

Abstract
Von Willebrand factor (VWF) plays a central role in hemostasis. Triggered by shear-stress, it adheres to platelets at sites of vascular injury. Inactivation of VWF has been associated to the shielding of its adhesion sites and proteolytic cleavage. However, the molecular nature of this shielding and its coupling to cleavage under shear-forces in flowing blood remain unknown. In this study, we describe, to our knowledge, a new force-sensory mechanism for VWF-platelet binding, which addresses these questions, based on a combination of molecular dynamics (MD) simulations, atomic force microscopy (AFM), and microfluidic experiments. Our MD simulations demonstrate that the VWF A2 domain targets a specific region at the VWF A1 domain, corresponding to the binding site of the platelet glycoprotein Ibα (GPIbα) receptor, thereby causing its blockage. This implies autoinhibition of the VWF for the binding of platelets mediated by the A1-A2 protein-protein interaction. During force-probe MD simulations, a stretching force dissociated the A1A2 complex, thereby unblocking the GPIbα binding site. Dissociation was found to be coupled to the unfolding of the A2 domain, with dissociation predominantly occurring before exposure of the cleavage site in A2, an observation that is supported by our AFM experiments. This suggests that the A2 domain prevents platelet binding in a force-dependent manner, ensuring that VWF initiates hemostasis before inactivation by proteolytic cleavage. Microfluidic experiments with an A2-deletion VWF mutant resulted in increased platelet binding, corroborating the key autoinhibitory role of the A2 domain within VWF multimers. Overall, autoinhibition of VWF mediated by force-dependent interdomain interactions offers the molecular basis for the shear-sensitive growth of VWF-platelet aggregates, and might be similarly involved in shear-induced VWF self-aggregation and other force-sensing functions in hemostasis.
Introduction
Von Willebrand Factor (VWF) is a giant extracellular protein playing a key adhesive role in blood clotting. Activated by shear-stress, this protein cross-links the extracellular matrix of the endothelium with blood platelets, at sites of vascular injury (1,2). It efficiently participates in the shear-induced reversible formation of biopolymer-colloid aggregates (3), and its malfunction leads to pathological bleeding and thromboembolic disorders (1).
Functional VWF is a linear multimer of tens of covalently linked monomers (4), extending up to 15 μm (5). Each monomer, with a length of 60 to 80 nm (2,6), comprises 2050 amino acids in domains of few nm in size (7). The large size in the μm range enables VWF multimers to sense changes in the shear flow of blood and to translate them into a mechanical stretching force along the protein chain (5,8,9). Shear-forces, by inducing a tumbling motion alternating between globular and extended states, facilitate the adhesion of VWF to the extracellular matrix (5,10) and to flowing platelets (3).
The VWF A1 and A2 domains are critical for the activation of VWF to bind platelets and for its deactivation by size control. These two domains are adjacent to each other and connected by a linker of 30 amino acids (Fig. 1 A). X-ray crystallography revealed that both domains adopt a stable Rossmann α/β-fold (11,12), stabilized by calcium in the case of A2 (13,14). Platelets bind through the glycoprotein Ibα (GPIbα) to a region of the A1 domain (15,16), in a shear-dependent manner (17–21). For size control, the A2 domain is cleaved by the metalloprotease ADAMTS13 (22), after exposure of the Y1605-M1606 (YM) cleavage site, because of shear-induced domain unfolding (23–27).
Figure 1.
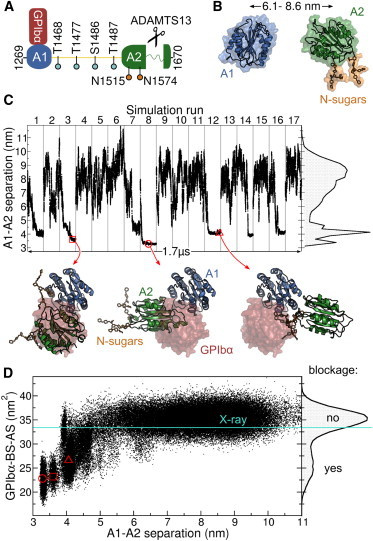
Blockage of the GPIbα binding site in the VWF revealed by MD simulations of the VWF A1 and A2 domains. (A) Scheme illustrating the human VWF-A1A2 fragment (residues 1269 to 1670). The A1 and A2 domains are connected by a 30 residue linker (yellow). GPIbα anchors platelets to VWF by binding to the A1 domain. VWF size is controlled by cleavage of the unfolded A2 domain by ADAMTS13. O-linked (cyan) and N-linked sugars (N-sugars, orange) are found within the fragment. (B) One of the multiple starting conformation used in the MD simulations (protein as cartoon and surface and sugars as sticks). The domain-domain center of mass (A1-A2) separation is indicated with the black arrow. (C) A1-A2 separation along the concatenated MD simulation time. Gray lines separate individual MD runs. The right plot shows the normalized histogram of the A1-A2 separation. Conformations at the bottom show examples with the two domains in contact (cartoon) contrasted to the region occupied by GPIbα when it binds to A1 (red surface), taken at the instants marked with the red symbols. (D) GPIbα binding site accessible surface (GPIbα-BS-AS) as a function of the A1-A2 separation (main panel) and its normalized histogram (right plot), recovered from MD simulations. Reduced GPIbα-BS-AS values indicate blockage of the GPIbα binding site. The GPIbα-BS-AS derived from the VWF A1-GPIbα complex x-ray structure (16) is depicted by the cyan line. The red symbols correspond to the conformations shown in (C). To see this figure in color, go online.
Under equilibrium or under low shear-stress conditions, VWF is incapable of binding platelets. This inactivation has been associated with a shielding of the GPIbα binding site of A1. Recent experiments revealed that, in addition to the D’D3 domains (28) and the linker connecting them to the A1 domain (29), isolated A2 domains modulate glycoprotein Ib (and thereby platelet) binding (30,31). However, electron microscopy (EM) images established the separation between these two domains, within the same VWF molecule, from 4.4 to 11 nm (6), challenging the inhibitory role of A2 on A1. Hence, little is known on how these two domains interact with each other, causing inhibition, and how sensitive this interaction is to shear-forces in flowing blood. It also remains unclear how VWF activation, through the release of the GPIbα binding site, and VWF deactivation, through unfolding of the A2 domain, are mechanically regulated to balance the propagation and attenuation of hemostasis. We addressed these questions by performing molecular dynamics (MD) simulations of the VWF A1 and A2 domains, under equilibrium and force-probe conditions, together with molecular docking calculations, atomic force microscopy (AFM) binding measurements, and microfluidic experiments. To our knowledge, our results suggest a novel mechanism for shear-dependent primary hemostasis, involving a force-sensitive autoinhibition state, in which platelets are incapable to bind to VWF because of direct (intra- or intermolecular) A1-A2 interactions precluding the A1-GPIbα interaction.
Materials and Methods
Equilibrium MD simulations
In the first simulation system, the A1 and A2 domains of the VWF were not covalently connected by their interdomain linker. They were either initially separated by distances from 6.1 to 8.6 nm to monitor association or already bound in conformations blocking the GPIbα binding site (obtained by docking, see below) for refinement. The second simulation system corresponded to the VWF-A1A2 fragment consisting of the A1 and A2 domains connected by a 30 amino acid linker, with an initial interdomain separation of 7.9 nm based on EM estimates (6). The most predominantly found sugars in the VWF glycome (32,33) were attached to the protein (Fig. 1 A and Fig. S1 in the Supporting Material). Simulations were carried out with the GROMACS package (4.5 version) (34–36). Sixteen or 17 runs, considering multiple interdomain initial orientations, were performed for each condition ( 82 ns per run) yielding a concatenated simulated time of 4.86 μs. The GPIbα binding site accessible surface (GPIbα-BS-AS) was computed by monitoring the amount of exposed surface of the GPIbα binding site in the A1 domain. A principal component analysis (PCA), consisting in the calculation and diagonalization of the covariance matrix of the atomic coordinates (37), was employed to monitor the interdomain orientations (Fig. 2). The solvent accessible hydrophobic surface (SAHS) reduction was estimated as [SAHS(A1A2) − SAHS(A1) − SAHS(A2)]/[SAHS(A1) + SAHS(A2), computing separately the surface for the complex (A1A2) and for the domains A1 and A2.
Figure 2.
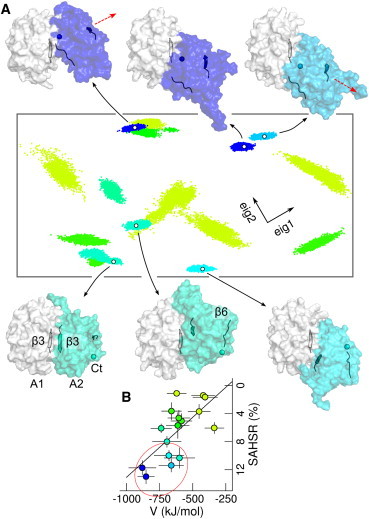
Orientational preferences of the VWF-A1A2 complex in the blocked state. (A) Principal component analysis (PCA) of the structures of the not-covalently linked VWF-A1A2 complex, with the GPIbα binding site blocked, predicted by molecular docking, yielded two main collective vectors (eig1 and eig2). MD trajectories (the last 50 ns) starting from these structures were projected onto the two-dimensional (2D) space created by these two vectors (projections in arbitrary units). Each dot, representing a simulation snapshot, reflects an adopted interdomain orientation. Each run is colored according to its interdomain potential interaction energy, V, and average solvent accessible hydrophobic surface reduction, SAHSR (see B). Representative orientations of runs with both high V and SAHSR (enclosed by the red circle in B) are displayed (A1 domain, white; A2 domain, color; β3 strands, cartoon; A2 β6 strand, ribbon, and A2 C terminus, sphere). The red arrows illustrate the change in orientation of A2 on horizontal changes in the 2D-PCA space. (B) SAHSR as a function of V (time-average standard deviation from the last 50 ns of each run). Colors indicate the projection along a linear fit (black line), with both V and SAHSR ranging from small (light green) to large (blue) values. To see this figure in color, go online.
Force-probe MD simulations
The A1 and A2 domains of the VWF were subjected to external harmonic forces on the N-terminus of the A1 domain and on the C terminus of the A2 domain (Fig. 3 A). Harmonic springs (with elastic constants of 500 kJmol–1nm–2) were attached to these termini and moved away from each other at a speed of 0.2 m/s. These simulations were started from 17 different starting conformations: one was extracted from an equilibrium MD run showing spontaneous binding (run number eight in Fig. 1 C) and the remaining 16 corresponded to representative conformations of the equilibrium simulations of the VWF-A1A2 complex (one conformation taken from each run presented in Fig. 2 A). Hence, starting conformations with high but also moderate stability were considered. The two monomers were not connected, first, to resemble dissociation of the A1-A2 complex either within or across VWF monomers (preventing from possible artifacts by the inclusion of the flexible linker for which the structure is unknown), and second, to have a direct comparison with our AFM experiments (also carried out with nonconnected domains, see below). Dissociation was assigned to the moment when the interdomain number of contacts was zero. Detachment of the A2-β5 strand from the core of the A2 domain was monitored by measuring the distance between V1625-P1627 (at β5) and V1604-Y1605 (at β4).
Figure 3.
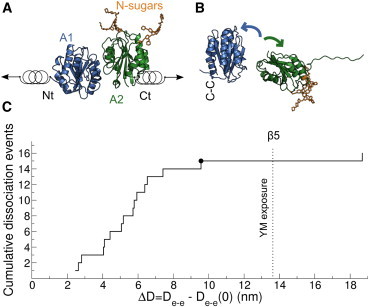
Force response of the VWF-A1A2 complex from force-probe MD simulations. (A) The N-terminus (Nt) of the A1 domain and the C terminus (Ct) of the A2 domain were pulled away from each other by harmonic springs. The domains were initially in contact but not connected by a linker (domains in cartoon and N-linked sugars in stick representation). (B) Snapshot illustrating a typical dissociation event of the VWF-A1A2 complex induced by the applied force (same representation as in A). Slight unfolding of the C-terminal part of the A2 domain was observed. The disulfide bond Cys1272-Cys1458 (C-C) prevented the A1 domain from unfolding. (C) Cumulative dissociation events (from 17 runs) as a function of the distance between the pulled N- and C termini at the moment of dissociation. Here, , subtracting the initial distance , is shown. The Y1605-M1606 (YM) ADAMTS13 cleavage site was exposed after separation of the A2 C-terminal β5 strand from the core of the protein (event indicated by the dotted line). The black circle corresponds to the dissociation event illustrated in (B). To see this figure in color, go online.
Molecular docking
To augment the MD-generated conformational ensemble of the VWF-A1A2 complex, with a blocked GPIbα binding site, we used molecular docking. Two independent docking approaches, either using Patchdock (38) with further refinement with Firedock (39) or using RosettaDock (40) were considered. Starting conformations of the MD simulations with the domains in contact were generated by Patchdock and Firedock (see selection criterion in Fig. S3).
Cloning, expression, and purification of VWF constructs
The cDNAs coding for either the full-length human VWF, or the A1, A2, and A3 domain, the latter three with 6x His-tag, were cloned into the mammalian expression vector pcDNA3 (41). A1-VWF and A2-VWF mutants were obtained by deleting either the A1 or the A2 domain from the full-length cDNA, by site-directed mutagenesis, employing the QuickChange kit (Stratagene, La Jolla, CA). All primers are available on request. Recombinant expression of VWF constructs in HEK293-EBNA cells was performed as described (42) and the His-tagged VWF domain constructs were purified employing the His-Pur Ni-NTA Resin (Thermo Scientific, Waltham, MA).
AFM
Force distance cycles (FDC) were acquired by approaching and retracting the VWF A1 domain (C-terminally linked to the AFM cantilever by maleimide-polyethylene glycol (PEG)-NHS -mPN- molecules) to VWF A2 domains (C-terminally immobilized on a mica surface by mPN linkers). The disulfide bond Cys1272-Cys1458, connecting the N- and C terminus of the A1 domain, ensured a high similarity of the pulling geometry in the force-probe MD simulations (pulling the N-terminus) and the AFM experiments (pulling the C terminus). Binding events were discerned from nonspecific adhesion by how much they differed in the approach and retraction force signals. To have an unbiased choice of binding events, FDC displaying a characteristic worm-like-chain-type force signal, as well as FDC not showing such behavior, were included for further analysis. To validate specific binding, control experiments were carried out either in the presence of 0.1 mg/ml soluble A2 domains or by replacing either the A1 or the A2 domain by VWF A3 domains. The latter case constitutes a critical control experiment, because A3 is a protein domain that is in the vicinity of A1 and A2 in physiological conditions, and also has the Rossmann topology. For each system, four cantilever tips were utilized. At least 1000 FDC were recorded for each of the tips at a pulling speed of 600 nm/s.
The elongation L corresponded to the extension of the A1 and A2 domains, together with the ones of the mPN linkers and 3-aminopropyltriethoxy silane (APTES) coating molecules. It was measured, during a binding event, as the distance in which the attraction and retraction force-distance curves differed minus the cantilever deflection CD (Fig. 4 A). In practice, was measured by fitting a second-order polynomial to the force curves, followed by the determination of the point in the retraction curve where the force abruptly returned back to zero. The cantilever deflection CD was determined as the applied stretching force F (extracted at the moment of rupture during the FDC) divided by the actual spring constant of the cantilever (30 pNnm–1). The expectation value of L (EV) was estimated as , with the measured probability to have an elongation of , summing over all the measured values. To account for the size of the A1 domain and the mPN linkers, was subtracted to each measured elongation L. The size of the A1 domain () was estimated as 2 its radius of gyration (1.6 nm, derived from MD simulations of the isolated A1 domain (43)). A worm-like-chain model was employed to compute the extension of the mPN linkers as a function of the force F. It reads as follows:
where P is the persistence length (0.38 nm (44)), is the mPN linker contour length (8.9 nm, considering 27 PEG units and 0.33 nm per unit), is the Boltzmann constant, and T is the temperature. The cumulative distributions of both the original elongation L and its theoretical reduction (accounting for the size of A1 and linkers) were shown.
Figure 4.
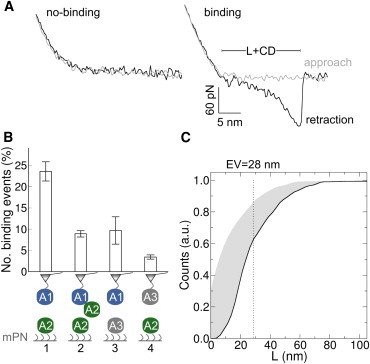
Force response of the VWF-A1A2 complex from AFM. (A) Typical approach-retraction force-distance profiles associated to no-binding and binding events. The elongation L, of the A1 and A2 domains, together with the mPN linkers and the 3-aminopropyltriethoxy silane coating molecules, summed to the cantilever deflection (CD) was determined by the difference between approach and retraction curves. (B) (1) Number of binding events between VWF A1 and A2 domains. A1 was connected to the tip of the AFM cantilever (triangle) using malemide-PEG-NHS (mPN) linkers. It was approached to and retracted from the surface carrying mPN-linked A2 domains. Force-distance cycles presented in (A) correspond to this situation. (2–4) Number of binding events measured in control AFM experiments, in which the A1 domain was blocked by soluble A2 domains (2), or either the A2 domains on the surface (3) or the A1 domain connected to the cantilever (4) were replaced by VWF A3 domain. (C) Cumulative distribution of L (black line) and its correction by subtracting the size of A1 and the mPN linkers (gray area). Dotted line indicates the expectation value (EV) of L. To see this figure in color, go online.
Microfluidic experiments
For distinct shear rate application, air-pressure driven microfluidic channels were coated with recombinant wild-type VWF, A2-VWF, or A1-VWF. For the functional characterization, the coated microfluidic channels were mounted onto an inverted fluorescence microscope and perfused, as previously published (45), with wild-type VWF, VWF with the A2 domain deleted, or VWF with the A1 domain deleted. Live cell fluorescence images were taken and analyzed at shear rates in the range of 500 s–1 to 4000 s–1. To track the motion of VWF-platelet fibers and aggregates, an image composition of 20 sequential frames (taken at a frequency of two frames/s) was implemented. Increasing number of frames was considered for the composition (from one to all 20 frames), subtracting identical pixels among frames. Dynamical monitoring allowed the exact determination of the critical shear rate for VWF-platelet fiber and aggregate formation.
See further details of the simulations and the experimental procedures in the Supporting Material.
Results
Blockage of the VWF GPIbα binding site in A1 by A2
We first investigated whether the VWF A2 domain spontaneously binds to the A1 domain. To this end, we carried out 17 independent 100 ns equilibrium MD simulations, starting with these two domains separated by distances (between their center of masses) from 6.1 to 8.6 nm and adopting different orientations with respect to each other (Fig. 1 B). The linker connecting the two domains was not considered (in the following, this situation will be referred as not connected domains). The two domains spontaneously came into contact and remained stably bound in seven out of 17 simulation runs, as reflected by drops in their separation to values smaller than 5 nm (Fig. 1 C).
We next analyzed if the GPIbα binding site in the A1 domain was blocked upon binding of the A2 domain. We quantified the amount of blockage by computing the GPIbα binding site accessible surface (GPIbα-BS-AS) (Fig. 1 D). The GPIbα-BS-AS histogram recovered from our simulations revealed a major peak close to the value estimated from the x-ray structure of the VWF A1-GPIbα complex (16) (33.4 nm2), indicating no blockage. In addition, the histogram contained a tail extending to values smaller than 20 nm2, reflecting substantial blockage (of more than 40% of the x-ray GPIbα-BS-AS). Remarkably, blockage was found correlated with the separation between domains, with the GPIbα binding site fully accessible (large GPIbα-BS-AS) only for large interdomain separations, whereas completely blocked (small GPIbα-BS-AS) when the A2 domain approached the A1 domain. Thus, from our simulations, A2 binding to A1 implies blockage of the VWF-GPIbα interaction site.
We also tested the blockage of the GPIbα binding site within a VWF-A1A2 fragment, with the A1 and A2 domains connected by the linker. We simulated the dynamics of such fragment, in 16 independent MD runs of 82 to 100 ns, with initial interdomain separations ( 7.9 nm) and linker extensions ( 6.0 nm) taken from EM estimates (6) (Fig. S2 A). The fragment populated the lower range of separations measured in the EM experiments (6) (Fig. S2 B). Again, the MD-generated conformations included several instances of direct A1-A2 interactions (Fig. S2 B). The presence and involvement of the O-linked glycosylated linker now alleviated the strong correlation between A1-A2 binding and blockage of the GPIbα binding site as observed for not connected domains (Fig. S2 C).
Orientational preferences in the blocked state
Our simulations raised the question on the most-favorable conformation of the two domains with GPIbα binding blocked. We addressed this by performing molecular docking followed by MD refinement. We generated a set of conformations by docking the A2 domain to the A1 domain. From this set, we selected representative conformations with both the GPIbα binding site blocked and high docking score as starting positions of 16 MD simulations of 100 ns each (see Fig. S3 and the Supporting Material for the selection criterion). Similar conformations presenting blockage were predicted by two independent docking approaches (Fig. S4). Furthermore, an enrichment of blocked conformations over random conformations was observed, because of their large interdomain shape complementarity and favorable protein-protein interactions, thus justifying our selection criterion of only blocked and high-docking-score structures (Fig. S5 and the Supporting Material).
During the simulations the domains remained bound causing blockage, while maintaining their internal structure almost intact (backbone root-mean-square deviation to the initial structure below 1.5 Å for A1 and 2.3 Å for A2), but accommodating with respect to each other in multiple orientations. To capture the extent of stable blocking interdomain orientations we carried out a PCA of the conformations predicted by docking (yielding two main collective eigenvectors covering 68% of the possible interdomain orientations), followed by projections of the MD trajectories onto the two-dimensional (2D) space generated by these two vectors (Fig. 2 A). Furthermore, we narrowed the orientations to those with high interdomain potential energy, V, and substantial solvent accessible hydrophobic surface reduction (SAHSR) (Fig. 2 B). Remarkably, in all orientations with large V and SAHSR contributions, the A2 domain was found directly obstructing the A1-domain β3 strand (the one connecting with GPIbα (15,16)) and displaying only small orientational deviations (small point clouds in the 2D-PCA projections), indicating high structural integrity. Within this preferred set of VWF-A1A2 complexes, the A2 domain oriented in two main modes: either with its C terminus in proximity to the A1 domain or—on ∼180° relative rotation—with its β3 strand in proximity, almost forming a stable interdomain β-sheet in the latter case (compare top with bottom projections and snapshots in Fig. 2 A). The residues Arg1668 and Asp1587, both in A2, were found to strongly interact with A1: Arg1668, when the C terminus was in proximity to A1, and Asp1587, when the β3-strand was in vicinity. Destabilizing mutations Arg1668Asp and Asp1587Lys are thus potential candidates to detect the most favored conformation of the complex among the two observed orientational modes. In addition, replacement of Val1548 located directly at the β3 strand of A2, for instance by a bulky polar residue such as serine or asparagine, would further distort the orientational mode that features a quasi interdomain β-sheet.
We validated the observed orientational preferences by comparing this with our previous set of simulations (Fig. S6). The docking-MD refined region was also sampled during the MD simulations starting from separated domains, with the A2 domain located directly in front of the β3 strand of the A1 domain. However, the conformational ensemble in the blocked state was further broadened presumably because of the sugars and also the linker between A1 and A2.
VWF-A1A2 complex under force: activation versus cleavage
Induced by shear-forces, the release of the GPIbα binding site in the A1 domain would allow platelet-binding activation, whereas exposure of the YM catalytic site after unfolding of the A2 domain would enable cleavage and degradation. We studied how a stretching force balances these two processes. For this purpose we performed 17 independent force-probe MD simulations, starting from a diverse set of conformations of the two domains, not connected, forming a complex, and with the GPIbα binding site obstructed (Fig. 3 A). We pulled the N-terminus of the A1 domain and the C terminus of the A2 domain away from each other, until dissociation of the complex (and thereby unblocking of the GPIbα binding site) occurred (Fig. 3 B). The A2 domain slightly unfolded in its C-terminal part, while the A1 domain remained folded because of its Cys1272-Cys1458 disulfide bond (Fig. 3 B).
We quantified the extent of unfolding of the C terminus of the A2 domain by monitoring the increase in the distance between the pulled termini, , with respect to the initial distance . Exposure of the YM cleavage site, as an initial requirement for ADAMTS13 cleavage, occurred after the detachment of the β5 strand ( nm). In comparison, dissociation of the fragment, as needed for activation, occurred before YM exposure, in 15 of the 17 runs (88% of the cases) (Fig. 3 C).
We next probed the physical interaction between A1 and A2 and the coupling between dissociation and unfolding, as suggested by our simulations, at the single-molecule level by using AFM (Fig. 4). FDC were acquired by approaching the A1 domain (linked to the AFM cantilever) to A2 domains (immobilized on a surface) and retracting it again. A retracting force signal differing from the approaching one, with an abrupt drop to zero at dissociation, was used as an indicator for a binding event (Fig. 4 A). It was observed in ∼23% of the cycles (1 in Fig. 4 B). In contrast, a substantially reduced number of binding events (less than 10%) was observed in the presence of soluble A2 domains, presumably because of the blocking of the A1 domain at the cantilever (2 in Fig. 4 B). As a control, reduction in the number of binding events was also observed when replacing either the A2 domains at the surface (3 in Fig. 4 B) or the A1 domain at the cantilever (4 in Fig. 4 B) by VWF A3 domains. This implies that binding events are exclusively through A1-A2 interactions, thus confirming the observation from our MD simulations and from previous binding assays (30) that the VWF A1 and A2 domains specifically interact.
To further investigate the coupling between dissociation of the VWF-A1A2 complex and unfolding of the A2 domain, we measured the elongation of the complex (together with linkers and coating molecules) before dissociation by AFM (Fig. 4 C). The measured expectation value of the elongation (∼28 nm) was substantially lower than the extension of a fully stretched unfolded A2 domain (∼80 nm (23–27)). In fact, in all FDC, the elongation remained below those levels of extension. Although the noise in the length distribution is expected to be large because of the tip and surface chemistry, our AFM data speak against full unfolding of A2 before dissociation. Instead, it suggests a small extent of unfolding of A2 before dissociation.
Functional characteristics of VWF with the A2 domain deleted in shear-induced fiber formation
We next examined if the A2 domain inhibits VWF-platelet binding in a shear-dependent manner, by performing microfluidic experiments, in wild-type VWF-coated channels, under replacement of the plasmatic wild-type VWF by recombinant VWF with the A2 domain deleted (A2-VWF), and in a wide shear range. In the presence of wild-type VWF in the perfusion medium, above a critical shear rate of 4000 s–1, large aggregates of VWF and platelets were observed to roll along the surface coated with VWF (Fig. 5, top right). At lower shear rates, rolling VWF-platelet aggregates were absent. Here, we only observed either rolling of single platelets along the microfluidic channel (at 500 s–1, Fig. 5, top left) or reversibly formed platelet-decorated VWF fibers, which stayed attached to the channel surface (at 2500 s–1, Fig. 5, top middle). Instead, in the presence of A2-VWF in the perfusion medium, the critical shear rate for rolling aggregate formation was decreased to 2500 s–1, indicating a gain of function for the VWF by deletion of its A2 domain (Fig. 5, middle, and Movie S1). Identical results were obtained using A2-VWF instead of wild-type VWF for coating of the microfluidic channels (data not shown). In a multimer analysis, similar VWF size distributions were observed for the mutants and for the wild-type VWF, just slightly shifted down because of the deletions in the mutant proteins (Fig. S7). Changes in the VWF distribution size are thus discarded as the reason for the gain in function of the A2-VWF mutant. As expected, neither fibers nor VWF-platelet aggregates were formed in the presence of VWF with an A1-domain deletion (Fig. 5, bottom). Furthermore, coating with A1-VWF led to a complete absence of both single platelet rolling and the formation of rolling VWF-platelet aggregates independent of the VWF present in the perfusion medium.
Figure 5.
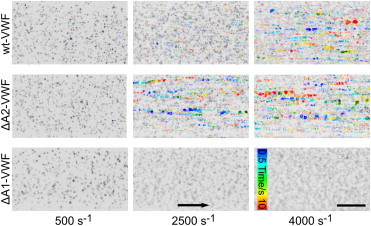
Changes in shear-induced fiber and aggregate formation on deletion of the VWF A2 domain. Live-cell fluorescence images of platelet-decorated VWF fibers and platelet-VWF aggregates observed in microfluidic experiments at the indicated shear rates (different columns). Microfluidic channels were perfused with plasmatic wild-type VWF (wt-VWF, top row), VWF with the A2 domain deleted (A2-VWF, middle row), or VWF with the A1 domain deleted (A1-VWF, bottom row). A static image is presented as background, displaying platelets, fibers, and aggregates in black. Moving fibers and aggregates are highlighted in color. Their positions were tracked during 10 s after taking the static image. Flow direction is indicated with the arrow and the line corresponds to 100 μm. To see this figure in color, go online.
Discussion
Blockage of the GPIbα binding site mediated by A1-A2 interactions implies autoinhibition
Our extensive set of simulations (in the μs time range) demonstrates that the GPIbα binding site of VWF (located in the A1 domain) can be significantly blocked, upon spontaneous binding of the A2 to the A1 domain (Fig. 1). In addition, the binding of these two domains was further confirmed at the single-molecule level by AFM (Fig. 4). The increase in blockage with reducing interdomain separation observed in our simulations suggests that the A2 domain does not recognize a random region in the A1 domain but instead it specifically targets the GPIbα binding site. This observation was further supported by our docking calculations, which showed enrichment toward blocked conformations over random conformations, by enhanced shape complementarity and favorable protein-protein interactions (Fig. S5). With the GPIbα binding site blocked, platelets are prevented to bind and thus the VWF remains inactive. Our results, together with the experimentally observed platelet-binding modulation in the presence of A2 domains (30), thus imply an autoinhibition mechanism for the binding of platelets to the VWF mediated by A1-A2 interactions.
Additional simulations, this time with the two domains connected (also in the μs time range), revealed broad dynamics of the VWF-A1A2 fragment (Fig. S2). Although the A2 domain (bound to the A1 domain) was sometimes observed causing no shielding, presumably stabilized by the connecting linker, it was also found in many other times substantially blocking the GPIbα binding site. This indicates that not only not connected, but also vicinal, covalently linked, A1 and A2 domains can interact with each other causing blockage, further supporting the hypothesis of VWF autoinhibition because of A1-A2 interactions.
Our simulations of connected domains sampled a range from compact to extended conformations, covering the lower region of the interdomain separations measured by EM (6). In fact, compact conformations are expected from a direct A1-A2 interaction, as established in previous assays (30) and confirmed in our AFM experiments. Also, the presence of a third domain (e.g., D’D3 or A3) or deposition on the surface may favor more extended conformations in the EM experiments compared with the ones sampled in our simulations.
Autoinhibition driven by A1-A2 interactions provides a molecular picture of the shielding of the GPIbα (platelet) binding site, crucial to maintain the VWF inactive under equilibrium conditions. This is a complementary scenario to previous shear-dependent models (18,20) for GPIbα binding, but is the only one reconciling previous inactivation experiments (30).
Main orientational modes of the autoinhibited state
From our simulations, the minimum structural requirement to block GPIbα binding is to have A1-A2 binding and this is effectively achieved by the A2 domain specifically targeting the GPIbα binding site in A1. Our docking calculations and further extensive MD refinement narrowed the interdomain conformational variability to two main orientational modes of blockage, stabilized by an attractive interdomain potential energy and a reduction in the amount of solvent accessible hydrophobic surface (Fig. 2). A2 located either with its C terminus or with its β3 strand in proximity to the β3 strand of A1, resulting in a quasi-extended cross-domain β-sheet in the latter case. Notably, as a general feature, the A2 domain obstructs the A1-domain β3 strand (which connects to GPIbα (15,16)), thus suggesting drastic VWF autoinhibition. Direct blockage of the main interaction partner of GPIbα in the A1 domain (the β3 strand) was also observed in our simulations started from unbiased positions, with the domains separated, further supporting our proposed mode of autoinhibition. In addition, the agreement between our force-probe MD simulations and AFM experiments (see below) stresses on the validity of the chosen conformations from docking, followed by MD refinement, and the robustness of the MD simulation results. Our structural predictions are anticipated to motivate future structural studies aiming at determining the structure of the A1-A2 complex, in the nonconnected and connected situations, both of physiological relevance. Mutants Arg1668Asp, Asp1587Lys, and Val1548Ser(Asn) may serve as initial candidates for mutagenesis studies to discern among the two proposed modes of blockage.
In the simulations started from separated domains, additional blocking orientations were observed. Here, the presence of the N-linked sugars or the O-glycosylated linker may also play stabilization roles. An additional stabilization of the blocked (autoinhibited) state of the VWF by the sugars is consistent with recent microfluidic experiments that showed an increase in platelet adhesion when the VWF was N-deglycosylated (46).
Force unblocks the GPIbα-binding site before exposure of the ADAMTS13 cleavage site, ensuring VWF activation before cleavage
In our force-probe simulations, we induced the dissociation of the complex formed by the A1 and A2 domains by applying an external stretching force. In complex, the VWF A2 domain showed only marginal unfolding, which proceeded from the C terminus, in line with the unfolding mechanism previously observed for this domain in isolation (with different force fields) (23,47). Dissociation was found to occur before exposure of the ADAMTS13 cleavage site in the A2 domain with a very high probability (∼0.88) (Fig. 3). This is consistent with our AFM measurements, which yielded in the majority of the binding events small elongations of the VWF-A1A2 complex at rupture (Fig. 4). Our simulations and AFM thus support that a stretching force unblocks the GPIbα-binding site, by detaching the A1 and A2 domains, and that this process is coupled to the exposure of the ADAMTS13 cleavage site in the A2 domain after its unfolding. The stretching force ensures, however, that the VWF is activated for platelet binding predominantly before deactivation through cleavage. In this respect, the interactions between A1 and A2 may also serve to clarify the role of ristocetin, coupling platelet binding and ADAMTS13 cleavage (48).
Deletion of the A2 domain results in a VWF with a gain of function
Our microfluidic experiments showed a reduction of the critical shear-rate for the formation of VWF-platelet fibers and rolling aggregates, when the A2 domain was deleted. This implies a VWF with a gain in function (Fig. 5). Our results in consequence expand the experiments by Martin et al. (30), proving that not only present in solution but also within the VWF molecule, the A2 domain critically influences platelet binding in a shear-dependent manner. In addition, our combined computational and experimental results suggest that the A2 domain stabilizes a VWF inactive state, by direct A1-A2 interactions, either within or across VWF monomers. However, additional inhibitory mechanisms must be at play, because the A2-VWF mutant still requires intermediate shear rates for the formation of rolling aggregates (2500 s–1 for A2-VWF instead of 4000 s–1 for the wild-type VWF). We speculate that the exposure of the GPIbα binding site requires both a global globule-to-stretch transition, eventually involving other—specific or nonspecific—domain-domain interactions (e.g., between D’D3 and A1 (28)), and VWF-A1A2 dissociation.
Conclusions
In this study, we examined the inactivation of VWF for platelet binding, induced by a specific domain-domain interaction, and its coupling to VWF cleavage degradation driven by force, by using MD simulations, molecular docking, AFM, and microfluidic experiments. We demonstrate that under equilibrium conditions the VWF A1 and A2 domains bind to each other, with the A2 domain specifically targeting the GPIbα binding site in the A1 domain, thus blocking the binding of GPIbα (and thereby of platelets) to VWF. This implies autoinhibition of the VWF mediated by A1-A2 interactions. We identified two main orientational blocking modes, which have the shielding of the A1 β3 strand, the site critical for GPIbα binding, in common. Detachment of the two domains, induced by a stretching force, unblocked the GPIbα binding site most predominantly before exposure of the cleavage site in the A2 domain. This suggests that A2 blocks GPIbα binding in a force-dependent manner, but guaranteeing that the VWF is ready for activation before cleavage, to mechanically balance the propagation and attenuation of hemostasis. Deletion of the A2 domain enhanced platelet binding, corroborating the key autoinhibition role of this domain. In summary, our results suggest, to our knowledge, a new interdomain-mediated autoinhibition mechanism that explains the inactivation of VWF under equilibrium conditions while allowing shear-sensitive growth of blood coagulates. This mechanism reconciles previous and can be tested by future experiments. It will be highly interesting to investigate if this or other domain-domain interactions are a common regulatory mechanism, not only for the shear-sensitive binding of VWF to its partners, but also potentially for the shear-dependent self-aggregation of VWF.
Author Contributions
C.A.-S. performed the MD simulations and docking calculations with Patchdock and Firedock. C.A.-S., C.B., and F.G. analyzed the computational results. V.H., S.G., and S.W.S. performed the microfluidic experiments. S.P. and P.H. carried out the AFM experiments. A.K.B. performed the docking calculations with RosettaDock. M.A.B., T.O., and R.S. generated the VWF constructs. C.B. and F.G. conceived the project. All authors discussed the results. C.A.-S., V.H., S.P., A.K.B., M.A.B., C.B., and F.G. wrote the manuscript.
Acknowledgments
This study was supported by research funding from the German Research Foundation, to the Research Group FOR1543: “Shear flow regulation of hemostasis—bridging the gap between nanomechanics and clinical presentation” (C.A.-S., V.H., S.P., S.G., M.A.B., T.O., R.S., P.H., S.W.S., C.B., and F.G.), the Klaus Tschira Stiftung (F.G.), and the BIOMS program of the Heidelberg University (A.K.B.). We thank the Jülich Supercomputing Centre (J.S.C.) (HHD24 and HHD25 projects). We thank Gesa König for technical assistance with protein purification.
Editor: Nathan Baker
Contributor Information
Carsten Baldauf, Email: baldauf@fhi-berlin.mpg.de.
Frauke Gräter, Email: frauke.graeter@h-its.org.
Supporting Material
(Minute 1) Shear is maintained constant at 500 s−1. Here, rolling of single platelets is observed for both proteins. (Minutes 2–4) Shear is kept fixed at 2500 s−1. Large rolling aggregates, formed by platelets and VWF, are observed for the VWF lacking the A2 domain (lower channel), while only reversibly formed platelet-decorated VWF fibers, which stayed attached to the channel surface, are observed for wild-type VWF (upper channel). (Minute 5 to end of the movie) Shear is retained at a value of 4000 s−1. Large aggregates are formed for both proteins. Frames were taken at a frequency of two frames per second. Time is indicated in seconds the first minute and in minutes after. The white bar corresponds to 100 μm. Platelets and VWF are shown in white.
Supporting citations
References (49–74) appear in the Supporting Material.
References
- 1.Schneppenheim R., Budde U. von Willebrand factor: the complex molecular genetics of a multidomain and multifunctional protein. J. Thromb. Haemost. 2011;9(Suppl. 1):209–215. doi: 10.1111/j.1538-7836.2011.04324.x. [DOI] [PubMed] [Google Scholar]
- 2.Springer T.A. Biology and physics of von Willebrand factor concatamers. J. Thromb. Haemost. 2011;9(Suppl. 1):130–143. doi: 10.1111/j.1538-7836.2011.04320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen H., Fallah M.A., Alexander-Katz A. Blood-clotting-inspired reversible polymer—colloid composite assembly in flow. Nat. Commun. 2013;4:1333. doi: 10.1038/ncomms2326. [DOI] [PubMed] [Google Scholar]
- 4.Lippok S., Obser T., Rädler J.O. Exponential size distribution of von Willebrand factor. Biophys. J. 2013;105:1208–1216. doi: 10.1016/j.bpj.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schneider S.W., Nuschele S., Schneider M.F. Shear-induced unfolding triggers adhesion of von Willebrand factor fibers. Proc. Natl. Acad. Sci. USA. 2007;104:7899–7903. doi: 10.1073/pnas.0608422104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou Y.-F., Eng E.T., Springer T.A. A pH-regulated dimeric bouquet in the structure of von Willebrand factor. EMBO J. 2011;30:4098–4111. doi: 10.1038/emboj.2011.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou Y.-F., Eng E.T., Springer T.A. Sequence and structure relationships within von Willebrand factor. Blood. 2012;120:449–458. doi: 10.1182/blood-2012-01-405134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alexander-Katz A., Schneider M.F., Netz R.R. Shear-flow-induced unfolding of polymeric globules. Phys. Rev. Lett. 2006;97:138101. doi: 10.1103/PhysRevLett.97.138101. [DOI] [PubMed] [Google Scholar]
- 9.Sing C.E., Alexander-Katz A. Elongational flow induces the unfolding of von Willebrand factor at physiological flow rates. Biophys. J. 2010;98:L35–L37. doi: 10.1016/j.bpj.2010.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sing C.E., Selvidge J.G., Alexander-Katz A. Von Willlebrand adhesion to surfaces at high shear rates is controlled by long-lived bonds. Biophys. J. 2013;105:1475–1481. doi: 10.1016/j.bpj.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Emsley J., Cruz M., Liddington R. Crystal structure of the von Willebrand factor A1 domain and implications for the binding of platelet glycoprotein Ib. J. Biol. Chem. 1998;273:10396–10401. doi: 10.1074/jbc.273.17.10396. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Q., Zhou Y.-F., Springer T.A. Structural specializations of A2, a force-sensing domain in the ultralarge vascular protein von Willebrand factor. Proc. Natl. Acad. Sci. USA. 2009;106:9226–9231. doi: 10.1073/pnas.0903679106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou M., Dong X., Ding J. A novel calcium-binding site of von Willebrand factor A2 domain regulates its cleavage by ADAMTS13. Blood. 2011;117:4623–4631. doi: 10.1182/blood-2010-11-321596. [DOI] [PubMed] [Google Scholar]
- 14.Xu A.J., Springer T.A. Calcium stabilizes the von Willebrand factor A2 domain by promoting refolding. Proc. Natl. Acad. Sci. USA. 2012;109:3742–3747. doi: 10.1073/pnas.1121261109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huizinga E.G., Tsuji S., Gros P. Structures of glycoprotein Ibalpha and its complex with von Willebrand factor A1 domain. Science. 2002;297:1176–1179. doi: 10.1126/science.107355. [DOI] [PubMed] [Google Scholar]
- 16.Dumas J.J., Kumar R., Mosyak L. Crystal structure of the wild-type von Willebrand factor A1-glycoprotein Ibalpha complex reveals conformation differences with a complex bearing von Willebrand disease mutations. J. Biol. Chem. 2004;279:23327–23334. doi: 10.1074/jbc.M401659200. [DOI] [PubMed] [Google Scholar]
- 17.Chen Z., Lou J., Schulten K. Flow-induced structural transition in the beta-switch region of glycoprotein Ib. Biophys. J. 2008;95:1303–1313. doi: 10.1529/biophysj.108.132324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lou J., Zhu C. Flow induces loop-to-beta-hairpin transition on the beta-switch of platelet glycoprotein Ib α. Proc. Natl. Acad. Sci. USA. 2008;105:13847–13852. doi: 10.1073/pnas.0801965105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zou X., Liu Y., Schulten K. Flow-induced beta-hairpin folding of the glycoprotein Ibalpha beta-switch. Biophys. J. 2010;99:1182–1191. doi: 10.1016/j.bpj.2010.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim J., Zhang C.-Z., Springer T.A. A mechanically stabilized receptor-ligand flex-bond important in the vasculature. Nature. 2010;466:992–995. doi: 10.1038/nature09295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blenner M.A., Dong X., Springer T.A. Structural basis of regulation of von Willebrand factor binding to glycoprotein Ib. J. Biol. Chem. 2014;289:5565–5579. doi: 10.1074/jbc.M113.511220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sadler J.E. A new name in thrombosis, ADAMTS13. Proc. Natl. Acad. Sci. USA. 2002;99:11552–11554. doi: 10.1073/pnas.192448999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baldauf C., Schneppenheim R., Gräter F. Shear-induced unfolding activates von Willebrand factor A2 domain for proteolysis. J. Thromb. Haemost. 2009;7:2096–2105. doi: 10.1111/j.1538-7836.2009.03640.x. [DOI] [PubMed] [Google Scholar]
- 24.Chen W., Lou J., Zhu C. Molecular dynamics simulated unfolding of von Willebrand factor A domains by force. Cell Mol. Bioeng. 2009;2:75–86. [Google Scholar]
- 25.Zhang X., Halvorsen K., Springer T.A. Mechanoenzymatic cleavage of the ultralarge vascular protein von Willebrand factor. Science. 2009;324:1330–1334. doi: 10.1126/science.1170905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu T., Lin J., Zhu C. Force-induced cleavage of single VWFA1A2A3 tridomains by ADAMTS-13. Blood. 2010;115:370–378. doi: 10.1182/blood-2009-03-210369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ying J., Ling Y., Shao J.-Y. Unfolding the A2 domain of von Willebrand factor with the optical trap. Biophys. J. 2010;98:1685–1693. doi: 10.1016/j.bpj.2009.12.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ulrichts H., Udvardy M., Deckmyn H. Shielding of the A1 domain by the D’D3 domains of von Willebrand factor modulates its interaction with platelet glycoprotein Ib-IX-V. J. Biol. Chem. 2006;281:4699–4707. doi: 10.1074/jbc.M513314200. [DOI] [PubMed] [Google Scholar]
- 29.Auton M., Sowa K.E., Cruz M.A. N-terminal flanking region of A1 domain in von Willebrand factor stabilizes structure of A1A2A3 complex and modulates platelet activation under shear stress. J. Biol. Chem. 2012;287:14579–14585. doi: 10.1074/jbc.M112.348573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martin C., Morales L.D., Cruz M.A. Purified A2 domain of von Willebrand factor binds to the active conformation of von Willebrand factor and blocks the interaction with platelet glycoprotein Ibalpha. J. Thromb. Haemost. 2007;5:1363–1370. doi: 10.1111/j.1538-7836.2007.02536.x. [DOI] [PubMed] [Google Scholar]
- 31.Lenting P.J., Denis C.V. von Willebrand factor A1 domain: stuck in the middle. J. Thromb. Haemost. 2007;5:1361–1362. doi: 10.1111/j.1538-7836.2007.02573.x. [DOI] [PubMed] [Google Scholar]
- 32.Canis K., McKinnon T.A.J., Dell A. The plasma von Willebrand factor O-glycome comprises a surprising variety of structures including ABH antigens and disialosyl motifs. J. Thromb. Haemost. 2010;8:137–145. doi: 10.1111/j.1538-7836.2009.03665.x. [DOI] [PubMed] [Google Scholar]
- 33.Matsui T., Titani K., Mizuochi T. Structures of the asparagine-linked oligosaccharide chains of human von Willebrand factor. Occurrence of blood group A, B, and H(O) structures. J. Biol. Chem. 1992;267:8723–8731. [PubMed] [Google Scholar]
- 34.Van Der Spoel D., Lindahl E., Berendsen H.J.C. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 35.Hess B., Kutzner C., Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 36.Pronk S., Páll S., Lindahl E. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Amadei A., Linssen A.B.M., Berendsen H.J.C. Essential dynamics of proteins. Proteins Struct. Funct. Bioinformatics. 1993;17:412–425. doi: 10.1002/prot.340170408. [DOI] [PubMed] [Google Scholar]
- 38.Schneidman-Duhovny D., Inbar Y., Wolfson H.J. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33:W363–W367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mashiach E., Schneidman-Duhovny D., Wolfson H.J. FireDock: a web server for fast interaction refinement in molecular docking. Nucleic Acids Res. 2008;36(Suppl. 2):W229–W332. doi: 10.1093/nar/gkn186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lyskov S., Gray J.J. The RosettaDock server for local protein-protein docking. Nucleic Acids Res. 2008;36(Suppl. 2):W233–W238. doi: 10.1093/nar/gkn216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schneppenheim R., Michiels J.J., Budde U. A cluster of mutations in the D3 domain of von Willebrand factor correlates with a distinct subgroup of von Willebrand disease: type 2A/IIE. Blood. 2010;115:4894–4901. doi: 10.1182/blood-2009-07-226324. [DOI] [PubMed] [Google Scholar]
- 42.Schneppenheim R., Budde U., Oldenburg J. Expression and characterization of von Willebrand factor dimerization defects in different types of von Willebrand disease. Blood. 2001;97:2059–2066. doi: 10.1182/blood.v97.7.2059. [DOI] [PubMed] [Google Scholar]
- 43.Grässle S., Huck V., Schneider S.W. von Willebrand factor directly interacts with DNA from neutrophil extracellular traps. Arterioscler. Thromb. Vasc. Biol. 2014;34:1382–1389. doi: 10.1161/ATVBAHA.113.303016. [DOI] [PubMed] [Google Scholar]
- 44.Kienberger F., Pastushenko V.P., Hinterdorfer P. Static and dynamical properties of single poly(ethylene glycol) molecules investigated by force spectroscopy. Single Molecules. 2000;1:123–128. [Google Scholar]
- 45.Brehm M.A., Huck V., Schneppenheim R. von Willebrand disease type 2A phenotypes IIC, IID and IIE: a day in the life of shear-stressed mutant von Willebrand factor. Thromb. Haemost. 2014;112:96–108. doi: 10.1160/TH13-11-0902. [DOI] [PubMed] [Google Scholar]
- 46.Fallah M.A., Huck V., Schneider M.F. Circulating but not immobilized N-deglycosylated von Willebrand factor increases platelet adhesion under flow conditions. Biomicrofluidics. 2013;7:044124. doi: 10.1063/1.4819746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Interlandi G., Ling M., Thomas W.E. Structural basis of type 2A von Willebrand disease investigated by molecular dynamics simulations and experiments. PLoS ONE. 2012;7:e45207. doi: 10.1371/journal.pone.0045207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen J., Ling M., Chung D.W. Simultaneous exposure of sites in von Willebrand factor for glycoprotein Ib binding and ADAMTS13 cleavage: studies with ristocetin. Arterioscler. Thromb. Vasc. Biol. 2012;32:2625–2630. doi: 10.1161/ATVBAHA.112.254144. [DOI] [PubMed] [Google Scholar]
- 49.Woods Group. 2005–2013. GLYCAM Web. In R. J. Woods, editor. Complex Carbohydrate Research Center, University of Georgia, Athens, GA.
- 50.Schrödinger, L. L. C. 2010. The PyMOL Molecular Graphics System, Version 1.3r1.
- 51.Hornak V., Abel R., Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins Struct. Funct. Bioinformatics. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Best R.B., Hummer G. Optimized molecular dynamics force fields applied to the helix-coil transition of polypeptides. J. Phys. Chem. B. 2009;113:9004–9015. doi: 10.1021/jp901540t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lindorff-Larsen K., Piana S., Shaw D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinformatics. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kirschner K.N., Yongye A.B., Woods R.J. GLYCAM06: a generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 2008;29:622–655. doi: 10.1002/jcc.20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 56.Joung I.S., Cheatham T.E., 3rd Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J. Phys. Chem. B. 2008;112:9020–9041. doi: 10.1021/jp8001614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hess B., Bekker H., Fraaije J.G.E.M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. [Google Scholar]
- 58.Feenstra K.A., Hess B., Berendsen H.J.C. Improving efficiency of large timescale molecular dynamics simulations of hydrogen-rich systems. J. Comput. Chem. 1999;20:786–798. doi: 10.1002/(SICI)1096-987X(199906)20:8<786::AID-JCC5>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 59.Miyamoto S., Kollman P.A. Settle: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992;13:952–962. [Google Scholar]
- 60.Hockney R.W., Eastwood J.W. Hilger; Bristol, UK: 1988. Computer Simulation Using Particles. [Google Scholar]
- 61.Darden T., York D., Pedersen L. Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 62.Essmann U., Perera L., Pedersen L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 63.Bussi G., Donadio D., Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 64.Berendsen H.J.C., Postma J.P.M., Haak J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 65.Parrinello M., Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 1981;52:7182–7190. [Google Scholar]
- 66.Connolly M.L. Analytical molecular surface calculation. J. Appl. Cryst. 1983;16:548–558. [Google Scholar]
- 67.Budde U., Schneppenheim R., Peake I. Detailed von Willebrand factor multimer analysis in patients with von Willebrand disease in the European study, molecular and clinical markers for the diagnosis and management of type 1 von Willebrand disease (MCMDM-1VWD) J. Thromb. Haemost. 2008;6:762–771. doi: 10.1111/j.1538-7836.2008.02945.x. [DOI] [PubMed] [Google Scholar]
- 68.Budde U., Schneppenheim R., Zimmerman T.S. Luminographic detection of von Willebrand factor multimers in agarose gels and on nitrocellulose membranes. Thromb. Haemost. 1990;63:312–315. [PubMed] [Google Scholar]
- 69.Schneppenheim R., Plendl H., Budde U. Luminography—an alternative assay for detection of von Willebrand factor multimers. Thromb. Haemost. 1988;60:133–136. [PubMed] [Google Scholar]
- 70.Johannes Kepler University Linz. Crosslinkers and protocols for AFM tip functionalization. Published online April 8, 2015. http://www.jku.at/biophysics/content/e257042.
- 71.Hutter J.L., Bechhoefer J. Calibration of atomic-force microscope tips. Rev. Sci. Instrum. 1993;64:1868–1873. [Google Scholar]
- 72.Ebner A., Hinterdorfer P., Gruber H.J. Comparison of different aminofunctionalization strategies for attachment of single antibodies to AFM cantilevers. Ultramicroscopy. 2007;107:922–927. doi: 10.1016/j.ultramic.2007.02.035. [DOI] [PubMed] [Google Scholar]
- 73.Zhu R., Howorka S., Hinterdorfer P. Nanomechanical recognition measurements of individual DNA molecules reveal epigenetic methylation patterns. Nat. Nanotechnol. 2010;5:788–791. doi: 10.1038/nnano.2010.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rickham P.P. Human experimentation: code of ethics of World Medical Association. Declaration of Helsinki. BMJ. 1964;2:177. doi: 10.1136/bmj.2.5402.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(Minute 1) Shear is maintained constant at 500 s−1. Here, rolling of single platelets is observed for both proteins. (Minutes 2–4) Shear is kept fixed at 2500 s−1. Large rolling aggregates, formed by platelets and VWF, are observed for the VWF lacking the A2 domain (lower channel), while only reversibly formed platelet-decorated VWF fibers, which stayed attached to the channel surface, are observed for wild-type VWF (upper channel). (Minute 5 to end of the movie) Shear is retained at a value of 4000 s−1. Large aggregates are formed for both proteins. Frames were taken at a frequency of two frames per second. Time is indicated in seconds the first minute and in minutes after. The white bar corresponds to 100 μm. Platelets and VWF are shown in white.