

Abstract
Tenascin-C (TNC), a multifunctional matricellular glyco-protein, is highly expressed in the majority of melanoma cell lines and has been implicated in the progression of melanoma. A growing body of evidence has implicated the role of TNC in the process of invasion and metastasis for melanoma. However, the mechanism and individual signaling pathways by which TNC drives melanoma progression have not been illuminated. Herein we provide perspectives from the investigation of TNC in other settings that may hint at the mechanistic role of TNC in this disease.
Keywords: invasion, melanoma, survival, Tenascin-C
Abbreviations
- TNC
Tenascin-C
- ECM
extracellular Matrix
- EGFL
epidermal growth factor-like
- FNIII
fibronectin type III
Introduction
Melanoma is the most lethal of skin cancers, originating in the pigment-producing melanocytes of the basal layer of the epidermis. Patients with melanoma can be cured by surgical resection if they are discovered early in the process of progression, at stages where the transformed melanocytes have invaded locally, and radially rather than vertically, where the risk of regional or distant disease dissemination rises considerably. The 5-year survival rate for melanoma declines dramatically once tumor cells have invaded vertically through the dermal matrix, and to distant organs. The likelihood of dissemination is directly proportional to the depth of invasion. Thus, vertical invasion represents a key step in progression, where the likelihood of morbidity due to relapse, and mortality due to vital organ metastasis rises with progression of melanoma.
During the past 2 decades, many studies have aimed to decipher the mechanisms by which melanoma cells disseminate from the primary tumor site, invading through the dermis, and finally to colonize distant vital organs. Two aspects have drawn attention – the initial invasiveness and the growth of melanoma at metastatic sites. (As a note, we will not be discussing aspects of melanomagenesis that involves mutations of known and putative oncogenes and tumor suppressors1). As the dermis is comprised mainly of extracellular matrix (ECM) by mass, there has been a focus on changes in matrix and on signals that drive the locomotion through this collagen I-rich barrier.2 At the sites of dissemination, work has delved into the signals that support melanoma cell survival and growth in what should be hostile microenvironment. In both situations, TNC is striking in its upregulation and aberrant expression.
TNC Signaling Germane to Melanoma Invasion and Survival
TNC, the best described member of the tenascin family of matricellular proteins that consists of 4 tenascin proteins –C, -X, -R and –W, is a homodimer of homotrimers in which each monomer has a molecular weight ranging between 180 and 330 kDa depending on the extent of glycosylation at its 23 potential sites and alternative splicing of domains.3 Each TNC subunit is comprised of a N-terminal rod-like assembly domain, a domain consisting of 14.5 epidermal growth factor-like (EGFL) repeats of 30–50 amino acids in length for each repeat, a domain composed of up to 17 fibronectin type III-like (FNIII) repeats, and a carboxyl terminus homologous to fibrinogen4-7 (Fig. 1). Each domain imparts select behaviors to the cells adherent to them. Thus the cell responses to TNC are dependent on the receptor repertoire present on the cell surface.
Figure 1.
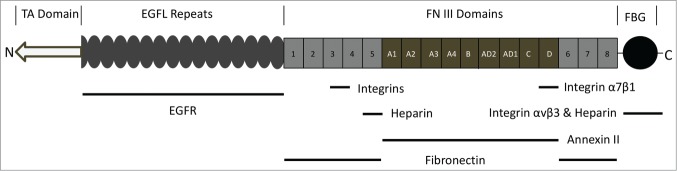
Schematic representation of TNC monomer. Listed above the structure are the domains: TA, TNC Assembly domain allows the assembly of hexabrachion. EGFL, Epidermal Growth Factor-Like; FN III, FibroNectin type III homology repeats; FBG, FiBrinogen Globe. Below the structure are known interacting receptors and matricellular proteins. Information on specific TNC binding ligands/receptors is based on.3
The FNIII domains and the fibrinogen domain of TNC are considered adhesive.8-13 However, the complete molecule is considered anti-adhesive. This function has been mapped primarily to the EGFL component of TNC. This anti-adhesive aspect of cell interaction with TNC can shift the adhesion/contractile ratio to a setpoint that promotes cell migration in a mesenchymal state14 or reversion to an amoeboid state for penetration of a denser matrix.7,15-17
The EGFL of TNC functions uniquely as an ultralow affinity ligand for the EGF receptor18,19 and laminins as well as possibly SPARC also present such matrikine or matricryptin signaling.11,20,21 The tethering of TNC within the insoluble matrix converts the select EGFR-binding low affinity EGFL into high avidity ligands due to limited diffusion and multimeric clustering. This results in predominantly cell surface signaling that is preferential for motility over proliferation22 due to the preference for PLCgamma and ERK m-calpain activation over Ras-triggered pathways to the nucleus.23-25
More recently, it has been noted that restriction of EGFR signaling to the cell membrane, by tethering ligands, provides for increased cell survival in the face of death inducing cytokines (Fig. 2).6,26 As TNC EGFL domains resemble a physiological correlate of EGF, TNC may also protect stem cells from apoptosis.6 Thus, the presence of TNC can serve 2 functions in melanoma progression. First, it can promote migration and invasion through the dermis. Second, in the hostile ectopic microenvironment of the distant target organ, it could support survival of the disseminated melanoma cells.
Figure 2.

Schematic representation of survival signaling vs. proliferative signaling from the EGF receptor based on surface restriction of EGFR activation. The TNC EGF-like repeats provide for less efficient activation of the Ras-Raf-ERK and PI3kinase-Akt pathways in a persistent staccato manner; this leads to survival signaling. However, soluble EGF ligands drive EGFR internalization with the endosomal active EGFR efficiently signaling via the Ras-Raf-ERK pathway to drive mitogenesis.53 Not shown are the motogenic pathways (PLCgamma, ERK-calpain II, and PKCdelta) that are preferentially signaled from surface restricted EGFR.
It is of interest that the effects of TNC EGFL on cell migration and survival were noted first on mesenchymal stem cells/multipotent stromal cells (MSC) that have very low levels of EGF receptors (3000–7000 per cell versus the ∼105 in stromal cells).27 Low levels of EGFR are important for these responses, as cells with high levels of EGFR experience excessive signaling. In most adult cells, tonic EGFR signaling becomes anti-adhesive to the point of inducing anoikis. Melanoma cells also express low levels of EGFR protein, at steady state, even though they show increased levels of message RNA and even gene copy number (functionality is shown by the cells being dependent on EGFR signaling for proliferation in vitro28,29). All this suggests a high flux through the system secondary to autocrine receptor activation/downregulation. However, the lower level of EGFR protein would modulate the anti-adhesive effects to bring the adhesion/contractility ratio to the range that enhances migration rather than driving anoikis.14
Regulation of TNC Expression in Melanoma
The expression of TNC is tightly regulated, and largely present in appreciable quantities during organogenesis/embryogenesis and wound repair and cancer invasion; in adult skin and other tissues there is only a trace of TNC (reviewed in3,30). During wound healing, TNC is dramatically upregulated during the regenerative phase of repair that is marked by rapid angiogenesis, migration of fibroblasts into the wounded area and re-epithelialization of migrating keratinocytes; TNC is then suppressed and removed so that during the resolving phase little if any TNC expression persists as the tissue undergoes quiescence and the excess vessels and stromal cells involute. This is recapitulated in part during melanoma progression.
Previous and recent studies revealed that the melanoma biopsies and cell lines present an elevated level of TNC.7,31-34 The increased expression of TNC accompanies the transformation of melanocytes into melanoma.35 Microarray analysis and immunohistochemical data performed by Hölttä's group showed that the increased expressions of TNC is associated with a switch from benign or a non-invasive phase into an invasive growth phase of melanoma and thus was considered as a potential biomarker of aggressiveness, and a potential therapeutic target for prevention of melanoma metastasis.36 Interestingly, coculture of melanoma cells with fibroblasts significantly enhanced the expression of TNC in fibroblasts which was mediated by cell-cell contact rather than secretion of any components by melanoma cells37 although the precise mechanism remains to be further determined. TNC is also detected in the sera of human melanoma patients with dramatically elevated level in patients with advanced stages compared to normal donors or patients with lower tumor stages.38,39 In light of these findings, it is important to note that expression of TNC is relevant to the progression of melanoma and is enriched in the environment of melanoma invasion and metastasis (Fig. 3A).
Figure 3.

TNC promotes both vertical invasion to allow for dissemination and then survival at the ectopic site. (A) TNC expression is upregulated as melanomas progress (see7). (B) Schematic of TNC roles during melanoma progression. RGP: radial growth phase; VGP: vertical growth phase; MGP: metastatic growth phase. TNC secreted from both melanoma cells and fibroblasts enhances the dissemination of melanoma cells from the primary tumor and invasion into the dermal matrix. In metastatic organ, TNC promotes the survival of melanoma cells from tumor environmental stress and chemotherapies.
The mechanism behind the upregulation of TNC is uncertain, as to the key cells that produce the matricellular protein or the intracellular mechanisms driving greater production. For the melanoma cells this may relate to mutations in B-Raf, as pathways downstream from oncogenic Ras upregulate TNC in mouse mammary epithelial cells.40 However, further studies are required to determine the relevance of B-Raf in triggering TNC expression by melanoma cells since only half of melanomas carry an activating mutation in B-Raf.41 On the other side, this pathway or others may increase TNC production by the dermal or immune cells, as inflammatory stimuli activate the PI3 kinase/AKT and NF-kB signaling pathway in myeloid cells.42 Another avenue for exploration involves tracking changes in TNC levels with response or resistance to generalized or targeted therapies. Still, these molecular targets needs to be better defined to develop new therapeutic approaches.
Function of TNC in Melanoma Cell Invasiveness
TNC contains 14.5 EGF-like repeats, of which at least 4 including the last full one bind as low-affinity/high-avidity ligands to EGFR, resulting in a sustained surface-restricted EGFR signaling.18,19,43 This mode of signaling is preferential for motility over proliferation as it activates the motogenic pathways of PLCgamma for hydrolysis of phosphoinositides at the front, and m-calpain cleavage of adhesion-related proteins for rear release.7,44
Most recently, our lab found that TNC, produced by melanoma cells in addition to fibroblasts, localizes in the front of melanoma cells invading into an ex vivo matrix.7 Thus, we queried whether this drove invasion. Interestingly, overexpression of a TNC fragment which consisted of the N-terminal assembly domain and the full EGFL repeats in melanoma cells resulted in reduced cell migration speed and persistence on a 2D in vitro wound healing area; this parameter is usually correlated with increased invasion. Of note, there was also delayed cell attachment and spreading when plated onto collagen, suggesting a lessened adhesiveness. This impaired adhesion of melanoma cells expressing TNC EGFL is at least in part due to the increased Rho-associated kinase (ROCK) activity and myosin light chain 2 as the dual phosphorylation of myosin light chain 2 at Thr-18 and Ser-19 is more constant compared to cells expressing an empty vector. Thus when tested in matrix invasion, expression of TNC EGFL in melanoma cells resulted in amoeboidal morphology concomitant with an increased movement into the matrix. The amoeboid morphology is consistent with other barrier penetrating tumor cells.15,45 This finding suggests that the TNC EGFL promotes vertical invasion through the underlying dermis from primary melanoma. Thus, this behavior, of amoeboidal migration could be a novel target to limit melanoma invasion and dissemination.7
Role of TNC in Melanoma Cell Survival
It is plausible that TNC also promotes the survival of melanoma cells based on the above cited data on MSC.6 This would be beneficial at the metastatic site where tumor cells face an ectopic microenvironment that lacks the normal trophic factors and that the mere onset of invasion triggers a non-specific foreign body response of death promoting cytokines.46 This is consistent with the upregulation of TNC throughout metastatic nodules,47,48 though this may represent persistent expression by the invasive cells that attained the distant site rather than an adaptive expression to promote tumorigenic behaviors in the metastatic site.
For directives as to which, there are data on the presumed cancer stem cell phenotype49,50 being modulated by TNC. Melanoma has been suggested to contain such stem cell-like populations.51 In line with this finding, Herlyn's group recently reported that expression of TNC in melanoma cells that grew in a 3D spheres, in which stem-like cells are enriched, is significantly upregulated compared to adherent cells.52 As a consequence, highly expressed TNC created a specific environment for stem-cell like melanoma cells to promote tumor growth and evade conventional therapy. Downregulation of TNC in melanoma cells by shRNA dramatically inhibited the growth of melanoma sphere and lowered their resistance to doxorubicin treatment. This finding implicates that TNC plays an important role in maintaining stem cell-like population and may extend to small clusters and nodules in ectopic sites. However, this role of TNC remains speculative even if strongly correlative, lacking solid experimental validation.
Conclusion or Perspective
Reports implicate the likelihood that TNC plays important roles both for invasion through the dermis at the primary site, and for survival at distant sites in melanoma metastasis (Fig. 3B). These behaviors relate in large part to the EGFL of TNC signaling via the EGFR in a unique manner to promote both motility and survival rather than proliferation.6,18,22 Still, while the findings both in human melanoma biopsies and from the laboratory experiments are highly suggestive, an experimental demonstration supporting this hypothesis is still lacking. Thus, future efforts should focus on isolating such behaviors in ex vivo organotypic microphysiological systems and animal models.
A second, but critical aspect of these implications of TNC is how to approach the patients afflicted with melanoma now. Obviously if the foundation model is borne out experimentally, new therapies could be developed and tested based on key trigger points such as the induction of TNC expression or downstream signals in the melanoma cells. This would take time, but offer a new avenue of intervention. More immediately, the approach to melanoma can be altered upon appreciation that TNC upregulation protects melanoma cells from current general cytotoxic therapies and even targeted biologics such as the B-Raf and MEK inhibitors (via secondary pathways from EGFR via AKT). One concept may be to directly evaluate this possibility, and to target this mechanism of survival advantage, possibly using approved EGFR inhibitors, in conjunction with current therapies to improve their efficacy. As TNC upregulation is a general feature of invasive/metastatic solid tumors, and not limited to melanoma, such a combinatorial approach could be useful in other cancers. As such our knowledge of the basic tumor biology of TNC and tumor progression along with chemoresistance may be more rapidly applicable to the bedside using already-existing tools.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed
Funding
Preparation of this article was supported by grants from National Institutes of Health (NIGMS), the VA Merit Award Program, the SPORE P50 CA121973–07. We thank the Wells lab, and Drs. Lisa Butterfield and Dorothea Becker for helpful discussions.
References
- 1. Sullivan RJ, Fisher DE. Understanding the Biology of Melanoma and Therapeutic Implications. Hematol Oncol Clin North Am 2014; 28:437-53; PMID:24880940; http://dx.doi.org/ 10.1016/j.hoc.2014.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Elenbaas B, Weinberg RA. Heterotypic signaling between epithelial tumor cells and fibroblasts in carcinoma formation. Exp Cell Res 2001; 264:169-84; PMID:11237532; http://dx.doi.org/ 10.1006/excr.2000.5133 [DOI] [PubMed] [Google Scholar]
- 3. Midwood KS, Orend G. The role of tenascin-C in tissue injury and tumorigenesis. J Cell Commun Signal 2009; 3:287-310; PMID:19838819; http://dx.doi.org/ 10.1007/s12079-009-0075-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Engel J. EGF-like domains in extracellular matrix proteins: localized signals for growth and differentiation? FEBS Lett 1989; 251:1-7; PMID:2666164; http://dx.doi.org/ 10.1016/0014-5793(89)81417-6 [DOI] [PubMed] [Google Scholar]
- 5. Aukhil I, Joshi P, Yan Y, Erickson HP. Cell- and heparin-binding domains of the hexabrachion arm identified by tenascin expression proteins. J Biol Chem 1993; 268:2542-53; PMID:7679097 [PubMed] [Google Scholar]
- 6. Rodrigues M, Yates CC, Nuschke A, Griffith L, Wells A. The matrikine tenascin-C protects multipotential stromal cells/mesenchymal stem cells from death cytokines such as FasL. Tissue Eng Part A 2013; 19:1972-83; PMID:23541003; http://dx.doi.org/ 10.1089/ten.tea.2012.0568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Grahovac J, Becker D, Wells A. Melanoma cell invasiveness is promoted at least in part by the epidermal growth factor-like repeats of tenascin-C. J Invest Dermatol 2013; 133:210-20; PMID:22951722; http://dx.doi.org/ 10.1038/jid.2012.263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Saito Y, Imazeki H, Miura S, Yoshimura T, Okutsu H, Harada Y, Ohwaki T, Nagao O, Kamiya S, Hayashi R, et al. . A peptide derived from tenascin-C induces beta1 integrin activation through syndecan-4. J Biol Chem 2007; 282:34929-37; PMID:17901052; http://dx.doi.org/ 10.1074/jbc.M705608200 [DOI] [PubMed] [Google Scholar]
- 9. Joshi P, Chung CY, Aukhil I, Erickson HP. Endothelial cells adhere to the RGD domain and the fibrinogen-like terminal knob of tenascin. J Cell Sci 1993; 106(Pt 1):389-400; PMID:7505785 [DOI] [PubMed] [Google Scholar]
- 10. Bourdon MA, Ruoslahti E. Tenascin mediates cell attachment through an RGD-dependent receptor. J Cell Biol 1989; 108:1149-55; PMID:2466038; http://dx.doi.org/ 10.1083/jcb.108.3.1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grahovac J, Wells A. Matrikine and matricellular regulators of EGF receptor signaling on cancer cell migration and invasion. Lab Invest 2014; 94:31-40; PMID:24247562; http://dx.doi.org/ 10.1038/labinvest.2013.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fowler WE, Erickson HP. Trinodular structure of fibrinogen. Confirmation by both shadowing and negative stain electron microscopy. J Mol Biol 1979; 134:241-9; PMID:537064; http://dx.doi.org/ 10.1016/0022-2836(79)90034-2 [DOI] [PubMed] [Google Scholar]
- 13. Hancox RA, Allen MD, Holliday DL, Edwards DR, Pennington CJ, Guttery DS, et al. . Tumour-associated tenascin-C isoforms promote breast cancer cell invasion and growth by matrix metalloproteinase-dependent and independent mechanisms. Breast Cancer Res 2009; 11:R24; PMID:19405959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kharait S, Hautaniemi S, Wu S, Iwabu A, Lauffenburger DA, Wells A. Decision tree modeling predicts effects of inhibiting contractility signaling on cell motility. BMC Syst Biol 2007; 1:9; PMID:17408516; http://dx.doi.org/ 10.1186/1752-0509-1-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zaman MH, Trapani LM, Sieminski AL, Mackellar D, Gong H, Kamm RD, Wells A, Lauffenburger DA, Matsudaira P. Migration of tumor cells in 3D matrices is governed by matrix stiffness along with cell-matrix adhesion and proteolysis. Proc Natl Acad Sci U S A 2006; 103:10889-94; PMID:16832052; http://dx.doi.org/ 10.1073/pnas.0604460103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Friedl P, Maaser K, Klein CE, Niggemann B, Krohne G, Zanker KS. Migration of highly aggressive MV3 melanoma cells in 3-dimensional collagen lattices results in local matrix reorganization and shedding of alpha2 and beta1 integrins and CD44. Cancer Res 1997; 57:2061-70; PMID:9158006 [PubMed] [Google Scholar]
- 17. Friedl P, Sahai E, Weiss S, Yamada KM. New dimensions in cell migration. Nat Rev Mol Cell Biol 2012; 13:743-7; PMID:23072889; http://dx.doi.org/ 10.1038/nrm3459 [DOI] [PubMed] [Google Scholar]
- 18. Swindle CS, Tran KT, Johnson TD, Banerjee P, Mayes AM, Griffith L, Wells A. Epidermal growth factor (EGF)-like repeats of human tenascin-C as ligands for EGF receptor. J Cell Biol 2001; 154:459-68; PMID:11470832; http://dx.doi.org/ 10.1083/jcb.200103103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Iyer AK, Tran KT, Borysenko CW, Cascio M, Camacho CJ, Blair HC, Bahar I, Wells A. Tenascin cytotactin epidermal growth factor-like repeat binds epidermal growth factor receptor with low affinity. J Cell Physiol 2007; 211:748-58; PMID:17311283; http://dx.doi.org/ 10.1002/jcp.20986 [DOI] [PubMed] [Google Scholar]
- 20. Schenk S, Hintermann E, Bilban M, Koshikawa N, Hojilla C, Khokha R, Quaranta V. Binding to EGF receptor of a laminin-5 EGF-like fragment liberated during MMP-dependent mammary gland involution. J Cell Biol 2003; 161:197-209; PMID:12695504; http://dx.doi.org/ 10.1083/jcb.200208145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schenk S, Quaranta V. Tales from the crypt; [ic] sites of the extracellular matrix. Trends Cell Biol 2003; 13:366-75; PMID:12837607; http://dx.doi.org/ 10.1016/S0962-8924(03)00129-6 [DOI] [PubMed] [Google Scholar]
- 22. Iyer AK, Tran KT, Griffith L, Wells A. Cell surface restriction of EGFR by a tenascin cytotactin-encoded EGF-like repeat is preferential for motility-related signaling. J Cell Physiol 2008; 214:504-12; PMID:17708541; http://dx.doi.org/ 10.1002/jcp.21232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Haugh JM, Huang AC, Wiley HS, Wells A, Lauffenburger DA. Internalized epidermal growth factor receptors participate in the activation of p21(ras) in fibroblasts. J Biol Chem 1999; 274:34350-60; PMID:10567412; http://dx.doi.org/ 10.1074/jbc.274.48.34350 [DOI] [PubMed] [Google Scholar]
- 24. Haugh JM, Schooler K, Wells A, Wiley HS, Lauffenburger DA. Effect of epidermal growth factor receptor internalization on regulation of the phospholipase C-gamma1 signaling pathway. J Biol Chem 1999; 274:8958-65; PMID:10085141; http://dx.doi.org/ 10.1074/jbc.274.13.8958 [DOI] [PubMed] [Google Scholar]
- 25. Glading A, Uberall F, Keyse SM, Lauffenburger DA, Wells A. Membrane proximal ERK signaling is required for M-calpain activation downstream of epidermal growth factor receptor signaling. J Biol Chem 2001; 276:23341-8; PMID:11319218; http://dx.doi.org/ 10.1074/jbc.M008847200 [DOI] [PubMed] [Google Scholar]
- 26. Fan VH, Tamama K, Au A, Littrell R, Richardson LB, Wright JW, Wells A, Griffith LG. Tethered epidermal growth factor provides a survival advantage to mesenchymal stem cells. Stem Cells 2007; 25:1241-51; PMID:17234993; http://dx.doi.org/ 10.1634/stemcells.2006-0320 [DOI] [PubMed] [Google Scholar]
- 27. Tamama K, Fan VH, Griffith LG, Blair HC, Wells A. Epidermal growth factor as a candidate for ex vivo expansion of bone marrow-derived mesenchymal stem cells. Stem Cells 2006; 24:686-95; PMID:16150920; http://dx.doi.org/ 10.1634/stemcells.2005-0176 [DOI] [PubMed] [Google Scholar]
- 28. Rakosy Z, Vizkeleti L, Ecsedi S, Voko Z, Begany A, Barok M, Krekk Z, Gallai M, Szentirmay Z, Adány R, et al. . EGFR gene copy number alterations in primary cutaneous malignant melanomas are associated with poor prognosis. Int J Cancer 2007; 121:1729-37; PMID:17594688; http://dx.doi.org/ 10.1002/ijc.22928 [DOI] [PubMed] [Google Scholar]
- 29. Timar J, Gyorffy B, Raso E. Gene signature of the metastatic potential of cutaneous melanoma: too much for too little? Clin Exp Metastasis 2010; 27:371-87; PMID:20177751; http://dx.doi.org/ 10.1007/s10585-010-9307-2 [DOI] [PubMed] [Google Scholar]
- 30. Tucker RP, Chiquet-Ehrismann R. The regulation of tenascin expression by tissue microenvironments. Biochim Biophys Acta 2009; 1793:888-92; PMID:19162090; http://dx.doi.org/ 10.1016/j.bbamcr.2008.12.012 [DOI] [PubMed] [Google Scholar]
- 31. Natali PG, Nicotra MR, Bartolazzi A, Mottolese M, Coscia N, Bigotti A, Zardi L. Expression and production of tenascin in benign and malignant lesions of melanocyte lineage. Int J Cancer 1990; 46:586-90; PMID:1698727; http://dx.doi.org/ 10.1002/ijc.2910460406 [DOI] [PubMed] [Google Scholar]
- 32. Tuominen H, Kallioinen M. Increased tenascin expression in melanocytic tumors. J Cutan Pathol 1994; 21:424-9; PMID:7532653; http://dx.doi.org/ 10.1111/j.1600-0560.1994.tb00284.x [DOI] [PubMed] [Google Scholar]
- 33. Brellier F, Martina E, Degen M, Heuze-Vourc'h N, Petit A, Kryza T, Courty Y, Terracciano L, Ruiz C, Chiquet-Ehrismann R. Tenascin-W is a better cancer biomarker than tenascin-C for most human solid tumors. BMC Clin Pathol 2012; 12:14; PMID:22947174; http://dx.doi.org/ 10.1186/1472-6890-12-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hood BL, Grahovac J, Flint MS, Sun M, Charro N, Becker D, Wells A, Conrads TP. Proteomic analysis of laser microdissected melanoma cells from skin organ cultures. J Proteome Res 2010; 9:3656-63; PMID:20459140; http://dx.doi.org/ 10.1021/pr100164x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hoek K, Rimm DL, Williams KR, Zhao H, Ariyan S, Lin A, Kluger HM, Berger AJ, Cheng E, Trombetta ES, et al. . Expression profiling reveals novel pathways in the transformation of melanocytes to melanomas. Cancer Res 2004; 64:5270-82; PMID:15289333; http://dx.doi.org/ 10.1158/0008-5472.CAN-04-0731 [DOI] [PubMed] [Google Scholar]
- 36. Kaariainen E, Nummela P, Soikkeli J, Yin M, Lukk M, Jahkola T, Virolainen S, Ora A, Ukkonen E, Saksela O, et al. . Switch to an invasive growth phase in melanoma is associated with tenascin-C, fibronectin, and procollagen-I forming specific channel structures for invasion. J Pathol 2006; 210:181-91; PMID:16924594; http://dx.doi.org/ 10.1002/path.2045 [DOI] [PubMed] [Google Scholar]
- 37. Adam B, Toth L, Pasti G, Balazs M, Adany R. Contact stimulation of fibroblasts for tenascin production by melanoma cells. Melanoma Res 2006; 16:385-91; PMID:17013087; http://dx.doi.org/ 10.1097/01.cmr.0000205022.25397.86 [DOI] [PubMed] [Google Scholar]
- 38. Herlyn M, Graeven U, Speicher D, Sela BA, Bennicelli JL, Kath R, Guerry D,. Characterization of tenascin secreted by human melanoma cells. Cancer Res 1991; 51:4853-8; PMID:1716515 [PubMed] [Google Scholar]
- 39. Burchardt ER, Hein R, Bosserhoff AK. Laminin, hyaluronan, tenascin-C and type VI collagen levels in sera from patients with malignant melanoma. Clin Exp Dermatol 2003; 28:515-20; PMID:12950343; http://dx.doi.org/ 10.1046/j.1365-2230.2003.01326.x [DOI] [PubMed] [Google Scholar]
- 40. Maschler S, Grunert S, Danielopol A, Beug H, Wirl G. Enhanced tenascin-C expression and matrix deposition during Ras/TGF-β-induced progression of mammary tumor cells. Oncogene 2004; 23:3622-33; PMID:15116096; http://dx.doi.org/ 10.1038/sj.onc.1207403 [DOI] [PubMed] [Google Scholar]
- 41. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. . Mutations of the BRAF gene in human cancer. Nature 2002; 417:949-54; PMID:12068308; http://dx.doi.org/ 10.1038/nature00766 [DOI] [PubMed] [Google Scholar]
- 42. Goh FG, Piccinini AM, Krausgruber T, Udalova IA, Midwood KS. Transcriptional regulation of the endogenous danger signal tenascin-C: a novel autocrine loop in inflammation. J Immunol 2010; 184:2655-62; PMID:20107185; http://dx.doi.org/ 10.4049/jimmunol.0903359 [DOI] [PubMed] [Google Scholar]
- 43. Tran KT, Griffith L, Wells A. Extracellular matrix signaling through growth factor receptors during wound healing. Wound Repair Regen 2004; 12:262-8; PMID:15225204; http://dx.doi.org/ 10.1111/j.1067-1927.2004.012302.x [DOI] [PubMed] [Google Scholar]
- 44. Wells A. Tumor invasion: role of growth factor-induced cell motility. Adv Cancer Res 2000; 78:31-101. [DOI] [PubMed] [Google Scholar]
- 45. Friedl P, Gilmour D. Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol 2009; 10:445-57; PMID:19546857; http://dx.doi.org/ 10.1038/nrm2720 [DOI] [PubMed] [Google Scholar]
- 46. Gunasinghe NP, Wells A, Thompson EW, Hugo HJ. Mesenchymal-epithelial transition (MET) as a mechanism for metastatic colonisation in breast cancer. Cancer Metastasis Rev 2012; 31:469-78; PMID:22729277; http://dx.doi.org/ 10.1007/s10555-012-9377-5 [DOI] [PubMed] [Google Scholar]
- 47. Talts JF, Wirl G, Dictor M, Muller WJ, Fassler R. Tenascin-C modulates tumor stroma and monocyte/macrophage recruitment but not tumor growth or metastasis in a mouse strain with spontaneous mammary cancer. J Cell Sci 1999; 112 (Pt 12):1855-64; PMID:10341205 [DOI] [PubMed] [Google Scholar]
- 48. O'Connell JT, Sugimoto H, Cooke VG, MacDonald BA, Mehta AI, LeBleu VS, Dewar R, Rocha RM, Brentani RR, Resnick MB, et al. . VEGF-A and Tenascin-C produced by S100A4+ stromal cells are important for metastatic colonization. Proc Natl Acad Sci USA 2011; 108:16002-7; PMID:21911392; http://dx.doi.org/ 10.1073/pnas.1109493108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature 2001; 414:105-11; PMID:11689955; http://dx.doi.org/ 10.1038/35102167 [DOI] [PubMed] [Google Scholar]
- 50. Wicha MS. Cancer stem cells and metastasis: lethal seeds. Clin Cancer Res 2006; 12:5606-7; PMID:17020960; http://dx.doi.org/ 10.1158/1078-0432.CCR-06-1537 [DOI] [PubMed] [Google Scholar]
- 51. Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature 2008; 456:593-8; PMID:19052619; http://dx.doi.org/ 10.1038/nature07567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fukunaga-Kalabis M, Martinez G, Nguyen TK, Kim D, Santiago-Walker A, Roesch A, Herlyn M. Tenascin-C promotes melanoma progression by maintaining the ABCB5-positive side population. Oncogene 2010; 29:6115-24; PMID:20729912; http://dx.doi.org/ 10.1038/onc.2010.350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rodrigues M, Griffith LG, Wells A. Growth factor regulation of proliferation and survival of multipotential stromal cells. Stem Cell Res Ther 2010; 1:32; PMID:20977782; http://dx.doi.org/ 10.1186/scrt32 [DOI] [PMC free article] [PubMed] [Google Scholar]