

Abstract
Most phosphoproteomics experiments rely on prefractionation of tryptic digests before online liquid chromatography-mass spectrometry. This study compares the potential and limitations of electrostatic repulsion–hydrophilic interaction chromatography (ERLIC) and anion-exchange chromatography (AEX). At a pH higher than 5, phosphopeptides have two negative charges per residue and are well-retained in AEX. However, peptides with one or two phosphate groups are not separated from peptides with multiple Asp or Glu residues, interfering with the identification of phosphopeptides. At a pH of 2, phosphate residues have just a single negative charge but Asp and Glu are uncharged. This facilitates the separation of phosphopeptides from unmodified acidic peptides. Singly phosphorylated peptides are retained weakly under these conditions, due to electrostatic repulsion, unless hydrophilic interaction is superimposed in the ERLIC mode. Weak anion-exchange (WAX) and strong anion-exchange (SAX) columns were compared, with both peptide standards and a HeLa cell tryptic digest. The SAX column exhibited greater retention at pH 6 than did the WAX column. However, only about 60% as many phosphopeptides were identified with SAX at pH 6 than via ERLIC at pH 2. In one ERLIC run, 12 467 phosphopeptides were identified, including 4233 with more than one phosphate. We conclude that chromatography of phosphopeptides is best performed at low pH in the ERLIC mode. Under those conditions, the performances of the SAX and WAX materials were comparable. The data have been deposited with the ProteomeXchange with identifier PXD001333.
The reversible phosphorylation of proteins on serine, threonine, and tyrosine residues is one of the most prominent regulatory mechanisms in biology. Consequently, analysis of this important post-translational modification with mass spectrometry (MS) is a central piece in the proteomics toolbox. Phosphopeptides tend to be of low abundance in complex tryptic digests. To achieve high coverage of the phosphoproteome, specific enrichment and prefractionation of phosphopeptides is required before online liquid chromatography-tandem MS (LC-MS/MS). Several protocols exploit the charge difference between phosphorylated and nonphosphorylated peptides. One such technique, AEX, is increasingly being used for the isolation of phosphopeptides from tryptic digests.1−5 However, at low pH AEX is unsuited for the isolation of singly phosphorylated tryptic peptides. The electrostatic attraction conferred by a single phosphate group does not suffice to compensate for the electrostatic repulsion of the two basic groups inherent to most tryptic peptides: the N-terminus and the side-chain amino group of the C-terminal amino acid. Consequently, most tryptic phosphopeptides elute from AEX columns in or near the void volume at low pH.6 Phosphopeptides that are retained under these conditions tend to contain additional acidic residues.2,7 Two different analytical strategies, electrostatic repulsion–hydrophilic interaction chromatography (ERLIC) and AEX at high pH, can be employed to address this limitation.
In the case of ERLIC, the mobile phase contains more than 60% acetonitrile (ACN), leading to additional hydrophilic interaction superimposed on the electrostatic effects.8 The combination of the hydrophilic interaction of phosphate groups plus their electrostatic attraction to anion-exchange material suffices to separate them from most of the unmodified peptides even in complex tryptic peptide mixtures. ERLIC has been used successfully for the isolation of phosphopeptides in a number of studies.9−17 When the same phosphopeptide enrichment (IMAC or titania) method is used for fractions from either SCX or ERLIC, then considerably more phosphopeptides are identified via ERLIC.18 Off-line enrichment employing either titania or IMAC material is important because acidic unmodified peptides tend to coelute with phosphopeptides and then suppress the ionization of the phosphopeptides in mass spectrometry. Getting rid of the acidic unmodified peptides increases dramatically the number of phosphopeptides identified.19,20 The problem with acidic unmodified peptides is minimized by performing the chromatography at pH values of around 2.0, leaving carboxyl groups mostly uncharged.
A competing strategy uses AEX material at a pH higher than 6. In this case, phosphate groups acquire a second negative charge. That increases their retention, making AEX of singly phosphorylated tryptic peptides practical. Some studies involved a pH in the range 7–8,1,4,21 while other protocols used initial pH values as high as 9–10.3,5 High pH conditions impose two constraints upon the chromatographic conditions. First, weak anion-exchange (WAX) materials lose overall charge density in a continuum between pH 5 and 9.522 while strong anion-exchange (SAX) materials maintain their charge density over that range. Accordingly, all reported separations at high pH conditions have been performed with SAX materials. Second, silica dissolves at pH higher than 8. Accordingly, all of the work in that range has involved polymeric materials. The most frequently reported material is POROS HQ, while many reports merely cite the use of a SAX material with no further details.
Although WAX materials exhibit lower retention times than SAX materials at high pH conditions, the retention may still be adequate for the purpose. Additionally, the selectivity for certain analytes could conceivably be better. This is important because the carboxyl groups of acidic peptides are fully charged under these conditions. While the majority of tryptic phosphopeptides have a single phosphate group, unmodified peptides may contain numerous Asp and Glu residues; therefore, phosphopeptide enrichment methods should at the least be able to separate singly phosphorylated peptides from peptides with 2–3 acidic residues. The ability to distinguish between these categories has been assessed to some extent with some of the combinations listed above2,3 but not in a comparison of WAX versus SAX. Also, while the particular electrolytes used in the mobile phase have been shown to have a major effect on selectivity for phosphopeptides in the ERLIC mode,8 this has not been studied in the AEX mode. The goal of this study is to systematically optimize the selective isolation of tryptic phosphopeptides for phosphoproteomic studies. Toward this end, we assess the retentivity and selectivity of SAX and WAX materials for phosphopeptide separations, with special attention to the separation of phosphopeptides from acidic nonphosphorylated peptides over a wide pH range. Finally, we address the effects of using different salts for the separations.
Materials and Methods
Materials
PolyWAX LP, a commercial product of PolyLC Inc. (Columbia, MD), was synthesized via an adsorbed, cross-linked coating of linear polyethylenimine largely as described.22 The material was made with the standard commercial coating applied to a silica that had been synthesized via a process that makes it resist attack at pH 9.0–9.5 in case it was necessary to study performance in that range. PolySAX LP, an experimental material, was prepared by quaternizing the PolyWAX LP material with methyl groups in a manner similar to that described by Regnier and co-workers.23,24
Reagents
The peptides WAGGDASGK and WAGG(pS)ASGK were purchased from California Peptide Research (Napa, CA). WWGSGPSGSGGSGGGK and its analogues phosphorylated on Ser were synthesized in-house by Mathias Madalinski (IMP, Vienna) or were a gift of Goran Mitulović (Medical University of Vienna). Analogues of WWGSGPSGSGGSGGGK with Asp substituted for Ser were purchased from United Biosystems (Cabin John, MD).
All other reagents were from Sigma-Aldrich (St. Louis).
Mobile Phase Preparation
Salt buffers were prepared by dissolving a known quantity of the acid in water and titrating with the base to attain the desired pH (example: dissolving high-performace liquid chromatography (HPLC)-grade H3PO4 in water and titrating with KOH or triethylamine). This was measured prior to addition of ACN, after which no measurement or adjustment of pH was performed.
HeLa Sample Preparation
HeLa Kyoto cells were grown in Dulbecco's modified Eagle's medium. Mitosis was arrested with nocodazol treatment overnight. They were then harvested and lysed with freshly prepared 8 M urea and the protein concentration was measured using a Bradford assay. The lysate was reduced and alkylated with dithiothreitol and iodoacetamide and then digested with Lys-C and Trypsin. Peptides were enriched from the digestion mixture using reversed-phase (RP) C18 Sep-Pak cartridges (Waters) and the eluate was dried to completion in a vacuum centrifuge.
Phosphopeptide Enrichment
HPLC separation was performed on an UltiMate 3000 (Dionex) equipped with a fraction collector. Eight hundred micrograms of the peptide mixture was fractionated on either a PolySAX LP or PolyWAX LP column (PolyLC; 4.6 mm inner diameter (i.d.) × 200 mm, 5 μm particle size, 300 Å) using a binary solvent system of solvent A (for ERLIC separations: 70% acetonitrile, 20 mM sodium methylphosphonate, pH 2.0; for AEX separation: 10% acetonitrile, 20 mM ammonium acetate, pH 6.0) and solvent B (10% acetonitrile, 300 mM triethylammonium phosphate, pH 2.0) delivered at 1 mL/min per the following linear gradient: 5 min at solvent A, then 43 min to 100% solvent B, and then 5 min at 100% solvent B. Fractions were collected every 1 min between 0 and 50 min. Every two adjacent fractions were pooled. ACN was removed by drying to half the original volume. This was followed by desalting using RP-C18 Sep-Pak cartridges. The eluates were dried to completion in a vacuum centrifuge.
LC-MS/MS Analysis
Nano-HPLC-MS/MS was performed on an UltiMate 3000 RSLCnano (Dionex) coupled online to a Q-Exactive (Thermo Fisher Scientific). Fractions were first loaded onto a RP-C18 trap column (Acclaim PepMap Nano-Trap, Dionex; 100 μm i.d. × 100 mm, 5 μm particle size, 300 Å pore size) using 25 μL/min solvent C (0.1% trifluoroacetic acid in water) and then separated on a RP C18 column (Acclaim PepMap RSLC, Dionex; 75 μm i.d. × 500 mm, 2 μm particle size, 100 Å pore size) using a binary solvent system of solvent D (0.1% formic acid in water) and solvent E (80% acetonitrile and 0.1% formic acid in water) delivered at 230 nL/min in one of the following linear gradients: 10 min at 2% solvent D, in 60 min (1 h gradient) or 120 min (2 h gradient) to 35% solvent D, in 5 min to 90% solvent D, 5 min at 90% solvent D. Peptides were ionized by electrospray using coated nanospray emitters (SilicaTip, New Objective; 10 μm tip i.d.) biased to 1.9 kV. The mass spectrometer was operated in data-dependent acquisition mode with one MS scan followed by 15 sequential MS/MS scans of the most intense precursors. Airborne dodecamethylhexacyclosiloxane was used as lockmass. The MS scan was acquired from 350 to 2000 m/z at a resolution of 70 000, an automatic gain control (AGC) target value of 1 000 000 and a maximal injection time (IT) of 50 ms. Precursor ions with a charge >1 and an intensity >100 000 were selected for MS/MS, which was performed with an isolation width of 2 m/z and a HCD normalized collision energy of 27. The MS/MS scan was acquired at a resolution of 17 500 with a dynamic m/z range, an AGC target value of 100 000, and a maximal IT of 200 ms. Fragmented precursor ions were excluded from fragmentation for 30 s.
Data Analysis
For peptide identification, the.RAW-files were loaded into Proteome Discoverer (Thermo Fisher Scientific; version 1.4.0.288). All MS/MS spectra created thereby were searched using MS Amanda25 against the Swiss-Prot human protein sequence database (www.uniprot.org; retrieved June 29, 2014; 20 581 entries). The following search parameters were used: carbamidomethylation on cysteine was set as a fixed modification and oxidation on methionine was set as a variable modification as was phosphorylation of serine, threonine, and tyrosine. Monoisotopic masses were searched within unrestricted protein masses for tryptic peptides. The peptide mass tolerance was set to ±8 ppm and the fragment mass tolerance to ±20 ppm. The maximal number of missed cleavages was set to 2. The result was filtered to 1% false discovery rate using the Percolator algorithm26 integrated in Proteome Discoverer. Numbers are reported for unique peptide sequences as well as for peptide spectrum matches. PhosphoRS was applied for phosphorylation site probability estimation for every phosphorylated peptide spectrum match.27 No threshold was defined since this may decrease identification of multiphosphorylated peptides. It is more difficult to localize their positions of phosphorylation, especially when multiple accessor sites are in close proximity. The focus in this paper was the chromatography behavior of peptides with one or more phosphate groups under different conditions and with different materials, not defining the exact position of phosphorylation of multiphosphorylated peptides.
Results
Generation of Column Materials
The silica used for this study was synthesized using a process that imparts some degree of resistance to attack in the pH range 9–9.5. The WAX material was prepared by adsorption of linear polyethylenimine to the surface with subsequent cross-linking of the adsorbed polymer to form a macromolecular network that envelopes the silica particle inside the pores and out. The lack of a silane coating increases the resistance to loss of capacity at elevated pH. The SAX material was formed by quaternization of the WAX material with methyl groups. Going by the data below, we estimate that about 70% of the amine residues in the resulting SAX material are quaternary (4°), the rest being some combination of primary (1°), secondary (2°), and tertiary (3°) amines. This is consistent with the silica-based materials prepared by Regnier and co-workers using a similar process.23,24 Titration data suggest that the POROS HQ material, widely used by groups currently isolating phosphopeptides, also has about 60% content of 4° amines,28 the rest being 1°, 2°, or 3°. This range of 60–70% for 4° amine residues appears to be a mark of SAX materials made by quaternization of WAX materials. SAX materials made through processes that can only result in a 4° amine (e.g., reaction of trimethylamine with an immobilized alkyl bromide) are readily distinguished by their titration curves.28,29 It is not evident that complete quaternization of the stationary phase is necessary or even desirable for this application. Drager and Regnier have reported that optimum selectivity for some oligonucleotide separations was obtained with materials that were 40–60% quaternized.24 That is not necessarily true for smaller solutes such as tryptic peptides that may have only one or two acidic residues. It is beyond the scope of the present study to ascertain the importance to peptide AEX of the extent of quaternization.
Impact of Salts on Selectivity
The importance of the choice of salt for the mobile phase is frequently underestimated. The nature of salt additives can have a critical effect on selectivity (Figure 1). Here, a peptide with a phosphoserine residue is far better retained than the corresponding peptide with an aspartyl residue when a phosphonate salt is used but not when a phosphate salt is used. As an alternative to solvents containing nonvolatile salts, it has been proposed that ERLIC be performed with solvents containing ammonium formate, weakly buffered in the range 2.0–2.2.30 Those conditions separated phosphopeptides from most unmodified peptides satisfactorily. The study did not consider the behavior of unmodified peptides that contained numerous acidic residues. Chien et al. subsequently noted19 that such peptides were so abundant in the fractions of retained peptides that they masked the phosphopeptides unless they were removed from the fractions by IMAC enrichment. Analysis of the current set of peptide standards with similar conditions demonstrates that the complaint by Chien et al. was justified; peptides containing 3 or 4 Asp residues were poorly resolved from the standard with one phosphate and peptides were retained in proportion to the number of acidic residues (Figure 2a). When methylphosphonate salt is used instead, then ionization of Asp residues is suppressed more effectively and the standards with 3–4 Asp residues elute well before the standard with 1 phosphate residue (Figure 2b).
Figure 1.
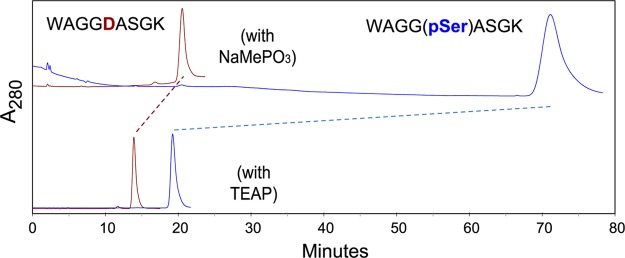
Effect of salt on selectivity for phosphopeptides. The peptides WAGGDASGK (red) and WAGG(pS)ASGK (blue) were separated using sodium methylphosphonate (Na-MePO3) [top] or triethylammonium phosphate (TEAP) [bottom] in the mobile phase. Column: PolySAX LP, 200 × 4.6 mm; 5 μm, 300 Å. Mobile phase: 20 mM of the relevant salt, pH 6.0, containing 70% ACN. Flow rate: 1 mL/min. Detection: 280 nm.
Figure 2.

Use of different salts for separation of tryptic peptides with either Asp or pSer at the same sites. Peak identities were confirmed by running the standards individually. Key to peptide standards: per insert. Column: PolyWAX LP, 200 × 4.6 mm; 5 μm, 300 Å. Flow rate: 1 mL/min. Detection: 280 nm. (A) ERLIC with ammonium formate: Mobile Phase A: 20 mM ammonium formate, pH 2.2, with 70% ACN. Mobile Phase B: 1 M ammonium formate, pH 2.2, with 10% ACN. Gradient schedule: 0–5′, 0% B; 5–35′, 0–100% B. (Note: Standard 2P was run separately from the others under identical conditions). (B) ERLIC with Na-MePO3: Mobile Phase A: 20 mM Na-MePO3, pH 2.0, with 70% ACN. Mobile Phase B: 300 mM TEAP, pH 2.0, with 10% ACN. Gradient schedule: 0–5′, 0% B; 5–48′, 0–100% B.
We then compared the retention of the same peptide standards under conditions typical of AEX (Figure 3), employing a gradient from ammonium acetate, pH 6, to 300 mM TEAP at pH 2 (both with 10% ACN). Any increase in retention afforded by the double ionization of phosphate groups is largely counteracted by the lack of retention from hydrophilic interaction. Of greater concern is the potential coelution with the singly and doubly phosphorylated peptides of the peptide standards with two or more ordinary acidic residues, which have full negative charge under these conditions. Retention times of most of the peptides are greater with the PolySAX LP column (Figure 3b). This is as might be expected in view of the greater charge density at pH 6 of a SAX material compared with a WAX material. However, retention of the standards with 3 or 4 phosphate groups or 4 Asp residues is as great or greater with the PolyWAX LP column (Figure 3a). With such highly charged analytes, the charge density of the stationary phase seems to be less critical to retention.
Figure 3.

High-pH AEX: Key to peptide standards, per Figure 2. Mobile Phase A: 20 mM ammonium acetate, pH 6.0, with 10% ACN. Mobile Phase B: 300 mM TEAP, pH 2.0, with 10% ACN. Gradient schedule: As in Figure 2b. Flow rate: 1 mL/min. Detection: 280 nm. (A) Column: PolyWAX LP, 200 × 4.6 mm; 5 μm, 300 Å. (B) Column: PolySAX LP, 200 × 4.6 mm; 5 μm, 300 Å.
Comparison of WAX vs SAX at Various pH Values with Isocratic Elution
Studies were performed using phosphate or phosphonate salts, which buffer across most of the range of interest. The buffering capacity is poor in some portions of the range. This seems less important than the consistency in selectivity coming from use of the same salt. The mobile phases contained either 10% ACN, for ordinary AEX operation, or 70% ACN, where a significant degree of hydrophilic interaction was superimposed upon the electrostatic effects. With 70% ACN the mode is ERLIC at an operating pH of 3, where carboxyl groups are substantially uncharged and electrostatic repulsion is significant. At higher pH values the mode could be termed AEX-HILIC. The concentration of electrolyte needed to form a reasonably complete double layer on the surface of a charged stationary phase is about 20 mM in the overall mobile phase.8,31 Accordingly, that concentration was used in this phase of the study.
We then compared retention on WAX and SAX columns with a mobile phase containing K-PO4 and 10% ACN (Figure 4A). With either column there is a modest increase in retention of standard WAGGDASGK over the range pH 3–5 as its carboxyl functional groups ionize. Retention of the phosphopeptides increases more significantly between pH 5–7, the range of the phosphate groups’ transition from one to two negative charges. The increase is more significant with the SAX column. Thereafter retention remains fairly constant with increasing pH values. The WAX column retains its capacity at higher pH to a surprising degree, in view of the progressive loss of charge density. We then performed separations on both columns using 70% ACN for the mobile phase, using TEAP instead of K-PO4 out of concern for solubility (Figure 4B). An increase in retention similar to the previous experiment (Figure 4A) was noted over pH 3–5. Retention times were appreciably longer than with 10% ACN. Presumably this is due to the superimposed hydrophilic interaction. However, retention fell off markedly on both columns above pH 5. Above pH 7 there was no significant difference in retention between the phosphorylated peptides and their unphosphorylated analogues.
Figure 4.

Effect of pH on isocratic retention times. WAX vs SAX: Columns and flow rate: As in Figure 3. Mobile Phase (isocratic): (A) 20 mM potassium phosphate, pH as noted, with 10% ACN. (B) 20 mM TEAP, pH as noted, with 70% ACN. (C) 20 mM Na-MePO3, pH as noted, with 10% ACN. (D) 20 mM Na-MePO3, pH as noted, with 70% ACN. Peptide standards: (1) WWGSGPSGSGGSGGGK (SAX, ⧫, red; WAX, ⧫, blue); (2) WWGSGPSGSGG(pS)GGGK (SAX, ■, red; WAX, ■, blue); (3) WAGGDASGK (SAX, ●, red; WAX, ●, blue); (4) WAGG(pS)ASGK (SAX, ▲, red; WAX,▲, blue).
When the electrolyte was changed to sodium methylphosphonate, with either 10% or 70% ACN, tryptic phosphopeptides were much better retained than tryptic unphosphorylated peptides, particularly over the pH range 6–7 for AEX (Figure 4C) and pH 5−6 for AEX-HILIC (Figure 4D). At a pH higher than 5, retention of phosphopeptides on the SAX column was nearly twice as great as that on the WAX column. However, retention fell off above pH 7 to about the same extent on both columns, as in Figure 4B. Retention of nonphosphorylated tryptic standard peptides increased modestly with pH or remained about the same throughout the range.
From these results one may draw the conclusions that, first, methylphosphonate affords the best selectivity for phosphopeptides, at any pH. Second, that at a pH higher than 5, retention of singly phosphorylated peptides is greater on the SAX material than on the WAX material. However, retention with WAX is still acceptable, and the selectivity between phosphopeptides and acidic unmodified peptides seems to be at least as good, if not better. Finally, depending on conditions, retention of phosphopeptides reaches a maximum in the pH range 5–7 and then declines on either material. Therefore, there is no benefit to performing AEX of phosphopeptides at a pH higher than 7. Judging from our data, a more appropriate pH is 6.
Analysis of a HeLa Cell Lysate
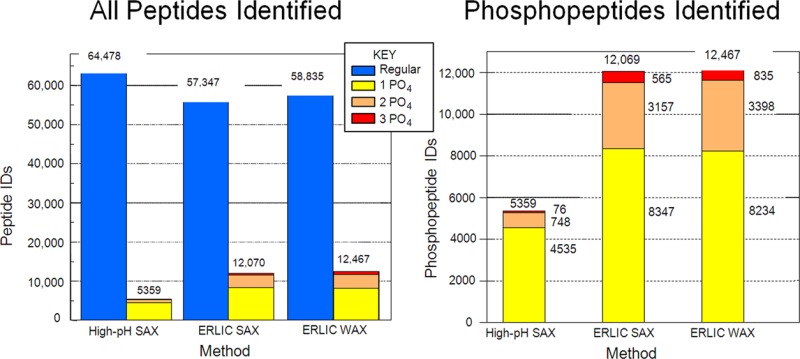
To test our optimized conditions on a biological sample, we chose tryptic peptides generated from a HeLa cell lysate. The performance of the different ERLIC conditions were again compared with AEX, this time by comparing the number and nature of the peptides identified via two-dimensional LC-MS/MS. First, a SAX column was eluted with conditions commonly used in the literature for AEX of phosphopeptides. A gradient to 0.3 M phosphate was used to ensure the elution of all phosphopeptides; however, this approach failed to separate phosphopeptides well from acidic unmodified peptides (Figure 5). The broad distribution of singly phosphorylated peptides could reflect variations in their content of Asp and Glu residues or the position of the phosphate group within the peptide.32 As expected from the results described above, the selectivity improved when changing to the ERLIC mode using the same column (Figure 6A) or a WAX column (Figure 6B). As a result, ERLIC identified significantly more phosphorylated peptides in the HeLa digest (Figure 7A). While the AEX method identified modestly more unmodified peptides, the two ERLIC runs each led to the identification of 2.1 times more total phosphopeptides and, specifically, 5.4 times the number of multiply phosphorylated peptides using either material (Figure 7B). The lower numbers of phosphopeptides identified in the AEX mode can be explained by their coelution with high numbers of unmodified peptides with 3 or more acidic residues, which suppressed their identification in LC-MS. By contrast, ERLIC effectively moved these unmodified peptides from the phosphopeptide-containing part of the gradient to the early part of the chromatogram. There was no serious difference in performance of the WAX and SAX materials under ERLIC conditions (Figure 7B).
Figure 5.
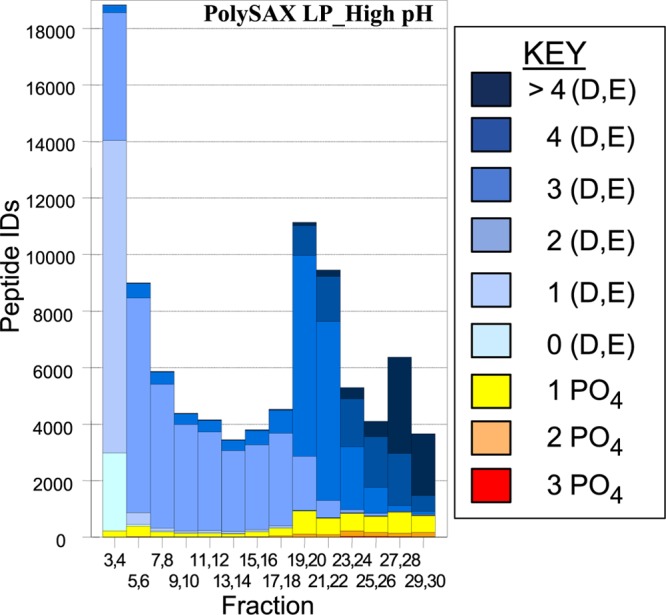
Number and type of peptides identified from a HeLa cell lysate via high-pH AEX. Column: PolySAX LP. Conditions: See Materials and Methods. See key for peptide categories.
Figure 6.

Number and type of peptides identified from a HeLa cell lysate via ERLIC. Column: (A) PolySAX LP; (B) PolyWAX LP. Conditions: See Materials and Methods. Key: See Figure 5.
Figure 7.
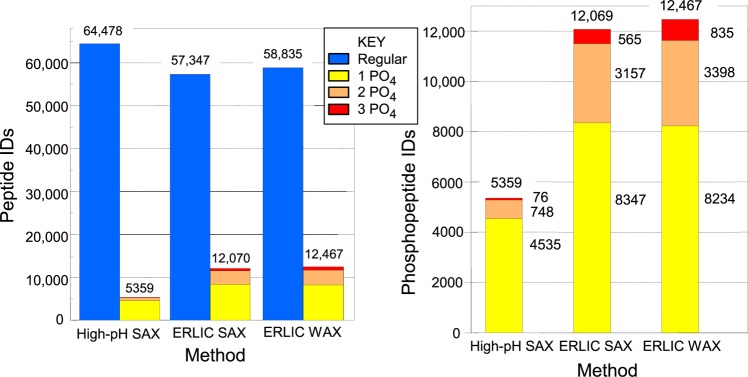
(A) Unmodified and phosphorylated peptides identified using SAX and WAX with ERLIC or AEX. The numbers are summations of the numbers in the various bars in Figures 5–7. (B) Phosphorylated peptides identified using SAX and WAX with ERLIC or AEX. This is an expansion of the bars for phosphopeptides in (A).
Conclusions
ERLIC is unequivocally superior to AEX for identification of tryptic phosphopeptides from complex mixtures. While the SAX material has greater retentivity than WAX material in the AEX mode at a pH higher than 5, both materials have full charge density at the low pH used for ERLIC. Their performance is then similar. Phosphopeptides are less well-retained at such low pH conditions than under AEX conditions, but are much better separated from acidic unmodified peptides. Consequently, many more phosphopeptides are identified using ERLIC. The use of methylphosphonate salts in the starting mobile phase affords superior selectivity for phosphopeptides; however, an additional desalting step is then required. That is no more troublesome than the extra affinity step that is needed, when any other salt is used, to separate phosphopeptides from acidic unmodified peptides. The subject of desalting ERLIC fractions has been explored in detail by Loroch et al.33,34 and Zarei et al.35
To the best of our knowledge, this is the first reported systematic study of the retention of peptides in AEX as a function of pH. It is not clear why retention decreases at pH values higher than 6 or 7, on the SAX as well as the WAX material. The effect is more pronounced when a significant amount of hydrophilic interaction is present. Gilar and Jaworski have calculated the retention coefficient of various residues in peptides when eluted from Atlantis silica HILIC columns.36 When electrostatic effects from charged amino acids and surface silanols are discounted, retention due to most neutral amino acids tends to increase from pH 3.0 to pH 4.5 and then decline at pH 10, roughly consistent with the trends observed here. It is possible that hydrophilic interaction decreases as the pH approaches an extreme.
Acknowledgments
This work was funded in part by Boehringer Ingelheim, the Austrian Academy of Sciences, the Austrian Science Fund (FWF) via the Special Research Program Chromosome Dynamics (SFB-F3402) and the European Commission via the FP7 projects MeioSys, PrimeXS, and MitoSys. We wish to thank Dr. Thomas Köcher for a critical reading and revision of the manuscript. A preliminary version of some of the data was presented at ASMS 2014 (Baltimore, June 2014), poster W091.
Accession Codes
The mass spectrometry proteomics data in this paper have been deposited with the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository37 with the support of PRIDE team EBI, with data set identifier PXD001333.
The authors declare no competing financial interest.
References
- Nühse T. S.; Stensballe A.; Jensen O. N.; Peck S. C. Mol. Cell. Prot. 2003, 2, 1234. [DOI] [PubMed] [Google Scholar]
- Han G.; Ye M.; Zhou H.; Jiang X.; Feng S.; Jiang X.; Tian R.; Wan D.; Zou H.; Gu J. Proteomics 2008, 8, 1346. [DOI] [PubMed] [Google Scholar]
- Dai J.; Wang L.-S.; Wu Y.-B.; Sheng Q.-H.; Wu J.-R.; Shieh C.-H.; Zeng R. J. Proteome Res. 2009, 8, 133. [DOI] [PubMed] [Google Scholar]
- Nie S.; Dai J.; Ning Z.-B.; Cao X.-J.; Sheng Q.-H.; Zeng R. J. Proteome Res. 2010, 9, 4585. [DOI] [PubMed] [Google Scholar]
- Ficarro S. B.; Zhang Y.; Carrasco-Alfonso M. J.; Garg B.; Adelmant G.; Webber J. T.; Luckey C. J.; Marto J. A. Mol. Cell. Prot. 2011, 10, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motoyama A.; Xu T.; Ruse C. I.; Wohlschlegel J. A.; Yates J. R. III Anal. Chem. 2007, 79, 3623.Fig. 4H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennrich M. L.; Groenewold V.; Kops G. J. P. L.; Heck A. J. R.; Mohammed S. Anal. Chem. 2011, 83, 7137. [DOI] [PubMed] [Google Scholar]
- Alpert A. J. Anal. Chem. 2008, 80, 62. [DOI] [PubMed] [Google Scholar]
- Gan C. S.; Guo T.; Zhang H.; Lim S. K.; Sze S. K. J. Proteome Res. 2008, 7, 4869. [DOI] [PubMed] [Google Scholar]
- Bennetzen M. V.; Larsen D. H.; Bunkenborg J.; Bartek J.; Lukas J.; Andersen J. S. Mol. Cell. Prot. 2010, 9, 1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razani B.; Zarnegar B.; Ytterberg A. J.; Shiba T.; Dempsey P. W.; Ware C. F.; Loo J. A.; Cheng G. Sci. Signaling 2010, 3123ra41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Guo T.; Li X.; Datta A.; Park J. E.; Yang J.; Lim S. K.; Tam J. P.; Sze S. K. Mol. Cell. Prot. 2010, 9, 635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T.; Lee S. S.; Ng W. H.; Zhu Y.; Gan C. S.; Zhu J.; Wang H.; Huang S.; Sze S. K.; Kon O. L. Cell. Mol. Life Sci. 2011, 68, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Wu D.; Zhao Y.; Wong B. H. C.; Guo L. J. Chromatogr. B 2011, 879, 25. [DOI] [PubMed] [Google Scholar]
- Hao P.; Guo T.; Sze S. K. PLoS One 2011, 6, e16884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarei M.; Sprenger A.; Gretzmeier C.; Dengjel J. J. Proteome Res. 2012, 11, 4269. [DOI] [PubMed] [Google Scholar]
- Tran T. H.; Hwang I.; Park J.-M.; Kim J. B.; Lee H. MS Lett. 2012, 3, 39. [Google Scholar]
- Kehasse A.Y.; Leymarie N.; McComb M. E.; Trinkaus-Randall V.; Costello C. E.60th ASMS Conference, 2012, poster MP 484.
- Chien K.-Y.; Liu H.-C.; Goshe M. B. J. Proteome Res. 2011, 10, 4041. [DOI] [PubMed] [Google Scholar]
- Chien K.-Y.; Blackburn K.; Liu H.-C.; Goshe M. B. J. Proteome Res. 2012, 11, 5663. [DOI] [PubMed] [Google Scholar]
- Wang F.; Han G.; Yu Z.; Jiang X.; Sun S.; Chen R.; Ye M.; Zou H. J. Sep. Sci. 2010, 33, 1879. [DOI] [PubMed] [Google Scholar]
- Alpert A. J.; Regnier F. E. J. Chromatogr. 1979, 185, 375. [Google Scholar]
- Kopaciewicz W.; Regnier F. E. Anal. Biochem. 1983, 133, 251. [DOI] [PubMed] [Google Scholar]
- Drager R. R.; Regnier F. E. Anal. Biochem. 1985, 145, 47. [DOI] [PubMed] [Google Scholar]
- Dorfer V.; Pichler P.; Stranzl T.; Stadlmann J.; Taus T.; Winkler S.; Mechtler K. J. Proteome Res. 2014, 13, 3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Käll L.; Canterbury J.; Weston J.; Noble W. S.; MacCoss M. J. Nat. Methods 2007, 4, 923. [DOI] [PubMed] [Google Scholar]
- Taus T.; Köcher T.; Pichler P.; Paschke C.; Schmidt A.; Henrich C.; Mechtler K. J. Proteome Res. 2011, 10, 5354. [DOI] [PubMed] [Google Scholar]
- Staby A.; Jensen I. H. J. Chromatogr. A 2001, 908, 149. [DOI] [PubMed] [Google Scholar]
- Staby A.; Jensen I. H.; Mollerup I. J. Chromatogr. A 2000, 897, 99. [DOI] [PubMed] [Google Scholar]
- Alpert A. J.; Mitulović G.; Mechtler K. HPLC 2008, poster P2412-W (link: http://www.polylc.com/downloads/HPLC_2008_poster_2412-W_ERLIC_of_phosphopeptides_slides.pdf).
- Guo Y.; Srinivasan S.; Gaiki S. Chromatographia 2007, 66, 223. [Google Scholar]
- Alpert A. J.; Petritis K.; Kangas L.; Smith R. D.; Mechtler K.; Mitulović G.; Mohammed S.; Heck A. J. R. Anal. Chem. 2010, 82, 5253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loroch S.; Zahedi R. P.; Sickmann A. Anal. Chem. 2015, 87, 1596. [DOI] [PubMed] [Google Scholar]
- Loroch S.; Schommartz T.; Brune W.; Zahedi R. P.; Sickmann A. Biochim. Biophys. Acta 2015, 18545460. [DOI] [PubMed] [Google Scholar]
- Zarei M.; Sprenger A.; Gretzmeier C.; Dengjel J. J. Proteome Res. 2013, 12, 5989. [DOI] [PubMed] [Google Scholar]
- Gilar M.; Jaworski A. J. Chromatogr. A 2011, 1218, 8890. [DOI] [PubMed] [Google Scholar]
- Vizcaíno J. A.; Côté R. G.; Csordas A.; Dianes J. A.; Fabregat A.; Foster J. M.; Griss J.; Alpi E.; Birim M.; Contell J.; O’Kelly G.; Schoenegger A.; Ovelleiro D.; Pérez-Riverol Y.; Reisinger F.; Ríos D.; Wang R.; Hermjakob H.. Nucl. Acids Res. 2013, 41, D1063–D1069; The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]