that of tenascin-C. J Cell Biol 1994; 125:483-93; PMID:7512972; http://dx.doi.org/ 10.1083/jcb.125.2.483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Geffrotin C, Garrido JJ, Tremet L, Vaiman M. Distinct tissue distribution in pigs of tenascin-X and tenascin-C transcripts. Eur J Biochem FEBS 1995; 231:83-92; http://dx.doi.org/ 10.1111/j.1432-1033.1995.0083f.x [DOI] [PubMed] [Google Scholar]
- 28. Fuss B, Wintergerst ES, Bartsch U, Schachner M. Molecular characterization and in situ mRNA localization of the neural recognition molecule J1-160/180: a modular structure similar to tenascin. J Cell Biol 1993; 120:1237-49; PMID:7679676; http://dx.doi.org/ 10.1083/jcb.120.5.1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Egging D, van Vlijmen-Willems I, van Tongeren T, Schalkwijk J, Peeters A. Wound healing in tenascin-X deficient mice suggests that tenascin-X is involved in matrix maturation rather than matrix deposition. Connect Tissue Res 2007; 48:93-8; PMID:17453911; http://dx.doi.org/ 10.1080/03008200601166160 [DOI] [PubMed] [Google Scholar]
- 30. Mackie EJ, Halfter W, Liverani D. Induction of tenascin in healing wounds. J Cell Biol 1988; 107:2757-67; PMID:2462568; http://dx.doi.org/ 10.1083/jcb.107.6.2757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Geffrotin C, Horak V, Créchet F, Tricaud Y, Lethias C, Vincent-Naulleau S, Vielh P. Opposite regulation of tenascin-C and tenascin-X in MeLiM swine heritable cutaneous malignant melanoma. Biochim Biophys Acta 2000; 1524:196-202; PMID:11113568; http://dx.doi.org/ 10.1016/S0304-4165(00)00158-6 [DOI] [PubMed] [Google Scholar]
- 32. Sakai T, Furukawa Y, Chiquet-Ehrismann R, Nakamura M, Kitagawa S, Ikemura T, Matsumoto K. Tenascin-X expression in tumor cells and fibroblasts: glucocorticoids as negative regulators in fibroblasts. J Cell Sci 1996; 109 (Pt 8):2069-77; PMID:8856503 [DOI] [PubMed] [Google Scholar]
- 33. Hasegawa K, Yoshida T, Matsumoto K, Katsuta K, Waga S, Sakakura T. Differential expression of tenascin-C and tenascin-X in human astrocytomas. Acta Neuropathol (Berl) 1997; 93:431-7; http://dx.doi.org/ 10.1007/s004010050636 [DOI] [PubMed] [Google Scholar]
- 34. Midwood KS, Orend G. The role of tenascin-C in tissue injury and tumorigenesis. J Cell Commun Signal 2009; 3:287-310; PMID:19838819; http://dx.doi.org/ 10.1007/s12079-009-0075-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bristow J, Carey W, Egging D, Schalkwijk J. Tenascin-X, collagen, elastin, and the Ehlers-Danlos syndrome. Am J Med Genet C Semin Med Genet 2005; 139C:24-30; PMID:16278880; http://dx.doi.org/ 10.1002/ajmg.c.30071 [DOI] [PubMed] [Google Scholar]
- 36. Veit G, Hansen U, Keene DR, Bruckner P, Chiquet-Ehrismann R, Chiquet M, Koch M. Collagen XII interacts with avian tenascin-X through its NC3 domain. J Biol Chem 2006; 281:27461-70; PMID:16861231; http://dx.doi.org/ 10.1074/jbc.M603147200 [DOI] [PubMed] [Google Scholar]
- 37. Egging D, van den Berkmortel F, Taylor G, Bristow J, Schalkwijk J. Interactions of human tenascin-X domains with dermal extracellular matrix molecules. Arch Dermatol Res 2007; 298:389-96; PMID:17033827; http://dx.doi.org/ 10.1007/s00403-006-0706-9 [DOI] [PubMed] [Google Scholar]
- 38. Minamitani T, Ikuta T, Saito Y, Takebe G, Sato M, Sawa H, Nishimura T, Nakamura F, Takahashi K, Ariga H, et al. Modulation of collagen fibrillogenesis by tenascin-X and type VI collagen. Exp Cell Res 2004; 298:305-15; PMID:15242785; http://dx.doi.org/ 10.1016/j.yexcr.2004.04.030 [DOI] [PubMed] [Google Scholar]
- 39. Elefteriou F, Exposito JY, Garrone R, Lethias C. Binding of tenascin-X to decorin. FEBS Lett 2001; 495:44-7; PMID:11322944; http://dx.doi.org/ 10.1016/S0014-5793(01)02361-4 [DOI] [PubMed] [Google Scholar]
- 40. Chiquet-Ehrismann R, Kalla P, Pearson CA, Beck K, Chiquet M. Tenascin interferes with fibronectin action. Cell 1988; 53:383-90; PMID:2452695; http://dx.doi.org/ 10.1016/0092-8674(88)90158-4 [DOI] [PubMed] [Google Scholar]
- 41. De Paepe A, Malfait F. The Ehlers-Danlos syndrome, a disorder with many faces. Clin Genet 2012; 82:1-11; PMID:22353005; http://dx.doi.org/ 10.1111/j.1399-0004.2012.01858.x [DOI] [PubMed] [Google Scholar]
- 42. Burch GH, Gong Y, Liu W, Dettman RW, Curry CJ, Smith L, Miller WL, Bristow J. Tenascin-X deficiency is associated with Ehlers-Danlos syndrome. Nat Genet 1997; 17:104-8; PMID:9288108; http://dx.doi.org/ 10.1038/ng0997-104 [DOI] [PubMed] [Google Scholar]
- 43. Schalkwijk J, Zweers MC, Steijlen PM, Dean WB, Taylor G, van Vlijmen IM, van Haren B, Miller WL, Bristow J. A recessive form of the Ehlers-Danlos syndrome caused by tenascin-X deficiency. N Engl J Med 2001; 345:1167-75; PMID:11642233; http://dx.doi.org/ 10.1056/NEJMoa002939 [DOI] [PubMed] [Google Scholar]
- 44. Voermans NC, Jenniskens GJ, Hamel BC, Schalkwijk J, Guicheney P, van Engelen BG. Ehlers-Danlos syndrome due to tenascin-X deficiency: muscle weakness and contractures support overlap with collagen VI myopathies. Am J Med Genet A 2007; 143A:2215-9; PMID:17702048; http://dx.doi.org/ 10.1002/ajmg.a.31899 [DOI] [PubMed] [Google Scholar]
- 45. Lindor NM, Bristow J. Tenascin-X deficiency in autosomal recessive Ehlers-Danlos syndrome. Am J Med Genet A 2005; 135:75-80; PMID:15793839; http://dx.doi.org/ 10.1002/ajmg.a.30671 [DOI] [PubMed] [Google Scholar]
- 46. Zweers MC, Bristow J, Steijlen PM, Dean WB, Hamel BC, Otero M, Kucharekova M, Boezeman JB, Schalkwijk J. Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers-Danlos syndrome. Am J Hum Genet 2003; 73:214-7; PMID:12865992; http://dx.doi.org/ 10.1086/376564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mao JR, Taylor G, Dean WB, Wagner DR, Afzal V, Lotz JC, Rubin EM, Bristow J. Tenascin-X deficiency mimics Ehlers-Danlos syndrome in mice through alteration of collagen deposition. Nat Genet 2002; 30:421-5; PMID:11925569; http://dx.doi.org/ 10.1038/ng850 [DOI] [PubMed] [Google Scholar]
- 48. Matsumoto K, Takayama N, Ohnishi J, Ohnishi E, Shirayoshi Y, Nakatsuji N, Ariga H. Tumour invasion and metastasis are promoted in mice deficient in tenascin-X. Genes Cells Devoted Mol Cell Mech 2001; 6:1101-11; http://dx.doi.org/ 10.1046/j.1365-2443.2001.00482.x [DOI] [PubMed] [Google Scholar]
- 49. Voermans NC, Verrijp K, Eshuis L, Balemans MC, Egging D, Sterrenburg E, van Rooij IA, van der Laak JA, Schalkwijk J, van der Maarel SM, et al. Mild muscular features in tenascin-X knockout mice, a model of Ehlers-danlos syndrome. Connect Tissue Res 2011; 52:422-32; PMID:21405982; http://dx.doi.org/ 10.3109/03008207.2010.551616 [DOI] [PubMed] [Google Scholar]
- 50. Minamitani T, Ariga H, Matsumoto K. Deficiency of tenascin-X causes a decrease in the level of expression of type VI collagen. Exp Cell Res 2004; 297:49-60; PMID:15194424; http://dx.doi.org/ 10.1016/j.yexcr.2004.03.002 [DOI] [PubMed] [Google Scholar]
- 51. Zou Y, Zwolanek D, Izu Y, Gandhy S, Schreiber G, Brockmann K, Devoto M, Tian Z, Hu Y, Veit G, et al. Recessive and dominant mutations in COL12A1 cause a novel EDS/myopathy overlap syndrome in humans and mice. Hum Mol Genet 2014; 23:2339-52; PMID:24334604; http://dx.doi.org/ 10.1093/hmg/ddt627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zweers MC, van Vlijmen-Willems IM, van Kuppevelt TH, Mecham RP, Steijlen PM, Bristow J, Schalkwijk J. Deficiency of tenascin-X causes abnormalities in dermal elastic fiber morphology. J Invest Dermatol 2004; 122:885-91; PMID:15102077; http://dx.doi.org/ 10.1111/j.0022-202X.2004.22401.x [DOI] [PubMed] [Google Scholar]
- 53. Margaron Y, Bostan L, Exposito J-Y, Malbouyres M, Trunfio-Sfarghiu A-M, Berthier Y, Lethias C. Tenascin-X increases the stiffness of collagen gels without affecting fibrillogenesis. Biophys Chem 2010; 147:87-91; PMID:20089348; http://dx.doi.org/ 10.1016/j.bpc.2009.12.011 [DOI] [PubMed] [Google Scholar]
- 54. Zweers MC, Hakim AJ, Grahame R, Schalkwijk J. Joint hypermobility syndromes: the pathophysiologic role of tenascin-X gene defects. Arthritis Rheum 2004; 50:2742-9; PMID:15457441; http://dx.doi.org/ 10.1002/art.20488 [DOI] [PubMed] [Google Scholar]
- 55. Mao JR, Bristow J. The Ehlers-Danlos syndrome: on beyond collagens. J Clin Invest 2001; 107:1063-9; PMID:11342567; http://dx.doi.org/ 10.1172/JCI12881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chiquet M, Birk DE, Bönnemann CG, Koch M. Collagen XII: protecting bone and muscle integrity by organizing collagen fibrils. Int J Biochem Cell Biol 2014; 53C:51-4; http://dx.doi.org/ 10.1016/j.biocel.2014.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jollymore A, Lethias C, Peng Q, Cao Y, Li H. Nanomechanical properties of tenascin-X revealed by single-molecule force spectroscopy. J Mol Biol 2009; 385:1277-86; PMID:19071135; http://dx.doi.org/ 10.1016/j.jmb.2008.11.038 [DOI] [PubMed] [Google Scholar]
- 58. Egging DF, van Vlijmen I, Starcher B, Gijsen Y, Zweers MC, Blankevoort L, Bristow J, Schalkwijk J. Dermal connective tissue development in mice: an essential role for tenascin-X. Cell Tissue Res 2006; 323:465-74; PMID:16331473; http://dx.doi.org/ 10.1007/s00441-005-0100-5 [DOI] [PubMed] [Google Scholar]
- 59. Bornstein P, Sage EH. Matricellular proteins: extracellular modulators of cell function. Curr Opin Cell Biol 2002; 14:608-16; PMID:12231357; http://dx.doi.org/ 10.1016/S0955-0674(02)00361-7 [DOI] [PubMed] [Google Scholar]
- 60. Meloty-Kapella CV, Degen M, Chiquet-Ehrismann R, Tucker RP. Avian tenascin-W: expression in smooth muscle and bone, and effects on calvarial cell spreading and adhesion in vitro. Dev Dyn Off Publ Am Assoc Anat 2006; 235:1532-42. [DOI] [PubMed] [Google Scholar]
- 61. Elefteriou F, Exposito JY, Garrone R, Lethias C. Cell adhesion to tenascin-X mapping of cell adhesion sites and identification of integrin receptors. Eur J Biochem FEBS 1999; 263:840-8; http://dx.doi.org/ 10.1046/j.1432-1327.1999.00563.x [DOI] [PubMed] [Google Scholar]
- 62. Fujie S, Maita H, Ariga H, Matsumoto K. Tenascin-X induces cell detachment through p38 mitogen-activated protein kinase activation. Biol Pharm Bull 2009; 32:1795-9; PMID:19801846; http://dx.doi.org/ 10.1248/bpb.32.1795 [DOI] [PubMed] [Google Scholar]
- 63. Goicoechea S, Orr AW, Pallero MA, Eggleton P, Murphy-Ullrich JE. Thrombospondin mediates focal adhesion disassembly through interactions with cell surface calreticulin. J Biol Chem 2000; 275:36358-68; PMID:10964924; http://dx.doi.org/ 10.1074/jbc.M005951200 [DOI] [PubMed] [Google Scholar]
- 64. Huang W, Chiquet-Ehrismann R, Moyano JV, Garcia-Pardo A, Orend G. Interference of tenascin-C with syndecan-4 binding to fibronectin blocks cell adhesion and stimulates tumor cell proliferation. Cancer Res 2001; 61:8586-94; PMID:11731446 [PubMed] [Google Scholar]
- 65. Lethias C, Elefteriou F, Parsiegla G, Exposito JY, Garrone R. Identification and characterization of a conformational heparin-binding site involving two fibronectin type III modules of bovine tenascin-X. J Biol Chem 2001; 276:16432-8; PMID:11278641; http://dx.doi.org/ 10.1074/jbc.M010210200 [DOI] [PubMed] [Google Scholar]
- 66. Alcaraz LB, Exposito J-Y, Chuvin N, Pommier RM, Cluzel C, Martel S, Sentis S, Bartholin L, Lethias C, Valcourt U. Tenascin-X promotes epithelial-to-mesenchymal transition by activating latent TGF-β. J Cell Biol 2014; 205:409-28; PMID:24821840; http://dx.doi.org/ 10.1083/jcb.201308031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Matsumoto K-I, Minamitani T, Orba Y, Sato M, Sawa H, Ariga H. Induction of matrix metalloproteinase-2 by tenascin-X deficiency is mediated through the c-Jun N-terminal kinase and protein tyrosine kinase phosphorylation pathway. Exp Cell Res 2004; 297:404-14; PMID:15212943; http://dx.doi.org/ 10.1016/j.yexcr.2004.03.041 [DOI] [PubMed] [Google Scholar]
- 68. Ikuta T, Ariga H, Matsumoto K. Extracellular matrix tenascin-X in combination with vascular endothelial growth factor B enhances endothelial cell proliferation. Genes Cells Devoted Mol Cell Mech 2000; 5:913-27; http://dx.doi.org/ 10.1046/j.1365-2443.2000.00376.x [DOI] [PubMed] [Google Scholar]
- 69. Wu MY, Hill CS. Tgf-beta superfamily signaling in embryonic development and homeostasis. Dev Cell 2009; 16:329-43; PMID:19289080; http://dx.doi.org/ 10.1016/j.devcel.2009.02.012 [DOI] [PubMed] [Google Scholar]
- 70. Massagué J. TGFbeta in Cancer. Cell 2008; 134:215-30; http://dx.doi.org/ 10.1016/j.cell.2008.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ten Dijke P, Arthur HM. Extracellular control of TGFbeta signalling in vascular development and disease. Nat Rev Mol Cell Biol 2007; 8:857-69; PMID:17895899; http://dx.doi.org/ 10.1038/nrm2262 [DOI] [PubMed] [Google Scholar]
- 72. Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, Sheppard D, Broaddus VC, Nishimura SL. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J Cell Biol 2002; 157:493-507; PMID:11970960; http://dx.doi.org/ 10.1083/jcb.200109100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lyons RM, Keski-Oja J, Moses HL. Proteolytic activation of latent transforming growth factor-beta from fibroblast-conditioned medium. J Cell Biol 1988; 106:1659-65; PMID:2967299; http://dx.doi.org/ 10.1083/jcb.106.5.1659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lyons RM, Gentry LE, Purchio AF, Moses HL. Mechanism of activation of latent recombinant transforming growth factor beta 1 by plasmin. J Cell Biol 1990; 110:1361-7; PMID:2139036; http://dx.doi.org/ 10.1083/jcb.110.4.1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev 2000; 14:163-76; PMID:10652271 [PMC free article] [PubMed] [Google Scholar]
- 76. D’Angelo M, Billings PC, Pacifici M, Leboy PS, Kirsch T. Authentic matrix vesicles contain active metalloproteases (MMP). a role for matrix vesicle-associated MMP-13 in activation of transforming growth factor-beta. J Biol Chem 2001; 276:11347-53; http://dx.doi.org/ 10.1074/jbc.M009725200 [DOI] [PubMed] [Google Scholar]
- 77. Ge G, Greenspan DS. BMP1 controls TGFbeta1 activation via cleavage of latent TGFbeta-binding protein. J Cell Biol 2006; 175:111-20; PMID:17015622; http://dx.doi.org/ 10.1083/jcb.200606058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wipff P-J, Hinz B. Integrins and the activation of latent transforming growth factor beta1—an intimate relationship. Eur J Cell Biol 2008; 87:601-15; PMID:18342983; http://dx.doi.org/ 10.1016/j.ejcb.2008.01.012 [DOI] [PubMed] [Google Scholar]
- 79. Dennis PA, Rifkin DB. Cellular activation of latent transforming growth factor beta requires binding to the cation-independent mannose 6-phosphate/insulin-like growth factor type II receptor. Proc Natl Acad Sci U S A 1991; 88:580-4; PMID:1846448; http://dx.doi.org/ 10.1073/pnas.88.2.580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Glinka Y, Stoilova S, Mohammed N, Prud’homme GJ. Neuropilin-1 exerts co-receptor function for TGF-beta-1 on the membrane of cancer cells and enhances responses to both latent and active TGF-beta. Carcinogenesis 2011; 32:613-21; PMID:21186301; http://dx.doi.org/ 10.1093/carcin/bgq281 [DOI] [PubMed] [Google Scholar]
- 81. Schultz-Cherry S, Murphy-Ullrich JE. Thrombospondin causes activation of latent transforming growth factor-beta secreted by endothelial cells by a novel mechanism. J Cell Biol 1993; 122:923-32; PMID:8349738; http://dx.doi.org/ 10.1083/jcb.122.4.923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Crawford SE, Stellmach V, Murphy-Ullrich JE, Ribeiro SM, Lawler J, Hynes RO, Boivin GP, Bouck N. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell 1998; 93:1159-70; PMID:9657149; http://dx.doi.org/ 10.1016/S0092-8674(00)81460-9 [DOI] [PubMed] [Google Scholar]
- 83. Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol 2012; 13:616-30; http://dx.doi.org/ 10.1038/nrm3434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Valcourt U, Kowanetz M, Niimi H, Heldin C-H, Moustakas A. TGF-beta and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol Biol Cell 2005; 16:1987-2002; PMID:15689496; http://dx.doi.org/ 10.1091/mbc.E04-08-0658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Thiery JP, Acloque H, Huang RYJ, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell 2009; 139:871-90; PMID:19945376; http://dx.doi.org/ 10.1016/j.cell.2009.11.007 [DOI] [PubMed] [Google Scholar]
- 86. Merke DP, Chen W, Morissette R, Xu Z, Van Ryzin C, Sachdev V, Hannoush H, Shanbhag SM, Acevedo AT, Nishitani M, et al. Tenascin-X haploinsufficiency associated with Ehlers-Danlos syndrome in patients with congenital adrenal hyperplasia. J Clin Endocrinol Metab 2013; 98:E379-87; PMID:23284009; http://dx.doi.org/ 10.1210/jc.2012-3148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Morissette R, Merke DP, McDonnell NB. Transforming growth factor-β (TGF-β) pathway abnormalities in tenascin-X deficiency associated with CAH-X syndrome. Eur J Med Genet 2014; 57(2-3):95-102. [DOI] [PMC free article] [PubMed] [Google Scholar]