

Abstract
A phase Ib/II trial was performed to evaluate safety, tolerability, recommended dose (RD) and efficacy of F16-IL2, a recombinant antibody-cytokine fusion protein, in combination with doxorubicin in patients with solid tumors (phase Ib) and metastatic breast cancer (phase II). Six patient cohorts with progressive solid tumors (n = 19) received escalating doses of F16-IL2 [5–25 Million International Units (MIU) of IL2 equivalent dose] in combination with escalating doses of doxorubicin (0–25 mg/m2) on day 1, 8 and 15 every 4 weeks. Subsequently, patients with metastatic breast cancer (n = 10) received the drug combination at the RD. Clinical data and laboratory findings were analyzed for safety, tolerability, and activity. F16-IL2 could be administered up to 25 MIU, in combination with the RD of doxorubicin (25 mg/m2). No human anti-fusion protein antibodies (HAFA) response was detected. Pharmacokinetics of F16-IL2 was dose-dependent over the tested range, with half-lives of ca. 13 and ca. 8 hours for cohorts dosed at lower and higher levels, respectively. Toxicities were controllable and reversible, with no combination treatment-related death. After 8 weeks, 57% and 67% disease control rates were observed for Phase I and II, respectively (decreasing to 43% and 33% after 12 weeks), considering 14 and 9 patients evaluable for efficacy. One patient experienced a long lasting partial response (45 weeks), still on-going at exit of study. F16-IL2 can be safely and repeatedly administered at the RD of 25 MIU in combination with 25 mg/m2 doxorubicin; its safety and activity are currently being investigated in combination with other chemotherapeutics, in order to establish optimal therapy settings.
Keywords: antibody, breast neoplasms, clinical trial phase I, immunocytokines, interleukin-2
Introduction
Interleukin-2 (IL2) is a proinflammatory cytokine, which is normally produced during an immune response, activating helper T-cells, cytotoxic T-cells, B-cells, natural killer (NK) cells, and macrophages.1 Human recombinant IL2 (Proleukin®, Novartis) is approved by US Food and Drug Administration (FDA) for the treatment of metastatic renal cell cancer,2 and metastatic melanoma.3 However, IL2 treatment is limited by its own toxicity profile; the most frequently reported side effects include capillary leak syndrome, resulting in hypotension, renal dysfunction with oliguria/anuria, pulmonary congestion, and mental status changes.2 These severe adverse events warrant a thorough clinical evaluation and the administration of human recombinant IL2 in a hospital setting under adequate medical supervision.
Cytokines do not preferentially localize to the tumor site after intravenous administration, resulting in serious side effects in vivo, which prevent dose escalation to the concentrations that are needed to achieve curative anti-tumor effects. A way to control the systemic side effects of cytokine administration is driving the cytokines to the tumor site by the antibody-mediated targeted delivery.4-6 With such approach, antibodies are used as modular components for the preparation of fusion proteins (“immunocytokines”), which allow the selective localization of the cytokine payload on neoplastic masses.
F16-IL2 is a noncovalent homodimeric recombinant fusion protein consisting of a human antibody fragment specific to the A1 domain of tenascin-C in the single chain fragment variable (scFV) format, named F16, and of the human cytokine IL2. Tenascin-C is a glycoprotein of the extracellular matrix. It comprises several fibronectin type 3 homology repeats that can be either included or omitted in the primary transcript by alternative splicing, leading to small and large isoforms that have distinct biological functions (Fig. 1).7,8 Whereas the small isoform is expressed in several tissues, the large isoform of tenascin-C exhibits a restricted pattern of expression. It is virtually undetectable in healthy adult tissues but is expressed during embryogenesis and is re-expressed in adult tissues undergoing tissue remodeling, including neoplasia. Its expression is localized around vascular structures in the tumor stroma of a variety of different tumors, including breast carcinoma,7 oral squamous cell carcinoma,9 lung cancer,10 prostatic adenocarcinoma,11 colorectal cancer,12 or astrocytoma and other brain tumors.13,14 Using MDA-MB-231xenograft model of human breast cancer, F16-IL2 has been shown to selectively deliver IL2 to the cancer sites by localizing to tumor tissues.15
Figure 1.
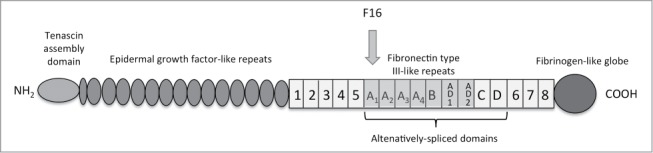
Domain structure of Tenascin- C
The clinical development of F16-IL2 was supported by preclinical studies performed in mice and toxicology studies completed in cynomolgus monkeys, which indicated that F16-IL2 is able to substantially increase the therapeutic efficacy of combined chemotherapy, and that did not raise any safety concerns.15
This is the first clinical study of F16-IL2 in combination with doxorubicin conducted in cancer patients. Doxorubicin is a well-characterized chemotherapeutic agent, which has been commonly used in the treatment of a wide range of cancers, including hematological malignancies, many types of carcinoma and soft tissue sarcomas.16 Because of known activity of F16-IL2 in preclinical models of breast cancer,15 and due to confirmed expression of the large isoform of Tenascin-C in this tumor type,17 breast cancer patients were included in the phase II part of the study, after definition of the dose in solid cancer patients. Moreover, additional clinical experimental evidence of the ability of the F16 antibody to selectively target breast cancer was provided by a radio-immunotherapeutic clinical study in patients with cancer (Supplementary Fig. 1).
Results
Patients’ characteristics
Nineteen patients with progressive solid tumors were enrolled in the phase Ib part of the study (Table 1) and treated with increasing doses of F16-IL2 (Fig. 2A). All patients were evaluable for safety, and 14 for anticancer activity.
Table 1.
Patients’ demographics
| Characteristics | Phase I (n) | Phase II (n) |
|---|---|---|
| Number of enrolled patients | 19 | 10 |
| Median (and range) age at study entry (years) | 63 (32–75) | 58 (40–77) |
| Male/Female | 9/10 | 0/10 |
| ECOG PS at baseline0/1 | 11/8 | 5/4 |
| Cancer diagnosis | ||
| Breast cancer | 3 | 10 |
| Colorectal cancer | 2 | |
| Endometrial cancer | 2 | |
| Pancreas cancer | 2 | |
| Prostate cancer | 2 | |
| Malignant vascular tumor | 2 | |
| Cancer of tongue | 1 | |
| Choroidal melanoma | 1 | |
| Pleural cancer | 1 | |
| Tyhmoma | 1 | |
| Thyroid cancer | 1 | |
| Cervix uteri cancer | 1 | |
| Prior Therapy | ||
| Surgery | 14 | 9 |
| Chemotherapy | 17a | 10a |
| Radiotherapy | 9 | 8 |
Median number of prior chemotherapy regimens.
a2 (range 1–12).
b3 (range 1–7).
Figure 2.

Study design and treatment schedule (A) Study design (B) Treatment schedule. Patients were screened up to 14 d prior to beginning of study. Each cycle of treatment comprises F16-IL2 administration on days 1, 8 and 15 (indicated as black arrows), and doxorubicin administrations on the same days (indicated as gray arrows), followed by 13 d rest (total duration of one cycle is 28 d). Patients could receive up to 6 treatment cycles. The tumor assessments (indicated by dotted lines) were performed according to RECIST: the lesions were measured at screening and at the end of cycle 2, 4 and 6.
Ten patients with metastatic breast cancer were enrolled in the phase II part of the study (Table 1) and treated with 25 MIU of F16-IL2 in combination with 25 mg/m2 of doxorubicin (Fig. 2A).
All patients were evaluable for safety, and 9 for anticancer activity. Two out of 3 breast cancer patients for phase I and 3 out of 10 patients for phase II were treated in the past with adjuvant anthracycline-based chemotherapy. A detailed list of patient demography data and characteristics, including cancer diagnosis and prior therapies, is given in Table 1.
Dose finding and safety
The safety study population includes all 29 patients who received at least one drug administration of F16IL2 (26 patients received at least one administration of doxorubicin). Four patients were recruited at the second dose level, as one discontinued treatment before completion of cycle 1 for an early progression. No F16-IL2 dose reduction was reported, while 11 patients overall (7/26 patients allocated to 25 mg/m2 doxorubicin: 4/16 for Phase I and 3/10 for Phase II) were treated with at least one reduced dosage of doxorubicin (i.e., inferior to 75% of the original dose, according to protocol), and one patient (1/26) received 80% of the originally administered doxorubicin dose, according to the investigator's judgment.
The complete list of treatment-related adverse events is depicted in Table 2; the most frequent adverse events included asthenia, pyrexia, and constipation.
Table 2.
Incidence of adverse events (AEs)
| Patients with related adverse events grade <3 and ≥3 by dose level |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5 MIU (n = 3) |
5 MIU 20 mg/m2 doxo (n = 4) |
5 MIU 25 mg/m2 doxo (n = 3) |
10 MIU 25 mg/m2 doxo (n = 3) |
15 MIU 25 mg/m2 doxo (n = 3) |
25 MIU 25 mg/m2 doxo (n = 13) |
Any Dose (n = 29) |
|||||||
| Related AEs by system organ class | G<3 | G≥3 | G<3 | G≥3 | G<3 | G≥3 | G<3 | G≥3 | G<3 | G≥3 | G<3 | G≥3 | All Grade |
| Blood and lymphatic system disorders | 1 | 1 | 1 | 1 | 10 | 7 | 14 | ||||||
| Cardiac disorders | 1 | 1 | 2 | ||||||||||
| Eye disorders | 1 | 1 | |||||||||||
| Gastrointestinal disorders | 1 | 2 | 2 | 1 | 9 | 1 | 15 | ||||||
| General disorders and administration site conditions | 2 | 2 | 1 | 1 | 1 | 2 | 10 | 3 | 19 | ||||
| Investigations | 1 | 1 | 2 | ||||||||||
| Metabolism and nutrition disorders | 1 | 1 | 2 | 2 | 4 | ||||||||
| Muscoskeletal and connective tissue disorders | 1 | 1 | |||||||||||
| Nervous system disorders | 1 | 1 | |||||||||||
| Renal and urinary disorders | 1 | 1 | |||||||||||
| Reproductive system and breast disorders | 1 | 1 | |||||||||||
| Respiratory, thoracic and mediastinal disorders | 1 | 1 | 2 | ||||||||||
| Skin and subcutaneous tissue disorders | 2 | 3 | 5 | ||||||||||
| Vascular disorders | 1 | 1 | 2 | ||||||||||
The toxicities were graded according to Common Terminology Criteria for Adverse Events v 3.0. The System Organ Class was coded using the Medical Dictionary for Regulatory Activities. Blood and lymphatic system disorders include anaemia, neutropenia, and leucopenia; cardiac disorders include pericardial effusion and tachycardia; eye disorders include conjunctivitis; gastrointestinal disorders include constipation, vomiting, nausea, abdominal pain, stomatitis, gastritis, and oesophagitis; general disorders and administration site conditions include asthenia, pyrexia, fatigue, flu-like syndrome, and mucosal inflammation; investigations include weight decrease and increase of alanine aminotransferase, aspartate aminotransferase; metabolism and nutrition disorders include anorexia and hypokalaemia; muscoloskeletal and connective tissue disorders include arthralgia; nervous system disorders include headache; renal and urinary disorders include hyperazotemia; reproductive system and breast disorders include pelvic pain; respiratory, thoracic and mediastinal disorders include dyspnoea, sinusitis; skin and subcutaneous tissue disorders include alopecia, palmar-plantar erythrodysaestesia syndrome, and nail disorders; vascular disorders include hypertension and phlebitis.
No dose-limiting toxicity (DLT) was observed in this trial and no expansion of the dose cohorts was needed to further explore the safety of the drug combination.
In a 76-year-old female patient with pancreatic cancer enrolled in the 4th cohort, grade 3 (G3) febrile neutropenia was observed; the patient later recovered and stopped the treatment; such event was assessed as serious and expected.
No case was considered a serious, unexpected suspected adverse event (SUSAR) and no deaths related to the combination therapy were reported during the study.
Pharmacokinetic and HAFA
Figure 3 shows the mean concentrations (and standard deviations) of F16-IL2 by dose group, during week 1 and week 2 of the first cycle of treatment. The maximum concentration (Cmax) of F16-IL2 increased dose-proportionally during week 1 but not during week 2. Cmax occurred almost within 1 hour from the start of the intravenous infusion. Table 3 shows Cmax and half-life time per groups of dose assuming a non-compartmental approach. After reaching Cmax, the F16-IL2 concentration decreased with a terminal half-life of ca. 13 hours for cohorts dosed at lower levels and ca. 8 hours for cohorts dosed at higher levels.
Figure 3.
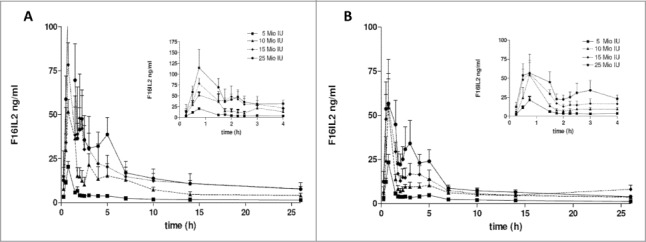
Pharmacokinetic analysis. Mean concentrations + standard deviation of F16-IL2 following administration of doxorubicin 25 mg/m2 and increasing doses of F16-IL2 (i.e., ♦ = 5 MIU, ○ = 10 MIU □ = 15 MIU ▪ = 25 MIU) are plotted versus time, by dose group; the dose is expressed in IL2 equivalents. Data were collected on day 1, corresponding to first F16-IL2 dose (A), and on day 8, corresponding to the administration of the second F16-IL2 dose (B), during the first cycle of treatment.
Table 3.
Cmax and T1/2 distribution by groups of dose
| Cmax (ng/mL) |
T1/2(hour) |
|||||||
|---|---|---|---|---|---|---|---|---|
| Dose MIU) | Nr. | Week | Min. | Median | Max. | Min. | Median | Max. |
| 5 | 10 | 1 | 7.52 | 22.04 | 35.56 | 3.91 | 13.93 | 23.15 |
| 10 | 2 | 9.32 | 16.67 | 46.60 | 3.40 | 13.36 | 24.33 | |
| 10 | 3 | 1 | 51.69 | 63.95 | 70.61 | 10.07 | 11.86 | 16.55 |
| 3 | 2 | 29.14 | 49.70 | 84.64 | 13.11 | 16.01 | 17.30 | |
| 15 | 3 | 1 | 56.64 | 78.52 | 100.05 | 8.93 | 11.06 | 11.36 |
| 3a | 2 | 39.62 | 58.02 | 103.81 | 8.86 | 11.35 | 13.84 | |
| 25 | 8 | 1 | 24.53 | 86.93 | 404.41 | 6.30 | 8.83 | 25.85 |
| 6 | 2 | 55.69 | 104.29 | 125.55 | 6.29 | 7.40 | 9.95 | |
Cmax and T1/2 minimum, maximum and median data by groups of dose and week of sampling. The T1/2 was calculated following a non-compartmental approach.
aThe T1/2 was calculated on a population of only 2 patients.
Immunogenic response against F16-IL2 induced after treatment of patients was investigated using ELISA methodology and Surface Plasmon Resonance (SPR), on Biacore®T100. A total of 46 serum samples from 19 patients treated repeatedly with F16-IL2 were tested for the presence of HAFA specific to F16-IL2.
All analyzed samples in the sandwich ELISA resulted negative. SPR detected in one patient a positive result for the end of treatment sample, with a very low signal correlated to presence of IgG antibodies, the subsequent time point at the follow up visit was negative.
Activity
Response was measured at week 8 after completion of 6 administrations of both F16-IL2 and doxorubicin. Patients with stable or responding disease received combination therapy for up to 6 months.
Patients with clinical diagnosis of progressive disease (PD) (e.g., symptomatic deterioration without proof of progressive disease) were withdrawn from the study treatment. Patients evaluated as PD at the first tumor assessment but still considered in good clinical condition continued treatment for 2 additional cycles at the Investigator's discretion.
Two out of 29 enrolled patients were replaced; one patient of the 2nd cohort had an early disease progression, and one patient enrolled during the phase II part of the study had a septic shock, possibly related to doxorubicin; both patients discontinued the treatment before completion of cycle 1 and could therefore be replaced, according to protocol. Three more patients discontinued the treatment after completion of cycle 1 but before reaching the first tumor assessment time point (i.e., end of cycle 2), and therefore could not be evaluated for activity.
After 2 cycles of treatment, the disease control rate was about 57% considering 14 evaluable patients during the phase Ib (14 patients with tumor assessment 8 weeks post baseline, 15 patients with at least one tumor assessment available post baseline), and about 67% considering 9 evaluable patients during the phase II part of the study. The disease control rates decreased to 43% and 33% after 12 weeks, respectively for Phase I and II. Among these latter 6 patients of phase II, one further improved into a long lasting partial response (up to 312 days). No complete response (CR) and partial response (PR) were observed in Phase Ib; 2/3 patient showed stable disease (SD) in the first cohort 5 MIU F16-IL2, 2/3 in the cohort 5 MIU F16-IL2 and 20 mg/m2 Doxorubicin, 1/2 in the cohort 5 MIU F16-IL2 and 25 mg/m2 Doxorubicin, 2/2 in the cohort 10 MIU F16-IL2 and 25 mg/m2 Doxorubicin, 0/2 in the cohort 15 MIU F16-IL2 and 25 mg/m2 Doxorubicin and 1/2 in the cohort 25 MIU F16-IL2 and 25 mg/m2 Doxorubicin.
In the phase Ib patients, median progression free survival (PFS) was 110 d [based on 15 patients with at least one tumor assessment post baseline, censored n = 1 (patient with SD in the last assessment after 193 d); 95% confidence interval (CI) 76–287 d]; while in the phase II patients, median PFS was 125 d [based on 9 patients, censored n = 4 (3 patients with SD and 1 patient with PR at the last assessment); 95% CI 54-upper limit not evaluable; first quartile 80 d, third quartile 206 d].
Median overall survival (OS) was 293 d [based on 18 patients, censored n = 4; 95% CI 144 – 646 d] and 351 d [based on 9 patients, censored n = 4; 95% CI 58 – upper limit not evaluable], for patients enrolled in the phase Ib and in the phase II part of the study, respectively.
A waterfall plot indicating the best response for target lesions compared with baseline experienced by evaluable patients according to the clinical centers’ assessment is shown in Figure 4A. The plot reports the maximum percentage of shrinkage of target lesions according to Response Evaluation Criteria In Solid Tumors (RECIST),18 by F16-IL2 dose group (i.e., cohort 1 to 3 received 5 MIU; cohort 4 received 10 MIU; cohort 5 received 15 MIU; cohort 6 and phase II patients received 25 MIU). Selected examples of target lesion shrinkages are shown in Figure 4B and C; panel 4B reports the partial response of a breast cancer patient (No. 21), whose target lesion diameters reduced up to -59%, and whose response lasted (45 weeks) until patient began a new anticancer treatment. Panel 4C displays a hilar lymph node, a precarinal lymph node, and several liver lesions of a breast cancer patients (No. 23), which were kept stable (−11%) by the F16-IL2/doxorubicin combination for up to 19 weeks, until she withdrew from the study for progression of bone metastasis and beginning of radiotherapy.
Figure 4.
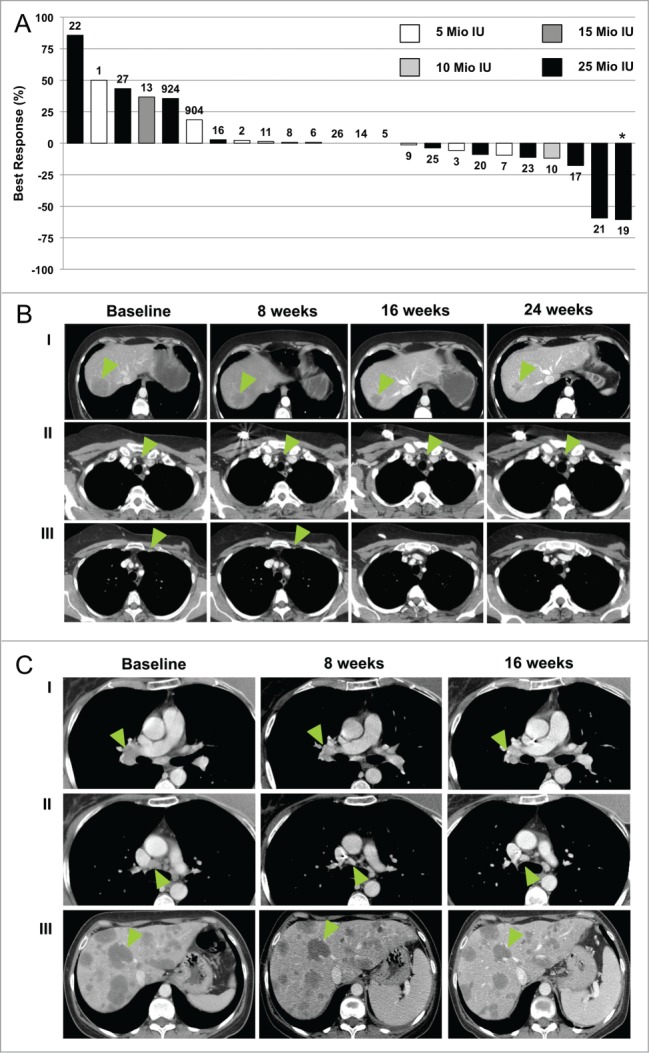
Waterfall Plot and radiological documentation of responses. (A) Best responses (defined as the largest shrinkage in the sum of diameters of target lesions at any moment in time, compared to baseline) of evaluable patients treated with the F16-IL2/doxorubicin combination are reported in plot. Patients 1–3 received 5 MIU IL2 equivalents of F16-IL2; patients 4–6 received 5 MIU of F16-IL2 plus 20 mg/m2 of doxorubicin; patients 7–9 received 5 MIU of F16-IL2 plus 25 mg/m2 of doxorubicin; patients 10–12 received 10 MIU of F16-IL2 plus 25 mg/m2 of doxorubicin; patients 13–15 received 15 MIU of F16-IL2 plus 25 mg/m2 of doxorubicin; patients 16–27 received 25 MIU of F16-IL2 plus 25 mg/m2 of doxorubicin. Patients No. 4, 12, 15, 18, and 24 are not reported in the graph since they were not evaluated for tumor response at any tumor assessment time point after baseline. Patient No. 4 discontinued treatment during cycle 1 due to worsening clinical conditions. He was replaced by Patient No. 904; Patient No. 12 withdrew from the study after cycle 1 for worsening of clinical condition; Patient No. 15 was assessed as progressive during cycle 1; Patient No. 18 discontinued treatment during the first 2 cycles due to severe adverse event (rectal hemorrhage); Patient No. 24 discontinued treatment after the second dose due to an adverse event and was replaced by Patient No. 924. Asterisks mark patients whose tumor shrinkage was accompanied by the development of new lesions, so that they were classified as PD. Computerized Tomography scan documentation of selected patients treated with the F16-IL2/doxorubicin combination are shown in (B and C). (B) CT images of a liver lesion (I), and 2 enlarged lymph nodes (II & III), indicated by the green arrows, of patient No. 21 (best response -59%) are shown. (C) CT images of the right hilar lymph node (I), a precarinal lymph node (II), and several liver lesions (III), of patient No. 23 (best response −11%), are shown. CT images were taken at baseline and at the following time-points indicated in figure.
Discussion
This is the ‘first-in-man’ study of the immunocytokine F16-IL2 combined with doxorubicin. The results of this phase Ib/II trial demonstrate that F16-IL2 can be safely administered at 25 MIU IL2 equivalents in combination with the RD of doxorubicin to patients with progressive solid tumors. The combination treatment was well tolerated and all reported toxicities were manageable and resolved completely. None of the 19 patients enrolled during phase Ib into the 6 planned cohorts experienced a DLT; this led to the administration, during the phase II part of the study, of the maximum administered dose (MAD; i.e., 25 MIU), without having formally defined the maximum tolerated dose (MTD).
The maximum dose of F16-IL2 to be administered in this trial was defined exploiting the analogy of this drug with the immunocytokine L19-IL2, which is composed of an antibody fragment specific to the tumor marker EDB domain of fibronectin and of human IL2.19,20 However, the comparison of the PK analysis of F16-IL2, presented in this trial, with the PK data collected in previous trials run with L19-IL2,19,20 highlighted few discrepancies between the 2 drugs, which could not be predicted on the basis of preclinical data, and which may lead to remarkable differences in the dose definition. Specifically, the Cmax values registered after infusion with the MAD of the 2 immunocytokines (i.e., 25 MIU IL2 equivalents for F16-IL2 and 22.5 MIU IL2 equivalents for L19-IL2) differ by a factor of approximately 10 (i.e., 100–120 ng/ml and 800–1200 ng/ml, for F16-IL2 and L19-IL2, respectively) indicating that the optimal effective dose of F16-IL2 might be significantly higher than the defined RD of L19-IL2.
In light of these safety and PK findings, this trial was prematurely stopped after the enrolment of 10 patients in the phase II part of the study (the original design foresaw the enrolment of 20 patient in the phase II), in order to refine the clinical development plan and in order to avoid treating patients with suboptimal F16-IL2 dosages.
Nevertheless, F16-IL2 has shown some preliminary signs of anti-cancer activity, when given in combination with doxorubicin, which may develop into even more striking effects once the optimal drug dosage is defined. Moreover, a similar trial which is currently ongoing, during which F16-IL2 is given in combination with paclitaxel to patients with solid tumors, is confirming the safety of the immunocytokine/chemotherapy combination and is showing even more pronounced benefits to the cancer patients in terms of both disease stabilization and tumor shrinkage.
In conclusion, the data presented here lay the foundation for further exploring the safety and activity of F16-IL2 and for future clinical trials with F16-IL2 in combination with approved anti-tumor drugs.
Materials and Methods
Study design and objectives
The trial here described was an open-label, non-randomized, phase I/II study, involving 4 Italian hospitals. During the dose escalation phase Ib part of the study, 3 to 6 patients were enrolled in sequential cohorts, corresponding to escalating doses of F16-IL2 (from 5 to 25 MIU of IL2 equivalent), in combination with escalating doses of doxorubicin (Ebewe Italia S.r.l., Rome, Italy; from 0 to 25 mg/m2). Escalation schemes and treatment schedules are described in Figure 1A and B, respectively.
Clinical-grade F16-IL2 was provided by Philogen S.p.A. (Siena, Italy;15), as a liquid, sterile, apyrogenous preparation. The primary objective of phase Ib part of the study was to determine the RD of F16-IL2 in combination with doxorubicin, in patients with advanced solid tumors, while the secondary objectives included the investigation of the pharmacokinetic properties of F16-IL2, the analysis of human anti-fusion protein antibodies HAFA levels, and the study of early signs of anti-tumor activity of the investigational drug combination. For phase II part of the study, the primary objective was to investigate the anti-cancer activity of the drug combination as measured by confirmed objective response rate (ORR), while secondary objectives included the investigation of safety and tolerability, analysis of HAFA levels, and assessment of PFS, defined as the time from enrolment until objective tumor progression or death and median overall survival (mOS) defined as the time, calculated from enrolment until death, when 50% of the patients are dead.
During the phase Ib part of the study, the highest investigated dose level for which a DLT incidence was observed in not more than one out of 6 patients, was defined as RD. DLT was defined using Common Terminology Criteria for Adverse Events v3.0 (CTCAE),21 as described in the supplementary materials.
Study population and eligibility criteria
Inclusion and Exclusion Criteria are summarized in the supplementary materials. Briefly, solid cancer patients amenable for doxorubicin treatment were eligible for phase Ib (except cohort 1, where patients could not be amenable for doxorubicin treatment), whereas only breast cancer patients were recruited for the phase II part of the study. All enrolled patients (or their legally acceptable representatives, when applicable) signed an informed consent form before being admitted into the study. The study was conducted in accordance with the Declaration of Helsinki, and was approved by the Italian national competent authority and the hospitals’ ethic committees. The trial is registered in http://clinicaltrials.gov/ with the code NCT01131364.
Safety and efficacy assessments
At screening, demographic data and complete medical and surgical history were collected; moreover, ECG, Multi Gated Acquisition (MUGA) scan, assessment of baseline findings (according to Medical Dictionary for Regulatory Activities v10), assessment of concomitant medications, complete physical examination and vital signs, blood sampling for standard safety laboratory examination, thyroid function and HAFA, urine sampling, as well as disease assessment according to RECIST criteria18 were performed.
During the study, adverse events and toxicity were graded as per CTCAE;21 disease status was assessed every 2 cycles (corresponding to 6 F16-IL2 infusions and 8 weeks) and at study discontinuation according to RECIST criteria.18
All patients with at least one drug intake were analyzed for safety evaluation. Patients who discontinued prior to having an assessment at week 8 were considered non-responders for the efficacy evaluation.
Each patient returned for an end-of-treatment visit when disease was assessed as progressive, after discontinuation of treatment or at week 24 (last week of treatment). The follow-up visits were performed starting approximately 5 weeks from last administration of the study drug, and every 8 weeks until disease progression, beginning of another anti-cancer therapy, or completion of 12 months.
The final analysis for PFS and OS was conducted when all patients had experienced the event of interest or had been followed for at least 12 months after enrolment.
Pharmacokinetics and HAFA
For pharmacokinetics (PK) analyses, venous blood samples (6 ml) were collected repeatedly from all phase I study patients and 5 phase II patients during week 1 and week 2 of cycle 1. Serum was frozen and stored at -80°C until analyzed. F16-IL2 serum concentration was assessed using a validated method: a ligand-binding assay where the antigen tenascin C A1 domain was captured onto ELISA plates, and the analyte (F16-IL2) bound to immobilized antigen was detected by a mouse anti-human IL2 antibody and a goat anti-mouse IgG peroxidase conjugate.
The immunogenic potential of F16-IL2 was monitored by the analysis of HAFA formation. Serum samples for HAFA analysis were collected from all patients before F16-IL2 administration on day one, at day 56, at the end of study drug treatment and at the end of follow-up period. The immunogenicity of F16-IL2 was evaluated at Philogen S.p.A., Siena, Italy, using a validated ELISA method and orthogonal technology on SPR using a Biacore®T100 instrument.
Supplementary Material
Disclosure of Potential Conflicts of Interest
D.N. is a cofounder and shareholder of Philogen, the company which owns F16-IL2 and develops the product in clinical trials.
Acknowledgments
Philogen S.p.A contributed to the study design, the collection, analysis and interpretation of data, the writing of the manuscript, and the decision to submit the manuscript for publication.
References
- 1. Kovacs JA, Vogel S, Albert JM, Falloon J, Davey RT, Jr, Walker RE, Polis MA, Spooner K, Metcalf JA, Baseler M, et al. Controlled trial of interleukin-2 infusions in patients infected with the human immunodeficiency virus. N Engl J Med 1996; 335:1350-6; PMID:8857018; http://dx.doi.org/ 10.1056/NEJM199610313351803 [DOI] [PubMed] [Google Scholar]
- 2. Schmidinger M, Hejna M, Zielinski CC. Aldesleukin in advanced renal cell carcinoma. Expert Rev Anticancer Ther 2004; 4:957-80; PMID:15606326; http://dx.doi.org/ 10.1586/14737140.4.6.957 [DOI] [PubMed] [Google Scholar]
- 3. Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, Abrams J, Sznol M, Parkinson D, Hawkins M, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol 1999; 17:2105-16; PMID:10561265 [DOI] [PubMed] [Google Scholar]
- 4. Helguera G, Morrison SL, Penichet ML. Antibody-cytokine fusion proteins: harnessing the combined power of cytokines and antibodies for cancer therapy. Clin Immunol 2002; 105:233-46; PMID:12498805; http://dx.doi.org/ 10.1006/clim.2002.5302 [DOI] [PubMed] [Google Scholar]
- 5. Schliemann C, Neri D. Antibody-based targeting of the tumor vasculature. Biochim Biophys Acta 2007; 1776:175-92; PMID:17920773 [DOI] [PubMed] [Google Scholar]
- 6. Schrama D, Reisfeld RA, Becker JC. Antibody targeted drugs as cancer therapeutics. Nat Rev Drug Discov 2006; 5:147-59; PMID:16424916; http://dx.doi.org/ 10.1038/nrd1957 [DOI] [PubMed] [Google Scholar]
- 7. Borsi L, Carnemolla B, Nicolo G, Spina B, Tanara G, Zardi L. Expression of different tenascin isoforms in normal, hyperplastic and neoplastic human breast tissues. Int J Cancer J Int du Cancer 1992; 52:688-92; PMID:1385335; http://dx.doi.org/ 10.1002/ijc.2910520504 [DOI] [PubMed] [Google Scholar]
- 8. Murphy-Ullrich JE, Lightner VA, Aukhil I, Yan YZ, Erickson HP, Hook M. Focal adhesion integrity is downregulated by the alternatively spliced domain of human tenascin. J Cell Biol 1991; 115:1127-36; PMID:1720121; http://dx.doi.org/ 10.1083/jcb.115.4.1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hindermann W, Berndt A, Borsi L, Luo X, Hyckel P, Katenkamp D, Kosmehl H. Synthesis and protein distribution of the unspliced large tenascin-C isoform in oral squamous cell carcinoma. J Pathol 1999; 189:475-80; PMID:10629546; http://dx.doi.org/ 10.1002/(SICI)1096-9896(199912)189:4%3c475::AID-PATH462%3e3.0.CO;2-V [DOI] [PubMed] [Google Scholar]
- 10. Kusagawa H, Onoda K, Namikawa S, Yada I, Okada A, Yoshida T, Sakakura T. Expression and degeneration of tenascin-C in human lung cancers. Brit J Cancer 1998; 77:98-102; PMID:9459152; http://dx.doi.org/ 10.1038/bjc.1998.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Katenkamp K, Berndt A, Hindermann W, Wunderlich H, Haas KM, Borsi L, Zardi L, Kosmehl H. mRNA expression and protein distribution of the unspliced tenascin-C isoform in prostatic adenocarcinoma. J Pathol 2004; 203:771-9; PMID:15221936; http://dx.doi.org/ 10.1002/path.1589 [DOI] [PubMed] [Google Scholar]
- 12. Hauptmann S, Zardi L, Siri A, Carnemolla B, Borsi L, Castellucci M, Klosterhalfen B, Hartung P, Weis J, Stocker G, et al. Extracellular matrix proteins in colorectal carcinomas. Expression of tenascin and fibronectin isoforms. Lab Invest; J Tech Methods Pathology 1995; 73:172-82; PMID:7543628 [PubMed] [Google Scholar]
- 13. Carnemolla B, Castellani P, Ponassi M, Borsi L, Urbini S, Nicolo G, Dorcaratto A, Viale G, Winter G, Neri D, et al. Identification of a glioblastoma-associated tenascin-C isoform by a high affinity recombinant antibody. Am J Pathol 1999; 154:1345-52; PMID:10329587; http://dx.doi.org/ 10.1016/S0002-9440(10)65388-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Castellani P, Dorcaratto A, Siri A, Zardi L, Viale GL. Tenascin distribution in human brain tumours. Acta neurochirurgica 1995; 136:44-50; PMID:8748826; http://dx.doi.org/ 10.1007/BF01411434 [DOI] [PubMed] [Google Scholar]
- 15. Marlind J, Kaspar M, Trachsel E, Sommavilla R, Hindle S, Bacci C, Giovannoni L, Neri D. Antibody-mediated delivery of interleukin-2 to the stroma of breast cancer strongly enhances the potency of chemotherapy. Clin Cancer Res 2008; 14:6515-24; PMID:18927291; http://dx.doi.org/ 10.1158/1078-0432.CCR-07-5041 [DOI] [PubMed] [Google Scholar]
- 16. Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol 2013; 65:157-70; PMID:23278683; http://dx.doi.org/ 10.1111/j.2042-7158.2012.01567.x [DOI] [PubMed] [Google Scholar]
- 17. Brack SS, Silacci M, Birchler M, Neri D. Tumor-targeting properties of novel antibodies specific to the large isoform of tenascin-C. Clin Cancer Res 2006; 12:3200-8; PMID:16707621; http://dx.doi.org/ 10.1158/1078-0432.CCR-05-2804 [DOI] [PubMed] [Google Scholar]
- 18. Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, et al. New guidelines to evaluate the response to treatment in solid tumors. European organization for research and treatment of cancer, national cancer institute of the United States, national cancer institute of Canada. J Natl Cancer Inst 2000; 92:205-16; PMID:10655437; http://dx.doi.org/ 10.1093/jnci/92.3.205 [DOI] [PubMed] [Google Scholar]
- 19. Eigentler TK, Weide B, de Braud F, Spitaleri G, Romanini A, Pflugfelder A, Gonzalez-Iglesias R, Tasciotti A, Giovannoni L, Schwager K, et al. A dose-escalation and signal-generating study of the immunocytokine L19-IL2 in combination with Dacarbazine for the therapy of patients with metastatic melanoma. Clin Cancer Res 2011; 17:7732-42; PMID:22028492; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-1203 [DOI] [PubMed] [Google Scholar]
- 20. Johannsen M, Spitaleri G, Curigliano G, Roigas J, Weikert S, Kempkensteffen C, Roemer A, Kloeters C, Rogalla P, Pecher G, et al. The tumour-targeting human L19-IL2 immunocytokine: preclinical safety studies, phase I clinical trial in patients with solid tumours and expansion into patients with advanced renal cell carcinoma. Eur J Cancer 2010; 46:2926-35; PMID:20797845; http://dx.doi.org/ 10.1016/j.ejca.2010.07.033 [DOI] [PubMed] [Google Scholar]
- 21.Trotti A, Colevas AD, Setser A, Rusch V, Jaques D, Budach V, Langer C, Murphy B, Cumberlin R, Coleman CN, Rubin P. CTCAE v3.0: development of a comprehensive grading system for the adverse effects of cancer treatment. Seminars Rad Oncol, 2003; 13:176-81; PMID:12903007; doi: 10.1016/S1053-4296(03)00031-6. http://dx.doi.org/10.1016/S1053-4296(03)00031-6 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.