

Abstract
There is strong evidence that changes in the actin/spectrin-based cortical cytoskeleton of outer hair cells (OHCs) regulate their motile responses as well as cochlear amplification, the process that optimizes the sensitivity and frequency selectivity of the mammalian inner ear. Since a RhoA/protein kinase C (PKC)-mediated pathway is known to inhibit the actin-spectrin interaction in other cell models, we decided to investigate whether this signaling cascade could also participate in the regulation of OHC motility. We used high-speed video microscopy and confocal microscopy to explore the effects of pharmacological activation of PKCα, PKCβI, PKCβII, PKCδ, PKCε, and PKCζ with lysophosphatidic acid (LPA) and their inhibition with bisindolylmaleimide I, as well as inhibition of RhoA and Rho-associated protein kinase (ROCK) with C3 and Y-27632, respectively. Motile responses were induced in isolated guinea pig OHCs by stimulation with an 8 V/cm external alternating electrical field as 50 Hz bursts of square wave pulses (100 ms on/off). We found that LPA increased expression of PKCα and PKCζ only, with PKCα, but not PKCζ, phosphorylating the cytoskeletal protein adducin of both Ser-726 and Thr-445. Interestingly, however, inhibition of PKCα reduced adducin phosphorylation only at Ser-726. We also determined that LPA activation of a PKCα-mediated signaling pathway simultaneously enhanced OHC electromotile amplitude and cell shortening, and facilitated RhoA/ROCK/LIMK1-mediated cofilin phosphorylation. Altogether, our results suggest that PKCα-mediated signals, probably via adducin-mediated inhibition of actin-spectrin binding and cofilin-mediated depolymerization of actin filaments, play an essential role in the homeostatic regulation of OHC motility and cochlear amplification.
Introduction
Outer hair cells (OHCs) have the ability to continuously adjust their total length and the amplitude of their electrically induced somatic movements to optimize the sensitivity and frequency selectivity of the mammalian cochlea (1). We have used the term “mechanical homeostasis” to identify this process (2), and our previous studies provided strong evidence that it involves finely regulated cytoskeletal changes mediated by Rho GTPases (2–5).
The hypothesis that the cytoskeleton is the essential structure involved in the mechanical homeostasis of OHCs is supported by experiments showing that activation or inhibition of RhoA/Rho-associated protein kinase (ROCK)-mediated pathways have a profound influence on the motile response of OHCs (2,3,5). In particular, we demonstrated that actin depolymerization, regulated by activation/inhibition of a RhoA/ROCK/LIMK1/cofilin-mediated pathway, has a pivotal role in OHC motility (5). Among other findings, it was reported that ROCK activation by RhoA induced phosphorylation of the cytoskeletal protein adducin at Thr-445 and Ser-726 in isolated OHCs (2). In other cell populations, adducin was reported to promote actin-spectrin binding after being phosphorylated at Thr-445 by RhoA/ROCK-mediated signals (6), and to inhibit this binding after being phosphorylated at Ser-726 by RhoA/protein kinase C (PKC)-mediated signals (7,8). Thus, the simultaneous detection of adducin phosphorylation at Thr-445 and Ser-726 in OHCs suggests that the adducin-mediated continuous assembly and disassembly of the actin-spectrin meshwork could dynamically regulate the structure of the OHC cortical cytoskeleton (2).
In mammals, PKCs are a family of at least 12 isoenzymes divided into four structurally and functionally distinct subgroups with variable distribution among different tissues and cell types (9,10). PKCα, PKCβ, and PKCγ represent conventional PKCs. Members of this subgroup are activated by a combination of Ca2+, diacylglycerol (DAG), and phospholipid binding to specific domains. A subgroup of novel PKCs includes PKCδ, PKCε, PKCθ, and PKCη, which are similarly activated by DAG and phospholipids, but do not respond directly to Ca2+. The atypical PKCs PKCι and PKCζ do not depend on Ca2+ or DAG for activation, and instead are allosterically activated by an interaction with the partitioning defective 6 (PAR6)-CDC42 complex, which is involved in specifying cell polarity.
Adducin is a tetrameric protein that consists of α/β or α/γ heterodimers (where α, β, and γ are subunits originating from three different genes with several different splice variants). Adducin has been proposed to function as a crucial assembly factor for the cytoskeletal network because it promotes the binding of spectrin to actin, two molecules with a natural low affinity. When Ser-726 in adducin is phosphorylated by PKCs, its affinity for actin and spectrin is significantly reduced (7), destabilizing the actin-spectrin membrane-associated cytoskeleton. To our knowledge, our laboratory was the first to immunolocalize adducin in cochlear OHCs and to suggest it could have an important role in the dynamics of the OHC cortical cytoskeleton (2).
Although studies have provided evidence that stimulation with lysophosphatidic acid (LPA) would cause adducin phosphorylation, the potential involvement of PKCs in the regulation of OHC motility has never been explored. In this work, we stimulated isolated guinea pig OHCs with an external alternating electrical field (EAEF) and used video and confocal microscopy techniques to investigate this issue and to identify the putative PKC isoform involved in this process. Our results confirm that PKC-mediated signals regulate OHC motility, indicate that PKCα is the relevant isoform, and strongly suggest that two signaling cascades, RhoA/ROCK/PKCα/adducin and RhoA/ROCK/LIMK1/cofilin, work coordinately in the fine regulation of OHCs’ motile responses.
Materials and Methods
Isolation of OHCs
Cochleae were obtained from young guinea pigs (200–300 g) euthanized with CO2 in accordance with the recommendations of the 2013 Report of the American Veterinary Medical Association Panel on Euthanasia. Cochlear spirals were placed in Leibowitz L-15 medium (Gibco-Invitrogen, Grand Island, NY) containing 1 mg/mL collagenase (type IV; Sigma, St. Louis, MO), and incubated at 31°C for 3 min. The organ of Corti was removed from the bone with fine needles and OHCs were mechanically dissociated by reflux through the needle of a 50 μL syringe (Hamilton, Reno, NV). Isolated OHCs were then moved to a recording chamber (PCCS1; Bioscience Tools, San Diego, CA) filled with L-15, and observed with the use of Nomarski differential interference contrast optics on an Axiovert 135TV inverted microscope with a 63×/1.2 C-Apochromat objective (Carl Zeiss, Jena, Germany). Only live OHCs that met established health criteria (11) were used for motility measurements.
OHC motility measurements
Isolated OHCs were suspended in Leibovitz L-15 medium in a perfusion chamber on an Axiovert 135 inverted microscope stage. Continuous perfusion of an external solution consisting of L-15 adjusted to 300 mOsm with distilled water was provided at a rate of 0.3 mL/min using a syringe pump. A full description of the equipment and procedures used to measure OHC electromotility with an EAEF is available in the literature (12,13). Briefly, suspended OHCs were placed in the experimental dish between two silver wire electrodes (ϕ = 0.25 mm) with a tip distance of 0.8 mm. An 8 V/cm EAEF was applied between electrodes as 50 Hz bursts of square wave pulses (100 ms on/off). The cells’ longitudinal axes were kept parallel to the applied EAEF by rotating the recording chamber. Illumination provided by an LED light source (High Power LED System 36AD3500; Lightspeed Technologies, Campbell, CA), electrical stimulation, and image capture were digitally synchronized (12,13). The osmolarity of every solution used in these experiments was controlled and adjusted with distilled water or glucose to 300 ± 2 mOsmol with a μOsmette 5004 freezing-point osmometer (Precision Systems, Natick, MA).
Video analysis and data handling
Images of isolated OHCs were captured in AVI format at 1000 frames/s (fps) using the ultra-high-speed Photron Fastcam X 1024 PCI camera (Photron USA, San Diego, CA). Cell images were analyzed offline using ProAnalyst software (Xcitex, Cambridge, MA) and further processed using Excel software (Microsoft, Redmond, WA) (12,13). Statistical analysis with ANOVA techniques was performed using JMP 9 software (SAS Institute, Cary, NC), and p ≤ 0.05 was used as the criterion for statistical significance.
Immunolabeling
Excised cochlear spirals were incubated for variable periods in either L-15 alone (control) or with the following agents (alone or combined): 10 μM LPA (Sigma-Aldrich, Saint Louis, MO), 1 μM or 10 μM bisindolylmaleimide I (BIM-1; Calbiochem, La Jolla, CA), 1 μM 12-O-tetradecanoylphorbol-13-acetate (TPA; Sigma), 100 μg/mL C3 (Cytoskeleton, Denver, CO), 10 μM Y27632 (Upstate Biotechnology, Lake Placid, NY), and 50 nM or 1 μM of H-89 (EMD Millipore, Darmstadt, Germany). OHCs dissociated from cochlear spirals were moved to a petri dish with an uncoated glass bottom (MatTek, Ashland, MA), fixed in 4% paraformaldehyde (EMS, Fort Washington, PA) in phosphate-buffered saline for 2 h, and processed for confocal microscopy. Anti-PKCα (C-20), anti-phosphorylated PKCα (anti-p-PKCα; at Ser-657), anti-PKCβI (C-16), anti-PKCβII (C-18), anti-PKCζ (C-20), anti-PKCε (C-15), anti-PKCδ (C-20), anti-RhoA (26C4), anti-RhoC (G-12), anti-phosphorylated cofilin (anti-p-cofilin; at Ser-3), anti-phosphorylated adducin (anti-p-adducin; at Ser-726), and anti-p-adducin (at Thr-445) from Santa Cruz Biotechnologies (Santa Cruz, CA) were used as primary antibodies at 1:100/1:200 dilutions. Alexa 488 (Molecular Probes-Invitrogen, Eugene, OR) was used as secondary antibody at 1:500/1:1000 dilutions. Actin was stained with rhodamine phalloidin (Molecular Probes-Invitrogen, Eugene, OR). Samples were observed with a TSC-SP5 Broadband Spectra laser confocal microscope (Leica, Wetzlar, Germany) with an HCX-PL 63×/1.2 objective.
Results
LPA stimulates expression and activation of PKCα in isolated guinea pig OHCs
We used confocal microscopy to investigate the expression patterns of PKCα, PKCβI, PKCβII, PKCδ, PKCε, and PKCζ in guinea pig OHCs. As shown in Fig. 1 A, all of these PKC isoforms were found to be expressed in these cells. PKCα, PKCβI, and PKCε showed strong and relatively homogeneous cytoplasmic expression, whereas expression of PKCβII, PKCδ, and PKCζ was weaker and concentrated mostly at the subcuticular organ, the perinuclear region, and/or the infranuclear region (Fig. 1 A, arrows).
Figure 1.
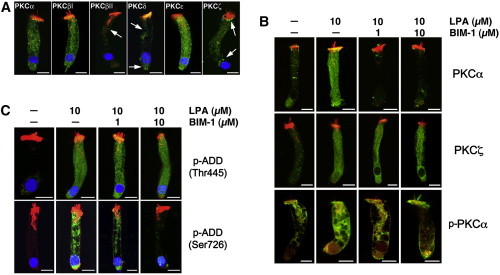
Immunolocalization of PKC isoenzymes and effect of BIM-1 on LPA-induced adducin phosphorylation in isolated guinea pig OHCs. (A) All PKC isoenzymes assayed were expressed in guinea pig OHCs (primary antibodies, green; DAPI, blue; rhodamine phalloidin, red). However, labeling was stronger for PKCα, PKCβI, and PKCε than for PKCβII, PKCδ, and PKCζ. In PKCβII and PKCδ, labeling was concentrated mostly at the subcuticular organ and the infranuclear region, whereas PKCζ immunoreactivity was frequently observed in the perinuclear region and below the cuticular plate (arrows). (B) LPA increased PKCα and PKCζ immunolabeling in the OHCs’ cytoplasm and nucleus. The effect on expression of PKCα, but not PKCζ, was prevented by pretreatment with BIM-1. LPA-induced increase in PKCα expression was associated with a similar increase in PKCα activation, as indicated by labeling with anti-p-PKCα at Ser-657, but only in the cytoplasm; no activation was observed in nuclear PKCα (primary antibodies, green; rhodamine phalloidin, red). (C) LPA also increased adducin phosphorylation of both Ser-726 and Thr-445 as indicated by reactivity to anti-p-adducin at Ser-726 and anti-p-adducin at Thr-445 antibodies (green; DAPI, blue; rhodamine phalloidin; red). However, treatment with BIM-1 reduced adducin phosphorylation only at Ser-726. In all images, scale bar = 10 μm.
Next, we investigated the effects of LPA, a well-known activator of Rho GTPases, and BIM-1, a potent cell-permeable and reversible pharmacological inhibitor of PKC proteins (14–18), on the expression of PKCs in OHCs. Stimulation of isolated OHCs with 10 μM LPA increased expression of PKCα and PKCζ in these cells (Fig. 1 B), whereas the immunoreactivity of other PKCs showed no changes (data not shown). Preincubation for 10 min before exposing OHCs to LPA with 1 or 10 μM of BIM-1 reduced PKCα expression in a dose-dependent manner, but the effects on PKCζ expression were only evident in the cell nucleus (Fig. 1 B). These results indicate that of all the PKC isoforms investigated in this study, PKCα was the only one whose expression was upregulated by LPA and downregulated by BIM-1 in the OHC cytoplasm. However, the increase or decrease in expression was not equal to the increased or decreased activation of the kinase. Therefore, we investigated the effect of LPA and BIM-1 on PKCα activation by labeling OHCs with antibodies targeting the phosphorylated form of the enzyme. As shown in Fig. 1 B, labeling of PKCα and p-PKCα in the cytoplasm of OHCs was correlated, with LPA enhancing and BIM-1 decreasing both PKCα expression and phosphorylation. Interestingly, LPA increased immunolabeling of PKCα, but not p-PKCα, in the cells’ nucleus. In the images of isolated OHCs in Fig. 1 B, we blocked the blue channel (DAPI) to make more evident the changes in nuclear expression induced by the treatment.
LPA stimulates adducin phosphorylation at both Thr-445 and Ser-726, but BIM-1 only inhibits phosphorylation at Ser-726
When we looked for additional effects of LPA treatment, we found a significant increase in adducin phosphorylation at Ser-726 and Thr-445 (Fig. 1 C), confirming a previous finding of our laboratory (2). However, we also found that preincubation with BIM-1 inhibited LPA-induced adducin phosphorylation only at Ser-726 (Fig. 1 C), which agrees with previous results from other laboratories indicating that adducin phosphorylation at Thr-445 by LPA would be a PKC-independent process (19). Thus, we concluded that PKCα is a mediator in the LPA-activated cascade that phosphorylates adducin at Ser-726.
LPA-induced PKCα activation and adducin phosphorylation would be mediated by RhoA and ROCK
We investigated the involvement of Rho GTPases in the signaling pathway activated by LPA, which induces PKCα-mediated adducin phosphorylation at Ser-726. For this purpose, in addition to BIM-1, we used the cell-permeable inhibitors C3 (an exoenzyme that selectively affects the highly homologous Rho isoforms RhoA, RhoB, and RhoC, but no other members of the family, such as Rac and Cdc42 (20)), and Y27632 (a specific inhibitor of the Rho kinase ROCK (21)). We found that both C3 (100 μg/mL) and Y27632 (10 μM) reduced the LPA-induced increase in PKCα expression as well as adducin phosphorylation at Ser-726 (Fig. 2 A). These results suggest that the signals that enhance PKCα expression and adducin phosphorylation at Ser-726 are mediated by Rho (A, B, or C) and ROCK.
Figure 2.
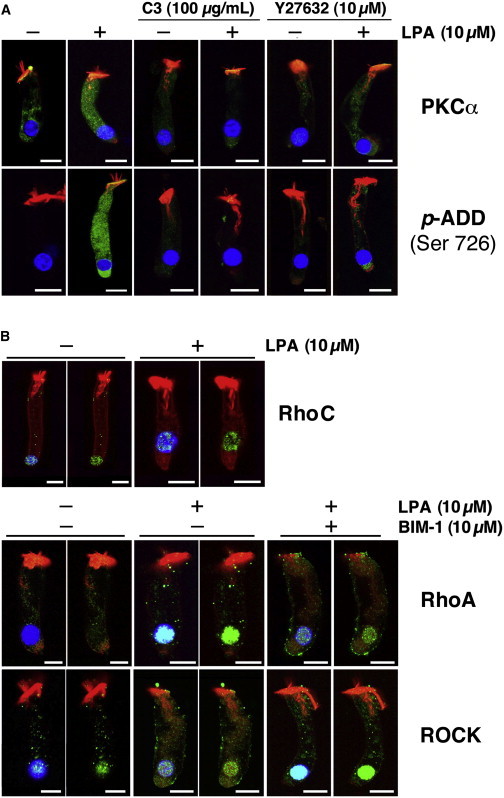
LPA-induced PKCα activation and adducin phosphorylation require RhoA and ROCK activation. Isolated guinea pig OHCs were stimulated with 10 μM LPA alone or preincubated with C3 (100 μg/ml), Y27632 (10 μM), or BIM-1 (10 μM) for 10 min before LPA stimulation. (A) The RhoA inhibitor C3 and the ROCK inhibitor Y27632 reduced PKCα expression and adducin phosphorylation at Ser-726. (B) OHCs express RhoA and RhoC, but only RhoA is stimulated by LPA. LPA increased the reactivity to RhoA antibodies in the OHCs’ nucleus, and this increase was reduced by pretreatment with BIM-1. In contrast, ROCK was expressed in the cytoplasm and nucleus of untreated OHCs, and preincubation with BIM-1 resulted in an evident increase in nuclear ROCK labeling. To better visualize the nuclear expression of RhoC, RhoA, and ROCK, the images are shown in duplicate with and without the blue (DAPI) channel. In all images, scale bar = 10 μm.
To identify the particular Rho GTPase involved in this signaling cascade, we used antibodies against RhoA, RhoB, and RhoC. We found that guinea pig OHCs express RhoA and RhoC (Fig. 2 B), but not RhoB (not shown). Interestingly, RhoC immunolabeling was more intense in the cells’ nucleus than in the cytoplasm, and expression levels were unchanged by treating the OHCs with 10 μM of LPA. In contrast, RhoA expression was similar in the cytoplasm and nucleus of untreated cells, but LPA dramatically increased the reactivity to RhoA antibodies in the OHCs’ nucleus, and this increase was visibly decreased by 10 μM of BIM-1 (Fig. 2 B). ROCK, on the other hand, was expressed in the cytoplasm and nucleus of untreated OHCs, and although LPA alone did not induce noticeable changes in ROCK immunoreactivity in the nucleus or cytoplasm, a 10 min preincubation with BIM-1 before LPA treatment resulted in an evident increase in nuclear ROCK labeling (Fig. 2 B). To better visualize the nuclear expression of RhoA and ROCK, the images of isolated OHCs in Fig. 2 B were duplicated with and without the blue (DAPI) channel. These results suggest that although RhoA and ROCK could indeed be upstream components of the signaling cascade activated by LPA that induces adducin phosphorylation at Ser-726 in OHCs, they could also be working downstream of PKCs (as suggested by experiments in other cell models (22)) in a pathway associated with RhoA expression in OHCs’ nucleus. However, the LPA-induced changes in nuclear expression of RhoA and ROCK would be not correlated with PKCα activation, since no significant increase in immunoreactivity to anti-p-PKCα was detected in the nucleus of OHCs that were either untreated or treated with LPA alone or with LPA plus BIM-1 (see Fig. 1 B).
BIM-1 inhibits the changes in OHC motility induced by LPA
Whereas the previous results indicate that LPA could activate a RhoA/ROCK/PKCα signaling cascade that induces adducin phosphorylation at Ser-726, they do not provide any evidence that this pathway could be involved in the regulation of OHC motility. To address this issue, we investigated changes in total cell length and EAEF-induced electromotile amplitude in OHCs treated with LPA for 15 min, with and without preincubation with BIM-1. As expected, our experiments showed that LPA induced a significant increase in the OHCs’ electromotile amplitude with respect to the control condition, which was diminished by preincubation with BIM-1 (Fig. 3, A and B). Moreover, BIM-1 alone (cells not treated with LPA) decreased the EAEF-induced electromotile amplitude in a dose-dependent manner (Fig. 3 C), suggesting that the RhoA/ROCK/PKCα pathway is continuously at work, even in the absence of external LPA stimulation, to regulate the motile response of cochlear OHCs. As described in previous studies (4,13), under the three experimental conditions the electromotile amplitude increased with time, reached a plateau ∼2 s after stimulation, and then remained stable. Here, we report data obtained from up to 3 s of electrical stimulation, the same final time point used in the previous studies.
Figure 3.
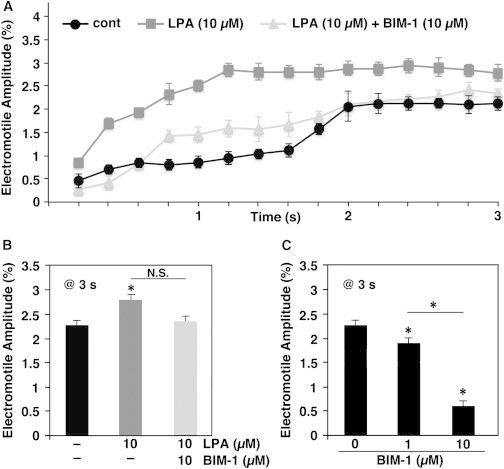
Effect of PKCα inhibition on OHC electromotile amplitude. OHCs were treated with 10 μM LPA alone for 15 min or preincubated with BIM-1 (1 and 10 μM) before LPA stimulation, and then electrically stimulated with an EAEF for 3 s. (A) Electromotile amplitude versus time in control (n = 5), LPA-treated (n = 5), and LPA+BIM-1-treated (10 μM, n = 5) OHCs. BIM-1 decreased the LPA enhancing effect on OHC electromotile amplitude. (B) Electromotile amplitude values (total cell length = 100%) at 3 s after the beginning of the stimulation (mean ± SE): control = 2.27% ± 0.11%, LPA = 2.78% ± 0.16% (p ≤ 0.008), LPA+BIM-1 (10 μM) = 2.34% ± 0.16% (p = N.S. with respect to both control and LPA). (C) Electromotile amplitude of untreated cells (n = 5) and cells exposed to BIM-1 (1 or 10 μM) only (n = 4): control = 2.27% ± 0.11%, BIM-1 (1 μM) = 1.87% ± 0.14%, BIM-1 (10 μM) = 0.60% ± 0.14% (p ≤ 0.02 and p ≤ 0.0001 re control, respectively, and p ≤ 0.001 between them).
We also found that BIM-1 modulated the EAEF-induced shortening of OHCs. Under our experimental conditions, control OHCs exposed to an EAEF contracted monotonically and showed a reduction of ∼1.5% in total length after 3 s. In contrast, in LPA-treated cells, cell length decreased by ∼2.2% in 1.6 s and then remained stable (Fig. 4 A). Preincubation with 10 μM of Bim-1 resulted in a response indistinguishable from that of control cells up to 2 s of stimulation, but at this point in time a sudden additional contraction drove the total decrease in cell length to ∼2.6% (Fig. 4, A and B). Moreover, BIM-1 alone (cells not treated with LPA) decreased OHC shortening in a dose-dependent manner (Fig. 4 C). These results suggest that the slow motility of OHCs is modulated by PKCα-mediated signals, but other pathways could also be controlling their slow somatic responses.
Figure 4.
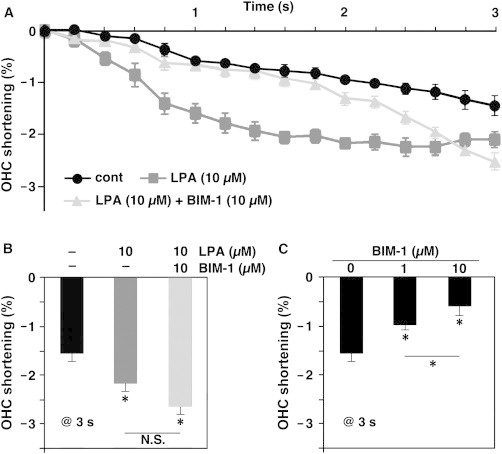
Effect of PKCα inhibition on OHC length. Electrically stimulated OHCs showed a decrease in total length (slow motility). (A) OHC shortening versus time in control (n = 5), LPA treated (n = 5), and LPA+BIM-1-treated (10 μM, n = 5) OHCs. Whereas LPA increased cell shortening, pretreatment with BIM-1 partially modified this response. (B) Cell shortening values (total cell length = 100%) at 3 s after the beginning of the stimulation (mean ± SE): control = 1.51% ± 0.18%, LPA = 2.18% ± 0.18% (p ≤ 0.01 re control), LPA+BIM-1 (10 μM) = 2.64% ± 0.18% (p ≤ 0.0008 re control, N.S. re LPA alone). (C) Shortening in untreated cells (n = 5) and cells exposed to BIM-1 (1 or 10 μM) only (n = 4): control = 1.51% ± 0.18%, BIM-1 (1 μM) = 1.02% ± 0.10%, BIM-1 (10 μM) = 0.64% ± 0.20% (p ≤ 0.02 and p ≤ 0.0001 re control, respectively, and p ≤ 0.01 between them).
Since BIM-I is also an inhibitor of protein kinase A (PKA), we wondered whether LPA’s effects on OHC motility could be associated with this kinase rather than with PKCα. To test this possibility, we used H-89, a potent inhibitor that is 500-fold more selective for PKA than for PKCs (23). Since its IC50 for PKA is 135 nM, we tested the effect of H-89 concentrations ranging from 50 nM to 1 μM on LPA-induced changes in OHC motility. We detected no significant influence of H-89 on either OHC electromotile amplitude or shortening (results not shown), indicating that BIM-1 effects are associated mainly with inhibition of PKCα.
PKCα influences cofilin phosphorylation
As shown in previous studies by our laboratory, LPA activates a RhoA/ROCK/LIMK1-mediated pathway that modulates actin filament dynamics by phosphorylating cofilin (5). Thus, it might be possible that the LPA effects described above as being associated with activation of PKCα are instead related to LPA-induced cofilin phosphorylation.
To address this issue, we used TPA, which stimulates PKCα activity but, unlike LPA, does not target RhoA or ROCK and therefore should not stimulate LIMK1-mediated cofilin phosphorylation. As expected, we confirmed that TPA treatment did not change ROCK expression and, importantly, did not induce cofilin phosphorylation (Fig. 5 A). This result was not surprising, since it is generally accepted that PKCs do not phosphorylate cofilin, and the typical site of cofilin phosphorylation, Ser-3, is not a consensus PKC phosphorylation site. Intriguingly, however, preincubation with BIM-1 decreased LPA-induced immunoreactivity to p-cofilin (Fig. 5 A), suggesting that activation of PKCα was required for the efficient phosphorylation of cofilin at Ser-3 induced by LPA. On the other hand, TPA stimulated PKCα and PKCζ expression and adducin phosphorylation at Thr-445 and Ser-726 just like LPA, and BIM-1 inhibited only PKCα activation and adducin phosphorylation at Ser-726 (Fig. 5 B).
Figure 5.
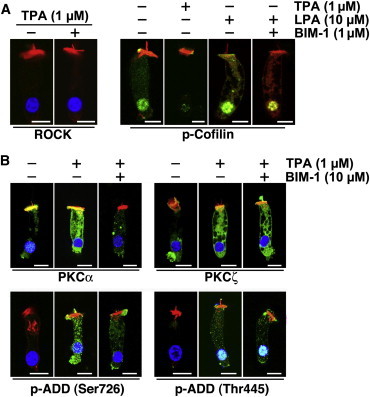
Effect of TPA on ROCK, PKCα, and PKCζ expression and adducin phosphorylation in OHCs. (A) Levels of ROCK expression were not changed, but labeling of nuclear p-cofilin decreased significantly in OHCs treated with TPA. LPA, in contrast, increased p-cofilin labeling in the cytoplasm and nucleus of OHCs, whereas BIM-1 significantly decreased LPA-induced cofilin phosphorylation. (B) Whereas TPA increased expression of both PKCα and PKCζ, BIM-1 inhibited this effect only for PKCα. Similarly, TPA induced adducin phosphorylation at both Ser-726 and Thr-445, but treatment with BIM-1 reduced adducin phosphorylation only at Ser-726. In all images, scale bar = 10 μm.
Thus, since BIM-1 inhibits PKCα and interferes with LPA-induced, RhoA/ROCK/LIMK1-mediated cofilin phosphorylation, we speculate that PKCα is part of a still unknown mechanism that facilitates cofilin phosphorylation by LIMK1 (Fig. 6).
Figure 6.
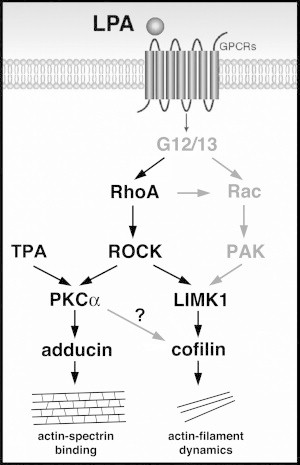
Cartoon representing LPA-activated signaling cascades that regulate OHC motility, as suggested by our results and previous studies (in black font), as well as potential mediators and interactions that require confirmation (gray font). LPA activation of the RhoA/ROCK/LIMK1/cofilin pathway would target actin-filament dynamics, enhancing actin depolymerization. In turn, activation of the RhoA/ROCK/PKCα/ adducin pathway would phosphorylate adducin at Ser-726, inhibiting actin-spectrin binding. However, these two signaling cascades would be working in parallel, with PKCα facilitating cofilin phosphorylation by a still unknown mechanism. The final result of the activation of these two signaling pathways would be the same, i.e., a decrease in the integrity of the OHC actin/spectrin-based cortical cytoskeletal network.
Discussion
OHC motility is the major mechanism underlying cochlear amplification, the process that increases the sensitivity and frequency discrimination of the mammalian cochlea (1,24,25). Our goal in this study was to obtain mechanistic insights into the regulation of OHC motility, and hence cochlear amplification, by the OHC cortical cytoskeleton. For about three decades, the prevalent technique for investigating OHC motility has been whole-cell voltage clamp. However, the patch pipette not only disrupts the plasma membrane but also affects the molecular organization and functional response of the cortical cytoskeleton, the specific structure whose influence on OHC motility we are trying to evaluate. Thus, in this study we used an EAEF. In this technique, cell movement is not constrained by any attachment to the rim of a suction micropipette that could influence the mechanical response of the OHC lateral wall, and the responses may be entirely attributed to the interaction of the electrical field with the cellular motile mechanism. Our findings indicate that activation of a RhoA/ROCK/PKCα signaling cascade with LPA results in adducin phosphorylation at Ser-726, enhancement of the OHC electromotile amplitude, and modulation of the changes in the total OHC length (slow motility). Since adducin phosphorylation at Ser-726 inhibits actin-spectrin binding (7,8), we speculate that the changes in OHC motile responses would be associated with a partial disintegration of the actin-spectrin network lining the cytoplasmic surface of the OHC lateral wall induced by LPA activation of a RhoA/ROCK/PKCα/adducin signaling cascade. Importantly, in a previous report (5) we presented evidence indicating that a regulatory mechanism of OHC motility relied, at least in part, on the control of the depolymerization rate of actin filaments by RhoA/ROCK/LIMK1/cofilin-mediated signals. The study presented here further extends those results, suggesting that both pathways (RhoA/ROCK/PKCα/adducin and RhoA/ROCK/LIMK1/cofilin) work together, with PKCα activation facilitating LPA-induced cofilin phosphorylation mediated by the RhoA/ROCK/LIMK1 signaling cascade (Fig. 6).
RhoA/ROCK-mediated pathways
The control of actin dynamics, cytoskeletal reorganization, cell motility, and migration in animal cells is concentrated in three members of the Rho family of small GTPases: Rho, Rac, and Cdc42 (26–28). Since in this study we used the specific cell-permeable Rho inhibitor C3, which inhibits Rho, but not Rac or Cdc42, we conclude that the LPA-mediated PKC activation pathway does not involve Rac or Cdc42.
The three Rho isoforms (RhoA, RhoB, and RhoC) have different expression and activation patterns, different binding activities and/or affinities for their target proteins, and different subcellular localizations (28,29). RhoB is a short-lived protein that, unlike RhoA and RhoC, is not constitutively expressed but rather is induced by both physical and chemical agents (30). Thus, we were not surprised that we could not detect RhoB in OHCs under our experimental conditions. The functions of RhoA and RhoC, on the other hand, are quite similar, although previous studies suggested that they have different effects on cell morphology, motility, and invasion because they act on different downstream targets (28,29). Although both RhoA and RhoC were found to be expressed in OHCs, this work as well as previous studies point to RhoA as the mediator of LPA-activated signals in these cells.
PKCα phosphorylates adducin at Ser-726 and regulates OHC motility
Our results indicate that guinea pig OHCs express all of the assayed PKCs (PKCα, PKCβI, PKCβII, PKCδ, PKCε, and PKCζ), although with different abundances and patterns of distribution. However, only the effects of LPA and TPA on PKCα are both inhibited by BIM-1 and correlated with adducin phosphorylation at Ser-726, supporting the idea that PKCα is the specific isoform involved in the LPA-activated RhoA/ROCK/PKC-mediated signaling cascade that regulates OHC motility.
We provide evidence that adducin phosphorylation at Ser-726 correlates with an enhancement of the OHC electromotile amplitude and changes in total cell length (slow motility). Although we are not demonstrating causality, the fact that phosphorylation of adducin at Ser-726 is a well-known promoter of actin-spectrin dissociation (7,8) strongly suggests that both events are mechanistically linked. We performed experiments on OHCs isolated from control and β-adducin knockout (KO) mice, but confocal microscopy experiments showed that β-adducin was replaced by γ-adducin in the KO animals, and we were unable to detect any significant difference in cell motility between the two groups (R. Kitani and F.K., unpublished results). This was not a completely surprising result, since it was previously reported that deletion of β-adducin results in its replacement by γ-adducin in red blood cells (RBCs), suggesting that α-adducin requires a heterologous binding partner for stability and function (31,32). Although the sequences of β and γ adducin have only 60–70% similarity, they both have an N-terminal globular head domain, a neck domain, and a C-terminal tail domain, with a highly conserved 22-residue MARCKS-related domain at the end of the C-terminal tail domain containing the major phosphorylation site for PKCs (7). Therefore, we can speculate that PKCs phosphorylate γ-adducin just like they phosphorylate β-adducin. However, RBCs in β-adducin KO mice were osmotically fragile and microcytic, and had the phenotype of hereditary spherocytosis, whereas no evident structural changes were observed in OHCs. We speculate that these different responses reflect the different structure of the cortical cytoskeleton in RBCs and OHCs. In RBCs the actin filaments are very short and interconnected by six to eight spectrin molecules in a spiderweb-like fashion (33). Therefore, even a partial malfunction of adducin in RBCs may result in extensive structural failure. In OHCs, in contrast, actin filaments are very long, nearly parallel, and interconnected by spectrin molecules spaced ∼30–40 nm along the actin cables (34). Thus, adducin malfunction in OHCs could be partially compensated for by just a low percentage of strong actin-spectrin connections between parallel actin fibers. Currently, we are investigating the motile responses of OHCs from β/γ-adducin double-KO mice to definitively confirm the involvement of adducin in the regulation of OHC motility.
Intriguingly, LPA also induced adducin phosphorylation at Thr-445, which promotes actin-spectrin binding, via a most likely PKCα-independent (not inhibited by BIM-1) pathway. We speculate that adducin phosphorylation at Thr-445 could be part of a safety mechanism aimed at preventing a massive disintegration of the actin-spectrin network that could compromise the OHC structure. This speculation is supported by our results showing that inhibition of PKCα in LPA-treated cells maintained OHC motile responses near control values, whereas PKCα inhibition in untreated cells induced a very significant and dose-dependent decrease in these responses. In previous studies (2,5) we speculated that acetylcholine (ACh) would be able to stimulate cofilin phosphorylation and OHC electromotility via a RhoA-dependent, ROCK-independent signaling cascade, and modulate OHC slow motility by inhibiting cofilin phosphorylation via the RhoA/ROCK pathway, exerting a profound influence on cochlear amplification. The potential interaction of ACh with the RhoA/ROCK/PKCα/adducin pathway is currently being investigated.
Together with previous studies showing the pivotal role of actin depolymerization, these results highlight the important role of the cortical cytoskeleton in the regulation of OHC motility.
Role of the RhoA/ROCK/PKCα/adducin pathway in the regulation of OHC motility
Our experiments showed that LPA induced a significant increase in the OHC electromotile amplitude, which was diminished by inhibiting PKCα (Fig. 3 B). Likewise, LPA increased OHC shortening (slow motility) and PKCα inhibition modulated, but did not prevent, the LPA effect (Fig. 4, A and B). Although a quantitative comparison of our results with previous findings regarding activation and inhibition of the RhoA/ROCK/LIMK1/cofilin pathway (5) is not possible because of the different techniques and experimental protocols used in the two studies (EAEF and patch-clamp stimulation, 10 min and 3 s long, respectively), they are clearly qualitatively similar. This is not a surprise, since both processes (enhancing actin depolymerization via RhoA/ROCK/LIMK1/cofilin and inhibiting actin-spectrin binding via RhoA/ROCK/PKCα/adducin) have a similar output, i.e., a decrease in the integrity of the OHC actin-spectrin cortical cytoskeleton. Interestingly, in cells not treated with LPA, PKCα inhibition decreased the EAEF-induced electromotile amplitude and OHC shortening in a dose-dependent manner (Figs. 3 C and 4 C), suggesting that the RhoA/ROCK/PKCα/adducin pathway is continuously at work to regulate the motile response of OHCs.
It has been suggested that in vascular smooth muscle cells, TPA-induced PKC activation leads to Rho downregulation and disassembly of actin filaments (22,35). Our experiments indicate that in OHCs, LPA-mediated activation of the RhoA/ROCK/LIMK1 signaling cascade requires PKCα activation to induce cofilin phosphorylation and hence disassembly of actin filaments. Thus, we speculate that in OHCs, the RhoA/ROCK/LIMK1/cofilin and RhoA/ROCK/PKCα/adducin pathways would work in parallel and show an active cross talk (Fig. 6). This could be an additional mechanism aimed at optimizing the homeostatic control of the OHCs’ mechanical responses. Interestingly, previous experimental evidence regarding the control of spontaneous phasic activities in rectal smooth muscles also suggested that PKC- and RhoA/ROCK-mediated pathways could be working in parallel, with an active cross talk (36). Moreover, a recent study on the mechanism of degranulation in rat basophilic leukemia-2H3 cells suggested a possible mechanistic explanation for our results (37). In that study, the authors used mass spectrometry and mutagenic analyses to reveal that PKCα phosphorylates cofilin at Ser-23 and/or Ser-24, but not at Ser-3. Other laboratories have not yet confirmed this result, but if PKCα indeed phosphorylates cofilin at Ser-23 and/or Ser-24, it could be inducing conformational changes in the cofilin molecule that facilitate the LPA-induced, LIMK-1-mediated cofilin phosphorylation at Ser-3. Experimental approaches to investigate this possibility in OHCs are currently being evaluated in our laboratory.
How does the cytoskeleton network influence OHC motor function?
A few years ago, we demonstrated that LPA stimulation has no direct effect on prestin performance (5). In a subsequent study, our laboratory provided evidence that the lateral wall of guinea pig OHCs consists of discrete structural microdomains that can move relative to each other and change their orientation with respect to the cell axis (4). Thus, we speculate that LPA-induced activation of the RhoA/ROCK/PKCα/adducin pathway, just like activation of the RhoA/ROCK/LIMK1/cofilin signaling cascade, does not directly affect prestin performance, but could be responsible for the rearrangement of microdomains in the plane of the plasma membrane and, probably, the organization or disorganization of the motor units (prestin single molecules, dimers or tetramers) inside the microdomains. In this way, the magnitude and/or direction of the vectorial component of the force generated by every molecular motor unit would depend on the cortical cytoskeleton.
Conclusions
Using an EAEF rather than the classical whole-cell, patch-clamp technique, which could influence the mechanical response of the OHC lateral wall, we found that activation of a RhoA/ROCK/PKCα signaling cascade with LPA resulted in adducin phosphorylation at Ser-726, enhancement of the OHC electromotile amplitude, and modulation of the changes in total OHC length (slow motility). We speculate that the changes in OHC motile responses are associated with a partial disintegration of the actin-spectrin network lining the cytoplasmic surface of the OHC lateral wall. We also found that activation of PKCα may enhance LPA-induced, RhoA/ROCK-mediated cofilin phosphorylation. Thus, our results support the idea that the cytoskeleton plays a crucial role in the regulation of OHC motility, and strongly suggest the existence of multiple control mechanisms that work coordinately in the fine regulation of these motile responses and cochlear amplification.
Author Contributions
C.P. performed the research. F.K. designed the research, analyzed data, and wrote the article.
Acknowledgments
This work was supported by NIDCD-NIH grants R01-DC010146 and R01-DC010397 and the Department of Head and Neck Surgery, David Geffen School of Medicine at UCLA. Its content is solely the responsibility of the authors and does not necessarily represent the official view of the National Institutes of Health or UCLA.
References
- 1.Ashmore J. Cochlear outer hair cell motility. Physiol. Rev. 2008;88:173–210. doi: 10.1152/physrev.00044.2006. [DOI] [PubMed] [Google Scholar]
- 2.Zhang M., Kalinec G.M., Kalinec F. ROCK-dependent and ROCK-independent control of cochlear outer hair cell electromotility. J. Biol. Chem. 2003;278:35644–35650. doi: 10.1074/jbc.M301668200. [DOI] [PubMed] [Google Scholar]
- 3.Kalinec F., Zhang M., Kalinec G. Rho GTPases mediate the regulation of cochlear outer hair cell motility by acetylcholine. J. Biol. Chem. 2000;275:28000–28005. doi: 10.1074/jbc.M004917200. [DOI] [PubMed] [Google Scholar]
- 4.Kitani R., Park C., Kalinec F. Microdomains shift and rotate in the lateral wall of cochlear outer hair cells. Biophys. J. 2013;104:8–18. doi: 10.1016/j.bpj.2012.11.3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matsumoto N., Kitani R., Kalinec F. Pivotal role of actin depolymerization in the regulation of cochlear outer hair cell motility. Biophys. J. 2010;99:2067–2076. doi: 10.1016/j.bpj.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fukata Y., Oshiro N., Kaibuchi K. Activation of moesin and adducin by Rho-kinase downstream of Rho. Biophys. Chem. 1999;82:139–147. doi: 10.1016/s0301-4622(99)00113-1. [DOI] [PubMed] [Google Scholar]
- 7.Matsuoka Y., Li X., Bennett V. Adducin is an in vivo substrate for protein kinase C: phosphorylation in the MARCKS-related domain inhibits activity in promoting spectrin-actin complexes and occurs in many cells, including dendritic spines of neurons. J. Cell Biol. 1998;142:485–497. doi: 10.1083/jcb.142.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matsuoka Y., Li X., Bennett V. Adducin: structure, function and regulation. Cell. Mol. Life Sci. 2000;57:884–895. doi: 10.1007/PL00000731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohno S., Nishizuka Y. Protein kinase C isotypes and their specific functions: prologue. J. Biochem. 2002;132:509–511. doi: 10.1093/oxfordjournals.jbchem.a003249. [DOI] [PubMed] [Google Scholar]
- 10.Zeng L., Webster S.V., Newton P.M. The biology of protein kinase C. In: Islam M.S., editor. Calcium Signaling, Advances in Experimental Medicine and Biology. Springer Science+Business Media; Dordrecht: 2012. pp. 639–661. [DOI] [PubMed] [Google Scholar]
- 11.Zajic G., Schacht J. Comparison of isolated outer hair cells from five mammalian species. Hear. Res. 1987;26:249–256. doi: 10.1016/0378-5955(87)90061-x. [DOI] [PubMed] [Google Scholar]
- 12.Kitani R., Kalinec F. Investigating outer hair cell motility with a combination of external alternating electrical field stimulation and high-speed image analysis. J. Vis. Exp. 2011;53:2965. doi: 10.3791/2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kitani R., Kakehata S., Kalinec F. Motile responses of cochlear outer hair cells stimulated with an alternating electrical field. Hear. Res. 2011;280:209–218. doi: 10.1016/j.heares.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Toullec D., Pianetti P., Loriolle F. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J. Biol. Chem. 1991;266:15771–15781. [PubMed] [Google Scholar]
- 15.Martiny-Baron G., Kazanietz M.G., Schächtele C. Selective inhibition of protein kinase C isozymes by the indolocarbazole Gö 6976. J. Biol. Chem. 1993;268:9194–9197. [PubMed] [Google Scholar]
- 16.Jacobson P.B., Kuchera S.L., Schrier D.J. Anti-inflammatory properties of Gö 6850: a selective inhibitor of protein kinase C. J. Pharmacol. Exp. Ther. 1995;275:995–1002. [PubMed] [Google Scholar]
- 17.Coultrap S.J., Sun H., Machu T.K. Competitive antagonism of the mouse 5-hydroxytryptamine3 receptor by bisindolylmaleimide I, a “selective” protein kinase C inhibitor. J. Pharmacol. Exp. Ther. 1999;290:76–82. [PubMed] [Google Scholar]
- 18.Wu-Zhang A.X., Newton A.C. Protein kinase C pharmacology: refining the toolbox. Biochem. J. 2013;452:195–209. doi: 10.1042/BJ20130220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fukata Y., Oshiro N., Kaibuchi K. Phosphorylation of adducin by Rho-kinase plays a crucial role in cell motility. J. Cell Biol. 1999;145:347–361. doi: 10.1083/jcb.145.2.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Just I., Huelsenbeck S.C., Genth H. Clostridium botulinum C3 exoenzyme: Rho-inactivating tool in cell biology and a neurotrophic agent. Open Toxinol. J. 2010;3:19–23. [Google Scholar]
- 21.Yamaguchi H., Miwa Y., Hakoshima T. Structural basis for induced-fit binding of Rho-kinase to the inhibitor Y-27632. J. Biochem. 2006;140:305–311. doi: 10.1093/jb/mvj172. [DOI] [PubMed] [Google Scholar]
- 22.Brandt D., Gimona M., Mischak H. Protein kinase C induces actin reorganization via a Src- and Rho-dependent pathway. J. Biol. Chem. 2002;277:20903–20910. doi: 10.1074/jbc.M200946200. [DOI] [PubMed] [Google Scholar]
- 23.Marunaka Y., Niisato N. H89, an inhibitor of protein kinase A (PKA), stimulates Na+ transport by translocating an epithelial Na+ channel (ENaC) in fetal rat alveolar type II epithelium. Biochem. Pharmacol. 2003;66:1083–1089. doi: 10.1016/s0006-2952(03)00456-8. [DOI] [PubMed] [Google Scholar]
- 24.Ashmore J., Avan P., Canlon B. The remarkable cochlear amplifier. Hear. Res. 2010;266:1–17. doi: 10.1016/j.heares.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dong W., Olson E.S. Detection of cochlear amplification and its activation. Biophys. J. 2013;105:1067–1078. doi: 10.1016/j.bpj.2013.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burridge K., Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167–179. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- 27.Jaffe A.B., Hall A. Rho GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- 28.Wheeler A.P., Ridley A.J. Why three Rho proteins? RhoA, RhoB, RhoC, and cell motility. Exp. Cell Res. 2004;301:43–49. doi: 10.1016/j.yexcr.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 29.Thumkeo D., Watanabe S., Narumiya S. Physiological roles of Rho and Rho effectors in mammals. Eur. J. Cell Biol. 2013;92:303–315. doi: 10.1016/j.ejcb.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 30.Rodriguez P.L., Sahay S., Whitehead I.P. ROCK I-mediated activation of NF-kappaB by RhoB. Cell. Signal. 2007;19:2361–2369. doi: 10.1016/j.cellsig.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gilligan D.M., Lozovatsky L., Peters L.L. Targeted disruption of the β adducin gene (Add2) causes red blood cell spherocytosis in mice. Proc. Natl. Acad. Sci. USA. 1999;96:10717–10722. doi: 10.1073/pnas.96.19.10717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sahr K.E., Lambert A.J., Peters L.L. Targeted deletion of the γ-adducin gene (Add3) in mice reveals differences in α-adducin interactions in erythroid and nonerythroid cells. Am. J. Hematol. 2009;84:354–361. doi: 10.1002/ajh.21427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gov N.S. Active elastic network: cytoskeleton of the red blood cell. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2007;75:011921. doi: 10.1103/PhysRevE.75.011921. [DOI] [PubMed] [Google Scholar]
- 34.Holley M.C., Kalinec F., Kachar B. Structure of the cortical cytoskeleton in mammalian outer hair cells. J. Cell Sci. 1992;102:569–580. doi: 10.1242/jcs.102.3.569. [DOI] [PubMed] [Google Scholar]
- 35.Brandt D.T., Goerke A., Mischak H. Protein kinase C δ induces Src kinase activity via activation of the protein tyrosine phosphatase PTP α. J. Biol. Chem. 2003;278:34073–34078. doi: 10.1074/jbc.M211650200. [DOI] [PubMed] [Google Scholar]
- 36.Singh J., Rattan S. Role of PKC and RhoA/ROCK pathways in the spontaneous phasic activity in the rectal smooth muscle. Am. J. Physiol. Gastrointest. Liver Physiol. 2013;304:G723–G731. doi: 10.1152/ajpgi.00473.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sakuma M., Shirai Y., Saito N. Novel PKCα-mediated phosphorylation site(s) on cofilin and their potential role in terminating histamine release. Mol. Biol. Cell. 2012;23:3707–3721. doi: 10.1091/mbc.E12-01-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]