

Abstract
Tuberculosis (TB) remains a major global health concern, and new therapies are needed to overcome the problems associated with dosing frequency, patient compliance, and drug resistance. To reduce side effects associated with systemic drug distribution and improve drug concentration at the target site, stable therapeutic nanocarriers (NCs) were prepared and evaluated for efficacy in vitro in Mycobacterium tuberculosis-infected macrophages. Rifampicin (RIF), a current, broad-spectrum antibiotic used in TB therapy, was conjugated by degradable ester bonds to form hydrophobic prodrugs. NCs encapsulating various ratios of nonconjugated RIF and the prodrugs showed the potential ability to rapidly deliver and knockdown intracellular M. tuberculosis by nonconjugated RIF and to obtain sustained release of RIF by hydrolysis of the RIF prodrug. NCs of the novel antibiotic SQ641 and a combination NC with cyclosporine A were formed by flash nanoprecipitation. Delivery of SQ641 in NC form resulted in significantly improved activity compared to that of the free drug against intracellular M. tuberculosis. A NC formulation with a three-compound combination of SQ641, cyclosporine A, and vitamin E inhibited intracellular replication of M. tuberculosis significantly better than SQ641 alone or isoniazid, a current first-line anti-TB drug.
Keywords: nanocarrier, SQ641, rifampicin, tuberculosis, nanoparticle, isoniazid, flash nanoprecipitation, block copolymer, prodrug
Introduction
Tuberculosis (TB), caused by Mycobacterium tuberculosis, is an intracellular disease infecting approximately 1 in 3 people throughout the world and causing over 1 million deaths annually.1 Schutz2 and Jindani et al.3 evaluated the susceptibility of M. tuberculosis to various antibiotics and established the principle that TB therapy should include multiple drugs. Effective drug regimens have been established, yet TB persists as a major societal challenge owing to the ease of transmission,4 patient noncompliance to a therapeutic course that requires adherence to frequent dosing over a 6 month period,5,6 and the high costs of overseeing treatment. The efficacy of anti-TB drugs can be hindered by poor drug permeability, solubility, and biodegradation,7,8 and undesirable side effects are caused by systemic drug distribution.9 Failed therapy has given rise to multidrug-resistant TB (MDR-TB) strains, which have become a global health concern. These strains are less responsive to traditional therapy and require second-line therapy with a 4-fold increase in duration as well as increased toxicity. Even with aggressive treatment, cure rates of only 60% are achieved,10 and there is now a growing prevalence of extensively drug-resistant (XDR-TB) strains, which do not respond to first- and second-line drugs.
While there has been growth in preclinical research investigating new ways of reducing the TB burden through the discovery of new therapeutic targets, antibiotics,11,12 and vaccines,13 drug delivery approaches have been explored to a lesser extent.10 Oral administration is preferred for anti-TB treatment, but the extended duration and aggressive nature of treatment for MDR-TB present an opportunity for innovative drug delivery strategies that can shorten the treatment cycle and improve patient outcomes. Particulate drug carriers have the potential to coencapsulate high payloads of multiple drugs, control release, and colocalize the drugs at the target site in vivo.
Several types of colloidal drug carriers have been reported with current anti-TB drugs, and a thorough and critical review of recent formulations was published by Blasi et al.10 Anisimovia et al. separately encapsulated isoniazid (INH), rifampicin (RIF), and streptomycin into acrylate nanoparticles and observed enriched intracellular drug concentrations in vitro and comparable or better efficacy of the nanoparticles compared to that of free drugs.14 A similar benefit was observed in a mouse model when INH and rifabutin in PLGA nanoparticles were administered by aerosol deposition and a 20-fold higher intracellular concentration of drug was achieved relative to the soluble drug.15 While numerous efficacy studies on PLGA, lectin-PLGA, alginate, and solid lipid nanoparticles produced by Khuller and co-workers over the past 10 years indicate the potential for colloidal formulations to reduce drug dosing frequency from daily to weekly administration,16 it remains unclear what mechanisms are responsible for the apparent extended depot effect that is observed in mice and guinea pigs, regardless of the vehicle or route of administration.
In addition to nanocarriers encapsulating and concentrating TB drugs intracellularly as reported previously, there is an opportunity to develop formulations that incorporate multiple functionalities into a single nanocarrier design, can extend the duration of therapy per dose with less frequent treatments, and target the drug(s) to the infected macrophages. A parallel effort in our group has been dedicated to developing nanocarriers (NCs) with terminal mannose groups on the PEG chains by premodification of block copolymer chains and subsequent directed assembly of nanocarriers.17 In this work, we have explored formulation strategies to encapsulate multiple anti-TB therapeutic agents for release profile modification or multiple drug delivery using the same directed assembly process. We investigate the formulation of RIF, currently a key component of anti-TB therapy,18 and SQ641, a novel anti-TB drug for which delivery of the drug is hindered by very poor aqueous solubility (<20 μg mL–1), low permeability, and rapid P-glycoprotein (P-gp) mediated efflux from cells.19 The combination of targeting infected macrophages, controlled release, and/or multiple drug delivery at the location of infection is a unique opportunity for NC delivery and may provide substantial benefits to MDR-TB infected patients treated in a hospital setting where IV administration is not a barrier if other advantages are substantial.
Anti-TB NCs of RIF, hydrophobic RIF prodrugs, the novel anti-TB drug SQ641,20 and multidrug cocktails are prepared at high loadings with tunable size, encapsulation, and stability by adjusting process parameters, such as solute ratios, solute concentration, and final solute solubility. In the case of RIF, we have established a prodrug route to forming stable RIF NCs with the potential for both immediate and controlled drug release profiles. For SQ641 delivery, improved activity in vivo has previously been achieved by using α-tocopheryl poly(ethylene glycol) 1000 succinate as a solubilizing agent.21 To improve drug load and enable the production of targeted carriers, we have developed colloidal drug NCs stabilized by block copolymers that coencapsulate SQ641 with cyclosporine A, a P-gp efflux inhibitor, to improve intracellular drug accumulation. The efficacies of these formulations were evaluated in vitro by incubation with M. tuberculosis-infected macrophages and confirm the effectiveness of these NCs for intracellular delivery of hydrophobic payloads.
Experimental Section
Experimental Reagents
Anhydrous dichloromethane (DCM), methanol (HPLC grade), tetrahydrofuran (THF, HPLC grade), N,N′-dicyclohexyl carbodiimide (DCC), 4-(dimethylamino)pyridine (DMAP, ReagentPlus, 99% purity), 1.000 N hydrochloric acid (HCl), RIF (95% purity), d(+)trehalose, α-tocopherol acid succinate (VES), and vitamin E (VE, 97% purity) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Anhydrous magnesium sulfate (MgSO4) was purchased from EMD chemicals INC (Gibbstown, NJ, USA). Cyclosporine A (CsA, >99% purity) was purchased from LC Laboratories (Woburn, MA, USA). SQ641 was synthesized by and received from Sequella Inc. (Rockville, MD, USA). Prior to use, water was purified via 0.2 μm filtration and four-stage deionization to a resistivity of 17.8 MΩ or greater (NANOpure Diamond, Barnstead International. Dubuque, IA, USA) and autoclaved to sterilize it. Pluronic F68 was from BASF Corporation (Parsippany, NJ, USA). Polylactide-b-poly(ethylene glycol) (PLA3.8k-b-PEG5k-OCH3) was synthesized under GMP conditions and was kindly provided by Evonik, Inc. (Birmingham, AL, USA). The syntheses of polylactide-co-glycolide-b-poly(ethylene glycol) (PLGA8k-b-PEG5k-OCH3) and poly(styrene)-b-poly(ethylene glycol) (PS1.5k-b-PEG5k–OH) have been previously reported.22,23
Two RIF prodrugs were synthesized by conjugation of RIF to (a) VES or (b) 2 kDa polycaprolactone (PCL) (Figure 1). The synthesis is described in the Supporting Information.
Figure 1.

Two rifampicin prodrugs were synthesized for this work, through esterification by DCC coupling, where the hydrophobic anchor was (a) vitamin E succinate or (b) 2 kDa polycapralactone.
Sterile NC Formulation in a Confined Impinging Jets Mixer (CIJ)
NC formulations for testing against macrophages infected with M. tuberculosis in vitro were prepared under sterile conditions using a CIJ Mixer24 (Figure 2a) designed for producing NCs by flash nanoprecipitation (FNP) with small solution volumes and as little as 0.5 mg of drug. The mixer was sterilized by soaking in 70% EtOH and operated in a sterile field. THF solutions (1 mL), with the formulations detailed in Tables 1 and 2, were loaded into a plastic syringe (5 mL, Norm-Ject) and mixed against 1 mL of water at 4 °C. Both syringes were driven manually, and the mixed stream was collected in 18 mL of stirred water at 4 °C. The syringes were emptied at the same rate and in less than 2 s to produce a mixing Reynolds number of Re ∼ 1300.25 The intensity-average particle size distribution was measured by dynamic light scattering of a dilute sample, using a ZetaSizer Nano ZS (Malvern Instruments. Worcestershire, UK). The deconvolution of the light scattering autocorrelation function was done using the Malvern analysis software in the normal resolution mode.
Figure 2.

NC formulation by flash nanoprecipitation. (a) Small volume sample preparation is performed in a CIJ mixer with manually driven flow, where the THF/water mixture is collected in water to dilute the THF. (b) Mixing is achieved for streams of unequal flow rates in a MIVM, allowing precise tuning of process parameters.
Table 1. THF Solutions Used in the Preparation of RIF NCs.
| THF
solution composition (mg mL–1) |
|||||
|---|---|---|---|---|---|
| formulation | PLA3.8k-b-PEG5k-OCH3 | RIF-PCL | RIF-VE | RIF | NC size (d.nm) |
| NC1 | 20 | 20 | 0 | 0 | 170 |
| NC2 | 20 | 0 | 20 | 0 | 60 |
| NC3 | 20 | 0 | 18 | 2 | 54 |
| NC4 | 20 | 0 | 10 | 10 | 36 |
Table 2. THF Solutions Used in the Preparation of SQ641 NCs.
| THF
solution composition (mg mL–1) |
||||||
|---|---|---|---|---|---|---|
| formulation | PS1.5k-b-PEG5k-OH | CsA | SQ641 | PCL | VE | NC size (d.nm) |
| NC5 | 60 | 0 | 20 | 0 | 40 | 97 |
| NC6 | 20 | 0 | 0 | 20 | 0 | 170 |
| NC7 | 20 | 20 | 0 | 0 | 0 | 135 |
| NC8 | 40 | 20 | 20 | 0 | 0 | 189 |
| NC9 | 80 | 20 | 20 | 0 | 40 | 100 |
The formulations in Table 1 were prepared with water containing Pluronic F68 (9.5 mg mL–1) and trehalose (12 mg mL–1). Aliquots of the NC suspension (1 mL) were frozen in standard 5 mL plastic cryovials immediately following precipitation in a dry ice/acetone bath and then lyophilized in a VirTis AdVantage 2.0 benchtop freeze dryer for 24 h at a pressure of <30 mTorr. To resuspend lyophilized NC for in vitro testing, sterile water was added to the dried powders, which were manually shaken for 25 s and then sonicated with a probe tip for 25 s in an ice-water bath (Supporting Information).
To remove 5% THF, formulations in Table 2 were dialyzed using a Spectra/Por dialysis membrane with a molecular weight cutoff (MWCO) of 6–8 kDa against 1 L of water, which was refreshed four times over 24 h. The suspensions were stored at 4 °C until used for in vitro tests.
Prodrug Release Experiments
The hydrolysis of dissolved RIF-VE prodrug was carried out by combining 0.1 mL of 10 mg mL–1 RIF-VE in 0.7 mL of THF and 0.2 mL of 10 mM PBS buffer at pH 7.4. Aliquots of the mixture were stored at 4 or 37 °C for 8 days. The relative molar ratio of free RIF and RIF-VE was determined by HPLC assay. The release and hydrolysis of RIF from the RIF-VE prodrug in the presence of serum was carried out in aqueous media by encapsulating the prodrug in NC form, as described above in the manually driven CIJ mixer. The organic solution was composed of 20 mg mL–1 RIF-VE and 20 mg mL–1 PLA3.8k-b-PEG5k-OCH3 in 1 mL of THF. This was mixed against 1 mL of water and collected in a stirred reservoir of 8 mL of water. The final suspension was dialyzed against 1 L of stirred water, which was refreshed three times. The NC suspension (0.8 mL) was added to fetal bovine serum (0.2 mL) and stored at 4 or 37 °C for 2 days. After 2 days, 0.5 mL aliquots were combined with 0.5 mL of THF and assayed by HPLC to determine the relative amounts of RIF and RIF-VE.
Preparation of NCs in a Multi-inlet Vortex Mixer (MIVM)
The MIVM is a high-intensity micromixing device that was used in this work to develop an optimized SQ641 NC formulation (Figure 2b).26 A THF stream containing SQ641 (10 mg mL–1), PLGA8k-b-PEG5k-OCH3 (10 mg mL–1), and VE (0, 5, 10, 15, or 20 mg mL–1) was fed into the MIVM by a digitally controlled syringe pump (Harvard PHD) at 12 mL min–1 against a total of 108 mL min–1 of water. Aliquots of each suspension (1 mL in duplicate) were immediately frozen and lyophilized for determination of the initial drug concentration. Then, 8–10 mL of the NC suspension was dialyzed to remove THF in a Spectra/Por dialysis membrane with a MWCO of 6–8 kDa against 1 L of water, which was refreshed four times over 24 h. After dialysis, each suspension was removed from the dialysis bags, and any volume change was noted. The suspension was filtered with a 5 μm PTFE syringe filter to remove precipitates, and aliquots of the filtrate were frozen and lyophilized to determine the final concentration of drug. The SQ641 encapsulation was calculated as the ratio of the concentration of SQ641 in the nanosuspension after dialysis/filtration to the initial SQ641 concentration after mixing. Changes in the suspension volume during dialysis were noted to account for dilution of the final SQ641 concentration. The lyophilized samples were dissolved in THF in an ultrasonic bath and filtered (0.2 μm), and the filtrate was analyzed to determine drug concentrations using HLPC (Supporting Information). The particle size distribution was measured by dynamic light scattering immediately following mixing, after dialysis and filtration, and after several days of storage to track the stability of the nanocarriers in each suspension (Supporting Information).
SQ641 Solubility and HPLC Analysis
The solubility of SQ641 in THF/water mixtures was determined by mixing aliquots of a saturated SQ641 solution in THF (170 mg mL–1) in various ratios with water. The vials were equilibrated at room temperature for 24 h. The solutions were filtered (0.2 μm), and the SQ641 concentration in the filtrate was determined by HPLC analysis, with dilution by THF when necessary. HPLC conditions are given in the Supporting Information.
In Vitro NC Efficacy Testing against Intracellular M. tuberculosis
The efficacy of the NC formulations against M. tuberculosis was evaluated following the protocol of Snewin et al.27 and Luna-Herrera et al.28 J774A.1 murine macrophage-like cells (ATCC, Manassas, VA, USA) were seeded in 24-well plates at 1 × 106 cells mL–1 well–1 in Dulbecco’s modified Eagles medium (DMEM) supplemented with 10% fetal bovine serum. After a 3 h incubation, the cells were washed 2× with Hanks’ saline. Less than 1 week old M. tuberculosis H37Rv luciferase reporter strain, pSMT1,27 grown in 7H9 broth, was washed 2× in DMEM, adjusted to 0.1 OD, and diluted 1/20 in DMEM. J774A.1 cells were infected with M. tuberculosis suspension for 3 h. At the end of 3 h, the cells were washed 3× with Hanks’ saline. The media was replaced with fresh DMEM containing four times the minimum inhibitory concentration (4× MIC) of each drug in Figure 4 (0.25 μg mL–1 RIF, 0.5 μg mL–1 INH) and 2× MIC of each drug in Figure 7 (8 μg mL–1 SQ641, 0.25 μg mL–1 INH). The cells were incubated at 37 °C in a 5% CO2 incubator. To dose the soluble drugs, RIF and SQ641 drug stock solutions were prepared in DMSO and INH was prepared in sterile water. The stock solutions were diluted in Hanks’ saline and the final concentrations were achieved by diluting with DMEM just before adding to macrophage cultures. The NC formulations were diluted directly in 2× DMEM. After 1 day of incubation with the drugs, the media was replaced with fresh drug-free medium, and the cells were incubated. On day 7, the media in the wells was removed, and the cells were lysed with 1 mL of 0.1% Triton X-100. From each well, 100 μL volumes, in quadruplicates, were transferred into 96-well plates, and 100 μL of 1% n-decyl aldehyde was dispensed. The relative light units (RLU) were read in a luminometer.27 The incubation time and drug concentrations for the in vitro efficacy comparison of the various formulations were determined to enable sensitive discrimination (Supporting Information) as well as to serve as a relevant model for drug delivery of NCs in circulation, where circulation half-times of 24 h have been reported for NCs formulated by FNP.29,30 The relative efficacies of the formulations were determined using a two-tailed Student’s t test for the difference between the two mean RLU values, with n = 3.31
Figure 4.

In vitro activity of RIF prodrug NC formulations against M. tuberculosis-infected macrophages compared to untreated macrophages and positive controls treated with soluble INH and RIF. Dosing was done at 4× MIC for each active. All anti-TB formulations have significantly better activity than control (p < 0.5), whereas only NC4 has equivalent activity to INH and RIF (p > 0.3). Inset: Particle size distributions for the four RIF NC formulations, detailed in Table 1, prior to freeze drying. RIF-PCL particles (NC1, ■) are significantly larger than the formulations with RIF-VE. As the weight fraction of RIF in the NC core is increased from 0% (NC2, □) to 10% (NC3, ●) and 50% (NC4, ○), relative to RIF-VE, the particle size distribution shifts to smaller sizes. Error bars represent the standard deviation of the mean of n = 3 replicates.
Figure 7.

In vitro activity of the SQ641 NCs and control formulations against M. tuberculosis-infected macrophages. Free SQ641 is less effective in culture than the soluble INH. By solubilizing the SQ641 in stable NCs, greater anti-TB activity is achieved. NC6, CsA, and NC7 do not have significant anti-TB efficacy, compared to the untreated control. All other formulations have significant activity, with NC5, SQ641 + CsA, and NC8 having equivalent activity (see Supporting Information). NC9 had the greatest efficacy of all formulations. Results of the statistical analysis are in the Supporting Information. Inset: Particle size distributions for NC formulations tested in vitro against M. tuberculosis-infected J774A.1 cells: NC5 (■), NC6 (□), NC7 (●), NC8 (○), and NC9 (▲). Error bars represent the standard deviation of the mean of n = 3 replicates.
Results and Discussion
RIF Conjugation, Prodrug Hydrolysis, and NC Formulation
RIF drug conjugates were prepared in order to stabilize the NCs by decreasing solute solubility and provide for controlled release33 via delayed dissolution and cleavage of the ester linkage. The conjugation reaction by DCC coupling was monitored by HPLC, which resolved peaks corresponding to RIF, the prodrugs, and VES, and monosubstitution was confirmed by LC-MS (Supporting Information).
To demonstrate reversibility of the conjugation and recovery of the parent compound under relevant physiological conditions, the RIF-VE prodrug was incubated in water and serum proteins in order to simulate possible in vitro and in vivo degradation routes. After incubation of the RIF-VE product in 1:4 PBS/THF, the fraction of free RIF increased from 9.9 to 46% (Figure 3a) after storage at 4 and 37 °C for 8 days. Due to the instability of serum proteins in organic media and the insolubility of the prodrug in aqueous media, the effect of serum proteins on conjugate degradation could not be assessed directly on the prodrug. NCs containing 1.6 mg mL–1 RIF-VE were incubated with 20% serum. The prodrug was chemically stable over 2 days 4 °C in aqueous media without serum, whereas at 37 °C, free RIF increased from less than 10 to 28% (Figure 3b). There was little water penetration into the NC core at 4 °C and hence low RIF release; however, at 37 °C, increased prodrug solubility results in partitioning into the aqueous media and subsequent hydrolysis. In the presence of serum proteins, the fraction of free RIF increased dramatically over the same time at 37 °C due to the additional mechanism of esterase-catalyzed hydrolysis. These results show that the NCs significantly exclude water while stored in aqueous media under refrigerated conditions, limiting hydrolysis, but when exposed to elevated temperatures and serum proteins, the rate of prodrug hydrolysis is increased, releasing the active drug RIF.
Figure 3.

Conversion of RIF-VE prodrug (gray bars) to free RIF (black bars) stored (a) in soluble form in mixed organic/aqueous media and (b) in encapsulated in NCs in aqueous media, in the presence of 20% serum (n = 2).
The two prodrugs were formulated with 50% prodrug and 50% PLA3.8k-b-PEG5k-OCH3 (Table 1) and gave rise to NCs of average particle sizes of 170 and 60 nm for RIF-PCL and RIF-VE prodrugs, respectively. The particle size distributions were narrow and monodisperse (Figure 4, inset). The significantly different NC particle sizes produced by FNP results from the difference in supersaturation during precipitation between the two formulations. Supersaturation depends on both the solubility of the prodrug and the number density of molecules (or molar concentration) injected at the fixed mass concentrations. For homogeneous precipitation, a higher nucleation rate, B, corresponds to the formation of a greater number of nuclei and smaller particles. The nucleation rate B is given by34
| 1 |
where K1 is a constant, k is Boltzmann’s constant, T is the absolute temperature, γ is the interfacial energy, and υ is the molar volume. In this equation, the supersaturation, S, is defined as the ratio of the concentration of drug after injected in the mixer divided by the equilibrium concentration of drug c∞ in the mixed solvent
| 2 |
The smaller size of RIF-VE NCs is due to a higher nucleation rate, which results when the same mass is distributed over a greater number of nuclei (c is constant). While the supersaturation is used to drive up nucleation rates35 and form NCs,34 it is expected that the supersaturation would be similar in the two systems due to the similarity in hydrophobicity and HPLC elution times at 26–28 min in the chromatograms (Supporting Information). The molar volume, υ, of the precipitating solute is a significant factor in the nucleation rate (eq 1). The molar volume of the RIF-VE prodrug is 3.5 times smaller than RIF-PCL (Supporting Information), which causes a substantial increase in the nucleation rate, relative to that of RIF-PCL, resulting in the significantly smaller particle size. Comparison of particle formation for these two prodrugs demonstrates the importance of hydrophobic anchor choice in determining the NCs’ ultimate physical properties such as particle size and drug loading.
NCs with mixtures of RIF and RIF-VE were prepared at increasing ratios of RIF to RIF-VE. The precursor THF solutions for NC3 and NC4 (Table 1) consisted of ratios of 10:1 and 1:1 of RIF-VE to RIF and a 1:1 ratio of core to the PLA3.8k-b-PEG5k-OCH3 stabilizer. Formulations NC3 and NC4 yielded particles with average particle diameters of 54 and 36 nm (Figure 4, inset) and RIF loadings of 33 and 42%, respectively. As the weight fraction of RIF-VE in the NC core is decreased from 100 to 90 to 50%, the particle size decreased. With two hydrophobic components in the system, the more hydrophobic RIF-VE controls heterogeneous nucleation and particle size distributions. This is similar to seeding in traditional crystallization processes, and hydrophobic macromolecules have been used previously in the place of seed crystals to reduce the activation energy for particle growth, to induce nucleation, and to control the number of nuclei.34 The prodrug, which has lower aqueous solubility and higher supersaturation than RIF, nucleates first and then induces heterogeneous nucleation and growth of the RIF. The resulting particle size decreases as the concentration of prodrug is decreased because the extent of primary nuclei growth prior to block copolymer stabilization is decreased as the prodrug becomes more dilute. Size reduction with a reduction in solute concentration has also been observed for VE NCs at constant block copolymer concentration.36
RIF Prodrug NC Efficacy against M. tuberculosis
The efficacy of the RIF NCs (NC1–NC4) was tested in vitro with J774A.1 cells infected with M. tuberculosis. After reconstitution of the freeze-dried powders (Supporting Information), all drugs were dosed at 4× MIC. RIF conjugated to PCL or VE was included in the calculation of the total RIF dosed; for example, in formulation NC4, of the 0.25 μg mL–1 of RIF dose, 38% of the RIF is conjugated to VES and 62% is free RIF. As a benchmark for efficacy, INH in saline and RIF in DMSO were administered in free form.20 The results are summarized in Figure 4 for the RLU counts after the single-day exposure to the drug, washing, and subsequent 6 day incubation period. The control, without treatment, had a RLU count of 4800. For the cultures exposed to 4× MIC of INH or RIF, there was 93–95% inhibition of growth. Treatment with formulation NC1, with a core of RIF-PCL, and NC2, with a core of RIF-VE, both resulted in a significant RLU reduction compared to that of the untreated control (p = 0.04 and 0.01, respectively). The difference between the two formulations was not statistically significant (p = 0.2). The RLU reduction achieved with 100% RIF-VE is 56%, with 90% RIF-VE, it is 85%, and with 50% RIF-VE, it is 93%. The efficacy of NC formulations NC3 and NC4 increased relative to that of NC2 (p < 0.01). Furthermore, for formulation NC4, 93% RLU reduction was achieved, which was not statistically different from the free RIF or INH (p > 0.3).
There are several significant implications of the data presented in Figure 4 regarding the efficacy of the RIF NC formulations. First, with 50% conjugated RIF and 50% free RIF, the effectiveness is equivalent to the effectiveness of the free RIF administered using DMSO. The efficacy of the dose of RIF in this assay has not changed by NC formulation, but obtaining equivalent efficacy with NC4, where 38% of RIF molecules are conjugated to VE compared to the DMSO RIF formulation, indicates the effectiveness of delivering high-payload nanocarriers at short times rather than relying on passive diffusion through cell membranes. In this context, inclusion of the conjugated RIF in the NC formation process has enabled the formation of NCs that would not be possible with the free drug alone.
Size may also play a role in the observed efficacy of the NC formulations. It is generally reported that smaller particles are more avidly internalized than larger particles since additional uptake pathways are available.37 For the RIF-VE series, the size decreases from 60 to 36 nm as the fraction of free RIF in the NC increases. Some of the greater efficacy observed for the NC formulation with a higher fraction of free RIF, which had an average diameter of 36 nm, may be due to a greater degree of cellular internalization. The RIF-PCL prodrug is less effective than the RIF-VE prodrug in NC form, and the RIF-PCL NCs were 170 nm, whereas the RIF-VE NCs were 60 nm. This trend would be what is expected for the effect of size on NC uptake, but, given the limited number of trials (i.e., 3), the difference between the two datum falls below statistical significance.
Finally, it is important to note that the in vitro assay we have used as an initial test for effectiveness of TB therapeutics fails to account for the potential advantages of extended circulation, delivery, and controlled release from prodrug NCs. The motivation for conjugation is to produce long-lasting delivery to reduce the frequency of administration and to enable higher drug concentrations to be administered without being limited by the maximum tolerated dose (MTD) of free drug. Sustained release of the conjugated RIF may confer extended efficacy to this formulation over times longer than 24 h, but verifying this hypothesis requires additional testing in vivo. This has been observed previously for nanocarrier encapsulation of nitric oxide prodrugs, which increased the circulation half-time and MTD relative to the administration of the free prodrug in vivo.38 Future in vivo animal studies are needed to test the hypothesis that extended release confers these advantages for the RIF prodrug NCs.
SQ641 NC Formulation
To aid in process development of NCs in the MIVM, the solubility of SQ641 was determined in mixed solvents. The volume fraction of THF, ϕ, is defined as the ratio of the volume of THF, QTHF, to the total combined volumes of THF and water, Qwater
| 3 |
The solubilities are plotted in Figure 5a as a function of ϕ and were used to calculate the supersaturation, S, that would be achieved for SQ641 in THF and water (Figure 5b). In the MIVM (Figure 2b), these conditions can be achieved by controlling the relative flow rates of THF and water fed into the mixing chamber. The system supersaturation, S, is calculated as
| 4 |
where cTHF is the concentration of SQ641 in the THF stream and c∞(ϕ) is the equilibrium solubility of SQ641 in the mixed solvent, as measured in Figure 5a. The product ϕcTHF corresponds to the initial concentration of dissolved drug in the system after mixing but before the onset of nucleation and growth. Changes in the density and excess molar volume deviate by 6% or less from ideal mixing at molar ratios of THF, xTHF, from 18 to 1.1%39 and are therefore neglected in this calculation. Values of the supersaturation calculated as a function of the volume fraction of THF, ϕ, are plotted in Figure 5b for cTHF = 10 mg mL–1, 20 mg mL–1, and saturated SQ641 in THF. S increases as the concentration of SQ641 in THF increases. When the concentration of SQ641 is 10 mg mL–1 in THF, the supersaturation is below 2 at ϕ = 0.5, and the supersaturation is not high enough to drive homogeneous nucleation and NC formation. The following process conditions were chosen to prepare NCs: ϕ = 0.1, cTHF = 10 mg mL–1, and S = 10. Working at a lower concentration of SQ641 allows for several formulations to be tested with less than 500 mg of drug; hence, the moderate supersaturation of 10 was chosen.
Figure 5.

(a) Solubility curve for SQ641 versus the volume fraction, ϕ, of THF in a water/THF mixture. The SQ641 solubility at ϕ = 0.1 and ϕ = 0.05 are 0.13 and 0.03 mg mL–1, respectively. (b) Supersaturation of SQ641 obtained by mixing THF solutions that are saturated (■), have 20 mg mL–1 (●), or 10 mg mL–1 (□) SQ641 with water in the MIVM, at different relative flow ratios, yielding the final vol % THF noted on the axis.
NCs with a target drug loading of 50 wt % were prepared using the MIVM to precipitate particles from a THF solution of PLGA8k-b-PEG5k-OCH3 (10 mg mL–1) and SQ641 (10 mg mL–1). Within the first 30 min following FNP, the particles were 70 nm in diameter; however, the drug recrystallized during dialysis and formed macroscopic precipitates (Figure 6a). The suspension was filtered with a 5 μm Teflon filter, and the filtrate was assayed for SQ641 by HPLC. It was found that only 10% of the drug remained solubilized in the NCs after dialysis (Figure 6b).
Figure 6.
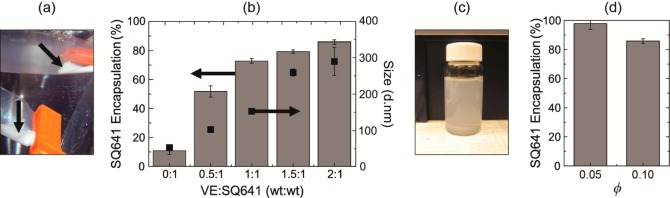
(a) Shortly after NC formation in the MIVM, SQ641 NCs stabilized by PLGA8k-b-PEG5k-OCH3 visibly precipitate in dialysis bags (black arrows) without the use of VE in the formulation. (b) As increasing amounts VE are incorporated in the NC formulation, there is a corresponding increase in drug encapsulation (gray bars, left axis) as well as the NC size (■, right axis). Error bars represent the mean of two replicates. (c) At a ratio of 2:1 VE/SQ641 in the formulation, a drug encapsulation of 86% is reached, but there is no drug recrystallization visibly observed. (d) In the MIVM, the relative flow rates of THF and water streams can be controlled to decrease residual drug solubility. By reducing the final solvent content from 10 to 5% THF, the drug encapsulation efficiency increases to 98%. Error bars correspond to the standard deviation of the mean of n = 2 samples.
To inhibit recrystallization and improve NC stability, VE was added to the formulation to solubilize the drug in the NC core. The concentrations of SQ641 and PLGA8k-b-PEG5k-OCH3 were held constant at 10 mg mL–1 each. As the ratio of VE to SQ641 was increased from 0:1 to 2:1, the fraction of SQ641 remaining in the NC after dialysis and filtration increased to 86% (Figure 6b). The stability of the NCs containing VE was tracked by DLS measurements over a week of storage at 4 °C. In these formulations, the primary particle size did not change (Supporting Information). At a 2:1 ratio of VE to SQ641, for cTHF = 10 mg mL–1 SQ641 and a final concentration in the mixed final aqueous phase of 1 mg/mL, the encapsulation efficiency was 86%, and there was no macroscopic drug recrystallization (Figure 6c). Precipitation of SQ641 during FNP ceases when the supersaturation ratio, S, is 1 (c = c∞) and c∞ = 0.13 mg mL–1 SQ641 in the mixed solvent ϕ = 0.1 (Figure 5a). This soluble fraction of SQ641 was dialyzed away against excess water.
The absolute mass loss during dialysis was determined by the solubility in the mixed solvent and by the volume of the solvent phase. However, the fractional loss was determined by the amount of SQ641 solid NC phase relative to the volume of the solvent phase. Therefore, to further improve the SQ641 encapsulation, the flow rates were changed from the initial protocol to QTHF = 8 mL min–1 and Qwater = 160 mL min–1, where ϕ = 0.05, cTHF = 20 mg mL–1, and S = 30. Due to the 2-fold further dilution of the THF stream by water, the cTHF was increased from 10 to 20 mg mL–1 so that the concentration of SQ641 in the system remained 1 mg mL–1 after mixing. The VE and block copolymer concentrations were also doubled so that the NC composition remained identical. The increased dilution of the THF stream decreases the solubility of the SQ641 in the final mixed solvent 4.4-fold to c∞ = 0.03 mg mL–1. Therefore, after mixing, there was less drug in solution to be lost during dialysis, and the drug encapsulation in NCs was 98% (Figure 6d). The final particle size was reduced from 300 to 150 nm. The reduction in the particle size at the same composition (1:2:1 SQ641/VE/polymer) resulted from the increase in supersaturation, from S = 10 to S = 30.
Formulation of Anti-TB Nanoscale Drug Cocktails
A series of NC formulations (Table 2) was prepared by FNP under sterile conditions in the manually driven CIJ mixer (Figure 2a). NC5 corresponds to the SQ641 NC formulation developed in the previous section, in which the core was composed of a 1:2 ratio of SQ641 to VE. The amount of stabilizer was increased so that the ratio of NC core (SQ641 + VE) to stabilizing polymer was 1:1. The block copolymer used for stabilizing these NCs was PS1.5k-b-PEG5k-OH, since parallel efforts to prepare mannose receptor-targeted NCs for TB drug delivery have been based on this same polymer.17 Under conditions of mixing 20 mg mL–1 SQ641, 40 mg mL–1 VE, and 60 mg mL–1 block copolymer, NCs with diameters of 97 nm were produced (Figure 7, inset). The decrease in size, from 150 nm particles produced in the MIVM, arose from increasing the amount of stabilizer in the formulation as well as the increased hydrophobicity of the PS hydrophobic block relative to the PLGA block.29 It was confirmed that NC5, which was formed in the CIJ and stabilized by PS1.5k-b-PEG5k-OH, had 98 ± 4% encapsulation of SQ641, despite the fact that mixing occurred at a 1:1 ratio of THF to water prior to further dilution in a stirred water reservoir. Nanocarriers with a hydrophobic polymer core were formed as a control (NC6) to establish any apparent in vitro anti-TB activity afforded by the NC structure or stabilizing polymers. This control formulation had an average particle diameter of 170 nm.
The three final NC formulations in Table 2 incorporated CsA, a poorly water-soluble P-gp efflux pump inhibitor.40 NCs with a CsA core and intensity-weighted average size of 135 nm were prepared to evaluate the efficacy of CsA NCs alone when incubated with cells infected with M. tuberculosis (NC7). In formulation NC8, a 1:1 ratio of SQ641 and CsA was encapsulated in the NCs, and the resulting particle size was 189 nm. The encapsulation of SQ641 was 85 ± 5% for this formulation. To improve the encapsulation of SQ641, formulation NC9 consisted of a core that maintained the 1:2 ratio of SQ641 to VE, along with incorporating CsA at a 1:1 ratio with SQ641. Hence, NC9 consists of a combination NC formulation with three hydrophobic components in the core: SQ641, CsA, and VE. The SQ641 encapsulation in NC9 did not improve over NC8, with 88 ± 3% of the SQ641 retained in the NC after dialysis and filtration. The encapsulation of CsA in formulations NC7–NC9 was consistent, with an average yield of ∼90%. Since the encapsulation efficiency for CsA was 90 ± 4% in NC7–NC9 and the encapsulation efficiency for SQ641 was 85 and 88% in NC8 and NC9, respectively, the ratio of SQ641 to CsA remained at 1:1, within experimental error. The particle size distributions for the three SQ641 formulations and the two control formulations are plotted in the inset of Figure 7.
Efficacy of Anti-TB Nanoscale Drug Combinations in Vitro
The efficacies of the NC formulations in Table 2 were evaluated by exposing J774A.1 cells infected with M. tuberculosis to the NC formulation. Drugs were dosed at 2× MIC, and the control NC formulation was dosed at equivalent polymer concentrations, where the core was homopolymer PCL. Drug cocktail NCs were loaded at 1:1 ratios of SQ641 to CsA, so CsA was dosed at 2× MIC of SQ641 in DMSO and in pure CsA NCs. The efficacy of all drugs and NC formulations was compared to a control sample, to which no drug was dosed.
In Figure 7, the measured RLU corresponding to the number of viable M. tuberculosis is plotted for each formulation tested. There was a significant reduction in TB proliferation at 2× MIC by INH (p = 0.028), with 60% RLU reduction, and SQ641 in DMSO (p = 0.054) produced 50% reduction. The proportion of viable M. tuberculosis quantified after exposure to the PCL NC control formulation was not significantly different from that of the untreated control (p = 0.6). Alone, there was no significant anti-TB activity for free CsA (DMSO) (p < 0.8) and NC7 (CsA) (p = 0.2) compared to control. Compared to SQ641 (DMSO), the activity of SQ641 was significantly enhanced in the presence of CsA, for both soluble SQ641 + CsA (DMSO) and for NC8 (SQ641 + CsA) doses (p < 0.01). There is no statistical difference in the activities of drug cocktails SQ641 + CsA (DMSO) and NC8 (SQ641 + CsA) and NC5 (SQ641 + VE) (p > 0.1). However, the formulation with SQ641 and the efflux pump blocker in the VE core, NC9 (SQ641 + CsA + VE), is the most efficacious formulation and is significantly better than all other SQ641 nanocarrier formulations (p < 0.04).
The RLU reduction achieved by the NC9 drug cocktail is 3.5 times higher than the SQ641 + CsA NC and 5 times more effective than the SQ641 + CsA administered in DMSO. It is important to note that while DMSO is a useful in vitro control it is not a viable delivery vehicle in vivo. The formulated SQ641 NCs outperformed the free form of SQ641 (p < 0.01) arising from the high payload internalized per particle (17 wt %) and the high intracellular concentrations that accumulate during the 1 day incubation. Since the RLU reduction observed after exposure to the drug free formulation (NC6) was insignificant, the improved efficacy of the NC5 SQ641 formulations is not due to the stabilizing polymers or NC toxicity to the cells. In addition, NC5, which is formulated with SQ641 and VE to improve colloidal stability, had better activity in vitro compared to INH.
To improve SQ641 intracellular accumulation, we also prepared formulations in order to codeliver SQ641 and CsA to enhance the activity of the drug by inhibiting drug efflux, a known limitation to SQ641 efficacy. Alone, CsA can enhance the antimycobacterial activity by inhibition of K+ pumps in the phagolysosome in macrophages, which are dependent on acidification and hydrolase activity inside of phagolysosomes and are inhibited by efflux pump inhibitors.41 In determining the appropriate incubation time and concentration conditions for these in vitro experiments, it was found that there was anti-TB activity for a 4 day incubation time with CsA alone, dosed in DMSO, but not for a 1 day incubation (Supporting Information). Therefore, soluble SQ641 + CsA in DMSO has improved anti-TB activity compared to SQ641 in DMSO, which is not due to activity of CsA alone but is specifically because of the combination of CsA with SQ641. In this case, the intracellular accumulation of SQ641 alone is limited due to efflux of drug from the cell, and the CsA efflux inhibition helps to maintain therapeutic levels. However, in NC form, there is no statistically significant difference in RLU reduction for SQ641 + VE (NC5) and SQ641 + CsA (NC8). With a calculated log P > 9, VE is highly lipophilic and helps to retain SQ641 in the NC core (NC5, Figure 6). In contrast, with a log P of 3.6, CsA is less lipophilic and less efficient in retaining SQ641 in the core (NC8). Even though we could prepare stable NCs with SQ641-CsA (NC8), upon dilution in culture medium and exposure to tissues, NC5 provides a more effective SQ641 reservoir, resulting in higher intracellular accumulation of the drug. In order to benefit from the inhibitory effect of CsA on P-gp mediated efflux of SQ641, both SQ641 and CsA must be colocated in the same cell. The less polar core of NC8 does not sufficiently retain the two drugs over the 24 h incubation period, and the inclusion of CsA in the NC8 formulation did not enhance the therapeutic effect. However, a ternary core of SQ641 + CsA + VE (NC9) results in a 3-fold enhanced RLU reduction compared to the SQ641 + VE or SQ641 + CsA NCs. It is unlikely that the VE in NC5 and NC9 is actively enhancing the activity, since VE is an antioxidant that can have an adverse effect on antibacterial peroxides and nitric oxide in the phagolysosome.42 Here, again, VE significantly decreases the polarity of the NC core and retains the two drugs in the NC through the incubation time, resulting in colocalization of the two drugs intracellularly and enhanced drug efficacy.
In previous studies where paclitaxel prodrugs were synthesized to stabilize NC formulations prepared by FNP, reduced drug in vitro activity was also observed,30 but equivalent or superior in vivo tumor size reduction by paclitaxel prodrugs has been reported.30,43 Future in vivo experiments will address this hypothesis. Therefore, this work provides preliminary evidence that NC formulations can improve the efficiency of drug delivery and may ultimately reduce the frequency of drug administered.
Conclusions
While effective therapies for bacterial infections such as tuberculosis (TB) exist, the success rate of therapy remains problematic, and the incidence of drug resistant strains poses a significant threat of increasing the global spread and mortality. In this study, the focus was on developing NCs to deliver and enhance the efficacy of both existing and novel anti-TB drugs. A new approach to controlling NC formation in flash nanoprecipitation (FNP) was demonstrated, using hydrophobic prodrugs to control nucleation and facilitate the precipitation of stable NCs loaded with RIF, with sizes from 34 to 170 nm. The NCs retained efficacy against M. tuberculosis in vitro, and NC formation may limit systemic distribution prior to payload delivery by sustained release in vivo. This work demonstrates the use of hydrophobic, cleavable prodrugs for use in therapeutic formulations and adds a dimension to their utility in showing their role in controlling NC formation. With the increasing challenge of MDR-TB, novel TB agents such as SQ641 are required. NC formulation of SQ641 with amphiphilic diblock copolymers resulted in higher intracellular activity than that of the free drug, but it did not achieve statistical significance. SQ641 was also formulated into stable suspensions with CsA, a P-gp efflux pump inhibitor. Co-formulation of SQ641, CsA, and VE resulted in the most potent formulation and presents an interesting avenue for further investigation of the role of the core matrix in NC efficacy in vitro. The potential to control release and limit systemic distribution of chemotherapeutic anti-TB drugs warrants further investigation of in vivo efficacy. Crucial factors for in vivo delivery include determining the clearance mechanisms of both the vehicle and the drug, both the prodrug release and cleavage rates, and potential vehicle accumulation as well as significantly improving the therapy’s efficacy over that of current therapies.
Acknowledgments
This work was supported by award no. R44AI066442 from the National Institute of Allergy and Infectious Diseases.
Supporting Information Available
Additional information on the prodrug synthesis and characterization, RIF NC redispersion, HPLC conditions, SQ641 NC stability, in vitro testing conditions, and significance testing. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
∥ (S.M.D.) Formulation Sciences, Merck & Co., Kenilworth, New Jersey 07033, United States.
Author Present Address
⊥ (Y.L.) Department of Chemical Engineering, University of Illinois at Chicago, Chicago, Illinois 60607, United States.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Global Tuberculosis Report; World Health Organization: Geneva, Switzerland, 2013.
- Schutz I. A comparison of rifampicin, ethambutol and PAS in short-term monotherapy. Preliminary results of the third cooperative study of the WATL. Acta Tuberc. Pneumol. Belg. 1969, 60, 437–41. [PubMed] [Google Scholar]
- Jindani A.; Aber V. R.; Edwards E. A.; Mitchison D. A. The early bactericidal activity of drugs in patients with pulmonary tuberculosis. Am. Rev. Respir. Dis. 1980, 121, 939–949. [DOI] [PubMed] [Google Scholar]
- Yoder M. A.; Lamichhane G.; Bishai W. R. Cavitary pulmonary tuberculosis: the Holy Grail of disease transmission. Curr. Sci. 2004, 86, 74–81. [Google Scholar]
- El-Sadr W. M.; Perlman D. C.; Denning E.; Matts J. P.; Cohn D. L. A review of efficacy studies of 6-month short-course therapy for tuberculosis among patients infected with human immunodeficiency virus: differences in study outcomes. Clin. Infect. Dis. 2001, 32, 623–632. [DOI] [PubMed] [Google Scholar]
- Pandey R.; Zahoor A.; Sharma S.; Khuller G. K. Nanoparticle encapsulated antitubercular drugs as a potential oral drug delivery system against murine tuberculosis. Tuberculosis 2003, 83, 373–378. [DOI] [PubMed] [Google Scholar]
- Gelperina S.; Kisich K.; Iseman M. D.; Heifets L. The potential advantages of nanoparticle drug delivery systems in chemotherapy of tuberculosis. Am. J. Resp. Crit. Care Med. 2005, 172, 1487–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azarmi S.; Roa W. H.; Loebenberg R. Targeted delivery of nanoparticles for the treatment of lung diseases. Adv. Drug Delivery Rev. 2008, 60, 863–875. [DOI] [PubMed] [Google Scholar]
- Yee D.; Valiquette C.; Pelletier M.; Parisien I.; Rocher I.; Menzies D. Incidence of serious side effects from first-line antituberculosis drugs among patients treated for active tuberculosis. Am. J. Resp. Crit. Care Med. 2003, 167, 1472–1477. [DOI] [PubMed] [Google Scholar]
- Blasi P.; Schoubben A.; Giovagnoli S.; Rossi C.; Ricci M. Fighting tuberculosis: old drugs, new formulations. Expert Opin. Drug Delivery 2009, 6, 977–993. [DOI] [PubMed] [Google Scholar]
- Koul A.; Arnoult E.; Lounis N.; Guillemont J.; Andries K. The challenge of new drug discovery for tuberculosis. Nature 2011, 469, 483–490. [DOI] [PubMed] [Google Scholar]
- Palomino J. C.; Ramos D. F.; da Silva P. A. New anti-tuberculosis drugs: strategies, sources and new molecules. Curr. Med. Chem. 2009, 16, 1898–1904. [DOI] [PubMed] [Google Scholar]
- Kaufmann S. H. E. How can immunology contribute to the control of tuberculosis?. Nat. Rev. Immunol. 2001, 1, 20–30. [DOI] [PubMed] [Google Scholar]
- Anisimova Y. V.; Gelperina S. I.; Peloquin C. A.; Heifets L. B. Nanoparticles as antituberculosis drugs carriers: effect on activity against Mycobacterium tuberculosis in human monocyte-derived macrophages. J. Nanopart. Res. 2000, 2, 165–171. [Google Scholar]
- Muttil P.; Wang C. C.; Hickey A. J. Inhaled drug delivery for tuberculosis therapy. Pharm. Res. 2009, 26, 2401–2416. [DOI] [PubMed] [Google Scholar]
- Pandey R.; Ahmad Z. Nanomedicine and experimental tuberculosis: facts, flaws, and future. Nanomedicine 2011, 7, 259–272. [DOI] [PubMed] [Google Scholar]
- D’Addio S. M.; Baldassano S.; Shi L.; Cheung L.; Adamson D. H.; Bruzek M.; Anthony J. E.; Laskin D. L.; Sinko P. J.; Prud’homme R. K. Optimization of cell receptor-specific targeting through multivalent surface decoration of polymeric nanocarriers. J. Controlled Release 2012, 168, 41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuberculosis. Current Topics in Microbiology and Immunology; Shinnick T. M., Ed.; Springer-Verlag: New York, 1996. [Google Scholar]
- Reddy V. M.; Bogatcheva E.; Protopopova M.. Enhancement of Intracellular Activity of Capuramycin (CM) Analogue SQ641 against M. tuberculosis (MTB), abstract C1-3851, 48th Interscience Conference on Antimicrobial Agents and Chemotherapy, Washington, DC, October 24–28, 2008.
- Reddy V. M.; Einck L.; Nacy C. A. In vitro antimycobacterial activities of capuramycin analogues. Antimicrob. Agents Chemother. 2008, 52, 719–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikonenko B. V.; Reddy V. M.; Protopopova M.; Bogatcheva E.; Einck L.; Nacy C. A. Activity of SQ641, a capuramycin analog, in a murine model of tuberculosis. Antimicrob. Agents Chemother. 2009, 53, 3138–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian H. T.; Wohl A. R.; Crow J. T.; Macosko C. W.; Hoye T. R. A strategy for control of “random”copolymerization of lactide and glycolide: application to synthesis of PEG-b-PLGA block polymers having narrow dispersity. Macromolecules 2011, 44, 7132–7140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S. Y.; Adamson D. H.; Prud’homme R. K.; Link A. J. Photocrosslinking the polystyrene core of block-copolymer nanoparticles. Polym. Chem. 2011, 2, 665–671. [Google Scholar]
- Han J.; Zhu Z.; Qian H.; Wohl A. R.; Beaman C. J.; Hoye T. R.; Macosko C. W. A simple confined impingement jets mixer for flash nanoprecipitation. J. Pharm. Sci. 2012, 10, 4018–4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson B. K.; Prud’homme R. K.. Engineering the Direct Precipitation of Stabilized Organic and Block Copolymer Nanoparticles as Unique Composites, The 226th ACS National Meeting, New York, NY, September 7–11, 2003.
- Liu Y.; Cheng C. Y.; Liu Y.; Prud’homme R. K.; Fox R. O. Mixing in a multi-inlet vortex mixer (MIVM) for flash nano-precipitation. Chem. Eng. Sci. 2008, 63, 2829–2842. [Google Scholar]
- Snewin V. A.; Gares M. P.; Gaora P. O.; Hasan Z.; Brown I. N.; Young D. B. Assessment of immunity to mycobacterial infection with luciferase reporter constructs. Infect. Immun. 1999, 67, 4586–4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna-Herrera J.; Reddy M. V.; Gangadharam P. R. J. In vitro activity of the benzoxazinorifamycin KRM-1648 against drug-susceptible and multidrug-resistant tubercle bacilli. Antimicrob. Agents Chemother. 1995, 39, 440–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Addio S. M.; Saad W.; Ansell S. M.; Squiers J. J.; Adamson D. H.; Herrera-Alonso M.; Wohl A. R.; Hoye T. R.; Macosko C. W.; Mayer L. D.; Vauthier C.; Prud’homme R. K. Effects of block copolymer properties on nanocarrier protection from in vivo clearance. J. Controlled Release 2012, 162, 208–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansell S. M.; Johnstone S. A.; Tardi P. G.; Lo L.; Xie S. W.; Shu Y.; Harasym T. O.; Harasym N. L.; Williams L.; Bermudes D.; Liboiron B. D.; Saad W.; Prud’homme R. K.; Mayer L. D. Modulating the therapeutic activity of nanoparticle delivered paclitaxel by manipulating the hydrophobicity of prodrug conjugates. J. Med. Chem. 2008, 51, 3288–3296. [DOI] [PubMed] [Google Scholar]
- Navidi W. C.Statistics for Engineers and Scientists, 1st ed.; McGraw Hill Higher Education: New York, 2006. [Google Scholar]
- Ansell S. M.; Johnstone S. A.; Tardi P. G.; Lo L.; Xie S.; Shu Y.; Harasym T. O.; Harasym N. L.; Williams L.; Bermudes D.; Liboiron B. D.; Saad W.; Prud’homme R. K.; Mayer L. D. Modulating the therapeutic activity of nanoparticle delivered paclitaxel by manipulating the hydrophobicity of prodrug conjugates. J. Med. Chem. 2008, 51, 3288–96. [DOI] [PubMed] [Google Scholar]
- D’Addio S. M.; Prud’homme R. K. Controlling drug nanoparticle formation by rapid precipitation. Adv. Drug Delivery Rev. 2011, 63, 417–426. [DOI] [PubMed] [Google Scholar]
- Mahajan A. J.; Kirwan D. J. Nucleation and growth kinetics of biochemicals measured at high supersaturations. J. Cryst. Growth 1994, 144, 281–290. [Google Scholar]
- Akbulut M.; Ginart P.; Gindy M. E.; Theriault C.; Chin K. H.; Soboyejo W.; Prud’homme R. K. Generic method of preparing multifunctional fluorescent nanoparticles using flash nanoprecipitation. Adv. Funct. Mater. 2009, 19, 718–725. [Google Scholar]
- Gratton S. E. A.; Ropp P. A.; Pohlhaus P. D.; Luft J. C.; Madden V. J.; Napier M. E.; DeSimone J. M. The effect of particle design on cellular internalization pathways. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 11613–11618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V.; Hong S. Y.; Maciag A. E.; Saavedra J. E.; Adamson D. H.; Prud’homme R. K.; Keefer L. K.; Chakrapani H. Stabilization of the nitric oxide (NO) prodrugs and anticancer leads, PABA/NO and double JS-K, through incorporation into PEG-protected nanoparticles. Mol. Pharmaceutics 2010, 7, 291–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belandria V.; Mohammadi A. H.; Richon D. Volumetric properties of the (tetrahydrofuran plus water) and (tetra-n-butyl ammonium bromide plus water) systems: Experimental measurements and correlations. J. Chem. Thermodyn. 2009, 41, 1382–1386. [Google Scholar]
- Liow J. S.; Lu S. Y.; McCarron J. A.; Hong J. S.; Musachio J. L.; Pike V. W.; Innis R. B.; Zoghbi S. S. Effect of a P-glycoprotein inhibitor, cyclosporin A, on the disposition in rodent brain and blood of the 5-HT1A receptor radioligand, [C-11](R)-(−)-RWAY. Synapse 2007, 61, 96–105. [DOI] [PubMed] [Google Scholar]
- Amaral L.; Martins M.; Viveiros M. Enhanced killing of intracellular multidrug-resistant Mycobacterium tuberculosis by compounds that affect the activity of efflux pumps. Antimicrob. Agents Chemother. 2007, 59, 1237–1246. [DOI] [PubMed] [Google Scholar]
- Rockett K. A.; Brookes R.; Udalova I.; Vidal V.; Hill A. V. S.; Kwiatkowski D. 1,25-Dihydroxyvitamin D-3 induces nitric oxide synthase and suppresses growth of Mycobacterium tuberculosis in a human macrophage-like cell line. Infect. Immun. 1998, 66, 5314–5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohl A. R.; Han J.; Guru B. R.; Panyam J.; Hoye T. R.; Macosko C. W. In Silicate Prodrug-Loaded Nanoparticles, AIChE Annual Meeting, Minneapolis, MN, October 16–21, 2011.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.