

Abstract
Peptide tags fused to proteins are used in a variety of applications, including as affinity tags for purification, epitope tags for immunodetection, or fluorescent protein tags for visualization. However, the peptide tags can disrupt the target protein function. When function is disrupted by fusing a peptide to either the N or C terminus of the protein of interest, identifying alternative ways to create functional tagged fusion proteins can be difficult. Here, we describe a method to introduce protein tags internal to the coding sequence of a target protein. The method employs in vitro Tn7-transposon mutagenesis of plasmids for random introduction of the tag, followed by subsequent Gateway cloning steps to isolate alleles with mutations in the coding sequence of the target gene. The Tn7-epitope cassette is designed such that essentially all of the transposon is removed through restriction enzyme digestion, leaving only the protein tag at diverse sites internal to the ORF. We describe the use of this system to generate a panel of internally epitope-tagged versions of the Saccharomyces cerevisiae GPI-linked membrane protein Dcw1 and the Candida glabrata transcriptional regulator Sir3. This internal protein tagging system is, in principle, adaptable to tag proteins in any organism for which Gateway-adapted expression vectors exist.
Keywords: Saccharomyces, epitope tag, protein tagging, transposition
RECOMBINANT fusion proteins are essential tools in molecular studies across all organisms. Fusing fluorescent proteins to a protein of interest (POI) allows for direct visualization in situ. Fusing peptide epitopes, for which antibodies have been developed, to the POI is fundamental to many techniques, including Western blots, immunofluorescence, co-immunoprecipitation, chromatin immunoprecipitation, and purification (Arnau et al. 2006; Young et al. 2012; Bell et al. 2013).
We describe an approach to add a protein or peptide epitope to a POI. One challenge to epitope tagging is choosing the location to attach the epitope to the POI. The standard design appends the epitope to the N- or C-terminus of the POI. However, in some instances, proteins cannot be tagged at the N or C terminus because a tag interferes with function, disrupting, for example, trafficking or post-translational modification. Tags can also interfere with protein folding or structure and disrupt protein–protein interactions.
Our efforts to study the localization and function of Dcw1 in Saccharomyces cerevisiae have been hampered because we cannot introduce a tag at either terminus. Dcw1 and its paralog Dfg5 are important in cell-wall structure and integrity in S. cerevisiae and other fungi (Kitagaki et al. 2002, 2004; Gonzalez et al. 2010; Maddi et al. 2012). Traditional methods of N- or C-terminally tagging Dcw1 are expected to fail due to interference with protein localization because Dcw1 localization at the cell membrane requires an N-terminal secretory signal sequence and a C-terminal signal sequence for addition of a glycosylphosphatidylinositol (GPI) anchor. Modeled after published constructs (Kitagaki et al. 2002), we initially engineered an HA-tag located at residue 26 in DCW1 (Dcw1-HA26), just downstream of the N-terminal signal sequence, but found that this tagged protein, while viable, is only partially functional, conferring a temperature-sensitive phenotype. This motivated the development of a transposon-based system to introduce epitope tags throughout DCW1; the resulting library of epitope (3× FLAG)-tagged alleles of DCW1 was then screened for function, permitting the isolation of functional internally tagged alleles of DCW1. To demonstrate the generality of the method, we mutagenized a second gene, SIR3 from Candida glabrata. SIR3 is not an essential gene but is absolutely required for subtelomeric transcriptional silencing in C. glabrata (De Las Penas et al. 2003). We used the transposon-based method to internally tag CgSIR3 with four different protein tags, including the fluorescent proteins GFP and mEOS2.
Transposons have been used as tools to introduce epitope tags and fluorescent proteins randomly into targets (Merkulov and Boeke 1998; Ross-Macdonald et al. 1999; Manoil and Traxler 2000; Kumar et al. 2002; Sheridan et al. 2002; Kumar et al. 2004; Osawa and Erickson 2005). In some cases, the transposons are used to identify regions of the target POI into which a tag may be inserted without disrupting target function, requiring later cloning steps to insert the epitope tag at permissive sites (Spreghini et al. 2003). In a recently described system, TAGIT, Tn5 transposition is used to introduce cassettes containing epitope tags internally into target genes (Gregory et al. 2010). Resulting insertions are screened to identify in-frame fusions in the target gene. Excision of the bulk of the transposon is done in vivo, using Cre recombinase.
We describe a method, related to TAGIT, using transposon Tn7 and in vitro mutagenesis to introduce epitopes into two different open reading frames (ORFs), generating functional internally tagged alleles. Tn7 mini transposons have been shown to have very little sequence bias, making them ideal tools for random mutagenesis (Biery et al. 2000; Seringhaus et al. 2006; Green et al. 2012). Our plasmid-based epitope-tagging system generates large libraries of internally tagged ORFs, which can be screened for function to identify useful fusion proteins.
Materials and Methods
Plasmids used in this study are listed in Supporting Information, Table S1. DNA primers are listed in Table S2.
Media
Escherichia coli was routinely grown at 37° in LB media containing appropriate antibiotics for selection. For any media including trimethoprim, Oxoid Isosensitest media was used instead of LB. Carbenicillin, not ampicillin, was used to select for plasmids marked with the ampicillin-resistance gene. Antibiotics were added at the following final concentrations: carbenicillin (Car; 100 μg/ml), kanamycin (Kan; 30 μg/ml), and trimethoprim (Tmp; 10 μg/ml). Solid media for E. coli growth was supplemented with 1.5% agar.
S. cerevisiae and C. glabrata strains were typically grown at 30° on YPD media (10 g/liter yeast extract, 20 g/liter peptone, 2% dextrose). All solid yeast media contained 2% agar. To maintain His- and Ura-marked plasmids, SD-His (1.7 g/liter yeast nitrogen base without amino acids or ammonium sulfate, 5 g/liter ammonium sulfate, 1.92 g/liter SC-His amino acid mixture, 2% dextrose) or SD-Ura media (1.7 g/liter yeast nitrogen base without amino acids or ammonium sulfate, 5 g/liter ammonium sulfate, 6 g/liter casamino acids, 2% dextrose) were used. To maintain plasmids with a nourseothricin (NAT) marker, YPD was supplemented with 50 μg/ml NAT in liquid media or 100 μg/ml in solid media. To select against Ura-marked plasmids, 5-FOA media (1.7 g/liter yeast nitrogen base without amino acids or ammonium sulfate, 5 g/liter ammonium sulfate, 6 g/liter casamino acids, 25 mg/liter uracil, 1 g/liter 5-FOA, 2% dextrose) was used.
Strains and transformation
All S. cerevisiae strains used in this study are listed in Table S3; all C. glabrata strains are listed in Table S4. Standard lithium acetate transformation protocols were used (Hill et al. 1991). DH10 E. coli cells were used for routine cloning. Strain BW23473 was used to maintain the Tn7 donor plasmids, which have a R6Kγ origin (Metcalf et al. 1996). DB3.1 (Life Technologies) was used to propagate Gateway destination vectors containing the ccdB cassette. Highly competent MegaX DH10B T1R cells (Life Technologies) were used to maintain large libraries of isolates throughout the mutagenesis procedure.
BY240 strain construction
DCW1 was deleted in a clean two-step loopout from the dfg5ΔG strain from the yeast knockout collection (Winzeler et al. 1999). Because DCW1 and DFG5 are synthetically lethal (Kitagaki et al. 2002), the dfg5ΔG yeast strain was transformed with a His-marked plasmid carrying a wild-type copy of DCW1, prior to deletion of DCW1. The intergenic regions immediately flanking DCW1 were amplified from genomic DNA using primers 1975–1978. The resulting fragments were cloned into YIPlac211 (URA3-marked). YIPlac211-DCW1 was linearized with a KpnI digest (separating the 5′ and 3′ flanking regions) and integrated at the DCW1 genomic locus. YIPlac211-DCW1 was replaced with a clean deletion construct, created by amplifying the flanking regions from YIPlac211 with primers 1975 and 1978. Clean deletions were identified by counterselection against the URA3 marker in YIPlac211-DCW1. This completely removes the DCW1 ORF from the genomic locus and leaves a KpnI scar in its place. A plasmid shuffle replaces the DCW1(His) plasmid with pCU-DCW1, creating BY240.
Tn7-FLAG donor plasmid construction
The transposon FLAG donor plasmid is pRZ101, which carries Tn7-FLAG and is based on the suicide plasmid backbone pJP5603 (Penfold and Pemberton 1992), which contains the R6Kγ origin of replication (ORI). We modified pJP5603 by removing the XbaI site from the polylinker by treatment with Klenow and religation. Overall, the transposon was assembled modularly in other vector backbones and then subcloned into the modified pJP5603 backbone. Tn7L was amplified as a 236-bp fragment from pIC6 (Castano et al. 2003) using primers 3201 and 3202. This PCR product introduces an FseI site at the distal end of Tn7L and was cloned as a BamHI-AscI fragment. The FLAG epitope is a 3× FLAG tag flanked by flexible linkers. It was synthesized by DNA2.0 and is flanked by AscI and XbaI restriction sites, allowing the FLAG tag to be easily subcloned into the Tn7 donor backbone. The dhfr gene was amplified from pAT-2 (Devine and Boeke 1994) cloned as an XbaI-PstI fragment. The Tn7R end is derived from Tn7R1-70* in pMCB64 (Biery et al. 2000) (which includes a PmeI site at the distal Tn7R end) and was synthesized as a PstI-EcoRI fragment (DNA2.0).
Other versions of the Tn7-tag donor vector were also constructed.The HA, biotinylation target sequence (bio), 6× His, and 3× myc epitope tags were all synthesized by DNA2.0. The mEOS2 epitope tag was PCR-amplified from pET28-ftsZ-mEOS (a gift of Jie Xiao) using primers 4724 and 4725. The GFP tag was PCR-amplified from pGRB2.3 (Zordan et al. 2013) using oligos 5300 and 5301 and then subcloned into the DNA2.0 backbone to position the GFP tag between AscI and XbaI sites in the backbone. All tags were subcloned into the Tn7-tag donor backbone (as described for pRZ101) with the AscI and XbaI sites flanking the epitope tag.
The following sequences for the Tn7-tag donor vectors are available from GenBank: pRZ49 = Tn7-mEOS2 (accession no. KP698385), pRZ98 = Tn7-biotin (accession no. KP698386), pRZ99 = Tn7-6His (accession no. KP698387), pRZ101 = Tn7-FLAG (accession no. KJ939358), pRA102 = Tn7-HA (accession no. KP698388), pRZ103 = Tn7-myc (accession no. KP698389), and pRZ106 = Tn7-GFP (accession no. KP698390).
DCW1 plasmid construction
pCU-DCW1:
This plasmid is used in BY240 to cover the synthetic lethality between DCW1 and DFG5 gene deletions. It is derived from p416GPD (Mumberg et al. 1995), containing the TDH3 (GPD) promoter, CYC1 transcription terminator, and CEN/ARS and URA3 markers for maintenance and selection in S. cerevisiae. The DCW1 ORF was PCR-amplified from S. cerevisiae genomic DNA using primers 1629 and 1630 and subcloned into p416GPD using BamHI and EcoRI.
DCW1-DONR vector (target):
The DCW1 gene was amplified from genomic DNA using oligos that appended standard attB1 (5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTA-3′) and attB2 (5′-GGGGACCACTTTGTACAAGAAAGCTGGGTA-3′) sites onto the forward and reverse oligos, respectively. The product was recombined into a Gateway entry vector pDONR201 using a standard Gateway BP clonase II reaction. The sequence of the plasmid was confirmed.
DCW1-destination vector:
The entire intergenic region upstream of the DCW1 ORF was PCR-amplified using primers 6333 and 6334 and cloned into a ccdB-containing backbone using SacI and XbaI restriction sites present in the primers. A SacI-XhoI digest was used to subclone the promoter and ccdB region into the p413GPD backbone (Mumberg et al. 1995), which contains the CYC1 transcriptional terminator and a HIS1 auxotrophic marker for selection in yeast.
Wild-type DCW1 expression vector:
Gateway LR clonase II (Invitrogen protocols) was used to recombine the DCW1-DONR vector and DCW1-destination vector, creating a wild-type, untagged version of the DCW1 expression vector pRZ160.
Dcw1-HA26 vector:
This vector is a derivative of pRZ160 with an HA tag inserted at amino acid position 26.
Verify DCW1 expression vector functions
Before beginning mutagenesis, it was important to test whether the destination vector and target gene would function in our system. We sequence-verified the wild-type DCW1 expression vector pRZ160 and then transformed it into BY240 and selected for growth on SC-His media (selecting for pRZ160). The resulting strain was grown on 5-FOA to verify that the strain could lose the original pCU-DCW1 plasmid and survive with the new DCW1(His) plasmid, pRZ160. This confirms that the DCW1 promoter and DCW1 ORF functioned in S. cerevisiae and would be suitable substrates for mutagenesis.
Transposition reaction and processing pool
A detailed transposition and processing protocol is in File S1. In short, TnsA, TnsB, and TnsCA225V enzymes (purified as described in Gamas and Craig 1992 and Choi et al. 2013) were used in an in vitro reaction, mobilizing the Tn7-tag cassette from a Tn7 donor vector into a target vector containing the DCW1 ORF or the CgSIR3 ORF. The mutagenized plasmids were recovered by transforming into E. coli MegaX cells and selecting for appropriate drug resistance. The DCW1 ORF was mobilized using Gateway recombination enzymes in a dedicated DCW1 destination vector, creating a mutagenized DCW1 expression pool; likewise, following mutagenesis, the CgSIR3 ORF was mobilized into a corresponding SIR3 destination vector. These expression pools were recovered in E. coli MegaX cells and selected in sequential rounds of Car and Tmp selection. The left end of Tn7 was removed by FseI digestion; the right end of Tn7 and the TmpR marker were removed by PmeI digestion.
Screening and analysis of Dcw1-FLAG alleles in S. cerevisiae
The final DCW1-FLAG plasmid pools were transformed into S. cerevisiae and grown on SD-His plates at 30° for 2 days. Transformants on the SD-His plates were replica-plated to 5-FOA plates and grown at 37° for 1 day and then re-replica-plated to 5-FOA plates and grown for 1 day at 37°. Only cells with functional DCW1-FLAG alleles will grow; those with nonfunctional DCW1-FLAG alleles will die on 5-FOA because of counterselection against the pCU-DCW1 plasmid.
We performed colony PCR on functional FOAR transformants to qualitatively determine where the FLAG was inserted within the DCW1 ORF. The PCR to determine FLAG insertion position used primers 2766 and 6244 or primers 5160 and M13F. We also performed PCR with 2766 and a primer (5626) that reads out of Tn7L; any isolates that gave a PCR product were eliminated from further study. A subset of isolates predicted to have different sites of insertion (based on PCR product size) were sequenced with primer 6244 to identify the exact placement of the FLAG tag. We screened 276 isolates by colony PCR, sent 48 isolates for sequencing, and found 10 unique DCW-FLAG alleles with internal insertions.
For the 10 unique isolates, functional DCW1-FLAG plasmids were isolated from S. cerevisiae and recovered by transformation into E. coli (Hoffman 2001). The plasmids were individually retransformed into BY240, and their growth phenotype was confirmed. These 10 clean functional strains were used for subsequent growth analysis and for Western blots. The 10 clean DCW1-FLAG strains, the DCW1-HA strain, and control strains were streaked onto YPD, and complementation was tested again on 5-FOA (at 39°, 37°, and 30°), SC-Ura, SC-His, and YPD plates at 30°. After growth for 2 days, pictures were taken on an Alpha Imager and growth was compared (Figure 4).
Figure 4.

Select DCW1-FLAG isolates rescue synthetic lethality of dcw1Δ dfg5Δ mutants. Each yeast strain was grown on 5-FOA at three temperatures to test for Dcw1 function. Strains were also grown on YPD to verify overall viability. The positions of each strain are shown in the schematic at the top. Three control strains, labeled here as N, C, and WT, were streaked onto each experimental plate. Strain “N” is a negative control: the parental strain BY240, a dcw1Δ dfg5Δ pCU-DCW1 strain derived from BY4742. Strain “V” is another negative control: BY240 transformed with an empty (His) vector, pRZ159. The “WT” strain is a positive control: strain BY240 transformed with a wild-type DCW1 (His) plasmid. BY4741 and BY4742 are DCW1 DFG1 ura3Δ strains used to derive other strains. dfg5Δ and dcw1Δ are deletion mutants derived from BY4741 from the yeast knockout collection (Winzeler et al. 1999). The strain BY240 transformed with a DCW1-HA26 (His) plasmid is in the position labeled “HA26.” The remaining strains are BY240 transformed with various DCW1-FLAG (His) plasmids and labeled to indicate the amino acid position of the FLAG insertion.
Construction of C. glabrata SIR3 Gateway vectors
The C. glabrata SIR3 destination vector was constructed by PCR-amplifying the regions flanking the SIR3 ORF from strain CBS138 (Dujon et al. 2004). Primers 3220 and 3221 were used to amplify the 5′ flanking region; primers 3222 and 3223 were used to amplify the 3′ flanking region. These PCR products were subcloned into a Gateway backbone on either side of a ccdB cassette, using the restriction sites present in the primers. This Gateway backbone contains replication origins and and an AmpR cassette for selection in E. coli, as well as a CEN/ARS and NatR marker for maintenance and selection in C. glabrata. The SIR3 entry vector was created by PCR-amplifying the SIR3 ORF from C. glabrata strain BG2 (Cormack and Falkow 1999) using primers 4470 and 4484 and subsequently introducing the ORF into the Gateway backbone pDONR201 using a Gateway BP reaction (Life Technologies). We note that there are three polymorphisms in the SIR3 ORF, relative to the CBS138 sequence, but these do not affect function, as a wild-type SIR3 expression vector generated from this ORF allele (pRZ47) complements a sir3Δ defect in C. glabrata. Schematic drawings of the SIR3 entry vector and destination vector are shown in Figure S2.
Microscopy of Sir3-GFP strains
Eight C. glabrata strains carrying various Sir3-GFP alleles, as well as a negative control strain carrying an untagged Sir3 vector, were grown to stationary phase in liquid YPD+Nat. Cells were washed in PBS and resuspended in PBS, and 5 μl of the cells was mounted on a slide. Images were taken using a Zeiss Axioskop microscope with a 100× objective. The captured images are automatically displayed with optimized brightness and contrast settings in Image J; these maximum and minimum values for the entire captured image are listed in Figure 8. Image J software was used to adjust the constrast to the same settings for all images, thereby allowing more direct comparison of GFP brightness across all images. The adjusted contrast images were converted to 8-bit images, cropped, and resized using Adobe Photoshop and Adobe Illustrator.
Figure 8.

Imaging of live C. glabrata strains carrying various Sir3-GFP alleles. Eight strains carrying different versions of Sir3-GFP, as well as an untagged Sir3 control strain, were grown to stationary phase in liquid culture. Live cells were imaged using differential interference contrast bright-field and fluorescence microscopy. GFP fluorescence is shown with both the contrast settings as captured (automated contrast) and adjusted so each strain has the same contrast levels (adjusted contrast). The maximum and minimum contrast settings for each strain are listed on the automated contrast panel. The adjusted contrast panels all have a minimum of 202 and a maximum of 727. Bar, 5 μm.
S. cerevisiae protein preparation and Westerns
S. cerevisiae strains carrying various DCW1-FLAG plasmids, or a wild-type DCW1 untagged plasmid (negative control), were grown in SD-His media to mid-log phase (OD600 between 0.1325 and 0.2325). Cells were pelleted by centrifugation and stored at −80°. Cell pellets were weighed on a microscale; these weights were used later to normalize loading between strains. Lysates were prepared similar to the method of Frieman and Cormack (2004).To prepare lysates, resuspend cell pellets in 50 mM Tris (pH 8.0) supplemented with protease inhibitors (Roche, 04693132001) and lyse with glass beads using a FastPrep (Bio101 Thermo Savant) (three times: 45 sec, 6.5 speed, ice 1 min between beatings). Supernatants were transferred to clean tubes, and beads were washed in 400 μl Tris (pH 8.0) + protease inhibitors and combined with an earlier fraction (800 μl lysate total). Lysates were clarified by centrifugation at 13,000 × g, 10 min, 4° in a microfuge. Supernatant was saved as the “cytoplasmic” fraction. The pellet was resuspended in 1 ml 50 mM Tris (pH 8.0) + 2% SDS and boiled for 20 min, vortexing to mix every 10 min. After centrifugation (13,000 × g, 10 min, 4°), the supernatant was saved as the “plasma membrane” fraction. The remaining pellet material was resuspended, washed in Tris+SDS, boiled, and centrifuged (as above) three additional times; supernatants were discarded. The pellet was washed four times (twice in 1 ml, twice in 500 μl) in 50 mM Tris + protease inhibitors and spun as before. The pellet was washed once in 500 μl 33 mM potassium phosphate and then resuspended in 100 μl 33 mM potassium phosphate + 60 mM β-mercaptoethanol; this is the “cell-wall” fraction.
Material from each fraction was loaded onto an SDS-PAGE gel; volumes were normalized across strains using pellet weights. Proteins were transferred to Immobilon-P membrane and immunoblotted using antibodies as indicated. Mouse monoclonal α-FLAG (F1804, Sigma) was used at 1:2000 diluted in TBS+3% milk; α-Pgk1 (A6457, Molecular Probes) was used at 1:1000 in TBS+3% milk. Both of these were used in conjunction with α-mouse HRP-linked (Cell Signaling) secondary antibody at a 1:2000 dilution. The α-Dcw1 polyclonal antibody was raised against the peptide VELDLDNYESLQ, representing amino acids 22–33 in the Dcw1 protein (Covance, Princeton, NJ). For Dcw1 detection, the α-Dcw1 primary antibody was diluted at 1:1000 in TBS+3% milk, and the secondary antibody was a 1:5000 dilution of α-rabbit HRP-linked antibody (Cell Signaling). Amersham’s ECL kit (RPN2132) was used for chemiluminescent detection; the α-FLAG and α-Pgk1 blots were exposed to film for 5–10 min; the α-Dcw1 blots were exposed for 45 min.
C. glabrata protein preparation and Westerns
C. glabrata strains carrying different Sir3-GFP plasmids or a wild-type SIR3 untagged plasmid (negative control) were grown in YPD+Nat media to log phase (OD600 between 0.5 and 0.635). Cells were pelleted by centrifugation and stored at −80°. Lysates were prepared in a urea lysis buffer (Ubersax et al. 2003), supplemented with protease inhibitors (Roche, 11836170001) . Proteins were separated on a 3–8% Tris–acetate SDS-PAGE gel (Nupage) and transferred to an Immobilon-P PVDF membrane. A polyclonal α-GFP primary antibody (Abcam, ab290) was used at 1:5000 dilution in PBST+5% milk; the α-rabbit HRP-linked secondary antibody (GE Healthcare, NA934V) was used at a 1:5000 dilution. Chemiluminescent detection was performed using the Amersham ECL kit (RPN2132).
Viability selection and sequence analysis of functional tagged SIR3 alleles in C. glabrata
SIR3 was mutagenized four separate times, using the Tn7-bio, Tn7-myc, Tn7-mEOS2, or Tn7-GFP donor vectors. After processing the mutagenized plasmid pools to remove leftover Tn7 sequence, the final Sir3-bio, Sir3-myc, Sir3-mEOS2, and Sir3-GFP pools were transformed into C. glabrata strain CGM293, and transformants were selected by growth on YPD+Nat plates for 2 days at 30°. Strain CGM293 carries URA3 integrated into a subtelomeric location, where it is subjected to Sir3-dependent transcriptional silencing. Only cells that carry a functional tagged version of Sir3 have intact subtelomeric silencing, thus silencing the URA3 gene at the telomere and permitting growth on plates containing 5-FOA. To select functional clones of Sir3, the YPD+Nat plates were replica-plated onto 5-FOA plates and grown for 2 days at 30°. For the Sir3-myc transformation the pool was outgrown in liquid YPD+Nat prior to plating. This pool showed skewed representation of particular insertion sites. Subsequently, for the other three pools, the transformations were plated directly onto YPD+Nat plates, and these pools show a more even representation of different functional alleles. As with the DCW1 screening, we performed colony PCR to qualitatively determine where the tag had inserted into SIR3. Judging from these PCR product sizes, plasmids carrying SIR3 with a range of insertion sites were sent for sequencing to determine the exact position of the tag within SIR3 (Table S6 and Table S7).
Results
Transposon design
The Tn7-FLAG transposon (Figure 1) is based off the miniTn7 design (Biery et al. 2000), using truncated Tn7 left and right (Tn7L and Tn7R) ends. The Tn7L region is 206 bp long, whereas the Tn7R region is 71 bp long. Previous research has shown the Tn7 ends can accommodate PmeI sites and still mobilize during in vitro transposition reactions (Biery et al. 2000). Here, we engineered FseI and PmeI sites at the distal Tn7 transposon ends to facilitate removal of the bulk of the transposon sequence using restriction digests after the mutagenesis occurs (see later description). These restriction enzymes have 8-bp restriction sites, which occur rarely in the DNA genome sequence. The Tn7-FLAG construct contains two FseI sites, which flank the Tn7L end; two PmeI sites flank a trimethoprim resistance (TmpR) cassette and the Tn7R end. PmFseI digestion, therefore, removes the left end of the transposon, while PmeI digestion removes the right portion of the transposon. The transposon contains a 3× FLAG epitope tag, located between the internal FseI and PmeI sites. The entire Tn7-FLAG construct is carried on the plasmid pRZ101, which serves as a Tn7 donor plasmid. The pRZ101 backbone includes a kanamycin resistance marker (KanR) and replicates using a R6Kγ ORI. This ORI functions only in E. coli cells containing the Π protein, encoded by the pir gene (Kolter et al. 1978; Haldimann et al. 1996; Metcalf et al. 1996).
Figure 1.
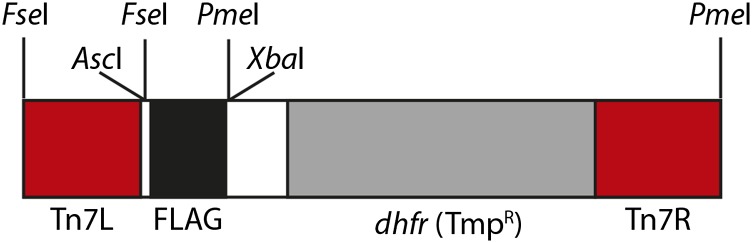
Schematic of the Tn7-FLAG construct. Areas indicated by red are Tn7 left and right ends. The 3×FLAG epitope tag is indicated by black. The gray area is the dhfr gene, which confers resistance to trimethoprim (TmpR). Restriction sites of interest are indicated above the schematic.
During Tn7 mutagenesis, the Tn7 transposase, containing TnsA and TnsB subunits, creates double-strand breaks at the Tn7 ends, releasing the Tn7-FLAG cassette from pRZ101. TnsCA225V mediates insertion of the Tn7-FLAG cassette into a variety of targets with essentially no sequence specificity (Green et al. 2012). During host-mediated repair of the insertion product, a 5-bp duplication is introduced at the insertion site. The epitope tag is positioned within Tn7-FLAG so that it will be translated correctly in only one reading frame after subcloning to excise transposon sequences. All other reading frames contain STOP codons. Thus, five of six transposon mutants are expected to be nonfunctional as a consequence of the epitope tag being out of frame and generating an internal stop codon. After all mutagenesis and processing of the transposon mutants, the Tn7-FLAG cassette introduces 159 bp into the target (including the 5-bp duplication), resulting in a 53-amino-acid insertion internally fused into the target protein.
Tn7-FLAG can be easily adapted to introduce other epitope tags. Unique AscI and XbaI sites flank the FLAG cassette, and these sites can be used to change the epitope tag to any other tag of interest. In our own work, we generated Tn7-6His, Tn7-myc, Tn7-GFP, Tn7-mEOS2, Tn7-biotin, and Tn7-3×HA donor plasmids, all of which have been confirmed to mobilize during in vitro transposition reactions (not shown).
Methodology overview
The in vitro mutagenesis method to introduce internal epitope tags is outlined schematically in Figure 2. To illustrate the utility of this system, we chose to internally tag the protein Dcw1, a GPI-anchored protein, which cannot be functionally tagged at either the N- or C-terminus. The method consists of three separable steps, all of which are carried out in vitro, with recovery of reaction products by transformation into E. coli. First, transposition is used to generate a pool of Tn7 insertions throughout the plasmid carrying the gene to be tagged. Second, Tn7 insertions within the ORF are isolated by a Gateway-mediated recombination step. Third, this pool of Tn7-mutated ORFs is treated with restriction enzymes to remove essentially all transposon sequences, leaving just the introduced protein tag.
Figure 2.

Overview of mutagenesis work flow. An in vitro mutagenesis reaction introduces the Tn7-FLAG transposon randomly throughout the target entry vector (here, DCW1). Mutagenized DCW1 plasmids are selected on the basis of TmpR and KanR. To isolate only those versions that have a Tn7-FLAG inserted in DCW1, and not in the backbone, a Gateway LR reaction is used to mobilize the DCW1 gene into a DCW1 destination vector. After transformation into MegaX E. coli cells and selection with Tmp and Car, only expression vectors that have a mutagenized DCW1 gene should remain. Restriction digests are used to remove the bulk of the transposon: FseI removes the Tn7L end; PmeI removes the TmpR cassette and Tn7R. After this series of digestions, the pool will consist of DCW1 expression vectors with a 3× FLAG epitope inserted randomly, in either direction, in the DCW1 gene.
Transposition:
Using a target plasmid containing the DCW1 ORF cloned into a Gateway entry vector, we performed an in vitro mutagenesis reaction with recombinant Tn7 transposase proteins. The reaction mobilizes the Tn7-FLAG cassette from the Tn7 donor plasmid into the DCW1 target plasmid. This creates a pool of mutagenized DCW1 entry vectors. This pool is transformed into highly competent E. coli DH10 cells and selected for both TmpR and KanR in liquid culture. The use of DH10 cells selects against the Tn7-FLAG donor plasmid, as this plasmid’s R6Kγ ORI will not replicate in DH10 cells. Double drug selection selects against unmutagenized DCW1 entry vectors. Thus, after growth overnight in Tmp and Kan, only cells carrying mutagenized DCW1 entry vectors should survive.
Isolation of mutagenized ORFs by Gateway recombination:
The pool consists of plasmids that have a transposon inserted in the DCW1 ORF as well as plasmids where the transposon is inserted in the plasmid backbone (and which have an unmutagenized ORF). To isolate only those ORFs that have been insertionally mutagenized, we used Gateway recombination to mobilize the DCW1 ORF from the entry vector into a DCW1 destination vector (Figure 2) to generate what we refer to as the expression pool. The DCW1 destination vector contains DCW1 promoter and terminator regions, flanking a ccdB cassette; it also contains replication origins and markers for propagation and selection in both E. coli and yeast. This DCW1 expression pool was recovered by transformation into E. coli DH10 cells. Expression pool plasmids, with the transposon-containing DCW1 ORF cassette, confer resistance to both Tmp and Car. We found that this double selection was maximally effective if we performed the drug selections sequentially (data not shown); accordingly, the transformed cells were initially selected only for carbenicillin resistance (CarR), which selects for all destination vectors carrying a mutagenized or unmutagenized DCW1 ORF. Entry vectors and destination vectors that did not recombine are selected against based on the CarR and ccdB counterselectable cassette, respectively. After selecting for CarR, plasmid DNA is recovered from the expression vector pool and transformed again into E. coli DH10 cells, this time selecting for TmpR. After growth in Tmp, only cells carrying DCW1 expression vectors with a Tn7-FLAG cassette remain. Note that the Tn7-FLAG may be inserted in any reading frame at this point, as the pool of mutants has not been screened for function in yeast yet.
Removal of transposon sequences:
Removal of the bulk of the transposon, leaving only the epitope tag, requires two restriction digests and ligation steps. These cloning steps are performed on the pooled DNA, allowing for easy processing of the entire insertion library. Digestion with FseI releases the Tn7L end from each plasmid, which is then recircularized by ligation. This “–Tn7L” pool of plasmid DNA is transformed into E. coli, and the liquid culture is treated with Tmp and Kan simultaneously. This second round of Tmp selection is possible since the Tn7R end and dhfr gene remain in the construct and selects against any residual unmutagenized DCW1 expression vectors that have escaped the previous selection step. Next, a PmeI digest is used to remove the dhfr (TmpR) gene and the Tn7R end (Figure 1). After intramolecular ligation, the plasmid pool is transformed into E. coli and selected for CarR. The DNA isolated from this pool contains mutagenized DCW1 expression vectors with the 59-aa FLAG epitope inserted in any of the six reading frames.
Monitoring pool sizes and complexity
Transposition efficiency was monitored by plating on various selective media. Transformation of our mutagenesis reaction resulted in 5 × 106 KanR colonies, representing all plasmids whether mutagenized or not. Of these, 2 × 105 (4%) contained transposon insertions (TmpRKanR) (Table 1). This represents a 47-fold coverage of all possible insertion sites for the plasmid (File S2).
Table 1. Sizes of mutagenized DCW1 pools.
| No. of colonies (calculated to full reaction size) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Pool | Processing step | TmpR KanR | TmpR | KanR | CarR | TmpR CarR | Fold Coverage | Fold coverage based on which drugs? |
| Mutagenesis | Mutagenesis of entry vector | 2.01E+05 | 2.45E+05 | 5.00E+06 | ND | ND | 47.5× | Tmp Kan |
| xpCa | Gateway LR to create expression pool | 1.20E+04 | ND | 8.50E+04 | 5.00E+06 | 2.5E+05a | 91× | Tmp Car |
| xpT | xpC, retransformed and selected for Tmp | 9.00E+03 | 4.00E+06 | 2.55E+05 | 1.05E+08 | 4.00E+06 | ∼91× | Tmp Car |
| xpT-Tn7L | After removal of Tn7L | 5.60E+03 | 3.06E+07 | 1.64E+04 | 3.10E+07 | 2.74E+07 | ∼91× | Tmp Car |
| xpT-Tn7L-Tn7R | after removal of Tn7R | 2.00E+02 | 1.87E+06 | 1.07E+05 | 5.40E+07 | 2.10E+06 | ∼91× | Car |
Colony counts, as plated after recovery during transformation, prior to drug selection in pool. Cells were diluted appropriately to prevent a lawn of growth and plated onto media as indicated. Numbers in the table are calculated to represent the number of colonies in the full transformation resistant to the given drug. ND, not determined.
The efficiency of Gateway LR mobilization, as well as the fraction of ORFs carrying the Tn7-flag, was estimated by scoring for Car and Tmp resistance (File S3). We determined that the initial expression pool (xpC) contained 2.5 × 105 TmpRCarR independent recombinants (Table 1). Thus, the majority of the pool complexity is maintained in the expression pool. For subsequent steps, FseI digestion and PmeI digestion, we recovered at least 4 × 106 transformants, again ensuring that pool complexity was maintained through different cloning steps.
Complexity of the pool was qualitatively assessed using restriction digests of pool DNA at each step of the processing (File S4). Additionally, the plasmids from a subset of the final mutagenized expression pool were isolated and sequenced with primer 6244 to assess the distribution of Tn7 insertion sites in the plasmid. From 48 sequenced isolates, we found 29 had FLAG inserted in the reverse orientation (representing 18 unique sites) and 19 had FLAG inserted in the forward direction (representing 18 unique sites). For two nucleotide positions, we isolated FLAG insertions in both the “forward” and the “reverse” orientations in different isolates.
Selection for functional clones in yeast
The final DCW1-FLAG pool was transformed in batch into S. cerevisiae to screen for functional clones. Mutations in DCW1 and the related gene DFG5 are synthetically lethal in S. cerevisiae (Kitagaki et al. 2002). We use the strain BY240, which is a dcw1Δ dfg5Δ strain carrying a wild-type copy of DCW1 on a URA3-marked plasmid (pCU-URA3). The plasmid backbone of the DCW1-FLAG expression pool contains a yeast CEN/ARS sequence and a HIS3 marker for selection. After transforming the DCW1-FLAG pool into BY240, we selected against the original wild-type pCU-URA3 plasmid by growth in the presence of 5- 5-FOA. Only strains transformed with a functional DCW1-FLAG allele will grow on 5-FOA plates. We performed the 5-FOA selection at 37°; the elevated temperature is a mild cell-wall stress and ensures that any cells that grow have a fully functional DCW1-FLAG allele. We screened ∼100,000 transformants, of which ∼5% had functional versions of DCW1-FLAG (data not shown).
Transformants were first screened for presence of a FLAG insertion using whole-cell PCR (data not shown). Products had a range of sizes, representing FLAG insertions across the DCW1 ORF. A subset of these was chosen for further characterization. Plasmids carrying functional DCW1-FLAG alleles were isolated from individual S. cerevisiae transformants and recovered in E. coli. The location of the FLAG insertion within the DCW1 ORF was determined by sequencing. A number of unique insertion sites were identified, as illustrated on the schematic shown in Figure 3. The majority of insertions were, as expected, in the ORF. In addition, several isolates were identified in which the FLAG was inserted just upstream or downstream of the DCW1 ORF in the sequence between the Gateway recombination sites and the ORF in the expression vectors (data not shown). We found that the Gateway attL1 and attL2 recognition sequences, required for mobilization of the DCW1 ORF into the destination vector, are partially permissive such that certain Tn7-disrupted attL1 and attL2 sequences still, surprisingly, function in the Gateway reaction. These few insertions, therefore, have the epitope outside of the ORF sequence and represent contamination in the overall library.
Figure 3.

Location of FLAG insertions in functional DCW1-FLAG alleles. Domain structure of Dcw1 is based on predictions from ELM. The locations of FLAG insertions within the functional DCW1-FLAG alleles used in subsequent experiments are indicated by hash marks above the diagram. Dcw1 is 499 amino acid residues in length.
Unique DCW1-FLAG plasmids were retransformed into S. cerevisiae strain BY240, and function was confirmed by testing for growth on 5-FOA plates (Figure 4). All internally tagged DCW1-FLAG isolates tested grew robustly on 5-FOA at temperatures up to 39°, confirming that the DCW1-FLAG alleles are fully functional. The DCW1-HA26 allele showed reduced growth on 5-FOA at 30° and no growth at 37° and 39°, indicating that it is a partially functional hypomorphic allele.
Detecting FLAG by Western blot
To verify that the FLAG epitope introduced into Dcw1 is detectable by Western analysis, we monitored Dcw1-FLAG protein levels in our strains carrying functional DCW1-FLAG clones. Cytoplasm, plasma membrane, and cell-wall fractions were isolated from log-phase cultures of each strain. Dcw1 is a membrane-bound GPI protein (Kitagaki et al. 2002), and abundant FLAG signal is easily detectable in the cell membrane fraction in 8 of the 10 strains expressing Dcw1-FLAG alleles, consistent with the fact that these are fully functional Dcw1-FLAG alleles (Figure 5). Untagged Dcw1, which would not be visible on the α-FLAG immunoblot, is predicted to have a molecular weight of 49.5 kDa. Dcw1-FLAG fusion proteins are expected to have a molecular weight of 55 kDa. However, mature Dcw1 is glycosylated, and previous studies have shown that Dcw1-HA has an apparent molecular weight of 80 kDa (Kitagaki et al. 2002), similar to what we observe in Figure 5. For three alleles, the FLAG insertion (at residue 5, 6, or 11) is predicted to disrupt the signal sequence (Figure 3). For two of these three, Dcw1-FLAG5 and Dcw1-FLAG6, the immunoblot showed no signal, and for one, Dcw1-FLAG11, the protein was detectable in the membrane fraction, but its molecular weight is lower than expected (∼60 kDa, instead of ∼80 kDa).
Figure 5.
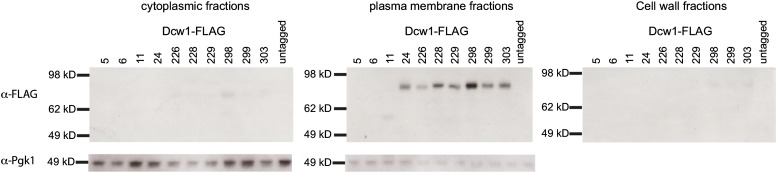
Western blot analysis of Dcw1-FLAG expression in S. cerevisiae. (Top) Anti-FLAG immunoblots of S. cerevisiae strains expressing Dcw1-FLAG alleles, representing the cytoplasmic, plasma membrane, and cell-wall fractions from each strain. Material from 7.5 × 105 cells is loaded for each strain in each fraction. Wells are labeled with the amino acid site of the FLAG tag within Dcw1. (Bottom) Anti-Pgk1 immunoblot of the cytoplasmic and plasma membrane fractions from strains carrying Dcw1-FLAG alleles. Cytoplasmic material from 3.75 × 105 cells is loaded for each strain. Membrane material from 3.5 × 105 cells was loaded for each strain
To confirm that the localization observed of the Dcw1-FLAG fusion proteins represents true localization of untagged Dcw1, we also performed immunoblots using these same cellular fractions with an α-Dcw1 antibody raised against a peptide portion of native Dcw1 (Figure S1). The signal is much fainter than what we observed using α-FLAG antibodies; importantly, the Dcw1-FLAG fusion proteins show similar size distributions using both the anti-Dcw1 and anti-FLAG antibodies.
Mutagenesis of a second target gene, C. glabrata SIR3
The demonstrate the generality of the tagging methodology, we used the same approach to mutagenize the SIR3 gene of Candida glabrata, which is required for silencing of sub-telomeric regions of the C. glabrata genome (De Las Penas et al. 2003). Schematic diagrams of the Sir3 Gateway constructs are shown in Figure S2. We used four different Tn7 derivatives (Tn7-myc, Tn7-GFP, Tn7-mEOS2, Tn7-biotin) to generate libraries of Tn7 insertions with between 3 and 6 × 105 independent insertions in each pool. Functional tagged alleles were identified by complementation of a sir3Δ strain. We sequenced the insertion sites for fully functional alleles, identifying tag insertion sites distributed across the ORF (Figure 6, Table S6, and Table S7). We chose to further characterize eight GFP-tagged SIR3 alleles, which complement for the telomeric silencing defect (Figure S3), representing insertions sites distributed across the ORF. By Western analysis, all eight were expressed at approximately similar levels (Figure 7). For all eight alleles, Sir3-GFP was localized in puncta, as has been reported previously for wild-type Sir3 protein in S. cerevisiae (Cockell et al. 1995) (Figure 8).
Figure 6.

Distribution of functional tagged alleles of C. glabrata SIR3. (Top) Linear representation of the C. glabrata Sir3 protein (1088 residues). The gray areas are predicted globular domains (by ELM), which have been found to have roles in associating with histones (N-terminal domain) and Sir3 dimerization (C-terminal domain). In the schematics below, a black line is located at the position for each unique insertion site identified from among the functional Sir3-bio, Sir3-myc, Sir3-mEOS2, and Sir3-GFP pools. (Bottom) Schematic overlays all sites at which a functional tagged Sir3 was identified. The color of the line indicates the number of mutagenesis pools in which the particular insertion site was identified; light blue indicates that the site was identified in only one pool; black indicates that it was found in all four mutagenesis pools.
Figure 7.
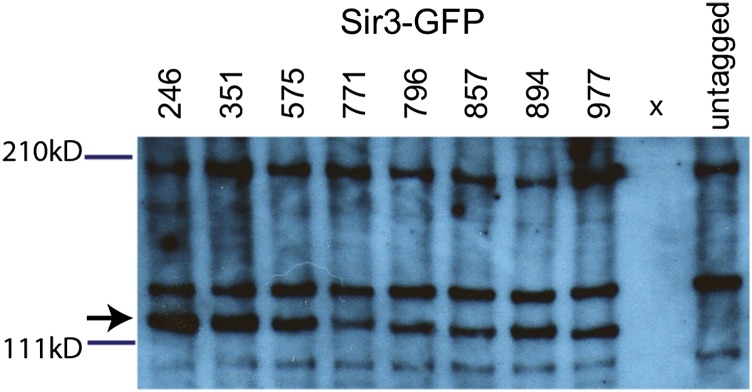
Western blot analysis of Sir3-GFP in C. glabrata. An anti-GFP immunoblot of C. glabrata strains carrying different Sir3-GFP alleles. Wells are labeled with the amino acid site of the GFP insertion within Sir3; “x” indicates an empty lane. The antibody cross-reacted with C. glabrata whole-cell lysates, as shown by the banding see in the untagged strain. The band representing Sir3-GFP is marked with an arrow.
Discussion
Here we describe a robust system for creating a library of internally tagged target protein constructs. This mutagenesis is carried out in vitro, and all subsequent steps of selecting for insertional mutants and removing excess Tn7 sequence are carried out on the bulk pool in vitro. This batch processing greatly reduces the effort in maintaining and screening the thousands of possible mutants.
As proof of principle, we mutagenized a target gene, DCW1, to saturation, introducing a 3× FLAG epitope throughout the ORF. Screening in yeast identified fully functional tagged DCW1-FLAG alleles, of which we chose 10 for further analysis. All 10 DCW1-FLAG alleles that we created proved to have wild-type levels of function when assayed for growth at elevated temperatures.
Monitoring the distribution of Dcw1 in fractionated cell lysates by Western blot illustrated that the available FLAG antibodies were more sensitive than antibodies directly raised against a peptide in Dcw1. The majority of alleles had a subcellular distribution similar to the wild-type protein—primarily in the membrane fraction, consistent with its proposed function (Kitagaki et al. 2002). Two alleles, Dcw1-FLAG5 and Dcw1-FLAG6, were not detectable in any cell fraction in the α-FLAG Western blots. We suspect that the FLAG tag, located in the signal sequence in these two alleles, may have been cleaved off during normal protein trafficking. In the case of Dcw1-FLAG11, the protein was detected in Western blots, but appeared smaller than the expected size (Figure 5). This apparent size difference could be due to truncation or incomplete post-translational glycosylation. We note that introduction of internal epitopes may still alter protein function. However, if multiple functional tagged alleles have the same phenotype and show the same subcellular distribution —as we observed for 7 of 10 alleles—this increases confidence that they reflect wild-type function.
We also mutagenized the C. glabrata SIR3 gene and isolated 126 functional, unique internally tagged alleles (four different tags). By Western analysis, GFP internally tagged proteins were expressed at similar levels, and all showed a punctate distribution similar to that documented for Sir3 in S. cerevisiae. Since the tranposon insertion is random and generates many nonfunctional insertions, the utility of the method depends on a robust screen for functional alleles. This can be an assay for viability (as for DCW1). For Sir3, the assay for function was not viability but growth in the presence of 5-FOA as a measure of transcriptional silencing of a subtelomeric URA3 gene. For DCW1 and SIR3, ∼5% of insertions were functional, suggesting that, for some proteins at least, functional alleles could also be identified by manual screening of a relatively small number of insertions.
For DCW1, the functional alleles had FLAG insertions clustered in a few regions of the gene, including in the central globular glycosyl hydrolase domain (identified by protein BLAST and ELM motif finding) (Kitagaki et al. 2002). For SIR3 as well, functional insertions for all four tags were found across the ORF, including insertions in N- and C-terminal predicted globular domains. It would have been difficult to predict that these regions would tolerate insertions solely based on bioinformatic modeling of the protein structure. Unlike other epitope-tagging systems (Khmelinskii et al. 2011; Ramsden et al. 2011), this Tn7-based system requires no prior knowledge of permissive sites to introduce tags in the POI. The efficient in vitro mutagenesis and cloning steps allow researchers to create and recover an unbiased set of tagged versions of a target gene, which can then be screened for function.
The system of Tn7-based epitope tagging that we describe should be broadly useful. We have described the application to epitope tagging of an essential yeast gene. However, several aspects of the methodology make it suitable to tagging of proteins in virtually any system. First, once Gateway vectors are constructed, mutagenesis, cloning, and screening (in yeast) takes 2–3 weeks total. Second, the mutagenesis is carried out in vitro, and Tn7 has minimal insertion sequence bias. Third, the insertions in the ORF are isolated without regard to function of those insertions. Fourth, the library of tagged proteins is generated in final form in E. coli, obviating the need to remove transposon sequences in the cell type where functional screening is done. Rather, the final library can simply be introduced into the appropriate cell type and screened directly.
Since a major use of transposon epitope-tagging methods is to generate fully functional tagged versions of the POI, it is imperative that the screening be done only on mutagenized ORFs since any untagged wild-type alleles in the pool would pass the functional screen. In the highly useful Tn5-based system, TAGIT, the selection for tagged alleles is by presence of the marker gene carried in the transposon. This allows introduction of the mutagenized library into the cell where screening is carried out; at this stage the transposon sequences are removed by expression in the cell of Cre recombinase. Our method provides an alternative to this approach. Insertions in the ORF are efficiently isolated from nonmutagenized ORFs by Gateway-mediated recombination. This step is highly efficient and can be carried out in batches, permitting construction of a library of tagged ORFs comprising tens or hundreds of thousands of independent insertion events. Subsequent efficient restriction digestions of the pool to remove the left and right ends of the transposon result in a complex pool of tagged ORFs in an appropriate expression vector. This library can then be directly screened for function as dictated by function of the POI, without the need to express recombinase enzymes in the target cell. As a caveat, we note that we recovered a small number of functional expression plasmids where the epitope had inserted between the Gateway recombination sites and the ORF (data not shown); it appears that the attL1 and attL2 sites are somewhat permissive for insertions. This leaves the target ORF unmutagenized, and our current plasmid architecture makes it impossible to eliminate these extraneous plasmids from the pool.
An advantage of this Tn7-based method over other transposon systems is the very limited amount of transposon-derived sequence left in the insertion site. In our method, the FseI and PmeI sites are engineered to be present within three bases of the transposon end, and following excision, there are only 22 nucleotides (essentially the FseI and PmeI sites) in addition to the epitope tag present at the insertion site. This increases, we would argue, the likelihood of identifying functional epitope insertions since it limits the amount of extraneous sequence shared by all insertions to a few amino acids.
Finally, the method in principle could be used for analysis of proteins in many different organisms. The mutagenesis of the cloned gene is done in vitro, and mobilization into the expression vector exploits Gateway recombination. This can be adapted most easily to any system for which Gateway-modified expression vectors exist. We suggest that the method will be useful in a range of systems, including model eukaryotic systems like S. cerevisiae as well as mammalian systems.
Supplementary Material
Acknowledgments
We thank Matthew Frieman for creating the dcw1Δdfg5Δ pCU-DCW1 strain BY240. We thank Irene Castaño for strain CGM293, Yuxia Ren for her assistance cloning the Tn7 donor vectors, and Jie Xiao for the gift of pET28-ftsZ-mEOS. R.E.Z. is supported by an American Cancer Society Postdoctoral Fellowship (PF-09-194-01-MPC). This work was supported by National Institutes of Health grants R01A1046223 (to B.P.C.) and RO1GM076425 (to N.L.C.). N.L.C. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Supporting information is available online at http://www.genetics.org/lookup/suppl/doi:10.1534/genetics.114.169482/-/DC1.
Sequence data from this article have been deposited with the GenBank Data Library under accession no. KP698385 (pRZ49 = Tn7-mEOS2); no. KP698386 (pRZ98 = Tn7-biotin); no. KP698387 (pRZ99 = Tn7-6His); no. KJ939358 (pRZ101 = Tn7-FLAG); no. KP698388 (pRA102 = Tn7-HA); no. KP698389 (pRZ103 = Tn7-myc); and no. KP698390 (pRZ106 = Tn7-GFP).
Communicating editor: O. Cohen-Fix
Literature Cited
- Arnau J., Lauritzen C., Petersen G. E., Pedersen J., 2006. Current strategies for the use of affinity tags and tag removal for the purification of recombinant proteins. Protein Expr. Purif. 48: 1–13. [DOI] [PubMed] [Google Scholar]
- Bell M. R., Engleka M. J., Malik A., Strickler J. E., 2013. To fuse or not to fuse: What is your purpose? Protein Sci. 22: 1466–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biery M. C., Stewart F. J., Stellwagen A. E., Raleigh E. A., Craig N. L., 2000. A simple in vitro Tn7-based transposition system with low target site selectivity for genome and gene analysis. Nucleic Acids Res. 28: 1067–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castano I., Kaur R., Pan S., Cregg R., Penas Ade L., et al. , 2003. Tn7-based genome-wide random insertional mutagenesis of Candida glabrata. Genome Res. 13: 905–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K. Y., Li Y., Sarnovsky R., Craig N. L., 2013. Direct interaction between the TnsA and TnsB subunits controls the heteromeric Tn7 transposase. Proc. Natl. Acad. Sci. USA 110: E2038–E2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockell M., Palladino F., Laroche T., Kyrion G., Liu C., et al. , 1995. The carboxy termini of Sir4 and Rap1 affect Sir3 localization: evidence for a multicomponent complex required for yeast telomeric silencing. J. Cell Biol. 129: 909–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormack B. P., Falkow S., 1999. Efficient homologous and illegitimate recombination in the opportunistic yeast pathogen Candida glabrata. Genetics 151: 979–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Las Penas A., Pan S. J., Castano I., Alder J., Cregg R., et al. , 2003. Virulence-related surface glycoproteins in the yeast pathogen Candida glabrata are encoded in subtelomeric clusters and subject to RAP1- and SIR-dependent transcriptional silencing. Genes Dev. 17: 2245–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine S. E., Boeke J. D., 1994. Efficient integration of artificial transposons into plasmid targets in vitro: a useful tool for DNA mapping, sequencing and genetic analysis. Nucleic Acids Res. 22: 3765–3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujon B., Sherman D., Fischer G., Durrens P., Casaregola S., et al. , 2004. Genome evolution in yeasts. Nature 430: 35–44. [DOI] [PubMed] [Google Scholar]
- Frieman M. B., Cormack B. P., 2004. Multiple sequence signals determine the distribution of glycosylphosphatidylinositol proteins between the plasma membrane and cell wall in Saccharomyces cerevisiae. Microbiology 150: 3105–3114. [DOI] [PubMed] [Google Scholar]
- Gamas P., Craig N. L., 1992. Purification and characterization of TnsC, a Tn7 transposition protein that binds ATP and DNA. Nucleic Acids Res. 20: 2525–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez M., Goddard N., Hicks C., Ovalle R., Rauceo J. M., et al. , 2010. A screen for deficiencies in GPI-anchorage of wall glycoproteins in yeast. Yeast 27: 583–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green B., Bouchier C., Fairhead C., Craig N. L., Cormack B. P., 2012. Insertion site preference of Mu, Tn5, and Tn7 transposons. Mob. DNA 3: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory J. A., Becker E. C., Jung J., Tuwatananurak I., Pogliano K., 2010. Transposon assisted gene insertion technology (TAGIT): a tool for generating fluorescent fusion proteins. PLoS ONE 5: e8731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldimann A., Prahalad M. K., Fisher S. L., Kim S. K., Walsh C. T., et al. , 1996. Altered recognition mutants of the response regulator PhoB: a new genetic strategy for studying protein-protein interactions. Proc. Natl. Acad. Sci. USA 93: 14361–14366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill J., Donald K. A., Griffiths D. E., 1991. DMSO-enhanced whole cell yeast transformation. Nucleic Acids Res. 19: 5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman, C. S., 2001 Preparation of yeast DNA. Curr. Protoc. Mol. Biol. Chapter 13: Unit13 (13.11-1-13.11.4). [DOI] [PubMed] [Google Scholar]
- Khmelinskii A., Meurer M., Duishoev N., Delhomme N., Knop M., 2011. Seamless gene tagging by endonuclease-driven homologous recombination. PLoS ONE 6: e23794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagaki H., Wu H., Shimoi H., Ito K., 2002. Two homologous genes, DCW1 (YKL046c) and DFG5, are essential for cell growth and encode glycosylphosphatidylinositol (GPI)-anchored membrane proteins required for cell wall biogenesis in Saccharomyces cerevisiae. Mol. Microbiol. 46: 1011–1022. [DOI] [PubMed] [Google Scholar]
- Kitagaki H., Ito K., Shimoi H., 2004. A temperature-sensitive dcw1 mutant of Saccharomyces cerevisiae is cell cycle arrested with small buds which have aberrant cell walls. Eukaryot. Cell 3: 1297–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolter R., Inuzuka M., Helinski D. R., 1978. Trans-complementation-dependent replication of a low molecular weight origin fragment from plasmid R6K. Cell 15: 1199–1208. [DOI] [PubMed] [Google Scholar]
- Kumar A., Agarwal S., Heyman J. A., Matson S., Heidtman M., et al. , 2002. Subcellular localization of the yeast proteome. Genes Dev. 16: 707–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A., Seringhaus M., Biery M. C., Sarnovsky R. J., Umansky L., et al. , 2004. Large-scale mutagenesis of the yeast genome using a Tn7-derived multipurpose transposon. Genome Res. 14: 1975–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddi A., Fu C., Free S. J., 2012. The Neurospora crassa dfg5 and dcw1 genes encode alpha-1,6-mannanases that function in the incorporation of glycoproteins into the cell wall. PLoS ONE 7: e38872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoil C., Traxler B., 2000. Insertion of in-frame sequence tags into proteins using transposons. Methods 20: 55–61. [DOI] [PubMed] [Google Scholar]
- Merkulov G. V., Boeke J. D., 1998. Libraries of green fluorescent protein fusions generated by transposition in vitro. Gene 222: 213–222. [DOI] [PubMed] [Google Scholar]
- Metcalf W. W., Jiang W., Daniels L. L., Kim S. K., Haldimann A., et al. , 1996. Conditionally replicative and conjugative plasmids carrying lacZ alpha for cloning, mutagenesis, and allele replacement in bacteria. Plasmid 35: 1–13. [DOI] [PubMed] [Google Scholar]
- Mumberg D., Muller R., Funk M., 1995. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156: 119–122. [DOI] [PubMed] [Google Scholar]
- Osawa M., Erickson H. P., 2005. Probing the domain structure of FtsZ by random truncation and insertion of GFP. Microbiology 151: 4033–4043. [DOI] [PubMed] [Google Scholar]
- Penfold R. J., Pemberton J. M., 1992. An improved suicide vector for construction of chromosomal insertion mutations in bacteria. Gene 118: 145–146. [DOI] [PubMed] [Google Scholar]
- Ramsden R., Arms L., Davis T. N., Muller E. G., 2011. An intein with genetically selectable markers provides a new approach to internally label proteins with GFP. BMC Biotechnol. 11: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Macdonald P., Coelho P. S., Roemer T., Agarwal S., Kumar A., et al. , 1999. Large-scale analysis of the yeast genome by transposon tagging and gene disruption. Nature 402: 413–418. [DOI] [PubMed] [Google Scholar]
- Seringhaus M., Kumar A., Hartigan J., Snyder M., Gerstein M., 2006. Genomic analysis of insertion behavior and target specificity of mini-Tn7 and Tn3 transposons in Saccharomyces cerevisiae. Nucleic Acids Res. 34: e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan D. L., Berlot C. H., Robert A., Inglis F. M., Jakobsdottir K. B., et al. , 2002. A new way to rapidly create functional, fluorescent fusion proteins: random insertion of GFP with an in vitro transposition reaction. BMC Neurosci. 3: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spreghini E., Davis D. A., Subaran R., Kim M., Mitchell A. P., 2003. Roles of Candida albicans Dfg5p and Dcw1p cell surface proteins in growth and hypha formation. Eukaryot. Cell 2: 746–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubersax J. A., Woodbury E. L., Quang P. N., Paraz M., Blethrow J. D., et al. , 2003. Targets of the cyclin-dependent kinase Cdk1. Nature 425: 859–864. [DOI] [PubMed] [Google Scholar]
- Winzeler E. A., Shoemaker D. D., Astromoff A., Liang H., Anderson K., et al. , 1999. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285: 901–906. [DOI] [PubMed] [Google Scholar]
- Young C. L., Britton Z. T., Robinson A. S., 2012. Recombinant protein expression and purification: a comprehensive review of affinity tags and microbial applications. Biotechnol. J. 7: 620–634. [DOI] [PubMed] [Google Scholar]
- Zordan, R. E., Y. Ren, S. J. Pan, G. Rotondo, A. De Las Penas et al., 2013 Expression plasmids for use in Candida glabrata. G3 (Bethesda) 3: 1675–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.