

Abstract
Clinical translation of tubulin inhibitors for treating melanoma is limited by multidrug efflux transporters, poor aqueous solubility and dose limiting peripheral toxicities. Tubulin inhibitors with efficacy in taxane resistant cancers are promising drug candidates and can be used as single agent or in conjunction with other chemotherapy. Systemic therapy of such a novel tubulin inhibitor, 2-(1H-Indol-5-yl) thiazol-4-yl) 3, 4, 5-trimethoxyphenyl methanone (abbreviated as LY293) is limited by its poor aqueous solubility. The objective of this study was to design a polymeric nanocarrier for systemic administration of LY293 to improve tumor accumulation and reduce side effects of tubulin inhibitor in a lung metastasis melanoma mouse model. Methoxy polyethylene glycol-b-poly (carbonate-co-lactide) (mPEG-b-P (CB-co-LA)) random copolymer was synthesized and characterized by 1H NMR and gel permeation chromatography (GPC). Polymeric nanoparticles were formulated using o/w emulsification method with a mean particle size of 150 nm and loading efficiency of 7.40%. Treatment with LY293 loaded nanoparticles effectively inhibited the proliferation of melanoma cells in vitro and exhibited concentration dependent cell cycle arrest in G2/M phase. Mitotic arrest activated the intrinsic apoptotic machinery by increasing the cellular levels of cleaved poly ADP ribose polymerase (PARP) and fraction of sub G1 cells. In vivo, LY293 loaded nanoparticles significantly inhibited the proliferation of highly aggressive metastasized melanoma in a syngeneic lung metastasis melanoma mouse model without toxicity to vital organs. In conclusion, we have designed a promising polymeric nanocarrier for systemic delivery of LY293 for treating metastatic melanoma while minimizing the toxicity associated with the administration of cosolvents.
Introduction
Melanoma is the fifth most common cancer in the Unites States and its incidence is increasing by 1.9% annually (1). Due to the high propensity of melanoma for metastasis to lungs, liver, brain and bones, its treatment remains a challenge and is associated with the poor survival rate. While surgery and radiation are the first line therapy for treating localized melanoma, systemic therapy is the cornerstone of clinical management of metastatic melanoma (2). Currently dacarbazine (DTIC) and kinase inhibitors are the FDA approved drugs for the treatment of metastatic melanoma. DTIC, an alkylating agent, exhibits modest efficacy and does not significantly prolong the survival benefit (3) (less than 5 % complete remission), whereas resistance develops invariably upon treatment with kinase inhibitors which restricts the prolonged systemic therapy with these inhibitors (4, 5).
Tubulin inhibitors like vinca alkaloids and taxanes such as paclitaxel have been clinically prescribed as a single agent or in combination with other chemotherapeutic agents (6, 7). Encouraged by the clinical utility of tubulin inhibitors, we have previously reported the synthesis of tubulin inhibitors with antiproliferative IC50 in nanomolar range for the treatment of metastatic melanoma (8, 9). LY293, a 5 indole derivative analog, binds to colchicine binding site and does not exhibit clinically prevalent drug resistance mechanism such as multidrug resistance (MDR) protein, breast cancer resistance protein (BCRP) and P-glycoprotein (P-gp) (9, 10). Like paclitaxel, poor aqueous solubility of LY293 necessitates the use of cosolvent for its systemic delivery (11). Due to poor aqueous solubility, we have previously administered PEG300 solution of LY293 structure analog through intraperitoneal route for efficacy studies in lung metastasis melanoma model (11). However, prolonged administration of drugs using cosolvents as a solubilizing agent is associated with hemolysis and acute hypersensitivity due to cosolvents which require administration of antihistamines and steroids (12, 13). Additionally, non-specific distribution of tubulin inhibitors like paclitaxel is associated with dose limiting toxicity of myelosuppression and peripheral neuropathy. Biodegradable polymeric nanoparticles are attractive formulation strategies for solubilizing the hydrophobic drug within their core and modulate the drug release (14–16). Modulation of drug release and preferential accumulation of nanoparticles at tumor through enhanced permeation and retention (EPR) effect reduces dose limiting toxicity like peripheral neuropathy and myelosuppression (17, 18). Amphiphillic poly(ethylene glycol) (PEG) based diblock polyester copolymers of lactide, glycolide and caprolactone have been synthesized for preparing nanoparticles for delivery of hydrophobic drugs (16). We have previously engineered the hydrophobic core of the polyester component of the copolymer into polyester/polycarbonate component to improve the drug loading of (2-(1H-Indol-5-yl) thiazol-4-yl) 3, 4, 5-trimethoxyphenyl methanone (LY293) (15, 19).
In this study, we have synthesized mPEG-b-P (CB-co-LA) copolymer for effective encapsulation of LY293 in nanocarriers as well as for enhanced tumor accumulation compared to free drug. Synthesized mPEG-b-P (CB-co-LA) diblock random copolymer of lactide and carbonate was characterized and utilized for nanoparticle formulation of LY293. Concentration dependent effect of LY293 loaded nanoparticles on cell cycle and mechanism of apoptosis was studied. Finally, efficacy of LY293 loaded nanoparticles in syngeneic B16F10 lung metastatic melanoma mouse model was evaluated against cosolvent solution of LY293.
Materials and Methods
Materials
2, 2-Bis (hydroxymethyl) propionic acid, methoxy poly (ethylene glycol) (mPEG, Mn = 5000), 8-diazabicycloundec-7-ene (DBU), 1, 5, 7-triazabicyclodecene (TBD) benzyl bromide, 2,2-bis(hydroxymethyl) propionic acid and L-lactide were purchased from Sigma-Aldrich (St. Louis, MO) and used as received. Dulbecco’s Modified Eagle Medium (DMEM) medium was purchased from Invitrogen (Carlsbad, CA). All primary antibodies were purchased from Cell Signaling (Beverly, MA). B16F10 mouse melanoma cell line and A375 human melanoma cell line was acquired from American Type Culture Collection and no authentication was performed by us. All other reagents were obtained from Sigma–Aldrich (St. Louis, MO) unless otherwise stated and were used as received. Cells were cultured in DMEM supplemented with 10% FBS (Atlanta Biologicals) and 1% antibiotic/antimycotic mixture (Invitogen).
Synthesis of mPEG-b-P (CB-co-LA)
Benzyl 2, 2-bis (methylol) propionate was synthesized as reported previously by reacting 2,2-bis(hydroxymethyl) propionic acid with benzyl bromide at 100°C for 15 h (15). Monomer 5-methyl-5-benzyloxycarbonyl-1, 3-dioxane-2-one (MBC) was synthesized by reacting the intermediate with triphosgene at −78°C in pyridine and dichloromethane. Random MBC and lactide block copolymer (mPEG-b-P (CB-co-LA)) of target molecular weight of 30,000 Da was obtained by ring opening polymerization of L-lactide and MBC using mPEG as macroinitator and 8-diazabicycloundec-7-ene (DBU) (2.5 mol relative to mPEG) catalyst (20, 21). Reaction was carried out in DCM at room temperature for 6 h, after which benzoic acid was added to quench the catalyst. The resulting solution was concentrated to approximately 50% of the initial volume and added dropwise into excess of cold isopropanol for polymer precipitation. Following synthesis, the copolymer was characterized by 1H NMR and GPC. For 1H NMR, chemical shifts were calibrated using tetramethylsilane as reference and reported as parts per million. The molecular weight and polydispersity index (PDI) of mPEG-b-P (CB-co-LA) polymers were determined by GPC using a Shimadzu GPC system equipped with a Styragel HR 4E GPC column and a differential refractive index detector. Dimethyl formamide (DMF) was used as an eluent at a flow rate of 0.5 mL/min. Narrow PEG standards (5,800–71,700 g/mol) from American polymer standards corp were used for generating standard curves and the data was processed using LC Solution ver.1.21 GPC option software.
Preparation and characterization of nanoparticles
Blank and LY293 loaded nanoparticles were prepared by o/w emulsification using 1% polyvinyl alcohol as an emulsifier (19). mPEG-b-P (CB-co-LA) and drug dissolved in dichloromethane and acetone mixture (1:1 ratio) were emulsified using a Misonix ultrasonic liquid processor (Farmingdale, NY) at amplitude of 20 for 2 min. Organic phase was removed by a rotavapor to obtain stabilized nanoparticles and concentrated by ultracentrifugation at 20,000 rpm for 30 min followed by washing with water to remove polyvinyl alcohol and unentrapped drug. Mean particle diameter and size distribution of drug-loaded nanoparticles were determined via dynamic light scattering (DLS) using a Malvern Zetasizer Nano Series (Worcestershire, UK). Surface morphology and particle size of nanoparticles was determined using a transmission electron microscope (TEM) (JEM-100S Japan). Suspension of nanoparticles on a copper grid was negatively stained with NanoVan®. The grid was visualized at 60 kV with magnifications ranging from 50, 000× to 100,000×.
Drug loading was estimated by reverse phase high performance liquid chromatography (RP-HPLC, Shimadzu) at 290 nm wavelength using a C18 column (250 mm × 4.6 mm, Alltech, Deerfield, IL). Mobile phase composed of 65:35 V/V of acetonitrile and water was used. LY293 concentration was calculated using the peak area from a standard curve with R2=0.9987 and lower quantification limit of 73.42 ng/ml. Drug loading was determined by the following formula
Cell viability assay
A375 and B16F10 cells were cultured in DMEM media supplemented with 10% fetal bovine serum (FBS) and 1% antibiotic–antimycotic at 37°C in humidified environment of 5% CO2. Cells were seeded overnight in 24 well plates at a density of 25,000 cells per well and thereafter treated with DMSO solution of LY293 and LY293 loaded nanoparticles for 72 h. At the end of 72 h, cell culture medium was replaced with 100 μL of 0.5 mg/mL 3-(4, 5-dimethyl-thiazol-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT) solution and incubated for 3 h at 37°C. Cell culture medium was removed and formazan crystals were dissolved in 500 μL of DMSO for measuring the absorbance. Absorbance was measured by a microplate reader at a wavelength of 560 nm. Cell viability was expressed as the percentage of the control group. DMSO control and blank nanoparticles were included in all experiments.
Propidium iodide staining and cell cycle analysis
A375 and B16F10 cells were cultured overnight in a 6 well plate followed by thymidine blockade for 12 h. Cells were then incubated with LY293 loaded nanoparticles for 48 h. Treated cells were trypsinized and fixed in 90% ice-cold ethanol overnight. After washing with PBS containing RNase (1 mg/mL), cell pellet was re-suspended in 5 μg/mL propidium iodide staining solution for 30 min at the room temperature. Cell cycle distribution was measured by flow cytometry (Becton, Dickinson, NJ, USA). Results from 10,000 fluorescent events were obtained for cell cycle analysis.
Western Blot Analysis
A375 cells cultured overnight in a 12 well plate were treated with LY293 for 24 h. Subsequently, cells were lysed using RIPA buffer (Sigma-Aldrich, St. Louis, MO) and protein concentration was measured with bicinchoninic acid (BCA) protein assay kit (Pierce, Rockford, IL). The lysate was boiled for 5 min, subjected to a 15 % SDS-PAGE and transferred to a PVDF membrane using iBlot™ system (Invitrogen, Carlsbad, CA). Membranes were blocked with 3% BSA in 1× Tris buffered saline (TBS) at room temperature for 1h and then incubated with primary antibodies at 4 °C overnight, followed by incubation with anti-goat or anti-rabbit IRDye 800CW secondary antibodies for 1h at room temperature. All the blots were re-probed with total β-actin antibody as control. The signal of target proteins was detected Li-COR Odyssey® infrared imaging system (Li-COR, Lincoln, NE).
In vivo anticancer efficacy study
All animal experiments were performed in accordance with the NIH animal use guidelines and protocol approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Nebraska Medical Center (UNMC, Omaha, NE). Lung metastasis in C57/BL6 mice (6–8 weeks old, weight around 20 g) was used as a tumor model. Mice were injected intravenously with 100 μL of B16F10 cell suspension containing 105 cells through tail vein. Post one week, mice were divided into three groups (n =6): control, LY293 in 35 % of cosolvent (composed of propylene glycol, cremophor EL and ethanol (50:30:20 ratio) and 65 % of dextrose solution and LY293 loaded nanoparticles. Formulations were injected into the tail vein of mice thrice a week at the dose of 15 mg/kg. Mice were sacrificed at the end of the treatment, and lungs were separated and expanded with 10% formalin buffer. The number of lung metastasis nodules was noted and tumor index was calculated using formula 1*a + 2*b + 3*c where a is the number of nodules with size <1 mm; b is the number of nodules with size between 1 and 2 mm; and c is number of nodules with size >2 mm. Animal mobility and body weight were monitored during the entire experiment period to assess any toxicity. In addition, tumor tissues were fixed in formalin buffer and sections were stained with hematoxylin and eosin (H&E). For immunohistochemistry (IHC) analysis, sections of 5–7 μm thickness were cut and immunostained with rabbit anti-Caspase-3, anti-Ki67 and anti-S100 primary antibodies, respectively. The immunoreactivity was detected using goat anti-rabbit IgG, H&L chain specific peroxidase conjugate and subsequent incubation with DAB substrate.
Statistical analysis
Statistical significance of the difference between the two groups was determined by an unpaired Student’s t-test and between several groups by one-way analysis of variance.
Results
Synthesis and characterization of mPEG-b-P(CB-co-LA)
DBU, an organic base with low nucleophilicity and mPEG as macroinitator was utilized for synthesis of mPEG-b-P(CB-co-LA) copolymer in solution at ambient temperature with high conversion rate. Synthesized mPEG-b-P(CB-co-LA) copolymer was characterized by 1H NMR and GPC. 1H NMR spectra was used to confirm the polymerization and calculation of molecular weight of the copolymer (Figure 1A). The emergence of the multiplet peak at δ: 4.25–4.35 ppm, δ: 7.3 ppm from the carbonate monomer and one methylene proton of lactide at δ= 5.12 ppm confirms the successful ring opening polymerization and formation of mPEG-b-P(CB-co-LA) copolymers. The relative molecular weight of the LA and MBC block was estimated based on the peak areas of three fifth of the 5.12 ppm signal and the five protons associated with the phenyl ring in the carbonate monomer at 7.3 ppm. From 1H NMR, calculated molecular weight of mPEG-b-P(CB-co-LA) was 31200 with 90% conversion rate for LA and 60% conversion rate for MBC (Table 1). As PEG was used as a macroinitator for ring opening polymerization, narrow PEG standards (5,800, 10,225, 14,500, 20000, 31,380, 50,450 and 71,700 g/mol) were used to generate standard curve for GPC analysis. We characterized mPEG-b-P(CB-co-LA) copolymer by GPC, which indicated a narrow molecular weight distribution with a PDI of 1.20 (Figure 1B and Table 1). The molecular weight values determined by GPC were 29671 for weight average molecular weight (Mw) and 24650 for number average molecular weight (Mn). NMR and GPC data confirmed the synthesis of copolymer with controlled molecular weight with narrow molecular weight distribution.
Figure 1. Synthesis and characterization of mPEG-b-P (CB-co-LA) copolymer.

(A) 1H NMR spectrum of mPEG-b-P(CB-co-LA) in DMSO-d6 and chemical structure of mPEG-b-P(CB-co-LA) copolymer. (B) Representative GPC chromatogram of mPEG-b-P(CB-co-LA).
Table 1.
Characterization of mPEG-b-P (CB-co-LA) copolymer by GPC for molecular weight and polydispersity index. Mw and Mn weight and number average molecular weights respectively as determined by GPC; PDI (Mw/Mn).
| Polymer | Mn (NMR) | Mw (GPC) | Mn (GPC) | PDI |
|---|---|---|---|---|
| PEG114-b-P(CB40-co-LA225) | 31200 | 29671 | 24650 | 1.20 |
Formulation, characterization and in vitro cytotoxicity of nanoparticles
The mean particle size of mPEG-b-P(CB-co-LA) nanoparticles was 150 nm with a PDI of 0.068 as determined by DLS. Drug loading and ultracentrifugation did not alter the particle size and its uniform distribution. TEM confirmed the uniform spherical particle of 50–70nm (Figure 2A). Drug loading calculated by HPLC was 2.5, 4.11 and 7.40 % at the theoretical loadings of 2.5, 5 and 10 %, respectively (Table 2). LY293 loaded nanoparticles inhibited the cancer cell proliferation as efficiently as DMSO solution of LY293 with an IC50 of 17.5 nM in A375 and B16F10 cells (Figure 3). Blank nanoparticles did not elicit any cytotoxicity to the cells as expected with biodegradable polymers.
Figure 2. Formulation and characterization of LY293 loaded mPEG-b-P (CB-co-LA) nanoparticles.

Transmission electron microscopy (TEM) of nanoparticles using methylamine vanadate staining.
Table 2.
Effect of theoretical loading on particle size and loading efficiency
| Theoretical loading | Drug loading | % Loading efficiency | Particle size (nm) | PDI |
|---|---|---|---|---|
| 2.5% | 2.5 ± 0.23% | 100.0 | 148 ± 10 | 0.090 |
| 5% | 4.11 ± 0.21% | 82.2 | 148 ± 10 | 0.080 |
| 10% | 7.40 ± 0.36% | 74.0 | 154 ± 6 | 0.10 |
Figure 3. Cell cytotoxicity of DMSO solution of LY293 and LY293 loaded nanoparticles.

(A) A375 and (B) B16F10 cell viability after 72h of treatment. Cell viability was determined by MTT Assay and expressed as % of control. Blank nanoparticles used as control for LY293 loaded nanoparticles group. Results are expressed as the mean ± SD (n = 4).
Effect of LY293 loaded mPEG-b-P(CB-co-LA) nanoparticles on cell cycle and apoptosis
Tubulin polymerization inhibitors bind to tubulin and arrest the cell cycle in G2/M phase. As shown in Figure. 4A&B, 48h treatment of A375 and B16F10 cells with LY293 loaded nanoparticles led to cell cycle arrest in G2/M phase while no effect was observed upon treatment with blank nanoparticles. Anti-mitotic activities of LY293 resulted in significant increase in G2/M phase from 14.13±0.60 % in the control group to 54.33±0.085% and 76.19±0.94 % at 15 nM and 250 nM of LY293 loaded nanoparticles group in A375 (p< 0.005).
Figure 4. Concentration dependent effect of LY293 loaded nanoparticles on cell cycle distribution.

Cells were treated with LY293 loaded nanoparticles (15 and 250 nM) for 48 h, stained with propidium iodide and analyzed on a flow cytometer (A) B16F10 (B) A375. Results are expressed as the mean ± SD (n=3). ***p<0.0005 for cells in G2/M phase using Student’s unpaired t test. (C) Histogram showing cell cycle distribution. Results were calculated from more than 10000 modeled events using ModFit LT V3.3.11(Win)
Disruption of the cell cycle progression and arrest of cells in G2/M activates apoptotic cell mechanism. Treatment with LY293 loaded nanoparticles significantly increased the protein levels of cleaved PARP with a corresponding decrease in the levels of total PARP levels (Figure 5A). In addition, the cells in sub G1 phase were also increased from 2.005±0.25% in control to 28.35± 0.92 % and 41.9 ±1.76% for 48 h at 15 and 250 nM, respectively (p<0.005). The increase of cell population in sub G1 phase indicated that apoptotic cells were increased as LY293 loaded nanoparticle treatment arrested cells in G2/M phase (Figure 5B and 5C).
Figure 5. Effect of treatment with LY293 nanoparticles on apoptosis and expression of apoptotic proteins.

(A) A375 were treated with LY293 loaded nanoparticles at 15 and 20 nm, protein was isolated at 24h. Total and cleaved PARP expression at protein level was determined using Western blot. Representative beta actin blot is shown. (B) A375 cells were treated with LY293 loaded nanoparticles for 24 h and quantitative analysis of apoptotic cells as determined by staining with propidium iodide and analyzed on a flow cytometer. Results are expressed as the mean ± SD (n=3). **p<0.005 using Student’s unpaired t test. (C) Histogram representation of flow cytometry modeled events for quantitative analysis of apoptotic cells.
In vivo lung metastasis model
Melanoma is highly metastatic, and lung is one of the major target organs for metastasis. We used an experimental lung metastasis mouse model established via tail vein injection of melanoma cells to test the efficacy of LY293 loaded nanoparticles in preventing tumor lung metastasis. After 2 weeks of experimental treatment, the tumor index was 84, 38, and 30 in control, the cosolvent solution of LY293 and LY293 loaded nanoparticle groups, respectively (Figure 6A). Tumor index was significantly different between the control and LY293 loaded nanoparticle treated groups (p<0.00005). Treatment with LY293 significantly reduced the nodule count and black cellular mass associated with the lungs (Figure 7). H&E staining of lung tissue confirmed the inhibition of proliferation of metastatic cells. As compared to the control, LY293 treated group exhibited widely patent alveoli (black arrow, Figure 7) and normal lung parenchyma with limited mass of metastatic growth (Solid arrow, Figure 7). In the control group, normal lung parenchyma was not visible and was completely covered with the dense metastatic tissue. Higher magnification images showed both pigmented (black spots as indicated by arrow) and non-pigmented dense metastatic growth in the control group. Significant inhibition of proliferation of melanoma cells by LY293 loaded nanoparticle treatment group was further confirmed by the reduced density of S100 immunostains (Figure 8). Cleaved caspase-3 staining showed significant apoptosis in LY293 loaded nanoparticle group as compared to the control group. Ki67 positive staining showed significant proliferation in control group as compared to LY293 treated group. We observed the reduction in body weight across all groups including control after treatment started. There was statistical difference for reduction in body weight between the control and LY293 loaded nanoparticle treatment group (p<0.05). Systemic administration of LY293 loaded nanoparticles and cosolvents did not exhibit any toxicity which was confirmed by the absence of necrosis or inflammation in H&E staining of liver and kidney sections (Figure 8).
Figure 6. Effect of systemic therapy of LY293 cosolvent solution and LY293 loaded nanoparticles on proliferation of lung metastasized melanoma.
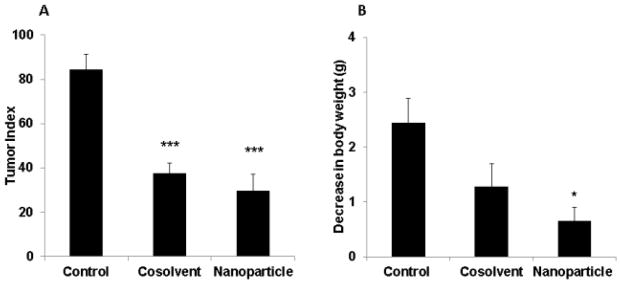
(A) Tumor index in B16F10 lung metastasis mouse melanoma model at two weeks after intravenous injection of different formulations. Tumor index was calculated using formula 1*a + 2*b + 3*c where a is the number of nodules with size <1 mm; b is the number of nodules with size between 1 and 2 mm; and c is number of nodules with size >2 mm. ***p<0.00005 using Student’s unpaired t test. (B) Change in body weight in of mice at the end of the study. Results are presented as mean ± SD (n = 6).
Figure 7. Morphological and histopathological examination of tumor nodule.

(A) Representative photos of lungs with melanoma nodules (black dots) on them (n= 06 per group) (B) H&E stained images of treated group showed normal lung parenchyma with limited mass of metastatic growth (Solid arrow) as compared to dense metastatic tissue in the control group (Solid arrow). White bar represents 200 μm. (C) 40 X magnified image showing pigmented (black spots as indicated by arrow) and non-pigmented dense metastatic growth in the control group. White bar represents 200 μm.
Figure 8. Immunohistochemistry staining of isolated lungs.

Representative IHC images for S100, cleaved caspase-3 and Ki67 staining of lung tissue sections after 2 weeks of treatment. White bar represents 200 μm. H&E staining of liver and kidney confirmed the absence of nanoparticle toxicity to vital organs.
Discussion
Prognosis of metastatic melanoma is poor and associated with a survival rate of less than 15 % at the end of five years (1, 22). Inherent ability of melanocytes to survive under duress as well as the development of resistance to cytotoxic drugs remains key challenges for poor prognosis of metastatic melanoma (23). For the last three decades since its initial FDA approval, dacarbazine remains the standard care of therapy for metastatic melanoma. However, treatment with dacarbazine leads to the development of resistance and has a poor response rate of approximately 7.5% (2). In spite of the clinical potential of paclitaxel and other tubulin inhibitors, ATP dependent efflux of these drugs by transporters contributes to the development of resistance (24, 25). To overcome MDR resistance and enhance the efficacy, we have previously synthesized potent tubulin inhibitor, phenyl thiazole (PAT) analogs with identical nanomolar efficacy in both parent and MDR overexpressing resistant cell lines (9). Although calcein AM efflux assay suggested that LY293 is a Pgp substrate (19) and should not have identical IC50 in MDR overexpressing cell lines, this anomaly can be attributed to the excellent permeability (33 × 10−6 cm/s) of LY293 analogs (26). Unlike paclitaxel, vinblastine and colchicine, LY293 has very high permeability and efflux by Pgp does not limit its intracellular accumulation (27, 28).
In addition to MDR resistance, poor solubility and non-specific distribution of tubulin inhibitors leading to the dose limiting toxicity are barriers for their systemic delivery. Nanodelivery systems have been designed to solubilize the poorly soluble drug and for effective tumor accumulation of drug through EPR effect. Enhanced tumor accumulation of nanoparticles will reduce the peripheral exposure and associated toxicity like peripheral neuropathy and myelosuppression. Additionally, nanoparticles undergo endocytic uptake and can overcome the P-gp mediated efflux. In this paper, we have synthesized random diblock copolymer of PEG block and block comprised of carbonate and lactide for systemic delivery of LY293. This copolymer consisted of mPEG (5000 Dalton) block that forms hydrophilic outer shell while the hydrophobic core is composed of lactide and carbonate. Previously, we have shown that modification of the core with carbonate monomer improved drug encapsulation and its release from nanocarriers (15). We have synthesized mPEG-b-P(CB-co-LA) copolymer by ring opening polymerization (ROP) of lactide and MBC using hydroxyl group of mPEG with N heterocyclic carbenes (NHCs). NHCs are strong organic bases, which allow ROP in solution at room temperature. Unlike organometallic catalyst which leads to the presence of trace amount of metal residues, there are no associated safety issues with ROP NHCs (29). In addition, ROP by NHCs is controlled reaction allowing polymers with desired molecular weight and narrow polydispersity index as compared to organometallic catalyst, stannous 2-ethylhexanoate (Sn(Oct)2). Previously, we have used Sn(Oct)2 as a catalyst for ROP which was associated with poor monomer conversion and low yield of polymers (19). Moreover, ROP by Sn(Oct)2 requires heating at elevated temperature for extended period of time which lead to transesterification and charring of the polymer (30). For the synthesis of polymer of desired molecular weight with low PDI, two different NHCs, DBU and TBD at molar ratios of 1, 2.5 and 5 relative to mPEG were utilized. Based on the GPC and NMR data (Figure 1A & 1B, Table 1), we found that DBU at 2.5 molar ratio for 6 h were the optimum reactions conditions. With TBD, we could synthesize the polymer of desired molecular weight but PDI was higher as compared to DBU catalyst. This can be attributed to the higher basicity of TBD which makes it highly reactive leading to transesterification and backbiting side reaction (31). Nederberg et al have reported ROP of lactide and carbonate monomer of desired degree of polymerization using 1 molar of DBU (20, 21). This difference in the molar ratio of a catalyst can be attributed to the initiator used for ROP and the targeted molecular weight of the polymer.
Nanoparticles were formulated by film evaporation, dialysis, nanoprecipitation and o/w emulsification method. However, nanoparticles formulated only by o/w emulsification method had acceptable size and drug loading. We were able to encapsulate LY293 with high efficiency (Figure 2 & Table 2), which can be attributed to the ability of the hydrophobic core to solubilize LY293. In addition to the appreciable drug loading, the hydrophilic mPEG shell could prevent the aggregation by steric hindrance and possibly reduce uptake by the reticuloendothelial system (RES) for longer blood circulation (32). We observed that an increase in initial drug loading led to decrease in the encapsulation efficiency of LY293 which can be attributed to the limited holding capacity of the hydrophobic core, reaching saturation at higher initial drug loading with a subsequent decrease in LY293 encapsulation (Table 2).
In vitro cytotoxicity of LY293 loaded nanoparticles showed no loss of LY293 activity against melanoma cancer cell lines (Figure 3). As a microtubule polymerization inhibitor, LY293 exerts its antiproliferative effect by targeting and destabilizing microtubules. The cell cycle analysis indicated that after treatment with LY293 loaded nanoparticles. LY293 loaded nanoparticles led to the concentration dependent arrest of A375 and B16F10 cells in G2/M phase (Figure 4). Tubulin polymerization is essential for rapid proliferation of cancer cells by mitotic division and the inhibition of tubulin polymerization results in the activation of intrinsic apoptotic pathway. Cell cycle analysis confirmed that the anticancer mechanism of LY293 is through the arrest of cell cycle in G2/M phase and subsequent activation of intrinsic apoptotic cellular machinery. In addition, LY293 loaded nanoparticle treatment resulted in significant increase in fraction of cells in sub-G1 phase (Figure 5B and 5C). This increase in cell population in sub-G1 phase indicated the fraction of cells undergoing DNA damage and apoptosis by LY293 loaded nanoparticles.
After demonstrating excellent efficacy of LY293 loaded nanoparticles against melanoma cell lines, we tested the in vivo efficacy of LY293 loaded nanoparticles in syngeneic B16F10 lung metastasis mouse model, which is an extremely aggressive metastatic model. Tumor burden as well as H&E data clearly showed that LY293 loaded nanoparticles inhibited the proliferation of metastatic B16F10 nodules (Figure 6 and 7). These results corroborate our previous findings where we have shown that intraperitoneal administration of structure analog of LY293 prevented the proliferation of metastasized melanoma. However, due to poor aqueous solubility daily intraperitoneal administration at higher dose of 25 mg/kg for two weeks was required. In the present study, there was no significant difference between cosolvent solution and LY293 loaded nanoparticles, which can be attributed to premature release of the encapsulated drug from the nanoparticle core. Rapid diffusion of the encapsulated drug across the hydrophobic core led to premature release during circulation and did not significantly alter the distribution of LY293 towards tumor. However, by designing the polymeric nanocarrier for LY293 administration, we were able to overcome the toxicity associated with the systemic infusion of cosolvents. To minimize the premature drug release and further improve the tumor accumulation, polymeric drug conjugates have been shown to be stable in the circulation resulting in enhanced tumor accumulation. For future studies, we plan to redesign the polymeric nanocarriers for allowing chemical conjugation of the drug analogs for preferential tumor accumulation and reduction of peripheral toxicity on systemic administration.
Acknowledgments
This work is supported by NIH/NCI grant R01CA148706 to WL. We would also like to thank Dr. Yuri Sheinin of the Department of Pathology & Microbiology of the University of Nebraska Medical Center for his assistance in interpreting the histopathology data.
Footnotes
Disclosure of Potential Conflicts of Interest
All authors declare that there are no potential conflicts of interest.
Animal studies
All institutional and national guidelines for the care and use of laboratory animals were followed.
References
- 1.http://seer.cancer.gov/statfacts/html/melan.html
- 2.Bhatia S, Tykodi SS, Thompson JA. Treatment of metastatic melanoma: An overview. Oncology (Williston Park) 2009;23:488–96. [PMC free article] [PubMed] [Google Scholar]
- 3.Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445:851–7. doi: 10.1038/nature05661. [DOI] [PubMed] [Google Scholar]
- 4.Dummer R, Flaherty KT. Resistance patterns with tyrosine kinase inhibitors in melanoma: New insights. Curr Opin Oncol. 2012;24:150–4. doi: 10.1097/CCO.0b013e32834fca92. [DOI] [PubMed] [Google Scholar]
- 5.Sun C, Wang L, Huang S, Heynen GJ, Prahallad A, Robert C, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508:118–22. doi: 10.1038/nature13121. [DOI] [PubMed] [Google Scholar]
- 6.Hersh EM, O’Day SJ, Ribas A, Samlowski WE, Gordon MS, Shechter DE, et al. A phase 2 clinical trial of nab-paclitaxel in previously treated and chemotherapy-naive patients with metastatic melanoma. Cancer. 2010;116:155–63. doi: 10.1002/cncr.24720. [DOI] [PubMed] [Google Scholar]
- 7.Chang W, Lee SJ, Park S, Choi MK, Hong JY, Kim YS, et al. Effect of paclitaxel/carboplatin salvage chemotherapy in noncutaneous versus cutaneous metastatic melanoma. Melanoma Res. 2013;23:147–51. doi: 10.1097/CMR.0b013e32835efd8d. [DOI] [PubMed] [Google Scholar]
- 8.Lu Y, Li CM, Wang Z, Ross CR, 2nd, Chen J, Dalton JT, et al. Discovery of 4-substituted methoxybenzoyl-aryl-thiazole as novel anticancer agents: Synthesis, biological evaluation, and structure-activity relationships. J Med Chem. 2009;52:1701–11. doi: 10.1021/jm801449a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu Y, Li CM, Wang Z, Chen J, Mohler ML, Li W, et al. Design, synthesis, and SAR studies of 4-substituted methoxylbenzoyl-aryl-thiazoles analogues as potent and orally bioavailable anticancer agents. J Med Chem. 2011;54:4678–93. doi: 10.1021/jm2003427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu Y, Chen J, Xiao M, Li W, Miller DD. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm Res. 2012;29:2943–71. doi: 10.1007/s11095-012-0828-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Z, Chen J, Wang J, Ahn S, Li CM, Lu Y, et al. Novel tubulin polymerization inhibitors overcome multidrug resistance and reduce melanoma lung metastasis. Pharm Res. 2012;29:3040–52. doi: 10.1007/s11095-012-0726-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.ten Bokkel Huinink WW, Eisenhauer E, Swenerton K. Preliminary evaluation of a multicenter, randomized comparative study of TAXOL (paclitaxel) dose and infusion length in platinum-treated ovarian cancer. canadian-european taxol cooperative trial group. Cancer Treat Rev. 1993;19(Suppl C):79–86. doi: 10.1016/0305-7372(93)90050-2. [DOI] [PubMed] [Google Scholar]
- 13.Hamaguchi T, Kato K, Yasui H, Morizane C, Ikeda M, Ueno H, et al. A phase I and pharmacokinetic study of NK105, a paclitaxel-incorporating micellar nanoparticle formulation. Br J Cancer. 2007;97:170–6. doi: 10.1038/sj.bjc.6603855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Danquah M, Li F, Duke CB, 3rd, Miller DD, Mahato RI. Micellar delivery of bicalutamide and embelin for treating prostate cancer. Pharm Res. 2009;26:2081–92. doi: 10.1007/s11095-009-9903-5. [DOI] [PubMed] [Google Scholar]
- 15.Danquah M, Fujiwara T, Mahato RI. Self-assembling methoxypoly(ethylene glycol)-b-poly(carbonate-co-L-lactide) block copolymers for drug delivery. Biomaterials. 2010;31:2358–70. doi: 10.1016/j.biomaterials.2009.11.081. [DOI] [PubMed] [Google Scholar]
- 16.Gaucher G, Marchessault RH, Leroux JC. Polyester-based micelles and nanoparticles for the parenteral delivery of taxanes. J Control Release. 2010;143:2–12. doi: 10.1016/j.jconrel.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 17.Danquah MK, Zhang XA, Mahato RI. Extravasation of polymeric nanomedicines across tumor vasculature. Adv Drug Deliv Rev. 2011;63:623–39. doi: 10.1016/j.addr.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 18.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J Control Release. 2000;65:271–84. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 19.Mundra V, Lu Y, Danquah M, Li W, Miller DD, Mahato RI. Formulation and characterization of polyester/polycarbonate nanoparticles for delivery of a novel microtubule destabilizing agent. Pharm Res. 2012;29:3064–74. doi: 10.1007/s11095-012-0881-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nederberg F, Lohmeijer BG, Leibfarth F, Pratt RC, Choi J, Dove AP, et al. Organocatalytic ring opening polymerization of trimethylene carbonate. Biomacromolecules. 2007;8:153–60. doi: 10.1021/bm060795n. [DOI] [PubMed] [Google Scholar]
- 21.Lohmeijer B, Pratt RC, Leibfarth F, Logan JW, Long DA, Dove AP, et al. Guanidine and amidine organocatalysts for ring-opening polymerization of cyclic esters. Macromolecules. 2006;39:8574–83. [Google Scholar]
- 22.Tas F. Metastatic behavior in melanoma: Timing, pattern, survival, and influencing factors. J Oncol. 2012;2012:647684. doi: 10.1155/2012/647684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soengas MS, Lowe SW. Apoptosis and melanoma chemoresistance. Oncogene. 2003;22:3138–51. doi: 10.1038/sj.onc.1206454. [DOI] [PubMed] [Google Scholar]
- 24.Frank NY, Margaryan A, Huang Y, Schatton T, Waaga-Gasser AM, Gasser M, et al. ABCB5-mediated doxorubicin transport and chemoresistance in human malignant melanoma. Cancer Res. 2005;65:4320–33. doi: 10.1158/0008-5472.CAN-04-3327. [DOI] [PubMed] [Google Scholar]
- 25.La Porta CA. Drug resistance in melanoma: New perspectives. Curr Med Chem. 2007;14:387–91. doi: 10.2174/092986707779941078. [DOI] [PubMed] [Google Scholar]
- 26.Li CM, Chen J, Lu Y, Narayanan R, Parke DN, Li W, et al. Pharmacokinetic optimization of 4-substituted methoxybenzoyl-aryl-thiazole and 2-aryl-4-benzoyl-imidazole for improving oral bioavailability. Drug Metab Dispos. 2011;39:1833–9. doi: 10.1124/dmd.110.036616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dahan A, Sabit H, Amidon GL. Multiple efflux pumps are involved in the transepithelial transport of colchicine: Combined effect of p-glycoprotein and multidrug resistance-associated protein 2 leads to decreased intestinal absorption throughout the entire small intestine. Drug Metab Dispos. 2009;37:2028–36. doi: 10.1124/dmd.109.028282. [DOI] [PubMed] [Google Scholar]
- 28.Dahan A, Amidon GL. Grapefruit juice and its constituents augment colchicine intestinal absorption: Potential hazardous interaction and the role of p-glycoprotein. Pharm Res. 2009;26:883–92. doi: 10.1007/s11095-008-9789-7. [DOI] [PubMed] [Google Scholar]
- 29.Kamber NE, Jeong W, Waymouth RM, Pratt RC, Lohmeijer BG, Hedrick JL. Organocatalytic ring-opening polymerization. Chem Rev. 2007;107:5813–40. doi: 10.1021/cr068415b. [DOI] [PubMed] [Google Scholar]
- 30.Bero M, Czapla B, Dobrzynski P, Janeczek H, Kasperczyk J. Copolymerization of glycolide and ε-caprolactone, 2. random copolymerization in the presence of tin octoate. Macromolecular Chemistry and Physics. 1999;200:911–6. [Google Scholar]
- 31.Seow WY, Yang YY. Functional polycarbonates and their self-assemblies as promising non-viral vectors. J Control Release. 2009;139:40–7. doi: 10.1016/j.jconrel.2009.05.028. [DOI] [PubMed] [Google Scholar]
- 32.Maeda H, Bharate GY, Daruwalla J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur J Pharm Biopharm. 2009;71:409–19. doi: 10.1016/j.ejpb.2008.11.010. [DOI] [PubMed] [Google Scholar]