

Abstract
Epigenetic control of gene expression in children remains poorly understood, but new technologies can help elucidate the relationship between expression and DNA methylation. Here, we utilized the nCounter Analysis System to characterise the expression of 60 genes in 69 9-year-old children from a cohort with a high prevalence of obesity. nCounter expression levels ranged broadly (from 3 to over 10000 messenger RNA counts) and were divided into four categories: high (>2000 counts), moderate (200–1000 counts), low (100–200 counts) and marginal (<100 counts). For a subset of five genes (ADIPOR1, PPARG1, GSTM1, PON1 and ACACA) from different expression level categories, we validated nCounter data using reverse transcription-polymerase chain reaction (RT-PCR), and expanded RT-PCR analysis of ADIPOR1 to include 180 children. Expression data from the two methodologies were correlated for all five genes included in the validation experiment, with estimates ranging from r s = 0.26 (P = 0.02) to r s = 0.88 (P < 5×10−6). ADIPOR1 and PPARG1 nCounter expression levels were negatively correlated (r = −0.60, P < 5×10−5), and this relationship was stronger in overweight children (r = −0.73, P < 5×10−5) than in normal weight children (r = −0.42, P = 0.016). Using methylation data from the Infinium HumanMethylation450 BeadChip (n = 180), we found eight CpG sites in ADIPOR1 and PPARG where methylation level was associated with expression by RT-PCR (P < 0.05). Hypomethylation of PPARG gene body site cg10499651 was associated with increased expression as measured by both RT-PCR and nCounter (P < 0.05). We found no statistically significant relationships between either expression or methylation of ADIPOR1 and PPARG and body mass index or waist circumference. In addition to demonstrating the validity of expression data derived from nCounter, our results illustrate the use of new technologies in assessing epigenetic effects on expression in children.
Introduction
Over the past three decades, childhood obesity rates in the USA have been steadily increasing (1–3). Although there is recent evidence that obesity prevalence in young children is declining (4), rates of childhood obesity in Mexican-American children remain considerably higher than in non-Hispanic whites (5–7). It is well accepted that obesity has a heritable component, yet genome-wide association studies have identified few genetic polymorphisms related to obesity (8,9). Although certain variants in genes such as LEP, LEPR, POMC, PCSK1 and MC4R can lead to obesity, these variants are rare and therefore explain only a fraction of obesity’s observed 40–70% heritability (8,10). It is possible that a portion of this unexplained heritability, an example of the ‘missing heritability problem’ (11), is due to epigenetic changes that alter the expression of genes involved in obesity. DNA methylation, the most commonly investigated epigenetic mark, may be a relevant biological mechanism which can influence obesity risk (12,13). DNA methylation refers to the addition of methyl groups to cytosine residues at specific locations in the genome, termed CpG sites, which are clustered in CpG-rich regions known as CpG islands and flanked on either side by larger spans of DNA called shores and shelves (14). CpG methylation can result in altered gene expression, which, in turn, is detectable with expression assays (9,15).
Although quantitative reverse transcription-polymerase chain reaction (RT-PCR) is a standard method utilised in gene expression analysis, there are a variety of powerful new technologies for the assessment of both gene expression and DNA methylation. The NanoString nCounter Analysis System is a digital single molecule count-based detection system that can assess the expression of many genes in a single multiplexed reaction (16,17). Recently, nCounter has been utilised for transcriptomics in studies investigating a variety of topics including cancer (18,19), infectious disease (20) and immunology (21), but has yet to be employed in the assessment of expression changes associated with obesity in humans. The nCounter Analysis System has a variety of advantages, especially in regards to genetic analyses using banked samples from epidemiological studies. nCounter is relatively affordable and allows for the simultaneous quantification of hundreds of targets in a single small-volume sample. The nCounter method of labelling transcripts with unique colour-barcodes allows for the generation of direct counts of RNA transcript, which is a more readily interpretable data format than the fluorescence values or cycle thresholds (Ct) that are obtained from other expression methodologies. Lastly, nCounter does not rely on a PCR amplification step, meaning it is not vulnerable to variability caused by differences in amplification efficiency between transcripts (22). One recent study reported that nCounter and expression microarray data were highly correlated, especially for genes with significant expression variability (23).
DNA methylation analysis in this study was performed through use of the Illumina Infinium HumanMethylation450 BeadChip. The 450 BeadChip offers genome-wide coverage of methylation, allowing for the interrogation of over 450000 CpG sites spanning 99% of RefSeq genes (24,25). Many of the CpG sites covered by the 450 BeadChip are located in CpG islands and are thought to be involved in transcriptional control. Although the 450 BeadChip does not achieve the same level of resolution as sequencing-based methylation analysis methods, it is more affordable and generates data that are comparatively easy to interpret (26). The 450 BeadChip has been utilised in epigenetic studies investigating a wide variety of topics, including cancer (20,24,27), arthritis (28), stress (29) and obesity (30).
Here, we employed both the NanoString nCounter Analysis System and the Infinium HumanMethylation450 BeadChip to assess the expression and methylation of 60 genes involved in metabolism and oxidative stress, with a focus on those that may play a role in obesity development. In particular, we investigated the relationships between CpG methylation and gene expression of ADIPOR1 and PPARG, two genes with well-known roles in maintaining metabolic homeostasis. ADIPOR1, which encodes one of the major skeletal muscle receptors for the protein hormone adiponectin, plays an important role in glucose and lipid metabolism (31). PPARG is alternatively spliced to form two mature messenger RNAs (mRNAs) (PPARG1 and PPARG2) that encode peroxisome proliferator-activated receptor (PPAR) gamma 1 and 2, nuclear receptors that act to control the expression of genes involved in adipogenesis, lipid metabolism and inflammation (32).
In order to assess relationships between expression and methylation, we used banked blood samples from children enrolled in the Center for the Health Assessment of Mothers and Children of Salinas, CA (CHAMACOS) longitudinal birth cohort study (33,34). CHAMACOS children have a particularly high rate of obesity (57% overweight or obese at age 9) (35), even compared to Mexican-American children in the National Health and Nutrition Examination Survey (36). Previously, CHAMACOS studies have shown associations between obesity and genetic susceptibility (37), adipokine hormone levels (38), perinatal factors (39) and environmental exposures (35,40,41). Here, we describe the use of new technologies in cohort studies and examine the relationship between the expression and methylation of obesity-related genes in children.
Materials and Methods
Study population
The CHAMACOS study is a longitudinal birth cohort that aims to assess the health effects of pesticides and other exposures on growth and development in Mexican-American children living in the agricultural region of Salinas Valley, CA (33,34). Mothers were enrolled during pregnancy in 1999–2000 and interviewed when their children were 6 months of age and repeatedly for the next 9 years. Developmental assessments of children, including anthropometrics, were conducted at birth and at the time of each maternal interview. Signed informed consent was obtained from all mothers in the study. The Committee for the Protection of Human Subjects at the University of California, Berkeley, approved all study procedures.
Anthropometric measurements
Children’s weights and heights were measured at the 9-year-visit using a calibrated electronic scale (Tanita Mother-Baby Scale Model 1582; Tanita Corp.) and stadiometer, respectively. Child height was measured in triplicate and the average of measurements was used. Child waist circumference was measured at the 9-year-visit with a tape placed above the crest of the ileum. Measurements were recorded to the nearest 0.1cm after the child exhaled, performed in triplicate and averaged. Body mass index (BMI) was calculated as mass in kilograms divided by the square of height in metres. Children were categorised as normal weight, overweight or obese using the sex- and age-specific BMI cut-offs (85th and 95th percentile, respectively) provided by the 2000 Centers for Disease Control and Prevention child growth data.
Blood collection
Whole blood was collected in BD vacutainers (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) containing either heparin as an anticoagulant or no anticoagulant. Aliquots of whole blood, plasma, buffy coat, red blood cells, serum and clot were stored at −80°C until isolation of RNA or DNA.
RNA and DNA isolation
Total RNA was isolated from randomly selected banked whole blood samples from CHAMACOS 9-year-old children using RiboPure Blood RNA Purification Kits (Life Technologies, Waltham, MA, USA) according to the manufacturer’s protocol. Briefly, RNA was purified from 100 μl of whole blood from each subject. Following cell lysis in guanidinium-based solution, RNA was extracted using acid-phenol:chloroform. Isolated RNA was purified by solid-phase extraction on glass fibre filter columns included in the RiboPure kit.
DNA was isolated from banked blood clot samples from the same CHAMACOS 9-year-old children using Clotspin Baskets and QIAamp DNA Blood Maxi Kits (Qiagen, Valencia, CA, USA) according to the manufacturer’s protocol, with minor modifications as previously described (42). Briefly, blood clots were de-clotted using Clotspin baskets, treated with lysis buffer and protease and incubated overnight in a 70°C water bath. DNA was then ethanol-extracted from lysed clots and purified using QIAamp DNA purification columns.
Quality and concentrations of RNA and DNA were assessed using the NanoDrop 2000 Spectrophotometer (Thermo Scientific, Waltham, MA, USA) and concentrations were normalized to 20, 1 and 55ng/μl before nCounter, RT-PCR or methylation analyses, respectively. RNA quality was also verified using the 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA). Purified RNA was stored up to 2 weeks at −80°C and DNA was stored at −20°C until analysis.
Analysis of gene expression
The nCounter Analysis System (NanoString Technologies, Seattle, WA, USA) was used to simultaneously assess the absolute expression of 60 genes of interest per subject (n = 80). Candidate genes of interest were selected based on previous findings in our own research (37,39) or because of their putative or published relationships with obesity or oxidative stress (10,43–45). Purified RNA samples were sent to NanoString Technologies for analysis. Inter- and intra-cartridge replicates were included in the assay. Normalisation of nCounter results was carried out using nSolver Analysis Software Version 1.1 (NanoString Technologies), according to the manufacturer’s guidelines. To address both sample and platform sources of variation, RNA counts were normalised using the expression of four reference genes (ACTB, OAZ, HUPO, HPRT) and positive spike-in controls included in each sample. Geometric means of positive control and reference gene concentrations were calculated for each lane. Geometric means for all lanes were averaged, and this average was divided by each lane’s geometric mean to determine lane-specific scaling factors for both reference gene and positive control expression.
Quality assessment and control were also carried out using nSolver Analysis Software, following the manufacturer’s recommendations. Samples were dropped from the dataset if (i) <75% of counts per field of view were successful, (ii) binding density was >2.25 or <0.05, (iii) positive control scaling factor was >3 or <0.3, (iv) reference gene scaling factor was >10 or <0.1, (v) correlation between known and measured positive control concentrations was less than R 2 = 0.9 or (vi) lowest level positive control was under two standard deviations higher than the average of all negative controls. Eleven samples were removed from the dataset due to poor performance, leaving a final subject count of n = 69.
To validate expression results from nCounter, relative expression of a subset of five genes of interest (ADIPOR1, PPARG1, GSTM1, PON1 and ACACA) spanning different ranges of expression was assessed via RT-PCR in the 69 subjects with nCounter results. The expression of ADIPOR1, the gene with the highest level of nCounter expression, was also assessed in an additional 111 subjects to maximise overlap with the 450 BeadChip. Expression of the reference gene ACTB was measured for use as an endogenous control. Analysis was completed using QuantiTect Primer Assays and Rotor-Gene SYBR Green RT-PCR Kits (Qiagen), according to the manufacturer’s protocol for one-step RT-PCR. Reactions were carried out on a Rotor-Gene 6000 (Qiagen, formerly Corbett Life Science). After verifying amplicon specificity via melt curve analysis, Ct were calculated for each gene for each sample. All reactions were performed in duplicate. Internal and negative controls were included in all experiments. Samples or genes with poor coefficients of variance (<5%) between replicates or abnormally high mean Ct (>36) were excluded from analysis.
Site-specific methylation analysis
Site-specific methylation analysis was conducted using the Infinium HumanMethylation450 BeadChip (Illumina, San Diego, CA) on DNA samples from the same 9-year-old CHAMACOS children that were included in expression analysis. Whole genome amplification, enzymatic fragmentation and purification were performed before DNA was applied to the 450 BeadChips according to the Illumina methylation protocol. BeadChips were processed with robotics and analysed using the Illumina Hi-Scan system. Sample data were extracted using Illumina GenomeStudio Software Version XXV2011.1, Methylation Module 1.9 (Illumina). All samples included in analysis met a quality threshold of having detection P values below 0.01 for 95% of sites. Sites mapping to probe single nucleotide polymorphisms in the Illumina annotation or having P values >0.01 for more than 95% of samples were excluded from analysis. Raw signal intensities were first background corrected, then normalised for colour-channel bias using the All Sample Mean Normalization method as described previously (46). Lastly, the BMIQ algorithm was applied to make interpretation between type I and type II probes comparable (47). Data were expressed as M values, which are calculated as the log2 ratio of the intensities of methylated probe to unmethylated probe (48). Negative M values therefore signify that the unmethylated form of a CpG site is more abundant than the methylated form.
Glutathione S-transferase (GSTM1 and GSTT1) genotyping
Study participants (n = 61 of the 69 included in nCounter) were also genotyped for GSTM1 and GSTT1 homozygous deletion polymorphisms in order to determine whether expression levels were substantially lower in subjects possessing deletions for one or both of these genes. Genotyping was accomplished using the Multiplex Polymerase Chain Reaction Kit (Qiagen) with some modifications (49). As an internal positive control to verify DNA amplification in double null subjects, a 212-bp section of the albumin gene was co-amplified. Gene fragments were simultaneously amplified by using a 96-Well GeneAmp PCR System 9700 (Applied BioSystems, Waltham, MA, USA). The null GSTM1 and GSTT1 genotypes were detected by the absence of a band at 267 and 434bp, respectively, after electrophoresis and visualisation on a 3.5% agarose gel stained with ethidium bromide.
Statistical analyses
In order to directly compare results from different gene expression methodologies, relative expression values were computed for both nCounter and RT-PCR. For nCounter data, relative values were calculated by taking the log2 of the ratio of normalised expression values of the gene of interest and ACTB, a reference gene. For RT-PCR data, the difference in Ct between the gene of interest and ACTB is directly interpretable as a 2-fold change, so no transformation is necessary. Therefore, each unit in the relative values from either platform represents a 2-fold difference in expression relative to ACTB.
Linear regression and Spearman correlation analysis were used to assess the relationships between relative values for each gene by method of analysis. If both technologies were unbiased estimators of gene expression relative to ACTB, linear regression models would be expected to have slopes of one and y-intercepts of zero. Significant deviation from these expected model parameters could suggest bias in either technology.
As a proof of principle, we assessed whether nCounter GSTM1 and GSTT1 expression levels differed between subjects with null or wild-type genotypes for these genes, as carriers of null genotypes were expected to have much lower levels of expression. Because expression results from nCounter were not normally distributed, the Wilcoxon rank-sum test was used to test for differences in gene expression between GSTM1 and GSTT1 null and wild-type subjects. Subjects were divided into two groups based on their GSTM1 or GSTT1 genotypes, and P values were calculated to assess the statistical significance of the differences in expression.
Gene expression results from either nCounter or RT-PCR were compared to methylation M values for ADIPOR1 and PPARG obtained by the 450 BeadChip. First, separate linear regressions were performed to assess the relationship of each individual CpG site with expression as measured by either nCounter or RT-PCR. Reverse stepwise elimination was used to build multivariable linear regression models for assessment of these relationships. Any CpG sites with methylation M values associated with gene expression at a significance level of P < 0.05 were selected for inclusion in the final model. Child sex was considered for inclusion as a variable in the model, but did not substantially affect the results. When RT-PCR data were used as the outcome, regression models were adjusted for the expression of the reference gene ACTB.
Since ADIPOR1 and PPARG1 both contribute to metabolic homeostasis and fat use and storage, Pearson correlation analysis was used to assess the existence of a relationship between log2-transformed expression values from nCounter for these two genes. Relationships between gene expression, methylation and obesity were assessed via several approaches. Both 9-year BMI and categorical overweight status were independently considered for inclusion in the methylation and expression models described above. The relationship between ADIPOR1 or PPARG1 expression and obesity was assessed via linear or logistic regression, depending on whether BMI, waist circumference or overweight status was used as the outcome variable.
The threshold for significance was set to 5% (α = 0.05) for all statistical tests. The Benjamini–Hochberg procedure was used to control the false discovery rate for nCounter and RT-PCR comparisons and for separate linear regressions for each CpG site. All analyses were conducted using statistical software packages Stata Version 12.0 (StataCorp, College Station, TX, USA) and R Version 3.0.2 (www.R-project.org).
Results
Subject characteristics
Subject characteristics for both the nCounter pilot subset (n = 69) and the full set of subjects included in ADIPOR1 RT-PCR (n = 180) are presented in Table 1. Similar to 9-year-old obesity prevalence levels in the full CHAMACOS cohort (35), over half (53%) of children were classified as overweight based on BMI above the 85th percentile. Of the 180 children included in this study, 42 and 15% carried homozygous deletions for GSTM1 and GSTT1, respectively.
Table 1.
Subject characteristics
| nCounter mean (SD) | RT-PCR mean (SD) | |
|---|---|---|
| Total subjects | 69 | 180a |
| Age (years) | 9.1 (0.1) | 9.1 (0.2) |
| % Female | 49.3% | 53.9% |
| Anthropometric measures | ||
| BMI (kg/m2) | 20.3 (4.4) | 20.7 (4.6) |
| Waist circumference (cm) | 73.3 (11.7) | 73.9 (11.8) |
| Normal weight (<85th percentile) | 46.4% | 46.7% |
| Overweight (>85th, <95th percentile) | 20.3% | 15.0% |
| Obese (>95th percentile) | 33.3% | 38.3% |
| GSTM1 and GSTT1 genotypes | ||
| GSTM1 nullb | 44.3% | – |
| GSTT1 nullb | 14.8% | – |
a n = 180 for ADIPOR1 and 69 for all other genes.
b n = 61 for GST genotypes.
nCounter expression analysis
Figure 1 shows the range of mean expression levels observed for the 60 genes of interest and four reference genes assessed by the NanoString nCounter Analysis System in whole blood samples. Table 2 summarises four broad categories of observed expression ranges. Only two genes (ADIPOR1 and IL8) were highly expressed, with normalised mean mRNA counts >2000. Thirty-three genes (including PPARG1, GSTM1 and PON1) were low to moderately expressed, with normalised mean mRNA counts >100. Twenty-five genes, including ACACA, had normalised mean mRNA counts <100 and were comparable with background level and therefore considered marginally expressed.
Figure 1.

NanoString nCounter expression results. Mean expression values observed for all genes included in NanoString nCounter. Arrows indicate genes validated by RT-PCR.
Table 2.
Summary of nCounter expression results
| Expression category | Number of genes | Mean count (SD) | Examples |
|---|---|---|---|
| High (>2000) | 2 | 2575.6 (1567.3) | ADIPOR1, IL8 |
| Moderate (200–1000) | 12 | 290.9 (108.0) | NFKB2, GPX1, HDAC1, GSTP1, PRDX1, RXRA, TNFA, CAT, IL1B |
| Low (100–200) | 21 | 140.6 (33.0) | PON1, GSTM1, PPARG1 |
| Marginal (<100) | 25 | 54.5 (27.2) | ACACA, APOE |
Comparison of RT-PCR and nCounter expression data
To validate findings from nCounter, expression analysis via RT-PCR was carried out for five genes (ADIPOR1, PPARG1, GSTM1, PON1 and ACACA) with a range of expression levels as shown in Figure 1 and Table 2. Summary statistics for RT-PCR Ct and nCounter RNA counts for these five genes are presented in Table 3.
Table 3.
Summary of RT-PCR and nCounter data
| Gene | Function | RT-PCR | nCounter | ||||
|---|---|---|---|---|---|---|---|
| Mean count (SD) | Min | Max | Median count (SD) | Min | Max | ||
| ADIPOR1 | Receptor for adiponectin, promotes fatty acid breakdown | 23.1 (2.1) | 20.2 | 29.8 | 3204.0 (2398.2) | 607.5 | 10535.6 |
| ACTB | Beta-actin, reference gene | 18.5 (1.1) | 16.3 | 21.9 | 3317.6 (1140.6) | 2087.9 | 8233.6 |
| PPARG1 | Nuclear receptor, stimulates lipid uptake and adipogenesis | 24.5 (0.9) | 22.3 | 25.9 | 59.3 (111.1) | 4.7 | 644.5 |
| GSTM1 | Glutathione S-transferase, catalyses phase II detoxification reactions | 21.3 (0.5) | 20.4 | 22.5 | 72.4 (82.7) | 2.9 | 423.8 |
| PON1 | Paraoxonase, multifunctional enzyme involved in oxidative stress and organophosphate metabolism | 30.9 (1.3) | 28.5 | 35.2 | 45.3 (105.1) | 2.9 | 563.9 |
| ACACA | Acetyl-CoA carboxylase, participates in fatty acid synthesis | 29.4 (1.6) | 26.4 | 33.7 | 8.3 (10.1) | 2.7 | 57.3 |
Results of the comparisons between nCounter and RT-PCR methodologies, assessed relative to the reference gene ACTB, are presented in Figure 2. Statistically significant correlation was observed between expression data measured by the two expression analysis methods for all five genes, with correlation estimates ranging from 0.26 to 0.88 (P < 0.05 for all). Genes with higher expression levels had stronger correlations between methodologies. For example, the strongest correlation was observed for ADIPOR1 (r s = 0.88, P < 5×10−6), the gene for which we observed one of the highest mean RNA counts. Fairly strong correlation was also observed for PPARG1 (r s = 0.62, P < 5×10−6), a gene with low to moderate expression. Somewhat unexpectedly, a low but statistically significant correlation between methodologies was observed for ACACA (r s = 0.26, P = 0.02), the gene with one of the lowest expression levels of all 60 genes analysed by nCounter. The remaining two genes (GSTM1 and PON1) had intermediate correlation estimates (r s = 0.31, P < 0.005 and r s = 0.47, P < 2×10−5, respectively) for the comparison between the two assays. All five correlations remained statistically significant after Benjamini–Hochberg adjustment.
Figure 2.
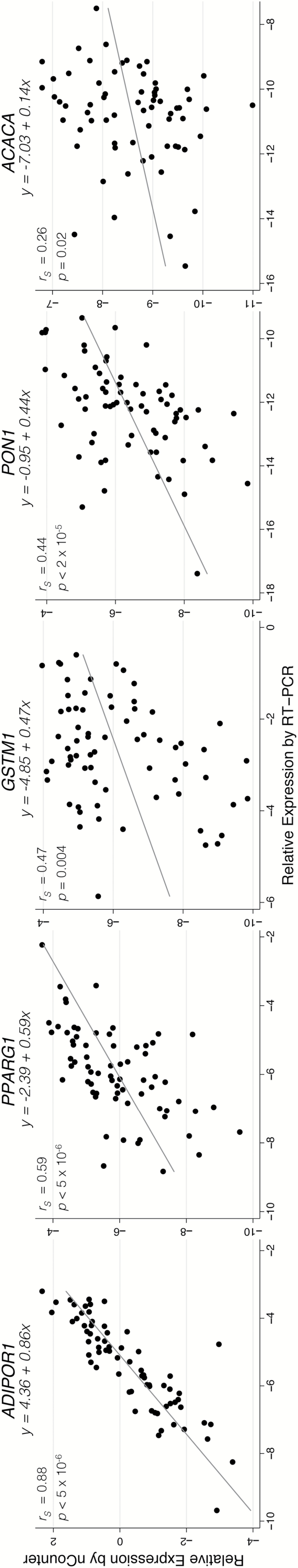
Relationship between nCounter and RT-PCR expression data for five genes. Results from nCounter and RT-PCR are expressed relative to the expression of ACTB, a reference gene, such that each unit represents a 2-fold difference from ACTB expression. Linear regression and Spearman correlation were used to assess the relationship between expression data from each platform.
In addition to correlation analysis, linear regression was also used to assess the consistency between the two methodologies. Because all values were expressed relative to a reference gene and on a log2 scale, regressions were expected to produce slope estimates of approximately one and y-intercept estimates of approximately zero for each comparison. Slope estimates ranged from 0.14 for ACACA to 0.86 for ADIPOR1, with all five comparisons having slope estimates lower than one (P < 0.05). PON1 was the only gene for which the y-intercept estimate did not differ significantly from zero (β 0 = −0.95, P = 0.43). The linear models for ACACA, GSTM1 and PPARG1 had y-intercepts significantly below zero (β 0 = −7.03, β 0 = −4.85, β 0 = −2.39, respectively), while the y-intercept for ADIPOR1 was greater than zero (β 0 = 4.36).
Glutathione S-transferase genotypes
Gene deletion variants that inhibit the production of functional protein are common for the phase II detoxification enzymes encoded by GSTM1 and GSTT1 (Table 1). Subjects homozygous for these deletion variants are expected to show little to no mRNA expression. In order to determine whether nCounter data were consistent with this expectation, expression results were compared between subjects with or without null (homozygous deletion) genotypes for either GSTM1 or GSTT1 (44 and 15% of subjects, respectively). Results for these comparisons are shown in Figure 3. As expected, subjects with the null genotype had substantially lower expression levels for both GSTM1 (null mean = 54.55, wild-type mean = 114.34, P < 5×10–5) and GSTT1 (null mean = 2.23, wild-type mean = 49.36, P < 5×10−5). The variability observed in wild-type subjects is most likely explained by the inclusion of both homozygous and heterozygous subjects in this group.
Figure 3.

Comparisons of glutathione S-transferase expression by genotype. ‘Null’ subjects possess homozygous deletions and do not produce functional enzyme. As expected, both GSTM1 (P < 5×10−5) and GSTT1 (P < 5×10−5) show significantly lower gene expression in GST-null subjects. P values were computed using the Wilcoxon rank-sum test.
CpG methylation of ADIPOR1 and PPARG
CpG methylation data from the Illumina Infinium HumanMethylation450 BeadChip included 17 CpG sites in ADIPOR1 and 21 CpG sites in PPARG after quality filtering. M value ranges, summary statistics and gene region information for each CpG site are presented in Figure 4 and supplementary Table 1, available at Mutagenesis Online. For ADIPOR1, the 450 BeadChip included 6 CpG sites in the gene promoter, 9 in the 5′ untranslated region (5′ UTR), 1 in the gene body and 1 in the 3′ UTR. Mean observed M values ranged from −8.80 to 3.58. For PPARG, the 450 BeadChip included 14 CpG sites in the gene promoter, 3 in the 5′ UTR, and 4 in the gene body. Mean observed M values ranged from −8.35 to 3.38. Both genes had groups of CpG sites where methylation levels were correlated. Correlation values for sites 2–3 and 14–17 in ADIPOR1 ranged from r = 0.49 to r = 0.70, and correlation values for sites 1–2 and 13–21 in PPARG ranged from r = 0.40 to r = 0.88. We observed a general pattern of decreasing methylation in the gene promoter as locations approached the transcription start sites of both genes, with a change to hypermethylation in gene bodies.
Figure 4.

Range of observed methylation M values for CpG sites in ADIPOR1 and PPARG. CpG site box plots are ordered according to their location in the genetic sequence. Horizontal bars indicate whether individual CpG sites are located within gene promoters or CpG islands.
Relationship between ADIPOR1 and PPARG1 expression and CpG methylation
To analyse the relationship between CpG methylation and gene expression, we first conducted individual linear regressions to assess the relationships between each CpG site in ADIPOR1 and PPARG and either nCounter or RT-PCR expression (supplementary Table 1, available at Mutagenesis Online). Next, we used reverse stepwise elimination to build multivariable linear regression models using only those sites that were statistically significantly associated with gene expression (Table 4). Regression models after stepwise elimination showed that three out of 17 ADIPOR1 CpG sites had methylation levels associated with gene expression as measured by RT-PCR (P < 0.05), but no sites were associated with expression measured by nCounter. For PPARG, four CpG sites had methylation levels associated with gene expression measured by RT-PCR only, and one CpG site (cg10499651) showed association between methylation and expression measured by both expression assays (P < 0.05). Moreover, for this PPARG gene body CpG site, both platforms showed the same direction of epigenetic effect, wherein hypermethylation was associated with reduced expression (lower RNA counts in nCounter and higher Ct values in RT-PCR).
Table 4.
Association between CpG methylation and expression
| CpG site | β | P value | Regulatory region |
|---|---|---|---|
| ADIPOR1 nCounter expression | |||
| No sites significantly associated with nCounter expression | |||
| ADIPOR1 RT-PCR expressiona | |||
| cg06525051 | 0.60 | 0.019 | 5′ UTR, island |
| cg15730680 | −0.18 | 0.012 | 5′ UTR, island |
| cg09157008 | 0.69 | 0.034 | Gene body |
| PPARG1 nCounter expression | |||
| cg10499651 | −59.25 | 0.049 | Gene body |
| PPARG1 RT-PCR expressiona | |||
| cg04748988 | 0.37 | 0.020 | Promoter, island |
| cg13518792 | 0.08 | 0.016 | Promoter, island |
| cg21859053 | −1.38 | 0.013 | Promoter, shore |
| cg04908300 | 0.87 | 0.009 | 5′ UTR, shelf |
| cg10499651 | 0.53 | 0.024 | Gene body |
aNote that higher Ct values and positive β estimates in RT-PCR are indicative of lower expression.
Gene expression, CpG methylation and obesity parameters
We observed a strong negative association between ADIPOR1 and PPARG1 nCounter gene expression (r = −0.60, P < 5×10−5). Interestingly, this correlation was substantially stronger in overweight children (r = −0.73, P < 5×10−5) than in normal weight children (r = −0.42, P = 0.016) (Figure 5).
Figure 5.

Relationship between ADIPOR1 and PPARG1 nCounter expression by overweight status. A strong negative correlation was observed between ADIPOR1 expression and PPARG1 expression (r = −0.60, P < 5×10−5). This relationship was substantially stronger in overweight children (r = −0.73, P < 5×10−5) than in normal weight children (r = −0.42, P = 0.0156).
Both 9-year child BMI and categorical overweight status were separately considered for inclusion in the expression and methylation stepwise models described above, but showed no evidence of statistically significant association. Using linear and logistic regression, we did not find any associations between ADIPOR1 or PPARG expression or methylation and BMI, waist circumference or odds of being overweight at 9 years of age (data not shown).
Discussion
nCounter RNA expression data were compared to Ct data generated by RT-PCR, the historically standard method, for five genes with a wide range of observed expression levels. For all genes tested, we observed statistically significant correlations between results (r s = 0.26–0.88), with much stronger correlation estimates for genes with higher expression in blood. Parameters of regression models indicate that although nCounter and RT-PCR results are correlated, one or both platforms may produce somewhat biased results. For example, RT-PCR is vulnerable to biases resulting from differences in PCR amplification efficiencies, which is not an issue in nCounter (22). However, the results of our RT-PCR validation of nCounter data suggest that both methodologies can be reliably used to compare gene expression between subjects within cohort or case–control studies (i.e. to determine which subjects are ‘high’, ‘medium’ or ‘low’ expressers). We also showed that subjects with homozygous GST deletion genotypes had substantially lower nCounter expression levels than wild-type subjects. The high expression levels observed for some GSTM1-null subjects may be attributable to compensation by another gene in the GST superfamily. For example, Bhattacharjee et al. (50) have reported that GSTM2 is 99% homologous to GSTM1 and capable of compensating for low enzyme expression in individuals with the GSTM1-null genotype. Altogether, our results augment existing nCounter validation efforts using both expression microarrays and RT-PCR (16,23), and confirm nCounter’s potential for use in population studies.
We focused our analyses of gene expression and CpG methylation on ADIPOR1 and PPARG, two important obesity-related genes. Adiponectin receptor 1 is one of the major receptors for the protein hormone adiponectin, and plays a significant role in glucose and lipid metabolism (31). PPARG is one member of a family of three PPARs that control the expression of a network of genes involved in adipogenesis, lipid metabolism, inflammation and the maintenance of metabolic homeostasis (32). Animal studies have demonstrated that upregulated expression of ADIPOR1 leads to increased AMPK and PPARα activation, thereby reducing gluconeogenesis, increasing levels of fatty acid oxidation and ameliorating diabetes (51,52). Conversely, the nuclear receptors PPARγ1 and PPARγ2 act to promote adipogenesis and lipid uptake when an excess of fat is present (53).
Consistent with the biological roles of these two genes, we observed a significant inverse relationship between ADIPOR1 and PPARG1 expression. Interestingly, this negative correlation was substantially stronger in overweight children than in normal weight children, possibly indicating that metabolism in overweight children is less balanced and more strongly polarised towards either lipid uptake or fatty acid oxidation. To our knowledge, this relationship between ADIPOR1 and PPARG1 expression in overweight children has not been previously reported. However, it has been shown that obese adults tend to have lower levels of PPARG1 expression (54) and higher levels of ADIPOR1 expression than non-obese individuals, possibly to compensate for the reduced levels of plasma adiponectin observed in obesity (55). Although it is not yet established in the literature, it is also possible that genetic polymorphisms (such as the pro12ala allele in PPARG, present in 10% of Mexican-Americans) may affect both obesity risk and the expression of these two genes (56–59). Since the expected number of variants would be low given our sample size, we did not genotype ADIPOR1 and PPARG polymorphisms in this study.
We were also interested in assessing whether expression or methylation of ADIPOR1 and PPARG were associated with obesity parameters such as BMI and waist circumference. It has already been established that changes in DNA methylation can affect the expression of genes involved in adipogenesis and metabolism, suggesting epigenetic dysregulation may be an essential mechanism for obesity development in children (60–62). In our study, however, we did not observe statistically significant associations between DNA methylation in these two genes and BMI, waist circumference or odds of being overweight at 9 years of age.
Finally, we demonstrated that specific CpG sites may be capable of altering ADIPOR1 and PPARG transcription. Using nCounter, RT-PCR, and the 450 BeadChip, we identified three CpG sites in ADIPOR1 and five CpG sites in PPARG that were significantly associated with gene expression. These sites spanned a variety of regions, including CpG islands, shores, 5′ and 3′ UTRs and gene bodies. Six of these CpG sites were associated with lower expression and two with higher expression. Though CpG island methylation is often considered to exert significant control over transcription, recent results from published studies indicate that CpG methylation in other gene regions may affect expression in unique ways (63–66), and the relationship between methylation and expression appears to be highly contextual (14). For one of the CpG sites in PPARG, we were able to validate our results across both nCounter and RT-PCR platforms. This particular CpG site is located in the gene body of PPARG, a gene region that has previously been shown to participate in splicing control (14). It is possible that this CpG site could be involved in PPARG splicing, since PPARG1 mRNA is one of two possible alternatively spliced PPARG gene products (32).
There are several strengths to the design of our study. It is one of few that directly assess the relationship between site-specific DNA methylation and gene expression, an area of epigenetics that is still poorly understood. Additionally, studies of this type seldom investigate minority or youth populations such as the CHAMACOS cohort. We also took advantage of data on the presence or absence of null genotypes for GSTM1 and GSTT1 in our subjects to assess the relationships between these genotypes and nCounter expression levels. Lastly, we further validated nCounter expression results using RT-PCR, the standard method for expression analysis.
Limitations of our study include relatively small sample size and selective analysis of CpG sites by the 450 BeadChip. Other CpG sites not interrogated by the 450 BeadChip may also regulate gene transcription or show association with obesity parameters; these sites can be assessed by targeted sequencing in future studies. The lack of observed association between methylation and obesity parameters in our study could also be attributed to the existence of longitudinal effects of methylation, important effects of other obesity-related genes or characteristics of this unique minority cohort. Additionally, we conducted this cohort study using DNA and RNA from blood because it is a readily accessible tissue and relatively non-invasive to collect, but we are mindful that methylation and expression variability may be tissue-specific (13,67,68). However, a wide variety of studies have successfully used blood as a biomarker of altered methylation patterns (69,70), and some analyses suggest that changes to the blood methylome can reflect broader changes across other tissues (71,72).
Conclusion
In this study, we employed two methods of gene expression analysis and a site-specific DNA methylation assay to investigate the relationship between methylation and expression in Mexican-American children. Using nCounter, RT-PCR and the 450 BeadChip, we reported several associations between DNA methylation and expression for the obesity-related genes ADIPOR1 and PPARG, and also showed that the expressions of these two genes are inversely correlated, especially among overweight children. Our findings suggest that individual CpG sites may be capable of either up- or down-regulating gene expression, and that sites in both gene promoter and gene body regions may be important in controlling transcription. These findings may contribute to understanding how DNA methylation may act through gene expression to influence metabolism and obesity development.
Supplementary data
Supplementary Table 1 is available at Mutagenesis Online.
Funding
National Institute of Environmental Health Sciences at the National Institutes of Health (1R01ES021369-01A1, 1R01ES023067-01A1, RO1 ES012503 and PO1 ES009605).
Supplementary Material
Acknowledgements
We are grateful to the participants and laboratory and field office staff of the CHAMACOS study and the Center for Environmental Research and Children’s Health for their contributions. We thank Dr Lisa Barcellos and Ms Hong Quach for their contributions to methylation analyses using the 450 BeadChip, and Dr Reuben Thomas and Mr Kelly Street for their assistance with data analysis. Ms Aurelia Cheng’s contribution to GST genotyping is appreciated.
Conflict of interest statement: None declared.
References
- 1. Kimm S. Y., Obarzanek E. (2002). Childhood obesity: a new pandemic of the new millennium. Pediatrics, 110, 1003–1007. [DOI] [PubMed] [Google Scholar]
- 2. Ogden C. L., Carroll M. D., Curtin L. R., Lamb M. M., Flegal K. M. (2010). Prevalence of high body mass index in US children and adolescents, 2007-2008. JAMA, 303, 242–249. [DOI] [PubMed] [Google Scholar]
- 3. Ogden C. L., Carroll M. D., Flegal K. M. (2008). High body mass index for age among US children and adolescents, 2003-2006. JAMA, 299, 2401–2405. [DOI] [PubMed] [Google Scholar]
- 4. Ogden C. L., Carroll M. D., Kit B. K., Flegal K. M. (2014). Prevalence of childhood and adult obesity in the United States, 2011-2012. JAMA, 311, 806–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Holub C. K., Lobelo F., Mehta S. M., Sánchez Romero L. M., Arredondo E. M., Elder J. P. (2014). School-wide programs aimed at obesity among Latino youth in the United States: a review of the evidence. J. Sch. Health, 84, 239–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. (2013). Health, United States, 2012: With Special Feature on Emergency Care. National Center for Health Statistics, Hyattsville, MD, USA. [PubMed] [Google Scholar]
- 7. (2013). Obesity and Hispanic Americans. Office of Minority Health, U.S. Department of Health and Human Services; http://minorityhealth.hhs.gov/omh/browse.aspx?lvl=4&lvlID=70 [Google Scholar]
- 8. Bell C. G., Walley A. J., Froguel P. (2005). The genetics of human obesity. Nat. Rev. Genet., 6, 221–234. [DOI] [PubMed] [Google Scholar]
- 9. Lee Y. S. (2013). Genetics of nonsyndromic obesity. Curr. Opin. Pediatr., 25, 666–673. [DOI] [PubMed] [Google Scholar]
- 10. Waalen J. (2014). The genetics of human obesity. Transl. Res., 164, 293–301. [DOI] [PubMed] [Google Scholar]
- 11. Slatkin M. (2009). Epigenetic inheritance and the missing heritability problem. Genetics, 182, 845–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Franks P. W., Ling C. (2010). Epigenetics and obesity: the devil is in the details. BMC Med., 8, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chadwick L. H., Sawa A., Yang I. V., et al. (2014). New insights and updated guidelines for epigenome-wide association studies. Neuroepigenetics. 10.1016/j.nepig.2014.10.004. [Google Scholar]
- 14. Jones P. A. (2012). Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet., 13, 484–492. [DOI] [PubMed] [Google Scholar]
- 15. Neeha V. S., Kinth P. (2013). Nutrigenomics research: a review. J. Food Sci. Technol., 50, 415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Geiss G. K., Bumgarner R. E., Birditt B., et al. (2008). Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat. Biotechnol., 26, 317–325. [DOI] [PubMed] [Google Scholar]
- 17. Payton J. E., Grieselhuber N. R., Chang L. W., Murakami M., Geiss G. K., Link D. C., Nagarajan R., Watson M. A., Ley T. J. (2009). High throughput digital quantification of mRNA abundance in primary human acute myeloid leukemia samples. J. Clin. Invest., 119, 1714–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chakravadhanula M., Ozols V. V., Hampton C. N., Zhou L., Catchpoole D., Bhardwaj R.D. (2014). Expression of the HOX genes and HOTAIR in atypical teratoid rhabdoid tumors and other pediatric brain tumors. Cancer Genet, 207, 425–428. [DOI] [PubMed] [Google Scholar]
- 19. Mairinger F. D., Walter R. F., Werner R., et al. (2014). Activation of angiogenesis differs strongly between pulmonary carcinoids and neuroendocrine carinomas and is crucial for carcinoid tumourigenesis. J. Cancer, 5, 465–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cheng S., Clancy C. J., Xu W., Schneider F., Hao B., Mitchell A. P., Nguyen M. H. (2013). Profiling of Candida albicans gene expression during intra-abdominal candidiasis identifies biologic processes involved in pathogenesis. J. Infect. Dis., 208, 1529–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stifano G., Affandi A. J., Mathes A. L., Rice L. M., Nakerakanti S., Nazari B., Lee J., Christmann R. B., Lafyatis R. (2014). Chronic Toll-like receptor 4 stimulation in skin induces inflammation, macrophage activation, transforming growth factor beta signature gene expression, and fibrosis. Arthritis Res. Ther., 16, R136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wong M. L., Medrano J. F. (2005). Real-time PCR for mRNA quantitation. Biotechniques, 39, 75–85. [DOI] [PubMed] [Google Scholar]
- 23. Richard A. C., Lyons P. A., Peters J. E., Biasci D., Flint S. M., Lee J. C., McKinney E. F., Siegel R. M., Smith K. G. (2014). Comparison of gene expression microarray data with count-based RNA measurements informs microarray interpretation. BMC Genomics, 15, 649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sandoval J., Heyn H., Moran S., Serra-Musach J., Pujana M. A., Bibikova M., Esteller M. (2011). Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics, 6, 692–702. [DOI] [PubMed] [Google Scholar]
- 25. Dedeurwaerder S., Defrance M., Calonne E., Denis H., Sotiriou C., Fuks F. (2011). Evaluation of the Infinium Methylation 450K technology. Epigenomics, 3, 771–784. [DOI] [PubMed] [Google Scholar]
- 26. Morris T. J., Beck S. (2014). Analysis pipelines and packages for Infinium HumanMethylation450 BeadChip (450k) data. Methods. 10.1016/j.ymeth.2014.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lindqvist B. M., Wingren S., Motlagh P. B., Nilsson T. K. (2014). Whole genome DNA methylation signature of HER2-positive breast cancer. Epigenetics, 9, 1149–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Glossop J. R., Emes R. D., Nixon N. B., Haworth K. E., Packham J. C., Dawes P. T., Fryer A. A., Mattey D. L., Farrell W. E. (2014). Genome-wide DNA methylation profiling in rheumatoid arthritis identifies disease-associated methylation changes that are distinct to individual T- and B-lymphocyte populations. Epigenetics, 9, 1228–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Boks M. P., Mierlo H. C., Rutten B. P., et al. (2015). Longitudinal changes of telomere length and epigenetic age related to traumatic stress and post-traumatic stress disorder. Psychoneuroendocrinology, 51, 506–512. [DOI] [PubMed] [Google Scholar]
- 30. Dick K. J., Nelson C. P., Tsaprouni L., et al. (2014). DNA methylation and body-mass index: a genome-wide analysis. Lancet, 383, 1990–1998. [DOI] [PubMed] [Google Scholar]
- 31. Jin Z., Pu L., Sun L., et al. (2014). Identification of susceptibility variants in ADIPOR1 gene associated with type 2 diabetes, coronary artery disease and the comorbidity of type 2 diabetes and coronary artery disease. PLoS One, 9, e100339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ahmadian M., Suh J. M., Hah N., Liddle C., Atkins A. R., Downes M., Evans R. M. (2013). PPARγ signaling and metabolism: the good, the bad and the future. Nat. Med., 19, 557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Eskenazi B., Harley K., Bradman A., Weltzien E., Jewell N. P., Barr D. B., Furlong C. E., Holland N. T. (2004). Association of in utero organophosphate pesticide exposure and fetal growth and length of gestation in an agricultural population. Environ. Health Perspect., 112, 1116–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Eskenazi B., Gladstone E. A., Berkowitz G. S., et al. (2005). Methodologic and logistic issues in conducting longitudinal birth cohort studies: lessons learned from the Centers for Children’s Environmental Health and Disease Prevention Research. Environ. Health Perspect., 113, 1419–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Harley K. G., Aguilar Schall R., Chevrier J., et al. (2013). Prenatal and postnatal bisphenol A exposure and body mass index in childhood in the CHAMACOS cohort. Environ. Health Perspect., 121, 514–20, 520e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ogden C. L., Carroll M. D., Kit B. K., Flegal K. M. (2012). Prevalence of obesity and trends in body mass index among US children and adolescents, 1999-2010. JAMA, 307, 483–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huen K., Harley K., Beckman K., Eskenazi B., Holland N. (2013). Associations of PON1 and genetic ancestry with obesity in early childhood. PLoS One, 8, e62565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Volberg V., Heggeseth B., Harley K., et al. (2013). Adiponectin and leptin trajectories in Mexican-American children from birth to 9 years of age. PLoS One, 8, e77964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Volberg V., Harley K. G., Aguilar R. S., et al. (2013). Associations between perinatal factors and adiponectin and leptin in 9-year-old Mexican-American children. Pediatr. Obes., 8, 454–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Volberg V., Harley K., Calafat A. M., Davé V., McFadden J., Eskenazi B., Holland N. (2013). Maternal bisphenol a exposure during pregnancy and its association with adipokines in Mexican-American children. Environ. Mol. Mutagen., 54, 621–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Warner M., Aguilar Schall R., Harley K. G., Bradman A., Barr D., Eskenazi B. (2013). In utero DDT and DDE exposure and obesity status of 7-year-old Mexican-American children in the CHAMACOS cohort. Environ. Health Perspect., 121, 631–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Holland N., Furlong C., Bastaki M., Richter R., Bradman A., Huen K., Beckman K., Eskenazi B. (2006). Paraoxonase polymorphisms, haplotypes, and enzyme activity in Latino mothers and newborns. Environ. Health Perspect., 114, 985–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. DeMenna J., Puppala S., Chittoor G., Schneider J., Kim J. Y., Shaibi G. Q., Mandarino L. J., Duggirala R., Coletta D. K. (2014). Association of common genetic variants with diabetes and metabolic syndrome related traits in the Arizona Insulin Resistance registry: a focus on Mexican American families in the Southwest. Hum. Hered., 78, 47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Go M. J., Hwang J. Y., Jang H. B., et al. (2014). A genome-wide association study identifies a LEPR gene as a novel predisposing factor for childhood fasting plasma glucose. Genomics, 104, 594–598. [DOI] [PubMed] [Google Scholar]
- 45. Kraja A. T., Chasman D. I., North K. E., et al. (2014). Pleiotropic genes for metabolic syndrome and inflammation. Mol. Genet. Metab., 112, 317–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yousefi P., Huen K., Schall R. A., Decker A., Elboudwarej E., Quach H., Barcellos L., Holland N. (2013). Considerations for normalization of DNA methylation data by Illumina 450K BeadChip assay in population studies. Epigenetics, 8, 1141–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Teschendorff A. E., Marabita F., Lechner M., Bartlett T., Tegner J., Gomez-Cabrero D., Beck S. (2013). A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics, 29, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Du P., Zhang X., Huang C. C., Jafari N., Kibbe W. A., Hou L., Lin S. M. (2010). Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics, 11, 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Block G., Shaikh N., Jensen C. D., Volberg V., Holland N. (2011). Serum vitamin C and other biomarkers differ by genotype of phase 2 enzyme genes GSTM1 and GSTT1. Am. J. Clin. Nutr., 94, 929–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bhattacharjee P., Paul S., Banerjee M., Patra D., Banerjee P., Ghoshal N., Bandyopadhyay A., Giri A. K. (2013). Functional compensation of glutathione S-transferase M1 (GSTM1) null by another GST superfamily member, GSTM2. Sci. Rep., 3, 2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yamauchi T., Nio Y., Maki T., et al. (2007). Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat. Med., 13, 332–339. [DOI] [PubMed] [Google Scholar]
- 52. Patel S. A., Hoehn K. L., Lawrence R. T., et al. (2012). Overexpression of the adiponectin receptor AdipoR1 in rat skeletal muscle amplifies local insulin sensitivity. Endocrinology, 153, 5231–5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mansour M. (2014). The roles of peroxisome proliferator-activated receptors in the metabolic syndrome. Prog. Mol. Biol. Transl. Sci., 121, 217–266. [DOI] [PubMed] [Google Scholar]
- 54. Leyvraz C., Verdumo C., Suter M., Paroz A., Calmes J. M., Marques-Vidal P. M., Giusti V. (2012). Changes in gene expression profile in human subcutaneous adipose tissue during significant weight loss. Obes. Facts, 5, 440–451. [DOI] [PubMed] [Google Scholar]
- 55. Felder T. K., Hahne P., Soyal S. M., Miller K., Höffinger H., Oberkofler H., Krempler F., Patsch W. (2010). Hepatic adiponectin receptors (ADIPOR) 1 and 2 mRNA and their relation to insulin resistance in obese humans. Int. J. Obes. (Lond)., 34, 846–851. [DOI] [PubMed] [Google Scholar]
- 56. Bermúdez V. J., Rojas E., Toledo A., et al. (2013). Single-nucleotide polymorphisms in adiponectin, AdipoR1, and AdipoR2 genes: insulin resistance and type 2 diabetes mellitus candidate genes. Am. J. Ther., 20, 414–421. [DOI] [PubMed] [Google Scholar]
- 57. Cecil J. E., Watt P., Palmer C. N., Hetherington M. (2006). Energy balance and food intake: the role of PPARgamma gene polymorphisms. Physiol. Behav., 88, 227–233. [DOI] [PubMed] [Google Scholar]
- 58. Berhouma R., Kouidhi S., Ammar M., Abid H., Ennafaa H., Benammar-Elgaaied A. (2013). Correlation of peroxisome proliferator-activated receptor (PPAR-γ) mRNA expression with Pro12Ala polymorphism in obesity. Biochem. Genet., 51, 256–263. [DOI] [PubMed] [Google Scholar]
- 59. Black M. H., Fingerlin T. E., Allayee H., et al. (2008). Evidence of interaction between PPARG2 and HNF4A contributing to variation in insulin sensitivity in Mexican Americans. Diabetes, 57, 1048–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lavebratt C., Almgren M., Ekström T. J. (2012). Epigenetic regulation in obesity. Int. J. Obes. (Lond)., 36, 757–765. [DOI] [PubMed] [Google Scholar]
- 61. Barres R., Zierath J. R. (2011). DNA methylation in metabolic disorders. Am. J. Clin. Nutr., 93, 897S–8900. [DOI] [PubMed] [Google Scholar]
- 62. Seki Y., Williams L., Vuguin P. M., Charron M. J. (2012). Minireview: epigenetic programming of diabetes and obesity: animal models. Endocrinology, 153, 1031–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Muers M. (2013). Gene expression: Disentangling DNA methylation. Nat. Rev. Genet., 14, 519. [DOI] [PubMed] [Google Scholar]
- 64. Wagner J. R., Busche S., Ge B., Kwan T., Pastinen T., Blanchette M. (2014). The relationship between DNA methylation, genetic and expression inter-individual variation in untransformed human fibroblasts. Genome Biol., 15, R37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gutierrez-Arcelus M., Lappalainen T., Montgomery S. B., et al. (2013). Passive and active DNA methylation and the interplay with genetic variation in gene regulation. Elife, 2, e00523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Greally J. M. (2013). Bidding the CpG island goodbye. Elife, 2, e00593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Reinius L. E., Acevedo N., Joerink M., Pershagen G., Dahlén S. E., Greco D., Söderhäll C., Scheynius A., Kere J. (2012). Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One, 7, e41361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. McKay J. A., Xie L., Harris S., Wong Y. K., Ford D., Mathers J. C. (2011). Blood as a surrogate marker for tissue-specific DNA methylation and changes due to folate depletion in post-partum female mice. Mol. Nutr. Food Res., 55, 1026–1035. [DOI] [PubMed] [Google Scholar]
- 69. Marsit C., Christensen B. (2013). Blood-derived DNA methylation markers of cancer risk. Adv. Exp. Med. Biol., 754, 233–252. [DOI] [PubMed] [Google Scholar]
- 70. Liu J., Chen J., Ehrlich S., Walton E., White T., Perrone-Bizzozero N., Bustillo J., Turner J. A., Calhoun V. D. (2014). Methylation patterns in whole blood correlate with symptoms in schizophrenia patients. Schizophr. Bull., 40, 769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Davies M. N., Volta M., Pidsley R., et al. (2012). Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol., 13, R43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ma B., Wilker E. H., Willis-Owen S. A., et al. (2014). Predicting DNA methylation level across human tissues. Nucleic Acids Res., 42, 3515–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.