

Abstract
Glioblastoma multiforme (GBM) is the most common and severe form of brain cancer. The median survival time of patients is approximately 12 months due to poor responses to surgery and chemoradiation. In order to understand the mechanisms involved in radioresistance, we conducted a genetic screen using an shRNA library to identify genes whose inhibition would sensitize cells to radiation. The results were cross-referenced with the Oncomine and Rembrandt databases to focus on genes that are highly expressed in GBM tumors and associated with poor patient outcomes. Spermidine/spermine-N1-acetyltransferase 1 (SAT1), an enzyme involved in polyamine catabolism, was identified as a gene that promotes resistance to ionizing radiation (IR), is overexpressed in brain tumors, and correlates with poor outcomes. Knockdown of SAT1 using shRNA and siRNA approaches in multiple cell and neurosphere lines resulted in sensitization of GBM cells to radiation in colony formation assays and tumors, and decreased tumorigenesis in vivo. Radiosensitization occurred specifically in G2/M and S phases, suggesting a role for SAT1 in homologous recombination (HR) that was confirmed in a DR-GFP reporter system. Mechanistically, we found that SAT1 promotes acetylation of histone H3, suggesting a new role of SAT1 in chromatin remodeling and regulation of gene expression. In particular, SAT1 depletion led to a dramatic reduction in BRCA1 expression, explaining decreased HR capacity. Our findings suggest that the biological significance of elevated SAT1 expression in GBM lies in its contribution to cell radioresistance and that SAT1 may potentially be a therapeutic target to sensitize GBM to cancer therapies.
Keywords: SAT1, polyamine, glioblastoma, radiation, histone acetylation
Introduction
GBM is the most common and aggressive of the gliomas, a group of tumors that derive from glia or their precursors in the central nervous system. Patients with GBM have a median survival time of ~12 months, and only 3–5% of the patients survive more than 3 years (1). GBM is characterized by a heterogeneous population of cells which are genetically unstable and infiltrative, and comprise some of the most challenging therapeutic targets due to their anatomic location, the blood barrier, and poor responses to conventional therapies (2). While the standard of care for newly diagnosed patients includes resection, followed by concurrent radiotherapy and temozolomide (3), the response to radiation and DNA damaging agents remains insufficient as tumors display resistance and a propensity to recur. Although several mechanisms have been proposed to explain the radioresistance in GBM (4–6), the molecular bases of radioresistance remain incompletely defined.
In order to identify novel mediators of radiation resistance in glioblastoma, we performed a genetic screen using an shRNA library on two GBM cell lines. The results showed a list of overlapping genes with a variety of disparate functions. Analyses of the genes using public databases revealed Spermidine/spermine-N1-acetyltranferase (SAT1) as a novel regulator of radiation response with no previously described association with DNA repair. SAT1 catalyzes the acetylation of polyamines spermidine and spermine to form acetyl derivatives and is considered a rate-limiting enzyme in polyamine catabolism, leading to degradation or excretion (7). Paradoxically, polyamines, ubiquitous cationic molecules, are known radioprotectors through their capacity to compact DNA (8–10), confounding speculation of how SAT1 could promote resistance to radiation.
Histone acetylation has been shown to have an essential role in DNA repair allowing critical proteins to be loaded at sites of damage (11), as well as altering gene expression by decompacting chromatin (12). Indeed, cells with DNA breaks maintain high levels of acetylation (13). In a parallel role to histone acetylation, we hypothesized that polyamine acetylation by SAT1 may have an integral role in DSB repair through alteration of chromatin and thereby contribute to radiation resistance. Our results indicate that SAT1 increases acetylation of histone H3, increasing BRCA1 expression, and allowing activation of the homologous recombination (HR) pathway to repair DNA damage. The findings support a novel role for SAT1 in histone acetylation and DNA repair, and suggest that the biological significance of SAT1 expression in GBM lies in its contribution to radio- and chemo-resistance. SAT1 may represent a therapeutic target to sensitize GBM to cancer therapies.
Materials and methods
Cell lines and reagents
U87MG and LN229 cells were obtained from ATCC. D54MG and D317MG lines were gifts of Dr. Jeremy Rich; MCF7 DR-GFP cells were from Dr. Junran Zhang. Neurosphere cell lines GBM 0821 and 0913 were a gift of Dr. Angelo Vescovi (to E. Bar). The lines were not independently authenticated. All lines were used in early passage. D-luciferin came from Byosynth International. Trichostatin A (TSA) was used at 250ng/ml (Sigma-Aldrich). Antibodies: BRCA1 (sc-6954, Santa Cruz Biotechnology), Mre11 (cs4847, Cell Signaling), NBS1 (cs3002), Rad51 (sc-8349), Histone H3 (sc-8654), acetyl-H3 (06-599, Upstate-Millipore), SAT1 (sc-67159). qRT-PCR was performed using 2XSYBR-Green Master mix (Roche) and normalized to β-actin. Primer sequences: SAT1 F-5’GCTGATCAAGGAGCTGGCTA 3’, R-5’CAACAATGCTGTGTCCTTCC 3’; BRCA1 F-5’TGGAAGAAACCACCAAGGTC 3’, R-5’ACCACAGAAGCACCACACAG 3’; Actin F-5’CATGTACGTTGCTATCCAGGC 3’, R-5’ CTCCTTAATGTCACGCACGAT3’; ODC F-5’ ACATCCCAAAGCAAAGTTGG 3’, R-5’ AGCTGACACCAACAACATCG 3’.
shRNA Screen/Knockdown
The Decode RNAi Screening Library was performed as directed (Open Biosystems). Microarray hybridizations were performed by the Stanford Microarray Facility (Stanford, CA). For siRNA, cells were transfected with DharmaFECT#1 and either 25nM control oligos or siSAT1 (Dharmacon). Stable SAT1 knockdown was performed with lentiviral shRNA pLKO.1 clones: TRCN0000035250 and TRCN0000035252 (Thermo-Fisher), shRNA GFP was used as control. Cells were selected with puromycin at 1µg/ml.
Colony Formation Assay
Radiation was performed with a 137Cs irradiator (Shepherd). 500–10,000 cells/plates were stained after 10 days with 0.1% crystal violet. Assays were done ≥ three times with individual samples in triplicate. Cell sorting was performed on live D54MG cells stained with Hoechst 33342 on an iCyt Reflection. Cells were then irradiated and plated. Clonogenic assays of neurosphere lines were performed in plates coated with 200 µl of 2mg/ml poly (2-hydroxyethyl methacrylate) (Sigma) in 95% ethanol. Wells were overlaid with 2ml of neuro stem cell (NSM) 1.5% methyl-cellulose media. 104 cells in NSM were mixed 1:4 with methyl cellulose, and fed every three days. Sphere formation was monitored and scored by light-microscopy after 12 days with Metamorph software, using a size cutoff of 100 µm.
Immunofluorescence
Cells were fixed in 3% formaldehyde and stained using standard procedures: BRCA1 antibody (sc-6954), secondary Alexa-Fluor 594 anti-mouse (A11032, Invitrogen). Slides were mounted in Vectashield with DAPI (Vector Laboratories). Immunofluorescence was observed at 100×, foci were counted from at least 50 cells.
Tumor formation assay
Eight-week-old athymic BALB/c mice were injected intacranially with 105 luciferase-expressing SAT1 or GFP knockdown GBM0821, GBM0913 neurosphere cells. Tumor growth was monitored twice weekly and quantified using bioluminescent imaging. Signal intensity was measured as photon counts within an ROI. Animal appearance, behavior and weight were monitored to evaluate tumor progression. Subcutaneous U87MG tumors were produced by injection of 5×106 cells in nude mice, and measured with calipers twice weekly. When tumors reached between 100–200 mm3, animals were randomized and injected intratumorally with 500 pmol of siRNA packaged in ECO as described (14). After 48 hours, half of the tumors were irradiated.
Chromatin Immunoprecipitation (ChIP)
ChIP was performed as described previously (15). The primers used were: BRCA1 F-5’ GGCAGGCATTTATGGCAAAC 3’, R-5’ TTCGGAAATCCACTCTCCCACG 3’. β-actin F-5’ CCGAAAGTTGCCTTTTATGG 3’, R-5’ CAAAGGCGAGGCTCTGTG 3’. Acetylated H3 antibody (Upstate-Millipore 06-599). Samples were normalized to input.
Polyamine levels
Polyamines were quantified using mass spectrometry. Proteins were precipitated with heptafluorobutyric acid, and supernatants containing polyamines were filtered through an ion-exchange membrane for salt removal. An internal standard, diethylspermine, was added into the samples that were injected into an Agilent 6420 triple quadrupole LC-MS system for analysis along with a 7-point standard curve. The chromatographic separations were achieved using a guarded Luna reversed phase CN column (3 mm×150 mm, Phenomenex, New Zealand). Concentrations were calculated from the standard curves. Three biological replicates were assayed for each sample.
Homologous recombination assays/Comet assays
Cells were transfected with 2µg of pcDNA or I-Sce plasmids using lipofectamine 2000 (Invitrogen), incubated 48 hours and analyzed by flow cytometry as described (16). Comet assays were performed as directed (Trevingen), and quantified with ComeScore software. At least 41 cells per sample were measured, and done in duplicate.
Statistical Analyses
Student’s t-tests were used throughout the study to test the significance of differences between samples. Survival analyses were performed by Log-rank tests in GraphPad Prism.
Results
shRNA screen identifies SAT1 as a mediator of radioresistance
In order to identify novel mediators of radiation resistance in GBM, we performed a genetic screen using a lentiviral-mediated shRNA library on two GBM lines. U87MG and LN229 were chosen based on known different genetic characteristics to maximize the application of hits. U87MG is p53 wild-type and PTEN-null; LN229 is p53 mutant and PTEN wild-type (17, 18). Thus, many classic DNA damage pathway genes will be excluded due to the differential status of p53. The library was comprised of roughly 30,000 barcoded shRNAs which target over 10,000 genes. GBM cells were infected with pools of lentiviruses at an estimated multiplicity of infection of 0.3 to ensure that cells were infected with only once. Following selection, cells were divided into treatment (2 Gy of IR) and control groups, maintained for 72 hours after irradiation, and lysed for genomic DNA. The screens were performed in duplicate.
Ostensibly, shRNA knockdown of genes that confer radioprotection should result in the sensitization of cells to radiation and depletion from the population. Knockdown of genes that promote sensitivity to radiation should result in protection and increased survival. Identification of shRNAs that altered the sensitivity of cells to radiation was performed by determining the relative abundances of the shRNA barcodes in the genomic DNA of the treatment group versus the control group. Barcodes were amplified by PCR, labeled with Cy3 or Cy5, and hybridized onto barcode-microarrays. The raw data of the U87MG screen are represented in Figure 1A: Log2 transformed ratios of barcode abundances in the irradiated sample divided by the control. Of 21,555 detected barcodes, 1,868 were decreased by 1.5-fold or more; 764 were decreased by 2-fold or more; and 126 were decreased by 4-fold or more (Figure 1B). As an internal positive control for the screen, 47 shRNAs targeting genes known to be involved in DNA repair were found (e.g. ATM, PARP1, RAD9, RAD51). When the results of the U87MG and LN229 screens were combined into one dataset, 79 shRNAs that were decreased 1.5-fold or more were identified, 10 were decreased 2-fold or more, and none were decreased by 4-fold. Genes with functions as broad ranging as synaptic nerve transmission to cell motility were included in the 10 genes (Supplementary Table 1; Microarray data are available in the ArrayExpress database, accession number E-MTAB-2861). Due to its metabolic role as a regulator of DNA compaction, we further investigated Spermidine/spermine N1-acetyltransferase 1 (SAT1).
Figure 1. Relevance of SAT1 to GBM.

A. Raw data of the U87MG shRNA screen; abundance of individual barcodes represented as Log2 transformed ratios of irradiated over non-irradiated cells, shaded by level of depletion. B. Quantification of barcode abundance ratios. 1,868 barcodes decreased ≥1.5-fold; 764 decreased ≥2-fold; and 126 decreased ≥4-fold. C. Oncomine data of SAT1 expression in brain tumors (N=normal, T=tumor). D. Rembrandt survival curves of glioma patients with high SAT1 versus low SAT1 expressing tumors.
SAT1 is overexpressed in GBM and correlates with poorer outcome
As a primary filter for relevance to GBM, expression levels of hits were queried in Oncomine. SAT1 expression was found elevated in a variety of brain and CNS cancers, derived from data from five studies of glioblastoma, two oligodendroglioma, one malignant glioma, one astrocytoma, and one oligoastrocytoma. The expression levels of SAT1 in cancers versus normal tissues in four of the studies are displayed in Figure 1C. In the Shai, Bredel, and TCGA studies of GBM samples, SAT1 was over expressed by factors of 3.11 (p<0.0001), 3.33 (p=0.0009), and 2.28 (p<0.0001), respectively; in the Pomeroy study of malignant gliomas, SAT1 was overexpressed by 3.33 (p=0.0042). We next looked at SAT1 Rembrandt, an NCI/NINDS database of gene expression and survival data from >340 brain tumor cases. Using SAT1 expression to categorize the samples, we observed that a 2-fold increase in SAT1 expression correlated with a significant decrease in survival time (603 vs. 450 days, p=0.0008, Log-Rank test) (Figure 1D). Levels of SAT1 could not identify patients with poorer or better outcomes among patients with GBM, but could delineate glioma patients with the poorest prognoses (i.e. patients with GBM) from the rest. Together, these data suggest SAT1 may play a role in GBM tumors and their radioresistant phenotype.
SAT1 knockdown sensitizes GBM cells to radiation
To verify SAT1 knockdown can sensitize cells to radiation, two unique lentiviral shRNAs were stably expressed first in U87MG cells, and clonogenic survival assays were performed with different doses of IR. Quantitative real-time polymerase chain reaction (qRT-PCR) and western blot were used to verify knockdown (Figure 2 insets, and Supplementary Figure 1). Importantly, the levels of knockdown consistently achieved were in the range of the levels found elevated in tumor samples, i.e. 3 to 5-fold. Both shRNAs sensitized U87MG cells, confirming the results of the screen (Figure 2A). We observed that the depletion of SAT1 also sensitized D317MG and D54MG cell lines to ionizing radiation, with dose modifying factors at 10% survival of 1.14, 1.32, and 1.36 for U87MG, D317MG, and D54MG, respectively (Figure 2B and 2C). To determine if transient knockdown was also sufficient to sensitize cells to radiation, we performed siRNA in the D54MG line and found similar sensitization (Figure 2D).
Figure 2. SAT1 knockdown sensitizes GBM cells to radiation.

A. Clonogenic survival of U87MG cells with two SAT1 shRNAs. B and C. Clonogenic survival of D317MG and D54MG with stable SAT1 knockdown. D. Clonogenic survival of D54 with transient SAT1 knockdown. E and F. Clonogenic survival of primary GBM neurosphere lines GBM0821 and GBM0913 with stable SAT1 knockdown. Error bars show standard deviations. *p≤0.001 (A); 0.002 (B); 0.013 (C); 0.005 (D); 0.019 (E); and 0.036 (F).
Tumors are comprised of cells with differing potentials to initiate or repopulate. The importance of such “tumor initiating cells” in cancer therapy has become of peak interest due to their tendencies to be particularly resistant (5). We next evaluated the effect of SAT1 knockdown on radiation response of primary GBM neurosphere cell lines (GBM0821, GBM0913) that are enriched in tumor initiating cells (19) in a 3-D neurosphere colony assay. We again observed sensitization to radiation following SAT1 depletion; GBM0821 by a factor of 1.70, and GBM0913 by a factor of 1.35 (Figure 2E, 2F). Together, the results show that inhibition of SAT1 is sufficient to sensitize multiple tumor cell lines to radiation.
SAT1 promotes GBM tumor growth and decreases survival in nude mice
Clonogenic capacity in vitro is often used as a surrogate for tumorigenic potential in vivo. Besides observing increased radiosensitivity in SAT1 depleted neurospheres, we also found decreased in clonogenic capacity absent of radiation. To determine if SAT1 affects tumor growth, nude mice were injected intracranially with luciferase-expressing SAT1 knockdown GBM0821 and GBM0913 neurospheres and compared to shGFP knockdown cells. Tumor growth was monitored using bioluminescent imaging. Over a 80–90 day period, we found that mice injected with control cells developed tumors more rapidly than mice injected with SAT1 knockdown cells from both neurosphere lines (Figure 3A and 3B). Kaplan-Meier curves demonstrated significantly increased survival of animals with shSAT1 tumors compared to controls (Figure 3C and 3D). In the GBM0913 line, control animals survived an average of 65.5 days post-injection, while the shSAT1 animals survived 83.0 days (p=0.0047). In the GBM0821 line, control animals survived an average of 47.0 days, and the shSAT1 animals survived 77.0 days (p=0.0064). To determine if eventual tumor growth in the shSAT1 neurosphere lines correlated with regained expression of SAT1, tumors were excised and subjected to qRT-PCR for SAT1. We found however that SAT1 knockdown was maintained in tumor samples over the course of the experiment, suggesting that SAT1 deficiency limited the growth of the tumor, rather than inducing a selective pressure against SAT1 knockdown (Supplementary Figure 2A and B). Thus elevated SAT1 expression in GBM promotes tumorigenesis in addition to reducing radiosensitivity.
Figure 3. SAT1 knockdown inhibits GBM tumor growth and increases survival in nude mice.

A and B. Bioluminescence of orthotopic tumor growth of SAT1 knockdown GBM0821 and GBM0913 neurospheres. C and D. Kaplan-Meier survival curves of GBM0821 and GBM0913 cells with SAT1 or GFP knockdown. GBM0913 controls (n=4) survived an average of 65.5 days post injection; shSAT1 (n=5) survived 82.0 days (p=0.0047). GBM0812 controls (n=5) survived an average of 47.0 days; shSAT1 (n=5) survived 77.0 days (p=0.0064). E U87MG tumor response curves following intratumoral siRNA and radiation. F. Kaplan-Meier survival curves of mice in E.
To test whether SAT1 depletion can sensitize tumors in vivo, transient knockdown in U87MG tumors was achieved by intratumoral injection of siRNA using cationic lipid-based nanoparticles (14). Subcutaneous U87MG tumors 100–200 mm3 were injected with 500 pmol of siSAT1 or RISC control packaged in 1-aminoethyl)iminobis[N-(oleicylcysteinyl-1-amino-ethyl)-propionamide] (ECO), and subsequently irradiated 48 hours later with 8 Gy. Tumors were measured twice-weekly, until the tumors reached ≥1.5 cm3. As can be seen in Figure 3E, while transient knockdown of SAT1 had no effect on tumor growth, and 8 Gy IR had a modest effect on tumor growth, the combination of siSAT and radiation led to the most significant delay on tumor growth. Likewise, Kaplan-Meier survival curves of the animals demonstrated a significant benefit to siSAT1 and radiation over radiation alone (Figure 3F). Together the data argue that the sensitization of GBM cells in vitro exists in vivo as well.
Global polyamine catabolism is not the mechanism of SAT1-mediated radioprotection
Polyamine-induced DNA compaction/aggregation is known to be a means of radioprotection (8, 10, 20). Thus, how SAT1, a rate-limiting enzyme of polyamine catabolism, would protect GBM cells from radiation is non-intuitive. To gain insight into the mechanism of SAT1 mediated radioprotection, we determined by mass spectrometry the levels of polyamines in control and SAT1 knockdown cells. Surprisingly, stable knockdown of SAT1 did not result in increases in steady-state levels of spermine or spermidine (Figure 4A). One explanation for this observation might be that stable knockdown cells acclimate to decreased SAT1 gene expression, as enzymes in the polyamine pathway are known to be tightly regulated by feedback mechanisms (21). To determine if decreased catabolic enzyme expression (i.e. SAT1) was accompanied by decreased anabolic enzyme levels, we measured the expression of the rate-limiting regulator of polyamine synthesis, ornithine decarboxylase 1 (ODC1) (21). We found that ODC1 expression mirrored SAT1, displaying significant repression with both SAT1 shRNAs (Figure 4B). Thus global alterations in polyamine levels are not evident in stable SAT1 knockdown cells, and therefore do not correlate with radiosensization following SAT1 depletion.
Figure 4. Polyamine levels are unchanged by stable knockdown of SAT1.
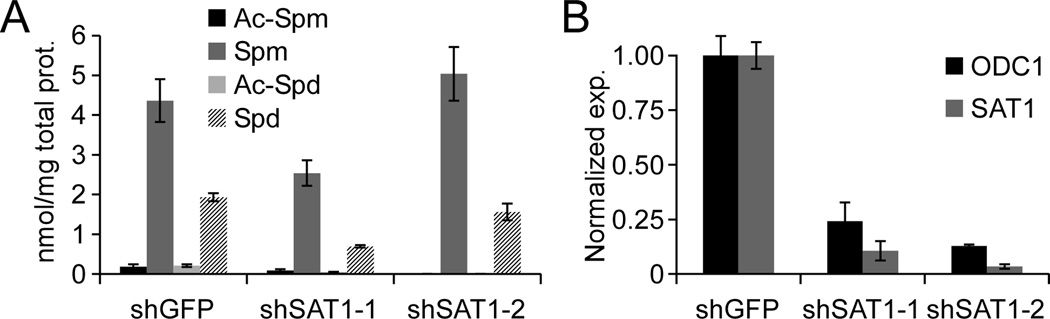
A. Acetyl-Spermine (Ac-Spm), Spermine (Spm), Acetyl-Spermidine (Ac-Spd) and Spermidine (Spd) were measured by MS in D54MG shGFP and shSAT1 cells. B. qRT-PCR of ODC and SAT1 mRNA in stable SAT1 knockdown cells. Error bars show standard deviations.
SAT1 protects GBM cells from irradiation by promoting HR
We next tested if alterations in the cell cycle could explain increased sensitivity to radiation by increasing the fraction of cells in radiosensitive phases (e.g. G1). We assessed the cell cycle distribution of D54MG cells following stable knockdown of SAT1, but found no significant differences (Figure 5A). In addition, SAT1 expression levels were not cell-cycle dependent (Figure 5B). In contrast, when we assessed the sensitivity of cells sorted by flow cytometry using Hoechst 33342 (Supplementary Figure 3), we found that S phase and G2/M phase cells were uniquely sensitized to radiation following knockdown, while G1 phase cells were unaffected (Figure 5C). S phase cells displayed a dose modifying factor of 1.90, at 10% survival; G2/M cells displayed a factor of 1.86.
Figure 5. SAT1 protects cells from IR by promoting HR.

A. Cell cycle profiles of D54MG shGFP and shSAT1 cells. B. SAT1 qRT-PCR in asynchronous and flow sorted cells normalized to β-actin. C. Clonogenic survival assays of flow-sorted D54MG shGFP or shSAT1 cells. *p values all ≤0.039 D. DR-GFP HR reporter assay in shSAT1 or shScram MCF7 cells. Mean efficiencies were 0.187% GFP positive cells for control, and 0.089% for shSAT1. E. Clonogenic survival of D54MG shGFP and shSAT1 cells with CPT. Error bars show standard deviations.
HR is the predominant DSB repair pathway in G2/M and S phases of the cycle due to the presence of homologous sister chromatids post-replication (22). Our findings of radiosensitization in G2/M and S phases led us to investigate the effect of SAT1 on HR using an established MCF7-based HR reporter assay (DR-GFP) (23). SAT1 deficient cells demonstrated a 54% decrease in HR compared to shGFP cells (p=0.012) (Figure 5D). To test whether SAT1 knockdown would sensitize cells to an S-phase specific DNA damaging agent, we treated D54MG cells with physiologically relevant concentrations of the topoisomerase I inhibitor Camptothecin (CPT). Chronic 10 nM CPT exposure reduced colony survival in control cells by 10-fold; in contrast, CPT exposure led to a 1000-fold reduction in SAT1 knockdown colony survival (Figure 5E). Thus SAT1 depletion sensitizes cells to IR and S-phase agents by inhibiting HR.
SAT1 depletion decreases BRCA1 foci and expression levels
To gain insight into the mechanism of regulation of HR by SAT1, we measured DNA damage after IR by comet assay in shGFP and shSAT1 D54MG cells. We found basal levels of damage and induced levels of damage after 10 Gy were similar. After 6 hours, however, SAT1 depleted cells demonstrated a marked decrease in the ability to repair compared to control (Figure 6A). The HR protein BRCA1 was then used to identify HR foci in U87MG and D54MG SAT1 knockdown and control cells at 0 or 6 hours following exposure to IR (Figure 6B). While control cells displayed a potent induction of BRCA1 foci, SAT1 knockdown cells from both the U87MG and D54MG lines exhibited diminished capacity to produce foci. Both the number of cells with foci (64% vs. 16% for U87MG shGFP vs. shSAT1; and 66% vs. 32% for D54 shGFP vs shSAT1), and the number of foci per cell were reduced (Figure 6C). As the proportion of cells in S and G2/M phases were unchanged between control and SAT1 knockdowns, the data suggest that SAT1 deficient cells have reduced HR signaling.
Figure 6. SAT1 depletion decreases BRCA1 levels.

A. Comet assay of shGFP and shSAT1 D54MG cells after 10 Gy IR at indicated times. B. Immunofluorescence of BRCA1 foci. C. Quantification of # foci/cell. D. Western blotting of HR proteins and acetylated or total histone H3 in shSAT1 cells versus controls. E. qRT-PCR of BRCA1 in shSAT1 cells versus controls. F. ChIP of acetylated H3 at the BRCA1 and β-actin promoters in shSAT1 GBM cells versus controls. G. Western analysis for acetylated H3 in D54MG shGFP and shSAT1 cells with TSA. H. qRT-PCR of BRCA1 in D54MG shGFP and shSAT1 cells with TSA. Error bars show standard deviations.
We next asked whether HR pathway components are altered in SAT1 knockdown cells. Western blotting revealed that BRCA1 itself is severely diminished in SAT1 depleted cells (Figure 6D). In contrast, expression levels of MRE11, NBS1, and Rad51 remained unchanged. As BRCA1 could be regulated at various points, we examined mRNA and found consistent reductions of expression of BRCA1 after SAT1 depletion, suggesting regulation of BRCA1 by SAT1 is at the transcriptional level (Figure 6E).
SAT1 knockdown reduces H3 acetylation at the BRCA1 promoter
Increased H3 acetylation at the BRCA1 promoter has been reported to regulate BRCA1 transcription (24). Recent studies have demonstrated a link between elevated levels of spermidine and histone deacetylation in aging yeast (25). We hypothesized that alterations in SAT1 levels may affect BRCA1 transcription through regulation of chromatin access via histone acetylation. We first assessed global histone H3 acetylation on cell lysates and found in both U87MG and D54MG cell lines that SAT1 knockdown resulted in decreased N-terminal H3 acetylation (Figure 6G). We next assessed the levels of acetyl-H3 at the BRCA1 promoter via chromatin immunoprecipitation (ChIP) and found similarly that the knockdown populations exhibited decreased levels of acetylated H3 localized to the BRCA1 promoter. In contrast, the β-actin promoter displayed mild increases in H3 acetylation (Figure 6F). To determine whether reversing the reduction in histone acetylation would restore BRCA1 levels, we treated D54MG shGFP and shSAT1 cells with the HDAC inhibitor TSA and found increased H3 acetylation and induction of BRCA1 mRNA (Figure 6H). Thus the data suggest that SAT1 promotes acetylation of histone H3 on the BRCA1 promoter, thereby inducing BRCA1 expression and facilitating HR repair.
SAT1 and BRCA1 expression correlate in Glioma
In order to validate the concept that SAT1 mediates poor outcome and potentially response to therapy through BRCA1 expression, we sought to determine if SAT1 and BRCA1 expression correlate in the Rembrandt data presented in Figure 1D. Indeed, SAT1 and BRCA1 expression correlate with statistical significance (p=0.0118) (Figure 7A), and BRCA1 expression revealed an outcome benefit for patients with lower expression as opposed to higher expression (p=0.0001) (Figure 7B). Together the clinical data support the importance of both SAT1 and BRCA1 in predicting patient outcomes, and suggest BRCA1 is at least partially responsible for the radioresistance of SAT1 overexpressing tumors.
Figure 7. SAT1 and BRCA1 expression correlate in Rembrandt.

A. Correlated expression of SAT1 and BRCA1 in Rembrandt data of all glioma. B. Survival of patients with glioma dichotomized by BRCA1 expression.
Discussion
In the present study, we have identified a novel mechanism of radioprotection in glioblastoma linked to the frequent overexpression of the polyamine catabolic enzyme SAT1. SAT1 is overexpressed almost uniformly in GBM tumor samples compared to normal brain, as assessed bioinformatically in the Oncomine and TCGA databases. Depletion of SAT1 radiosensitized multiple adherent cell lines and neurosphere lines, and tumors in mice. Mechanistically, we found that SAT1 promotes HR by controlling BRCA1 expression and BRCA1 foci following damage to DNA. We elucidated a novel function for SAT1 as a mediator of histone H3 acetylation controlling BRCA1 gene expression. Together our findings highlight a previously unappreciated role for the polyamine metabolic pathway in regulating radiation responses in tumors, and identify a potential target for radiosensitizers to improve therapeutic efficacy.
Polyamines, including putrescine, spermine and spermidine, are ubiquitous cellular constituents present in millimolar quantities. As positively-charged small molecules, polyamines bind negatively charged macromolecules throughout cells including nucleic acids and proteins, and regulate many processes (26). Polyamines promote protection to IR by inducing DNA compaction, producing a less susceptible target to direct radiation damage (8, 9, 27). SAT1, however, is the rate-limiting enzyme in polyamine catabolism, driving acetylation and subsequent degradation or excretion of polyamines. Indeed, acetylation has been shown to result in removal of polyamines from chromatin leading to chromatin relaxation and opening (28). Thus existing data do not provide a mechanistic explanation for the role of SAT1 in radioprotection.
A recent proteomics study drew a connection between polyamine acetylation in radioresponse, finding acetyl-spermidine as a blood-borne biomarker in mice exposed to whole-body radiation (29). Aged mice showed a diminished ability to induce acetyl-spermidine following exposure, suggesting that aging-related decreases in DNA repair correlate with reduced polyamine catabolism. While the mechanism behind the observations were not fully uncovered, the data intriguingly suggest acetylated polyamines may provide a non-invasive marker of radiation responses.
To assess the effect of SAT1 expression on homeostatic polyamine levels in our system, we quantified cellular polyamines and found negligible differences. While not surprising that cells acclimate to decreased SAT1 by reducing expression of ODC1 via described feedback loops (21), the data suggest that regulating global polyamine levels is not the mechanism of radioprotection by SAT1. Recent data have demonstrated however that controlling localized polyamine content is also an important function of SAT1. SAT1 has been found to modulate cell migration through an interaction with α9β1 integrin, localizing polyamine catabolism to membrane-bound potassium channels. Because spermine and spermidine are potassium channel blockers, the association of SAT1 with α9β1 in focal adhesions at the leading edges of migrating cells allows ion channel regulation (30). Thus, our data do not rule out that controlling localized polyamines at damage sites may be a mode by which SAT1 can affect radiation response.
Alternatively, the findings point to a novel mechanism of SAT1 in regulating histone acetylation. Excess polyamines have been recently shown to prolong lifespan via elevated autophagy due to alterations in histone acetylation and subsequent induction of autophagy genes (25). Further, depletion of polyamines led to hyperacetylation of histone H3, generation of reactive oxygen species, and ultimately necrosis. Conversely, spermidine treatment caused deacetylation of histone H3 through inhibition of histone acetyltransferases. Conceptually, the findings agree with ours, that by decreasing polyamines, SAT1 would promote acetylation of H3 and subsequently alter gene expression. In our studies, however, the importance of polyamines was unclear. As an acetyl-transferase, it is unknown whether SAT1 can also target proteins, as no specific targets have been identified beyond an autoacetylation activity (7). In yeast, the hpa1 and hpa2 genes, though structurally unrelated to SAT1, have been found to be capable of acetylating both polyamines and histones (31). In vitro we were unable to detect a direct acetylation activity of purified SAT1 on purified histone H3, however, leaving open the question of a direct or indirect effect of SAT1 on histones. Alternatively, the observation that the HDAC inhibitor reversed the acetylation status of H3 and rescued BRCA1 levels may suggest that SAT1 can function as an HDAC inhibitor. Future studies will be necessary to bear out this possibility.
Polyamine analogues have long been a focus of development of anticancer agents (32, 33). The mechanism of cell death by analogues is due to potent induction of SAT1 (>1000 fold), leading to polyamine depletion and excess production of reactive oxygen species (21). Ectopic overexpression of SAT1 is also toxic due to inhibition of general protein translation (34). In order to avoid complications of non-physiological alterations in SAT1 levels, we focused on shRNAs that reduced SAT1 levels to the range observed in normal brain (3–5 fold reductions in the GBM lines). In this context, our data suggest that decreasing SAT1 may have therapeutic benefit. Further, sensitizing brain tumors with an SAT1 inhibitor may have relevance in combination with Parp inhibitors, which take advantage of alterations in the BRCA1 pathway and have recently been shown to sensitize brain tumor initiating cells to radiation (35).
In summary, we have defined a novel function for the polyamine catabolic enzyme SAT1 in mediating HR repair in brain tumors through the epigenetic regulation of BRCA1. Our findings describe a new mechanism for SAT1 to regulate chromatin, and suggest that inhibition of SAT1 may sensitize brain tumors to radiation and increase therapeutic responses. Together the findings contribute to our understanding of radioresistance in GBM, and may have yet even broader implications due to the overexpression of polyamine metabolic enzymes in a variety of other tumors (36).
Supplementary Material
Acknowledgements
We thank Dr. Ravi Patel for colony-counting software. Core facilities of the Case Comprehensive Cancer Center, supported by P30CA43703, were used: Radiation Resources, Cytometry and Imaging Microscopy, Small Animal Imaging. Supported by 119999-IRG-91-022-18-IRG from the American Cancer Society.
Footnotes
Conflict of interest statement: The authors declare there are no conflicts of interest.
References
- 1.Krex D, Klink B, Hartmann C, von Deimling A, Pietsch T, Simon M, et al. Long-term survival with glioblastoma multiforme. Brain : a journal of neurology. 2007;130:2596–2606. doi: 10.1093/brain/awm204. [DOI] [PubMed] [Google Scholar]
- 2.Wen PY, Kesari S. Malignant gliomas in adults. The New England journal of medicine. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 3.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. The New England journal of medicine. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 4.Chakravarti A, Zhai GG, Zhang M, Malhotra R, Latham DE, Delaney MA, et al. Survivin enhances radiation resistance in primary human glioblastoma cells via caspase-independent mechanisms. Oncogene. 2004;23:7494–7506. doi: 10.1038/sj.onc.1208049. [DOI] [PubMed] [Google Scholar]
- 5.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 6.Hatanpaa KJ, Burma S, Zhao D, Habib AA. Epidermal growth factor receptor in glioma: signal transduction, neuropathology, imaging, and radioresistance. Neoplasia. 2010;12:675–684. doi: 10.1593/neo.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pegg AE. Spermidine/spermine-N(1)-acetyltransferase: a key metabolic regulator. Am J Physiol Endocrinol Metab. 2008;294:E995–E1010. doi: 10.1152/ajpendo.90217.2008. [DOI] [PubMed] [Google Scholar]
- 8.Chiu S, Oleinick NL. Radioprotection of cellular chromatin by the polyamines spermine and putrescine: preferential action against formation of DNA-protein crosslinks. Radiation research. 1998;149:543–549. [PubMed] [Google Scholar]
- 9.Warters RL, Newton GL, Olive PL, Fahey RC. Radioprotection of human cell nuclear DNA by polyamines: radiosensitivity of chromatin is influenced by tightly bound spermine. Radiation research. 1999;151:354–362. [PubMed] [Google Scholar]
- 10.Sy D, Hugot S, Savoye C, Ruiz S, Charlier M, Spotheim-Maurizot M. Radioprotection of DNA by spermine: a molecular modelling approach. International journal of radiation biology. 1999;75:953–961. doi: 10.1080/095530099139719. [DOI] [PubMed] [Google Scholar]
- 11.Murr R, Loizou JI, Yang YG, Cuenin C, Li H, Wang ZQ, et al. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nature cell biology. 2006;8:91–99. doi: 10.1038/ncb1343. [DOI] [PubMed] [Google Scholar]
- 12.Sterner DE, Berger SL. Acetylation of histones and transcription-related factors. Microbiology and molecular biology reviews : MMBR. 2000;64:435–459. doi: 10.1128/mmbr.64.2.435-459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Masumoto H, Hawke D, Kobayashi R, Verreault A. A role for cell-cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature. 2005;436:294–298. doi: 10.1038/nature03714. [DOI] [PubMed] [Google Scholar]
- 14.Gujrati M, Malamas A, Shin T, Jin E, Sun Y, Lu ZR. Multifunctional Cationic Lipid-Based Nanoparticles Facilitate Endosomal Escape and Reduction-Triggered Cytosolic siRNA Release. Molecular pharmaceutics. 2014;11:2734–2744. doi: 10.1021/mp400787s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krieg AJ, Hammond EM, Giaccia AJ. Functional analysis of p53 binding under differential stresses. Molecular and cellular biology. 2006;26:7030–7045. doi: 10.1128/MCB.00322-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kass EM, Helgadottir HR, Chen CC, Barbera M, Wang R, Westermark UK, et al. Double-strand break repair by homologous recombination in primary mouse somatic cells requires BRCA1 but not the ATM kinase. Proc Natl Acad Sci U S A. 2013;110:5564–5569. doi: 10.1073/pnas.1216824110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clark MJ, Homer N, O'Connor BD, Chen Z, Eskin A, Lee H, et al. U87MG decoded: the genomic sequence of a cytogenetically aberrant human cancer cell line. PLoS genetics. 2010;6:e1000832. doi: 10.1371/journal.pgen.1000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ishii N, Maier D, Merlo A, Tada M, Sawamura Y, Diserens AC, et al. Frequent co-alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain pathology. 1999;9:469–479. doi: 10.1111/j.1750-3639.1999.tb00536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer research. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 20.Balasundaram D, Tyagi AK. Polyamine--DNA nexus: structural ramifications and biological implications. Molecular and cellular biochemistry. 1991;100:129–140. doi: 10.1007/BF00234162. [DOI] [PubMed] [Google Scholar]
- 21.Casero RA, Pegg AE. Polyamine catabolism and disease. The Biochemical journal. 2009;421:323–338. doi: 10.1042/BJ20090598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barker CA, Powell SN. Enhancing radiotherapy through a greater understanding of homologous recombination. Seminars in radiation oncology. 2010;20:267–273. e3. doi: 10.1016/j.semradonc.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Litman R, Peng M, Jin Z, Zhang F, Zhang J, Powell S, et al. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer cell. 2005;8:255–265. doi: 10.1016/j.ccr.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 24.Rice JC, Futscher BW. Transcriptional repression of BRCA1 by aberrant cytosine methylation, histone hypoacetylation and chromatin condensation of the BRCA1 promoter. Nucleic acids research. 2000;28:3233–3239. doi: 10.1093/nar/28.17.3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eisenberg T, Knauer H, Schauer A, Buttner S, Ruckenstuhl C, Carmona-Gutierrez D, et al. Induction of autophagy by spermidine promotes longevity. Nature cell biology. 2009;11:1305–1314. doi: 10.1038/ncb1975. [DOI] [PubMed] [Google Scholar]
- 26.Pegg AE, Casero RA., Jr Current status of the polyamine research field. Methods in molecular biology. 2011;720:3–35. doi: 10.1007/978-1-61779-034-8_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ouameur AA, Tajmir-Riahi HA. Structural analysis of DNA interactions with biogenic polyamines and cobalt(III)hexamine studied by Fourier transform infrared and capillary electrophoresis. The Journal of biological chemistry. 2004;279:42041–42054. doi: 10.1074/jbc.M406053200. [DOI] [PubMed] [Google Scholar]
- 28.Liu B, Sutton A, Sternglanz R. A yeast polyamine acetyltransferase. The Journal of biological chemistry. 2005;280:16659–16664. doi: 10.1074/jbc.M414008200. [DOI] [PubMed] [Google Scholar]
- 29.Manna SK, Krausz KW, Bonzo JA, Idle JR, Gonzalez FJ. Metabolomics reveals aging-associated attenuation of noninvasive radiation biomarkers in mice: potential role of polyamine catabolism and incoherent DNA damage-repair. Journal of proteome research. 2013;12:2269–2281. doi: 10.1021/pr400161k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.deHart GW, Jin T, McCloskey DE, Pegg AE, Sheppard D. The alpha9beta1 integrin enhances cell migration by polyamine-mediated modulation of an inward-rectifier potassium channel. Proc Natl Acad Sci U S A. 2008;105:7188–7193. doi: 10.1073/pnas.0708044105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sampath V, Liu B, Tafrov S, Srinivasan M, Rieger R, Chen EI, et al. Biochemical characterization of Hpa2 and Hpa3, two small closely related acetyltransferases from Saccharomyces cerevisiae. The Journal of biological chemistry. 2013;288:21506–21513. doi: 10.1074/jbc.M113.486274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seiler N. Thirty years of polyamine-related approaches to cancer therapy. Retrospect and prospect. Part 2. Structural analogues and derivatives. Current drug targets. 2003;4:565–585. doi: 10.2174/1389450033490876. [DOI] [PubMed] [Google Scholar]
- 33.Seiler N. Thirty years of polyamine-related approaches to cancer therapy. Retrospect and prospect. Part 1. Selective enzyme inhibitors. Current drug targets. 2003;4:537–564. doi: 10.2174/1389450033490885. [DOI] [PubMed] [Google Scholar]
- 34.Mandal S, Mandal A, Johansson HE, Orjalo AV, Park MH. Depletion of cellular polyamines, spermidine and spermine, causes a total arrest in translation and growth in mammalian cells. Proc Natl Acad Sci U S A. 2013;110:2169–2174. doi: 10.1073/pnas.1219002110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Venere M, Hamerlik P, Wu Q, Rasmussen RD, Song LA, Vasanji A, et al. Therapeutic targeting of constitutive PARP activation compromises stem cell phenotype and survival of glioblastoma-initiating cells. Cell death and differentiation. 2014;21:258–269. doi: 10.1038/cdd.2013.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Casero RA, Jr, Marton LJ. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nature reviews Drug discovery. 2007;6:373–390. doi: 10.1038/nrd2243. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.