

Abstract
We establish structure activity relationships of extracellular nucleosides and nucleotides at G protein-coupled receptors (GPCRs), e.g. adenosine receptors (ARs) and P2Y receptors (P2YRs), respectively. We synthesize selective agents for use as pharmacological probes and potential therapeutic agents (e.g. A3AR agonists for neuropathic pain). Detailed structural information derived from the X-ray crystallographic structures within these families enables the design of novel ligands, guides modification of known agonists and antagonists, and helps predict polypharmacology. Structures were recently reported for the P2Y12 receptor (P2Y12R), an anti-thrombotic target. Comparison of agonist-bound and antagonist-bound P2Y12R indicates unprecedented structural plasticity in the outer portions of the transmembrane (TM) domains and the extracellular loops. Nonphosphate-containing ligands of the P2YRs, such as the selective P2Y14R antagonist PPTN, are desired for bioavailability and increased stability. Also, A2AAR structures are effectively applied to homology modeling of closely related A1AR and A3AR, which are not yet crystallized. Conformational constraint of normally flexible ribose with bicyclic analogues increased the ligand selectivity. Comparison of rigid A3AR agonist congeners allows the exploration of interaction of specific regions of the nucleoside analogues with the target and off-target GPCRs, such as biogenic amine receptors. Molecular modeling predicts plasticity of the A3AR at TM2 to accommodate highly rigidified ligands. Novel fluorescent derivatives of high affinity GPCR ligands are useful tool compounds for characterization of receptors and their oligomeric assemblies. Fluorescent probes are useful for characterization of GPCRs in living cells by flow cytometry and other methods. Thus, 3D knowledge of receptor binding and activation facilitates drug discovery.
Keywords: GPCR, Medicinal chemistry, Purines, X-ray structures, Nucleosides, Nucleotides, Polypharmacology
1. Introduction
A vast biology is associated with action at the G protein-coupled adenosine receptors (ARs) and P2Y receptors and at ionotropic P2X receptors, which is modulated by all of the nucleoside and nucleotide processing enzymes and transporters. This extensive signaling system is qualified to be considered part of the ‘purinome’ [1], a term already applied in the context of the > 3200 proteins that utilize purine cofactors, including intracellular kinases [2], as well as to describe the actions of extracellular and intracellular purines (and pyrimidines) in this collection of related receptors and enzymes.
The release of ATP, UTP and other nucleotides by various routes from cells results in a temporal sequence of activation of these three families of cell surface receptors [3]. The receptors activated initially are the fast P2X ion channels (ATP-responsive trimeric channels composed of seven distinct subunits) and some of the metabotropic P2Y receptors (i.e. P2Y2, P2Y4 and P2Y11Rs that respond fully to nucleoside 5′-triphosphates and P2Y14R that responds to UDP-sugars). Upon the sequential action of ectonucleotidases CD39 (ectonucleoside triphosphate diphosphohydrolase 1, ENTPD1) and CD73 (ecto-5′-nucleotidase, 5′-NT) [4], different sets of receptors are activated (i.e. P2Y1, P2Y6, P2Y12, P2Y13 and P2Y14Rs that respond to nucleoside 5′-diphosphates), followed by the activation of four AR subtypes (A1, A2A, A2B and A3ARs), which are not appreciably activated by any endogenous nucleotides. Naturally occurring dinucleotides, such as Up4A, are also known to activate various P2YRs [5].
A current challenge to medicinal chemists is to identify selective P2R agonist and antagonist ligands, which remains an unmet need for most of the P2XRs and many of the P2YRs. The effort to design purine receptor ligands is now aided by representative X-ray crystallographic structures in each of the three classes: A2AAR, P2Y12R and P2X4R [6–11]. Many more receptor structures in complex with different ligands will be needed to gain a detailed and broad knowledge of molecular recognition within these families. The high resolution G protein-coupled receptor (GPCR) structures and, to a lesser extent, homology models obtained so far have proven valuable for in silico screening campaigns [12,13]. Biophysical mapping of binding sites, lipophilic hotspots, explicit water networks and other techniques based on GPCR structures are now used for drug design [14]. Moro and coworkers validated a general pharmacophore hypothesis for the human A2AAR using an external test set of 29 newly synthesized antagonists [15]. Thus, we and others have demonstrated the predictive power of GPCR homology modeling and the value of applying newly determined X-ray structures to the medicinal chemistry of purine and pyrimidine GPCRs [11–16]. However, we are only at the beginning of exploring ligand design based on GPCR X-ray structures, and we are far away from predicting selectivity and function of ligands from such models.
In general, we establish detailed structure activity relationships in the purine receptor families, in order to provide selective agents as pharmacological probes and potential therapeutic agents. Our efforts to discover novel, selective ligands for purine receptors stem initially from the guidance and inspiration of John W. Daly, Ph.D. (1933–2008), a noted medicinal chemist and pharmacologist. He was one of those who defined the existence of receptors for adenosine and the biological effects of methylxanthines, by chemical and pharmacological means [17]. He emerged from the era in which many medicinal chemists were not yet accustomed to the idea of structure activity relationship (SAR), in which different functionality on a given molecule subserves distinct roles in receptor recognition. However, very early in the development of our field, Daly applied SAR analysis to the ARs to help define three of the four receptor subtypes and introduced important ligand tools, such as A1AR-selective N6-cycloalkyladenosines and 8-aryl- and 8-cycloalkyl-1,3-dipropylxanthines (with postdoctoral fellows R. F. Bruns and M. T. Shamim) [18,19]. Much of our study of the SAR of P2YRs has been in collaboration with T. Kendall Harden.
2. X-ray Structures of A2A and P2Y12 Receptors With Bound Agonists and Antagonists
2.1. Molecular Recognition at Adenosine Receptor Structures
The structure of adenosine consists of two chemically and conformationally distinct moieties, each of which is associated with separate roles in the AR orthosteric binding site(s). These two moieties can be divided into message (ribose) vs. address (adenine) portions. While adenine and similar flat, hydrophobic heterocycles often behave as AR antagonists, the addition of a ribose moiety at the appropriate position (adenine-9-ribosides or xanthine-7-ribosides) can confer the ability to activate the receptor, i.e. deliver the message by complementarity with the conformation of the AR protein required to induce its activation.
In some cases, the same substituents of the N6 and C2 positions of an isolated adenine AR antagonist maintain the same receptor subtype binding preferences that are found in riboside-bearing agonists (see A1 and A3AR ligands 1–10 in Table 1), suggesting a common mode of receptor binding. The affinity of the AR agonists is generally much greater than the corresponding adenines, because the ribose anchors and stabilizes the bound ligand. Although the AR subtype selectivity of the lone nucleobases has much commonality with the SAR of adenosine agonists, it is not identical [20–24]. For example, in a study of 2-, 6- and 8-substituted adenines, Klotz et al. [22] noted a pharmacological similarity between C2 substitution in adenosine and 8-substitution in adenine. No AR structures containing an unmodified adenine antagonist have been determined yet, but the positions of other AR antagonists in crystal structures so far indicate a similar hydrophobic binding region (either a close overlay of a 1,2,4-triazolo[1,5-a][1,3,5]triazine with the adenine of agonists or non-superimposed rings for xanthines) [6,25]. π–π Stacking of the nucleobase rings with a conserved Phe in extracellular loop (EL)2 and often H-bonding with a conserved Asn (6.55, using Ballesteros–Weinstein numbering [26]) are typically common to agonists and antagonists in the A2AAR structures and in models of the other AR subtypes.
Table 1.
Representative examples of the combination of address (adenine) and message (ribose or ribose-like) moieties in nucleoside ligands of various ARs. Binding affinity (Ki, nM) of corresponding adenosine agonist (I) and adenine antagonist (II) derivatives at three subtypes of ARs is shown, to examine the question whether SAR is parallel in the two series. The selectivity column refers to the complete nucleoside derivative (address + message). The affinity was measured for this study, using standard radioligand binding assays. The species is human unless noted (r, rat).
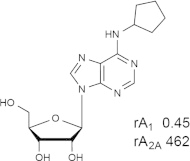
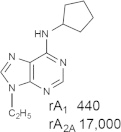
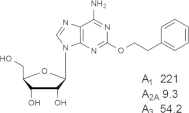
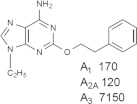
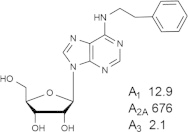
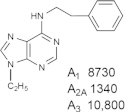
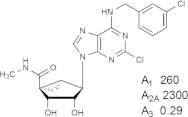
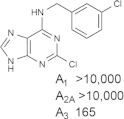
Because the ribose moiety constitutes the message portion of adenosine agonists, we have focused on its conformational characteristics at the ARs and also at P2X and P2YRs. From the X-ray structure of the human A2AAR containing a bound nucleoside (Fig. 1A) [7], we now understand that the ribose moiety fits in a deep subpocket of the ARs, where it activates the receptor by drawing hydrophilic residues in transmembrane helices (TMs) 3 and 7 toward it, like the tightening of a belt. The ribose of adenosine agonists is coordinated by H-bonding to conserved residues Thr (3.36), Thr or Ser (7.42) and His (7.43) [7,27,28]. At the same time, the binding of ribosides is driven entropically by the displacement of unstable water molecules in this region. Departure of unstable water molecules also can contribute to the binding of AR antagonists [14,29]. Although it lacks a G protein or G protein mimic, the agonist-bound A2AAR structure by Xu et al. [7] displays intracellular features that resemble other activated GPCR structures. This complex contains a bound 6-(2,2-diphenylethylamino)-9-((2R,3R,4S,5S)-5-(ethylcarbamoyl)-3,4-dihydroxytetrahydrofuran-2-yl)-N-(2-(3-(1-(pyridin-2-yl)piperidin-4-yl)ureido)ethyl)-9H-purine-2-carboxamide (UK432097), a failed experimental drug for treatment of chronic obstructive pulmonary disease. Thus, the agonist-induced contraction of the ribose-binding region of the A2AAR appears to be propagated to the cytoplasmic side of the receptor that is in contact with the Gs protein (as demonstrated structurally for the β2-adrenergic receptor [30]). This conformational adaptation to agonist is likely to be similar but not identical at the other AR subtypes.
Fig. 1.
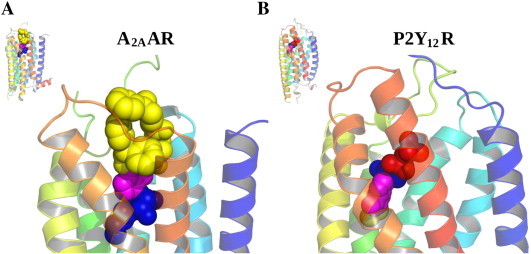
Agonist-bound crystallographic structures of A2AAR (PDB ID: 3QAK, bound to UK432097) [7] and P2Y12R (PDB ID: 4PXZ, bound to 2-MeSADP) [9]. Overall structure of the receptors and details of the binding sites. Transmembrane helices are colored from blue (TM1) to red (TM7). Ligands are shown in CPK representation with colors that indicate: the nucleobase (magenta), the ribose moiety (blue), the N6 and C2 substituents (yellow) and the phosphate groups (red). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Prior to the X-ray structural determination, we sought to define the receptor-preferred conformation of the ribose ring chemically by introducing steric constraints on the native, freely-twisting ribose moiety by fusing two small carbocyclic rings. The methanocarba (bicyclo[3.1.0]hexane) ring system (fused cyclopropyl and cyclopentyl rings) freezes opposite twists of a ribose-like moiety (either (N), North or (S), South envelope conformations) when incorporated into nucleoside/nucleotide analogues with the bond of ring fusion at either of two positions [31]. These modifications can enhance or reduce potency at different subtypes of purine receptors in comparison to the ribose moiety [31]. These ribose modifications, applied across the entire family of receptors, have been particularly potency-enhancing at certain subtypes, for example (N)-methanocarba at the A3AR [73]. Compounds 7 and 9 in Table 1 contain a potency- and selectivity -enhancing (N)-methanocarba ring. The effects of an (N)-methanocarba substitution on affinity at the A1 and A2AARs is variable, depending on other substitution of the nucleoside. Another effective means of enforcing a (N) or (S) conformation of the ribose was the addition of a methyl group to the 2′- or 3′-ribose carbon [32]. The X-ray structure of the agonist-bound A2AAR confirmed the presence of a (N)-ribose conformation [7]. Nevertheless, the general (N) conformation of ribose represents a range of geometric parameters [33], and the conformational requirements for ribose might differ slightly between AR subtypes. Without a crystal structure of the A3AR, we lack the detail of the ribose binding region needed to explain the greater preference of (N)-methanocarba analogues at the A3AR compared to other AR subtypes.
Fig. 2A shows a progression of structural changes from prototypical agonists 11–13 leading to enhanced A3AR selectivity, including (N)-methanocarba (15–18 and 23–25) and 4′-thio (14) [34] agonists. Either the reduction of H-bonding ability of the hydrophilic 5′-carboxamide (including by 4′-truncation for its complete removal, 20–22 and 26–28) or the introduction of rigidity of the ribose moiety, as in spirolactam 19 [35], tends to convert agonists into antagonists (or at least partial agonists) with the retention of A3AR selectivity.
Fig. 2.
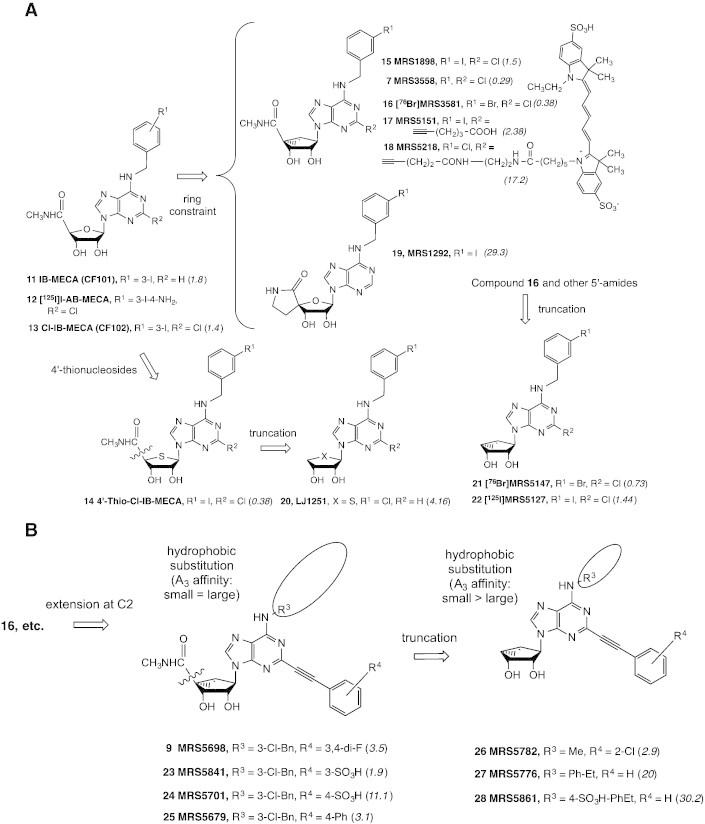
Progression of structural changes in adenosine derivatives leading to enhanced A3AR selectivity [27,28,35,36,43]. Binding Ki values at the human A3AR using 12 as radioligand are given in italics (nM). A. Successive structural changes leading to hypermodified (N)-methanocarba analogues. 4′-Truncation was performed in the ribose, thioribose and (N)-methanocarba series with the general observation that affinity but not efficacy at the A3AR was maintained. A3AR antagonist 19 was shown to be useful in lowering interocular pressure in glaucoma models [35]. Compounds 12 (nonselective) and 22 (A3AR-selective) are useful high affinity radioligands for routine binding assays. Compounds 16 and 21 are demonstrated to be useful radioligands for positron emission tomography (PET) [74]. B. Extension of the C2 position with rigid arylethynyl substituents.
Novel fluorescent derivatives of high affinity GPCR ligands are useful tool compounds for characterization of receptors and their oligomeric assemblies. Fluorescent probes are useful for characterization of GPCRs in living cells by microscopy or flow cytometry or in cell membranes. Fluorescent agonists but not antagonists are highly internalized, consistent with the general phenomenon of agonist-induced GPCR internalization. A (N)-methanocarba modification together with a 2-ethynyl group was incorporated in a highly selective A3AR agonist fluorescent probe, MRS5218 18 (KD 31 nM), which was useful for flow cytometry of whole cells expressing the A3AR [36]. This probe contains an amide-linked Cy5 (cyanine) fluorophore.
2.2. Molecular Recognition at P2YR Structures
In collaboration with Ray Stevens, Qiang Zhao, Christa Müller and colleagues [8,9], we analyzed the X-ray structure of the human P2Y12R (the first high resolution structure of any P2YR), an important antithrombotic target that is the site of action of three drugs already on the market. Three compounds used clinically: two irreversibly binding thienopyridines (clopidogrel and prasugrel), which are actually prodrugs of reactive thiols, and one reversible nucleotide-like antagonist, actually a nucleoside (tigcagrelor) [37]. The P2YRs belong on the δ-branch of GPCRs, which has only one other known structure for comparison, i.e. PAR1, another platelet receptor [38]. The antagonist-bound crystal structure of P2Y12R contained an experimental non-nucleotide antagonist from Astra Zeneca, AZD1283 29 (Fig. 3), an ethyl 6-aminonicotinate sulfonylurea [8,39]. A benzyl moiety on AZD1283 displaces outwardly the position of TM6 with respect to the agonist-bound P2Y12R structure (Figs. 1B and 3A) [9]. Several unusual features of the P2Y12R were noted, including a bifurcated binding pocket (Fig. 3B) and an extended and linear TM5. TM5 in most Family A GPCRs has a proline, which is associated with a kink in the helical structure. The extension and linearity of TM5 leads to the previously termed conserved disulfide bridge between TM3 and EL2 to be undefined in the AZD1283 structure and likely cleaved. Thus, it appears that this disulfide is dynamic in P2Y12R and may not be essential for the overall integrity of the receptor structure.
Fig. 3.
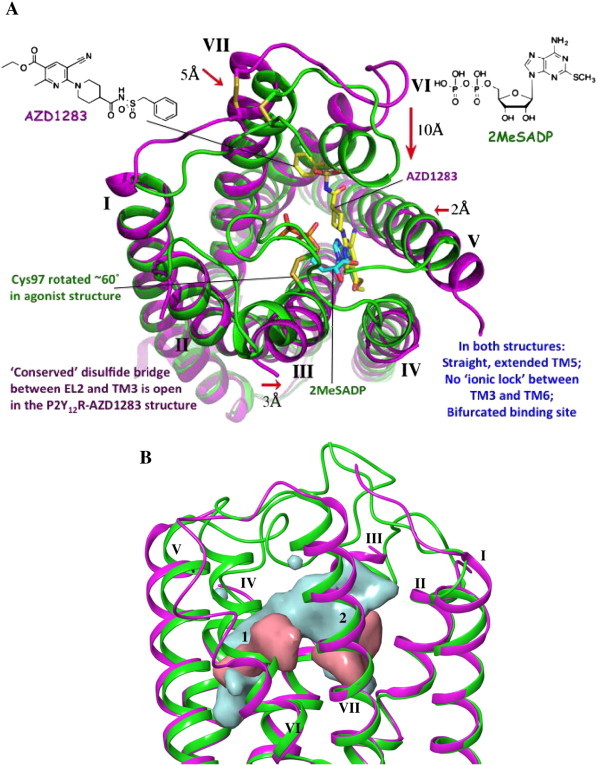
A. A comparison of nucleotide agonist-bound and non-nucleotide antagonist bound P2Y12R structures [8,9]. The binding site contracted on the extracellular side of the TMs in the P2Y12R–2MeSADP 30 structure (green) in comparison to the complex with antagonist AZD1283 29 (purple, PDB ID: 4NTJ). Only one nonnucleotide–P2Y12R complex has been determined so far; it is not known if this structure generalizes for other nonnucleotide antagonists. B. Side view of the superposed nucleotide agonist-bound and non-nucleotide antagonist-bound P2Y12R structures with surfaces showing the binding site shape. The P2Y12R–2MeSADP structure is shown in green ribbons with the associated binding site surface in cyan. The P2Y12R–AZD1283 structure is shown in magenta ribbons with the associated binding site surface in pink. The two subpockets of the bifurcated binding pocket are indicated as 1 (principle location of both ligands) and 2. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Also, the structures of P2Y12R complexes with a bound agonist 2-methylthioadenosine 5′-diphosphate (2-MeSADP 30) and a bound partial agonist (2-MeSATP) were determined [9]. The first antagonists identified for this receptor were nucleotides (2-MeSATP and other ATP derivatives), but it appears that some of these have agonist properties [40], which is consistent with the close overlay of 2-MeSATP with 2-MeSADP in the receptor structures. The only binding feature in common between AZD1283 and the nucleotides is π–π stacking of Tyr105 (3.33) with aromatic rings, a cyanopyridine and adenine, respectively.
A comparison of nucleotide-bound and nonnucleotide-bound complexes of the P2Y12R illustrates unprecedented divergence of structure for stable complexes of the same receptor. The negatively charged nucleotide ligands are surrounded by at least 7 cationic residues in the loop regions and the outer portions of the helices, which are highly positively charged. This draws the ELs and the top portions toward the nucleotide ligands, which are almost completely enclosed. The phosphate moiety of 30 is coordinated by cationic and H-bonding residues in the N-terminal segment (through water molecules), in TMs 3, 6 and 7 and in EL2. It is to be noted that this agonist-bound P2Y12R structure does not display typical intracellular features of activated GPCR structures, so it is considered an inactive state of the receptor. Nevertheless, the ligand binding pocket and the extracellular regions are informative concerning conformational adaptation of the structure to accommodate nucleotides. There are five other pairs of GPCRs in agonist-bound and antagonist-bound states [41], and the P2Y12R shows the greatest divergence of all these paired structures.
A cartoon representing the progressive opening of the P2Y12R structure depending on the nature of the bound ligand is shown in Zhang et al. [9], with the nucleotide agonists (closed) and the nonnucleotide antagonists (open) as the extremes. The nonnucleotide antagonist binds to a widely opened binding site (even in comparison to PAR1), which consequently is free of the steric constraints of the loops that are drawn toward the negatively-charged ligands. Some of the nucleotide-mimetic antagonists, such as Cangrelor [42], are predicted to bind with a similar conformation to 30 while other bulkier antagonist derivatives do not dock readily into the 2-MeSADP or 2-MeSATP complexes, because the binding site is too contracted. Thus, a possible displacement of parts of the TM region is proposed in order to fit these antagonists. We are currently modeling binding at the P2Y12R to establish energetically favorable docking modes of other known ligands, both agonists and antagonists.
The divergence of the A2AAR and P2Y12R structures emphasize that these are two very different receptor classes. In the nucleoside and nucleotide-bound structures of these two receptors, respectively, the adenosine moiety is flipped (Fig. 1). Thus, in the P2Y12R, the adenine base, not the ribose occupies a deeper portion of the binding site, which now has a hydrophobic character [7,9].
3. Structure-based Design of Novel A1, A2A and A3 Adenosine Receptor Agonists
3.1. Design of Ligands Based on the X-ray Structures
For the structure-based design of new AR nucleoside ligands, each portion of the adenosine molecule requires a different approach for derivatization; no single approach is general for all applications. A2AAR crystallographic structures are effectively applied to homology modeling of the closely related A1AR and A3AR, which are not yet crystallized. Based on the X-ray structure of the A2AAR [7], the C2 position is located in an approach path that easily frees itself from the constraints of the pharmacophore binding region to exit the receptor. This opening to the extracellular medium explains why the elongation of chains at the C2 position, for example by peptide or polymeric moieties, was found empirically to be allowed in receptor binding. The N6 region is slightly more constrained spatially than the C2. We have probed the tight fit of various hydrophobic N6 substituents at the ARs, thus explaining the early observations concerning stereochemistry at the α-carbon, for example that R-N6-(phenylisopropyl)adenosine (R-PIA) is more potent at ARs than S-PIA [17,43].
The 5′ position of AR agonists is unique in that it is situated in a small, partly hydrophilic region. There are no charged residues in this small subpocket, therefore attempts to introduce charged groups on the 5′ moiety were mostly unsuccessful. In silico fragment screening by Katritch and coworkers identified high scoring hits, of which we were able to synthesize 23 molecules [16]. Nearly all of the uncharged synthetic hits, including some novel 5′-amides that were not previously envisioned as AR ligands, bound to the A2AAR with micromolar affinity or better. Some of the hits were greatly enhanced in A1AR affinity, which was explained structurally.
3.2. A Novel A1 Agonist for Suppression of Seizures
More detail about the fit of a novel selective A1AR agonist in the N6 region is shown using small cycloalkyl rings on this substituent. We chose the 4′-truncated (N)-methanocarba series because of its compactness, steric constraint, and increased hydrophobicity for the design of novel A1AR agonists, and incorporated many hydrophobic N6 substituents that were known to be A1AR-enhancing in the ribose series. The only substituent that maintained A1AR selectivity and full efficacy in this truncated series was the dicyclopropylmethyl group [43]. This A1AR-selectivity in the truncated 2-chloro-(N)-methanocarba series was not general across all enhancing N6 groups identified in the riboside series; for example, the 4′-truncated equivalent of A1AR agonist 2-chloro-N6-cyclopentyladenosine (CCPA 31, structure not shown) was not A1AR-selective. In fact, a tight fit of the N6 group in the outer portion of the receptor surrounding the exocyclic amine seemed to be important for potency and efficacy, especially in the 4′-truncated series to compensate for the missing anchoring effect of the ribose 5′-region. Thus, MRS5474 37 (Fig. 4A), bearing a N6-dicyclopropylmethyl group, is a structural sweet spot with respect to the SAR in this series, because of its balance of full agonism and modest selectivity at the human A1AR (Ki 47.9 nM).
Fig. 4.
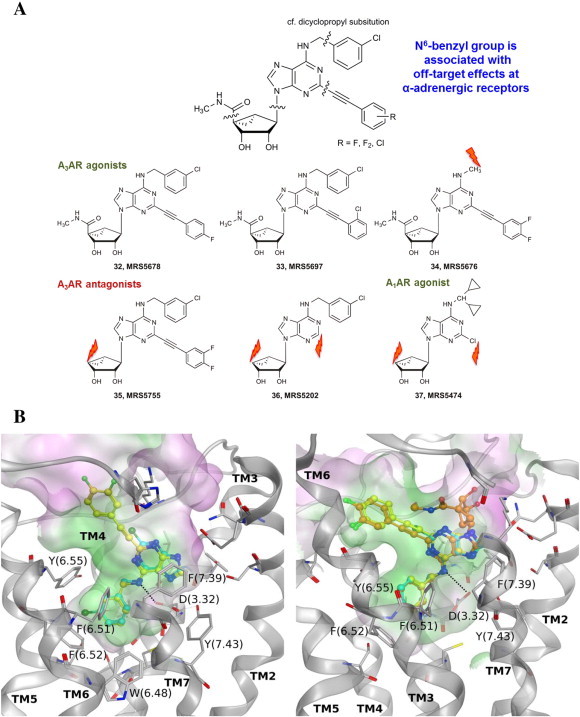
Representative data from a study of polypharmacology of otherwise selective AR ligands. A. Shown are six of the rigid congeners that are known to bind selectively to the A3AR [20] or in one case to the A1AR [43] and that were subjected to binding and structural analysis of off-target sites, including many GPCRs. Four other rigid congeners were included in this study: truncated compounds 7–10 in Table 1. Orange flash arrows indicate the position of truncation. B. As an example, binding affinity at three Gi-coupled α2 adrenergic receptors and structural analysis by docking to antagonist-bound receptor structures are shown [20]. Left side: adenines 8 (cyan carbons) and 10 (yellow carbons) at the α2B receptor; right side: adenines 8 (cyan carbons) and 10 (yellow carbons) and adenosine derivative 9 (orange carbons) at the α2C receptor. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
We wondered if the more drug-like physicochemical properties of MRS5474 37 (smaller size, fewer H-bonding donors and acceptors, less polar surface area) with respect to the prototypical A1AR agonists, such as CPA 1 and CCPA 31, would alter its in vivo activity, especially activity in the brain. Use of an A1AR agonist to control seizures has long been a goal of CNS research in the purinergic field, but progress has been hampered by the toxicity associated with the other agonists [45]. However, MRS5474 37 was found to reduce electrically-induced seizures in the 6 Hz minimal clonic seizure test in mice without the usual toxicity of A1AR agonists. In fact, a maximum tolerated dose (MTD) study in mice of this agonist revealed that doses up to 60 mg/kg (40-fold higher than the effective anti-seizure dose in mice) were not lethal.
3.3. A Rationally Designed A2A Agonist for Irreversible Inhibition of the Receptor
Irreversibly binding ligands of GPCRs are useful as pharmacological tool compounds and in some cases as therapeutic agents. We wondered if we could rationally design a chemically reactive agonist based on the position of a reactive group when the ligand was docked to the A2AAR X-ray structure. A series of active ester derivatives 38–40 was synthesized, in which a reactive o-nitro ester was incorporated on an elongated C2 chain (Fig. 5) [46]. The ester was intended to acylate a nucleophilic amino group in the extracellular regions of the receptor. This is also a useful structural probe, because the role of the ELs in receptor binding and activation is not entirely clear. The ELs are highly flexible regions of most GPCRs, and perhaps plasticity of these regions could induce or otherwise alter the activation of the receptor. Even though the business surface of the receptor for contacting the G protein (cytosolic sides of TMs 3, 5 and 6) or arrestin (cytosolic sides of TMs 1, 2 and 7) was clearly distinct from the ELs [41], conformational effects from the ELs could be propagated along the length of the helices to the intracellular side.
Fig. 5.
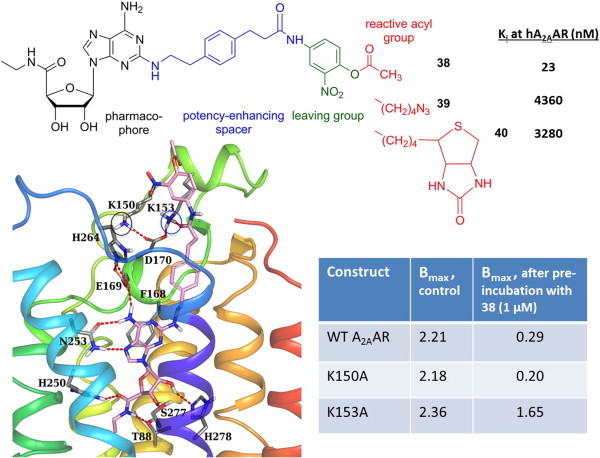
Targeted covalent modification of the A2AAR: Tethering of a reactive group to reach a distal site. Irreversible loss of receptor binding by MRS5854 38 was associated with the presence of K153, but not K150 (both circled). Bmax values are in pmol/mg protein [46].
Mutagenesis results indicated that the irreversible loss of receptor binding induced by acetyl ester MRS5854 38 was dependent on the presence of Lys153, but not Lys150 (both of which are in the region of EL2 where the active ester is predicted to be located). The amino group of Lys153 is predicted to lie closer to the reactive carbonyl of MRS5854 than Lys150 (Fig. 5). Thus, we have designed a chemically reactive agonist ligand based on structural insights.
3.4. Design of Highly Specific A3 Agonists Can Be Interpreted Structurally
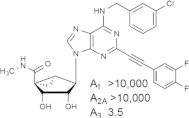
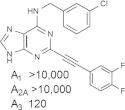
Some reports have detected dose-dependent activity of the prototypical agonists of the A3AR, such as 11 and 13, at other AR subtypes in vivo [47]. Thus, there is a need for the introduction of more highly selective A3AR agonists. The SAR of (N)-methanocarba nucleosides acting at the A3AR was explored, resulting in consistently high affinity and selectivity at the A3AR upon combination with favorable N6 groups, such as methyl or substituted benzyl, and a rigid C2-arylethynyl substituent. The combination of these substituents provides a general class of highly specific agonists, in which the A3AR is consistently in the nM range and the affinity at other AR subtypes is almost always > 10 μM. One example that had a Ki value of 3 nM at both human and mouse A3ARs, MRS5698 9 (Fig. 2B), contains difluoro substitution of the terminal phenyl ring intended to reduce oxidative metabolism in vivo [27].
In order to accommodate such rigid C2 substituents and maintain other conserved interactions of ribose and adenine with the receptor (Fig. 6), we used molecular modeling of the A3AR to propose an outward movement of TM2. This proposed receptor plasticity is not an arbitrary rearrangement; rather it is based on similarity to several other active-like state structures of GPCRs. Thus, a hybrid homology model was constructed, which was based on the active state of the A2AAR with respect to TMs 1 and 3–7 and on the active state of rhodopsin, i.e. opsin, with respect to TM2. The doubly extended biaryl derivative MRS5679 25 required an outward movement of TM2 by ~ 7 Å. The reason that a similar rearrangement of TM2 cannot occur in the A2AAR is that there are a total of 3 disulfide bridges that further constrain the movement on the outer loop region of the receptor.
Fig. 6.
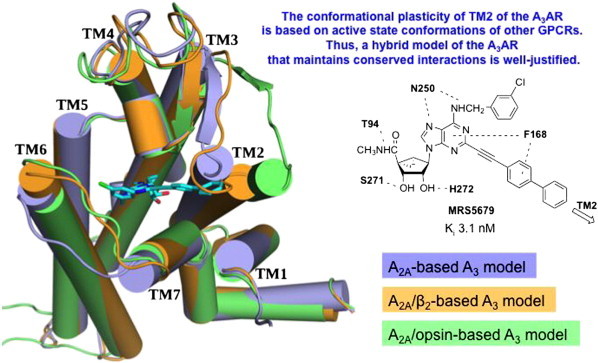
Predicted outward movement of TM2 of A3AR to accommodate a rigid aryl ethynyl extension at the adenine C2 position (example shown for a biaryl derivative, selective full agonist MRS5679 25 in cyan). A hybrid A3AR model containing TM2 based on the opsin structure was required for docking. The other TMs were based on the A2AAR agonist bound structure [7,27]. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Sulfonate groups are a means of preventing passage across biological membranes, because of a permanent negative charge at physiological pH. In 1994, we attempted empirically to introduce a sulfonate on the prototypical riboside analogues, such as IB-MECA 11. The effort failed; introduction of an m- or p-sulfonate group on the N6-benzyl group and greatly reduced the affinity at the rA3AR — from the nM to the μM level. Instead, we recently used a structure-based approach to design a pharmacological probe to distinguish central and peripheral effects of A3AR agonists [28]. Selectivity of > 3000-fold at both the human and mouse A3ARs was achieved with m-sulfophenyl analogue 23 (MRS5841, Fig. 2B), but a sulfonate in the p-position 24 produced a potent and selective mixed agonist of A1 and A3ARs. Thus, there is a strong dependence of the affinity on distal interactions between the ligand and the EL regions of the receptor, and we predicted specific interactions with polar and hydrophobic residues based on ligand docking to homology models of the human and mouse A3ARs.
3.5. Polypharmacology of AR Ligands
The analysis of off-target activities of new drug candidates is an essential component of drug discovery. With the design of rigid nucleoside congeners that bind to the ARs and now the availability of many GPCR structures, we have used this as a test case to quantify and possibly predict the likelihood of specific off-target (non-AR-related) activities in a systematic approach. In some cases these activities would be considered undesirable and would have to be eliminated, and other off-target activities that might synergize with the desired AR activity could be accentuated.
We analyzed the polypharmacology of otherwise ‘selective’ A3AR ligands at off-target GPCRs and other receptors [20]. Increased conformational rigidity of A3AR agonists and adenine antagonists allows the exploration of interaction of specific regions of the ligands with other GPCRs that have structural information (either by direct X-ray determination or by molecular modeling), such as biogenic amine receptors. A series of highly rigidified congeners (7–10, 32–37) was assembled for probing off-target effects experimentally and correlating these findings with GPCR structures (Fig. 4). The ten compounds submitted to the Psychoactive Drug Screening Program (PDSP) at the University of North Carolina are indicated, and Ki values of binding hits (defined as > 50% inhibition at 10 μM in the single point screen) were determined. The structural analysis was conducted at various GPCRs that turned up as hits in the screen. We present here the example of α2 adrenergic receptors, although a similar analysis was performed at β-adrenergic, serotonergic receptors and other GPCRs. The patterns of off-target binding often correlated with specific moieties on the molecules, which were predicted to be essential for binding at those receptors using molecular docking. As predicted, when the N6-benzyl group (viewed as a deep anchor in the α2 adrenergic receptors) was omitted in subsequent analogues, this off-target activity disappeared. The conserved Asp (3.32) that serves as the counterion for biogenic amines in their GPCRs, changed to a H-bonding role in the predicted recognition of adenosine congeners at the same receptors.
3.6. A3 Agonists for Inflammatory Diseases, Cancer and Chronic Neuropathic Pain
The action of A3AR agonists in disease models is pleiotropic [48], and diverse therapeutic applications are envisioned. Can-Fite Biopharma has advanced IB-MECA 11 to Phase 2 and 3 clinical trials for autoimmune inflammatory diseases, including psoriasis and rheumatoid arthritis, A3AR agonists effectively reduce tumor size in several animal models [49], and Cl-IB-MECA 13 has progressed to Phase 2 clinical trials for hepatocellular carcinoma [50]. Also, an imidazoquinolinamine allosteric enhancer of the A3AR [51] is being proposed for treatment of inflammatory diseases by Can-Fite.
The mechanisms involved in the actions of A3AR agonists in disease models have been summarized by Fishman et al. [49]. In cancer and inflammation, the A3AR is upregulated in the affected tissue. This pathological level of receptor expression, which can often be determined by measuring the A3AR level in peripheral blood mononuclear cells, is also predictive of a patient's response to an A3AR agonist. Activation of the A3AR appears to correct imbalances in the downstream signaling pathways leading to changes in transcription in the nucleus. In models of in vivo inflammation, A3AR agonists have an anti-inflammatory effect by reducing activation of NF-κB (in synoviocytes, neutrophils and other immune cells), in part by reducing the expression of TNF-α.
In cancer, an A3AR-agonist-induced decrease in expression or activity of NF-κB could reduce its antiapoptotic effect. Activation of the A3AR also corrects an imbalance in the Wnt signaling pathway. cAMP inhibition indirectly decreases phosphorylation (and therefore decreased inactivation) of the serine/threonine kinase GSK-3β. The resulting increased phosphorylation of β-catenin allows it to be removed from the cytoplasm by ubiquitination, and its reduced action in the nucleus leads to suppression of cyclin D1 and c-myc and consequently cell growth inhibition. Effects of A3AR activation on downstream signaling can also be in opposite directions. For example, its myeloprotective effect is accompanied by increased NF-κB in splenocytes.
Together with collaborator Daniela Salvemini, we have demonstrated that A3AR agonists, including prototypical and novel ligands that are highly selective for this receptor subtype, are effective in suppressing and preventing chronic neuropathic pain [52]. The chronic constriction injury (CCI) model of neuropathic pain in mice was used to screen dozens of analogues in vivo. The prototypical A3AR agonist IB-MECA and the hypermodified MRS5698 9 were both highly potent and efficacious in this model when administered intraperitoneally or orally, with effective doses well below 1 mg/kg in the mouse. There are both peripheral and central components to this action, as indicated using the nonpermeant MRS5841 23 [28]. Both reversal of mechanoallodynia and hyperalgesia were observed. The necessity for A3AR agonism was demonstrated using the selective AR antagonists and A3AR−/− mice. The potency of A3AR agonists was greater than other pain medications. Also, A3AR agonists do not develop analgesic tolerance [53].
4. Novel P2Y Receptor Ligands
There has been much recent progress in the design of selective P2YR ligands [42,54–60]. For example, BMS and Pfizer have reported diaryl urea derivatives as potent and selective nonnucleotide antagonists of the P2Y1R, a target for new antithrombotic drugs [58,59]. Nevertheless, there is still a large need for improved P2YR ligands of increased stability and bioavailability. Most of the P2YR agonists are still labile phosphate derivatives. Fischer et al. have demonstrated that introduction of a boranophosphate at the α-phosphate position of P2YR agonists greatly enhances the in vivo stability [60]. Other methods of increasing the stability of phosphate groups with varying success include methylenephosphonate bridges and thiophosphates. There is a need for additional uncharged and drug-like P2Y ligands, and the hope is that structure-based approaches will solve this need in the future.
In an effort to understand the conformational factors in recognition of P2YR ligands, we applied the steric constraints of the (N)-methanocarba modification of ribose in nucleotide agonists (and P2Y1R antagonists) to the eight subtypes of P2Y receptors [31,42]. In most cases, the (S)-methanocarba ring was introduced for comparison. This effectively defined the conformational preference at each of the P2Y1-like Gq coupled receptors, but uncertainty remains about which rigid ring would maintain affinity at the P2Y12-like Gi-coupled receptors. Among P2Y1-like Gq-coupled receptors, most receptors prefer the (N)-methanocarba modification over the (S). The potency of ATP at the hP2Y1R was enhanced 29-fold compared to native ATP by this (N) bicyclic ribose substitution [72]. At the hP2Y2 and hP2Y11Rs, (N)-methanocarba-ATP was roughly equipotent to ATP. (S)-methanocarba-ATP (racemic) was 5- and 44-fold less potent than ATP at the P2Y1 and P2Y2Rs, respectively. (N)-methanocarba-UTP was ~ 2-fold less potent than UTP at the P2Y2 and P2Y4Rs. Only the UDP-responsive Gq-coupled P2Y6R prefers the (S)-conformation of ribose; the preference is so strong that the (N)-methanocarba UDP analogue is completely inactive, while the (S)-methanocarba UDP analogue is 7-fold more potent than UDP. Thus, the identical ribose conformational preference is not maintained throughout the evolution of the P2YR family.
4.1. P2Y6 Agonists for Diabetes and Fluorescent P2Y6 Agonists
The protective effects of activation of P2Y6R against TNF-α were first shown in P2YR-expressing astrocytoma cells. P2Y6R activation protects pancreatic islet cells from apoptosis and stimulates glucose-dependent insulin release [61,62] and in insulin target tissues increases glucose uptake (unpublished). These actions involve the enzyme 5′-AMP-activated protein kinase (AMPK), a metabolic regulator that is a target for treatment of type 2 diabetes. We have used a potent and somewhat (12-fold vs. P2Y2R) selective P2Y6R dinucleotide agonist P1-(uridine 5′-)-P3-(N4-methoxycytidine 5′-)triphosphate (MRS2957, 41, structure not shown), which is more stable than most simple UDP analogues to the action of ectonucleotidases. In MIN6 β-islet cells, treatment with MRS2957 (500 nM) activated AMPK, which was blocked by P2Y6R-selective antagonist N,N″-1,4-butanediyl-bis-[N′-(3-isothiocyanatophenyl)]thiourea (MRS2578). Also, MRS2957 induced phosphorylation of acetylcoenzyme A carboxylase (ACC), a marker of AMPK activity [61]. We attenuated P2Y6R-mediated AMPK phosphorylation pharmacologically to reveal involvement of intracellular Ca2 + pathways. P2Y6R agonist induced insulin secretion at high glucose, which was also reduced by AMPK siRNA. The pharmacological agents used to block key steps in the P2Y6R-induced signaling cascade were: calcium chelator BAPTA-AM, calmodulin-dependent protein kinase kinase (CaMKK) inhibitor STO-069 and IP3 receptor antagonist 2-APB. In insulin target cells (C2C12 skeletal muscle cells and 3T3-L1 adipocytes), MRS2957 significantly increased glucose uptake compared to control, which was antagonized by MRS2578. MRS2957-treatment resulted in significant phosphorylation of AMPK in both cell lines, which was abolished by pre-incubation with MRS2578. Also, MRS2957 (30 min incubation) increased glucose transporter GLUT4 recruitment to the cell membrane (unpublished), which was blocked by MRS2578 or AMPK inhibitor (Compound C). Our results indicate that the P2Y6R is involved in controlling glucose metabolism at multiple levels, and this may be mediated through AMPK signaling.
P2Y6R activation by UDP was identified as an autocrine process by which insulin secretion is potentiated in response to glucose [63]. Nevertheless, the use of P2Y6R agonists for the treatment of diabetes is not yet a practical concept for pharmaceutical development. There are detrimental effects of P2Y6R receptor activation, such as increased interleukin (IL) 6 and other inflammatory mediators in the lung and elsewhere [64], vasoconstriction [65] and promotion of osteoclast formation in bone [66]. Moreover, all of the known P2Y6R agonist ligands are nucleotide derivatives and therefore of low oral bioavailability.
We designed fluorescent conjugates of functionalized congeners that display high P2YR affinity, for characterization of these GPCRs in living cells by flow cytometry and in cell membranes. Fluorescent agonists are mostly internalized consistent with agonist-induced GPCR internalization, and this labeling is attenuated by specific P2YR ligands. Examples are 5′-diphosphate derivative MRS4129 42 [54] and MRS4162 43 [55] (Fig. 7A), which are fluorescent pyrimidine nucleotides, respectively, selective agonist of the P2Y6R (EC50 9 nM, phospholipase C activation) and high affinity pan-agonist at P2Y2R, P2Y4R and P2Y6R (expressed in astrocytoma cells). At the end of the 60 min incubation period with astrocytoma cells expressing the P2Y6R, the fraction of fluorescence of MRS4129 associated with the internalized label was much greater (77% of the total binding) than the fraction on the cell surface (17% of the total binding).
Fig. 7.
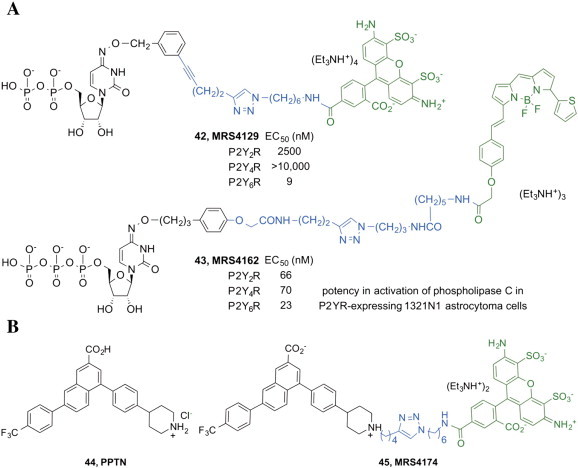
A. Fluorescent agonist ligands 42 and 43 (as salts) with high affinity, variably, for P2Y2R, P2Y4R and P2Y6R [54,55]. The colors range from the charged phosphate (brown) to the green nucleoside moiety, to the spacer (blue) and the fluorophore (violet). B. High affinity P2Y14R fluorescent antagonist ligand 45 (Ki = 80 pM, antagonist activity measured in hP2Y14R-expressing CHO cells) [70] in comparison with its structural lead PPTN 44 (as HCl salt). The colors indicate the pharmacophore (black), a spacer (blue) and the fluorophore (green). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The point of chain extension from which to tether the fluorophore was a 4-alkyloxoimino group on the pyrimidine ring. This group preserves the double bond character of the 4 substituent of the nucleobase, which favors receptor recognition by analogy to the 4-carbonyl group of uracil. A previous attempt to extend substituents through a pyrimidine 4-thioether failed to maintain P2YR activity (e.g. a 4-hexylthio-UDP equivalent was 10-fold less potent than UDP at the P2Y6R) [67]. In both cases, the fluorophores were conjugated by [2 + 3] click cycloaddition chemistry between an alkynyl group on the pharmacophore and an azide on the fluorophore (AlexaFluor488 for MRS4129 and BODIPY for MRS4162).
4.2. Receptor Docking and Chemical Modification of a P2Y14 Antagonist
Both UDP-glucose (UDPG) and UDP are cognate agonists of the Gi-coupled P2Y14R [68]. In collaboration with Harden, Vsevolod Katritch and others, we explored the SAR of P2Y14R antagonists based on potent and highly selective 4-((piperidin-4-yl)-phenyl)-(7-(4-(trifluoromethyl)-phenyl)-2)-naphthoic acid (PPTN 44, KB = 0.434 nM in antagonizing the cAMP effects of UDPG) [57], which was originally reported by Merck [56]. Unfortunately, PPTN displays low oral bioavailability, and additional P2Y14 antagonists will be needed. The lack of effect up to 10 μM PPTN at the other Gq-linked P2YRs was shown [57]. At Gi-coupled P2YRs, there was no inhibitory effect of PPTN up to 1 μM. This 2-naphthoic acid derivative inhibited UDPG-promoted chemotaxis in HL-60 cells and in human neutrophils, suggesting application of such antagonists in inflammation. Activation of the P2Y13R and the P2Y14R in mast cells promotes degranulation, suggesting application of P2Y antagonists for the treatment of asthma [69].
We designed the first fluorescent ligand of the P2Y14R, i.e. the AlexaFluor488-labeled antagonist MRS4174 45 (Fig. 7B) [70]. This compound contained a functionalized chain at the secondary nitrogen of the piperidine ring, a site that was chosen based on ligand docking at a P2Y14R homology model. An azide-bearing fluorophore was conjugated to an alkyne-functionalized antagonist intermediate by [2 + 3] click cycloaddition chemistry. We modeled the hP2Y14R based on the recent 2-MeSADP-bound hP2Y12R X-ray structure and simulated antagonist docking, which suggested that the piperidine N of PPTN is accessible for tethering fluorophores. Click chemistry was used to conjugate functionalized PPTN alkyne derivatives and azide-bearing fluorophores. Flow cytometry showed high specific P2Y14R binding of MRS4174 45 with exceptionally high affinity (Ki 80 pM, in antagonizing UDPG-induced cAMP inhibition in P2Y14R-expressing CHO cells). Known P2Y ligands inhibited cell labeling consistently with affinity order. Thus, the utility of MRS4174 has been validated initially, but additional methods optimization will be needed to utilize this compound in a routine assay.
5. Conclusions
Numerous therapeutic concepts are associated with selective modulation of ARs and P2YRs. We used chemical synthesis and molecular modeling based on X-ray structures to explore novel ligand discovery at adenosine receptors and P2Y receptors. X-ray structures of agonist-bound and antagonist-bound P2Y12R establish binding modes and detect conformational changes involved in activation, especially in the extracellular loop region. Thus, the 3D knowledge of receptor binding and activation is facilitating drug discovery at GPCRs that respond to extracellular nucleosides and nucleotides. However, its use as a primary tool in predicting the pharmacological profile of proposed new ligands is not yet realized. Future X-ray structures to reveal the structures of other receptors or the already crystallized receptor with novel ligands and the development of more systematic in silico methods will be required.
The ARs are well established as medicinal chemical targets with many selective agonists and antagonists. A3AR agonists have proven more beneficial in disease models (such as autoimmune inflammatory diseases and cancer) than A3AR antagonists. The A3AR selectivity of prototypical agonists has been improved using a structure-based design approach. A constrained ribose substitution in the North (N) conformation enhances A3AR affinity and selectivity, which leads to a general class of highly specific agonists. Off-target activity was probed structurally. Conformational plasticity of the A3AR is predicted to accommodate rigid C2 extension, leading to C2 arylethynyl analogue MRS5698, which protects against chronic neuropathic pain. At the antiepileptic A1AR, a compact, truncated selective agonist MRS5474 that does not produce the toxicity associated with prototypical A1AR agonists is suggested for reducing seizures. With the multiplicity of effects and sites of distribution of the P2Y receptors, their application to disease treatment (with the exception of antithrombotic P2Y12R antagonists) is still more exploratory. P2Y6R agonists have beneficial effects in the pancreas and in insulin target tissues, suggestive of possible application to diabetes. We reported the first fluorescent ligand of a P2Y receptor: MRS4129, which is selective and potent nucleotide agonist of the P2Y6R. Fluorescent agonists MRS5218 (A3AR), MRS4129 and MRS4162 (P2Y6R and other subtypes) and fluorescent antagonist MRS4174 (P2Y14R) are useful probes to characterize specific receptors expressed in living cells using flow cytometry and other methods.
Acknowledgments
We acknowledge support from the NIDDK, NIH Intramural Research Program (Z01 DK031117-27) and the NIGMS Postdoctoral Research Associate (PRAT) Program.
Footnotes
Presented by K. A. Jacobson on July 24, 2014 at Purines 2014 — Nucleotides, Nucleosides and Nucleobases, International Conference on Signalling, Drugs and Targets, Bonn, Germany.
References
- 1.Jacobson K.A., Zablocki J., Bhagwat S. Preface: special issue on medicinal chemistry of purines. Purinergic Signal. 2009;5:125. doi: 10.1007/s11302-008-9120-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murray J.M., Bussiere D.E. Targeting the purinome. In: Jacoby Edgar., editor. Chemogenomics. Vol. 275. 2009. pp. 47–92. (Methods Mol Biol). Chap. 3. [DOI] [PubMed] [Google Scholar]
- 3.Burnstock G. Introduction and perspective, historical note. Front Cell Neurosci. 2013;7:227. doi: 10.3389/fncel.2013.00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zimmermann H., Zebisch M., Sträter N. Cellular function and molecular structure of ectonucleotidases. Purinergic Signal. 2012;8:437–502. doi: 10.1007/s11302-012-9309-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jankowski V., Van Der Giet M., Mischak H., Morgan H., Zidek W., Jankowski J. Dinucleoside polyphosphates: strong endogenous agonists of the purinergic system. Br J Pharmacol. 2009;157:1142–1153. doi: 10.1111/j.1476-5381.2009.00337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jaakola V.P., Griffith M.T., Hanson M.A., Cherezov V., Chien E.Y., Lane J.R. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu F., Wu H., Katritch V., Han G.W., Jacobson K.A., Gao Z.G. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang K., Zhang J., Gao Z.G., Zhang D., Zhu L., Han G.W. Structure of the human P2Y12 receptor in complex with an antithrombotic drug. Nature. 2014;509:115–118. doi: 10.1038/nature13083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang K., Zhang J., Gao Z.G., Paoletta S., Zhang D., Han G.W. Agonist-bound structure of the human P2Y12R receptor. Nature. 2014;509:119–122. doi: 10.1038/nature13288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kawate T., Michel J.C., Birdsong W.T., Gouaux E. Crystal structure of the ATP-gated P2X4 ion channel in the closed state. Nature. 2009;460:592–598. doi: 10.1038/nature08198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Congreve M., Andrews S.P., Dore A.S., Hollenstein K., Hurrell E., Langmead C.J. Discovery of 1,2,4-triazine derivatives as adenosine A2A antagonists using structure based drug design. J Med Chem. 2012;55:1898–1903. doi: 10.1021/jm201376w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katritch V., Jaakola V.P., Lane J.R., Lin J., IJzerman A.P., Yeager M. Structure-based discovery of novel chemotypes for adenosine A2A receptor antagonists. J Med Chem. 2010;53:1799–1809. doi: 10.1021/jm901647p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carlsson J., Yoo L., Gao Z.G., Irwin J., Shoichet B., Jacobson K.A. Structure-based discovery of A2A adenosine receptor ligands. J Med Chem. 2010;53:3748–3755. doi: 10.1021/jm100240h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mason J.S., Bortolato A., Weiss D.R., Deflorian F., Tehan B., Marshall F.H. High end GPCR design: crafted ligand design and druggability analysis using protein structure, lipophilic hotspots and explicit water networks. In Silico Pharmacol. 2013;1:23. [Google Scholar]
- 15.Bacilieri M., Ciancetta A., Paoletta S., Federico S., Cosconati S., Cacciari B. Revisiting a receptor-based pharmacophore hypothesis for human A2A adenosine receptor antagonists. J Chem Inf Model. 2013;53:1620–1637. doi: 10.1021/ci300615u. [DOI] [PubMed] [Google Scholar]
- 16.Tosh D.K., Phan K., Gao Z.G., Gakh A., Xu F., Deflorian F. Optimization of adenosine 5′-carboxamide derivatives as adenosine receptor agonists using structure-based ligand design and fragment-based searching. J Med Chem. 2012;55:4297–4308. doi: 10.1021/jm300095s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Daly J.W., Padgett W.L., Secunda S.I., Thompson R.D., Olsson R.A. Structure–activity relationships for 2-substituted adenosines at A1 and A2 adenosine receptors. Pharmacology. 1993;46:91–100. doi: 10.1159/000139033. [DOI] [PubMed] [Google Scholar]
- 18.Fredholm B.B., Jacobson K.A. John W. Daly and the early characterization of adenosine receptors. Heterocycles. 2009;79:73–83. doi: 10.3987/COM-08-S(D)Memoire-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shamim M.T., Ukena D., Padgett W.L., Hong O., Daly J.W. 8-Aryl- and 8-cycloalkyl-1,3-dipropylxanthines: further potent and selective antagonists for A1-adenosine receptors. J Med Chem. 1988;31:613–617. doi: 10.1021/jm00398a020. [DOI] [PubMed] [Google Scholar]
- 20.Paoletta S., Tosh D.K., Salvemini D., Jacobson K.A. Structural probing of off-target G protein-coupled receptor activities within a series of adenosine/adenine congeners. PLoS ONE. 2014;9:e97858. doi: 10.1371/journal.pone.0097858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cristalli G., Lambertucci C., Marucci G., Volpini R., Dal Ben D. A2A adenosine receptor and its modulators: overview on a druggable GPCR and on structure–activity relationship analysis and binding requirements of agonists and antagonists. Curr Pharm Des. 2008;14:1525–1552. doi: 10.2174/138161208784480081. [DOI] [PubMed] [Google Scholar]
- 22.Klotz K.N., Kachler S., Lambertucci C., Vittori S., Volpini R., Cristalli G. 9-Ethyladenine derivatives as adenosine receptor antagonists: 2- and 8-substitution results in distinct selectivities. Naunyn Schmiedeberg's Arch Pharmacol. 2003;367:629–634. doi: 10.1007/s00210-003-0749-9. [DOI] [PubMed] [Google Scholar]
- 23.Camaioni E., Costanzi S., Vittori S., Volpini R., Klotz K.N., Cristalli G. New substituted 9-alkylpurines as adenosine receptor ligands. Bioorg Med Chem. 1998;6:523–533. doi: 10.1016/s0968-0896(98)00007-8. [DOI] [PubMed] [Google Scholar]
- 24.Thompson R.D., Secunda S., Daly J.W., Olsson R.A. N6,9-disubstituted adenines: potent, selective antagonists at the A1 adenosine receptor. J Med Chem. 1991;34:2877–2882. doi: 10.1021/jm00113a029. [DOI] [PubMed] [Google Scholar]
- 25.Dore A.S., Robertson N., Errey J.C., Ng I., Hollenstein K., Tehan B. Structure of the adenosine A2A receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure. 2011;19:1283–1293. doi: 10.1016/j.str.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ballesteros J.A., Weinstein H. Integrated methods for the construction of three-dimensional models of structure–function relations in G protein-coupled receptors. Meth Neurosci. 1995;25:366–428. [Google Scholar]
- 27.Tosh D.K., Deflorian F., Phan K., Gao Z.G., Wan T.C., Gizewski E. Structure-guided design of A3 adenosine receptor-selective nucleosides: combination of 2-arylethynyl and bicyclo[3.1.0]hexane substitutions. J Med Chem. 2012;55:4847–4860. doi: 10.1021/jm300396n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paoletta S., Tosh D.K., Finley A., Gizewski E., Moss S.M., Gao Z.G. Rational design of sulfonated A3 adenosine receptor-selective nucleosides as pharmacological tools to study chronic neuropathic pain. J Med Chem. 2013;56:5949–5963. doi: 10.1021/jm4007966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Higgs C., Beuming T., Sherman W. Hydration site thermodynamics explain SARs for triazolylpurines analogues binding to the A2A receptor. ACS Med Chem Lett. 2010;1:160–164. doi: 10.1021/ml100008s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rasmussen S.G., Devree B.T., Zou Y., Kruse A.C., Chung K.Y., Kobilka T.S. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tosh D.K., Jacobson K.A. Methanocarba ring as a ribose modification in ligands of G protein-coupled purine and pyrimidine receptors: synthetic approaches. Med Chem Commun. 2013;4:619–630. doi: 10.1039/C2MD20348K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Franchetti P., Cappellacci L., Vita P., Petrelli R., Lavecchia A., Kachler S. N6Cycloalkyl-and N6-bicycloalkyl-C5′(C 2′)-modified adenosine derivatives as high-affinity and selective agonists at the human A1 adenosine receptor with antinociceptive effects in mice. J Med Chem. 2009;52:2393–2406. doi: 10.1021/jm801456g. [DOI] [PubMed] [Google Scholar]
- 33.Marquez V.E., Siddiqui M.A., Ezzitouni A., Russ P., Wang J., Wagner R.W. Nucleosides with a twist. Can fixed forms of sugar ring pucker influence biological activity in nucleosides and oligonucleotides? J Med Chem. 1996;39:3739–3747. doi: 10.1021/jm960306+. [DOI] [PubMed] [Google Scholar]
- 34.Kim G.D., Oh J., Jeong L.S., Lee S.K. Thio-Cl-IB-MECA, a novel A3 adenosine receptor agonist, suppresses angiogenesis by regulating PI3K/AKT/mTOR and ERK signaling in endothelial cells. Biochem Biophys Res Commun. 2013;437:79–86. doi: 10.1016/j.bbrc.2013.06.040. [DOI] [PubMed] [Google Scholar]
- 35.Yang H., Avila M.Y., Peterson-Yantorno K., Coca-Prados M., Stone R.A., Jacobson K.A. The cross-species A3 adenosine-receptor antagonist MRS 1292 inhibits adenosine-triggered human nonpigmented ciliary epithelial cell fluid release and reduces mouse intraocular pressure. Curr Eye Res. 2005;30:747–754. doi: 10.1080/02713680590953147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kozma E., Gizewski E.T., Tosh D.K., Squarcialupi L., Auchampach J.A., Jacobson K.A. Characterization by flow cytometry of fluorescent, selective agonist probes of the A3 adenosine receptor. Biochem Pharmacol. 2013;85:1171–1181. doi: 10.1016/j.bcp.2013.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gachet C. P2Y12 receptors in platelets and other hematopoietic and non-hematopoietic cells. Purinergic Signal. 2012;8:609–619. doi: 10.1007/s11302-012-9303-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang C., Srinivasan Y., Arlow D.H., Fung J.J., Palmer D., Zheng Y. High-resolution crystal structure of human protease-activated receptor. Nature. 2012;492:387–392. doi: 10.1038/nature11701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bach P., Boström J., Brickmann K., van Giezen J.J., Groneberg R.D., Harvey D.M. Synthesis, structure–property relationships and pharmacokinetic evaluation of ethyl 6-aminonicotinate sulfonylureas as antagonists of the P2Y12 receptor. Eur J Med Chem. 2013;65:360–375. doi: 10.1016/j.ejmech.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 40.Schmidt P., Ritscher L., Dong E.N., Hermsdorf T., Cöster M., Wittkopf D. Identification of determinants required for agonistic and inverse agonistic ligand properties at the ADP receptor P2Y12. Mol Pharmacol. 2013;83:256–266. doi: 10.1124/mol.112.082198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tautermann C.S. GPCR structures in drug design, emerging opportunities with new structures. Bioorg Med Chem Lett. 2014;24:4073–4079. doi: 10.1016/j.bmcl.2014.07.009. [DOI] [PubMed] [Google Scholar]
- 42.Jacobson K.A., Jayasekara M.P.S., Costanzi S. Molecular structure of P2Y receptors: mutagenesis, modelling and chemical probes. WIREs Membr Transp Signal. 2012;1:815–827. doi: 10.1002/wmts.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tosh D.K., Paoletta S., Deflorian F., Phan K., Moss S.M., Gao Z.G. Structural sweet spot for A1 adenosine receptor activation by truncated (N)-methanocarba nucleosides: receptor docking and potent anticonvulsant activity. J Med Chem. 2012;55:8075–8090. doi: 10.1021/jm300965a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gao Z.G., Blaustein J., Gross A.S., Melman N., Jacobson K.A. N6-substituted adenosine derivatives: selectivity, efficacy, and species differences at A3 adenosine receptors. Biochem Pharmacol. 2003;65:1675–1684. doi: 10.1016/s0006-2952(03)00153-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Knutsen L.J., Lau J., Petersen H., Thomsen C., Weis J.U., Shalmi M. N-substituted adenosines as novel neuroprotective A1 agonists with diminished hypotensive effects. J Med Chem. 1999;42:3463–3477. doi: 10.1021/jm960682u. [DOI] [PubMed] [Google Scholar]
- 46.Moss S.M., Jayasekara P.S., Paoletta S., Gao Z.G., Jacobson K.A. Structure-based design of reactive nucleosides for site-specific modification of the A2A adenosine receptor. ACS Med Chem Lett. 2014;5:1043–1048. doi: 10.1021/ml5002486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun W.C., Moore J.N., Hurley D.J., Vandenplas M.L., Linden J., Cao Z. Adenosine A2A receptor agonists inhibit lipopolysaccharide-induced production of tumor necrosis factor-alpha by equine monocytes. Vet Immunol Immunopathol. 2008;121:91–100. doi: 10.1016/j.vetimm.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 48.Baraldi P.G., Preti D., Borea P.A., Varani K. Medicinal chemistry of A3 adenosine receptor modulators: pharmacological activities and therapeutic implications. J Med Chem. 2012;55:5676–5703. doi: 10.1021/jm300087j. [DOI] [PubMed] [Google Scholar]
- 49.Fishman P., Bar-Yehuda S., Liang B.T., Jacobson K.A. Pharmacological and therapeutic effects of A3 adenosine receptor (A3AR) agonists. Drug Discov Today. 2012;17:359–366. doi: 10.1016/j.drudis.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gessi S., Merighi S., Sacchetto V., Simioni C., Borea P.A. Adenosine receptors and cancer. Biochim Biophys Acta. 2011;1808:1400–1412. doi: 10.1016/j.bbamem.2010.09.020. [DOI] [PubMed] [Google Scholar]
- 51.Gao Z.G., Verzijl D., Zweemer A., Ye K., Göblyös A., IJzerman A.P. Functionally biased modulation of A3 adenosine receptor agonist efficacy and potency by imidazoquinolinamine allosteric enhancers. Biochem Pharmacol. 2011;82:658–668. doi: 10.1016/j.bcp.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen Z., Janes K., Chen C., Doyle T., Tosh D.K., Jacobson K.A. Controlling murine and rat chronic pain through A3 adenosine receptor activation. FASEB J. 2012;26:1855–1865. doi: 10.1096/fj.11-201541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Janes K., Wahlman C., Little J., Doyle T., Tosh D., Jacobson K. Spinal neuroimmmune activation is independent of T-cell infiltration and attenuated by A3 adenosine receptor agonists in a model of oxaliplatin-induced peripheral neuropathy. Brain Behav Immun. 2014 doi: 10.1016/j.bbi.2014.08.010. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jayasekara P.S., Barrett M.O., Ball C.B., Brown K.A., Kozma E., Costanzi S. 4-Alkyloxyimino-cytosine nucleotides: tethering approaches to molecular probes for the P2Y6 receptor. Med Chem Commun. 2013;4:1156–1165. doi: 10.1039/C3MD00132F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jayasekara P.S., Barrett M.O., Ball C.B., Brown K.A., Hammes E., Balasubramanian R. 4-Alkyloxyimino derivatives of uridine-5′-triphosphate: distal modification of potent agonists as a strategy for molecular probes of P2Y2, P2Y4 and P2Y6 receptors. J Med Chem. 2014;57:3874–3883. doi: 10.1021/jm500367e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gauthier J.Y., Belley M., Deschênes D., Fournier J.F., Gagné S., Gareau Y. The identification of 4,7-disubstituted naphthoic acid derivatives as UDP-competitive antagonists of P2Y14. Bioorg Med Chem Lett. 2011;21:2836–2839. doi: 10.1016/j.bmcl.2011.03.081. [DOI] [PubMed] [Google Scholar]
- 57.Barrett M.O., Sesma J.I., Ball C.B., Jayasekara P.S., Jacobson K.A., Lazarowski E.R. Inhibition of UDP-glucose-promoted chemotaxis of human neutrophils by a selective high affinity antagonist of the P2Y14 receptor. Mol Pharmacol. 2013;84:41–49. doi: 10.1124/mol.113.085654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chao H., Turdi H., Herpin T.F., Roberge J.Y., Liu Y., Schnur D.M. Discovery of 2-(phenoxypyridine)-3-phenylureas as small molecule P2Y1 antagonists. J Med Chem. 2013;56:1704–1714. doi: 10.1021/jm301708u. [DOI] [PubMed] [Google Scholar]
- 59.Pfefferkorn J.A., Choi C., Winters T., Kennedy R., Chi L., Perrin L. P2Y1 receptor antagonists as novel antithrombotic agents. Bioorg Med Chem Lett. 2008;18:3338–3343. doi: 10.1016/j.bmcl.2008.04.028. [DOI] [PubMed] [Google Scholar]
- 60.Haas M., Ginsburg-Shmuel T., Fischer B., Reiser G. 5-OMe-uridine-5′-O-(α-boranodiphosphate), a novel nucleotide derivative highly active at the human P2Y6 receptor protects against death-receptor mediated glial apoptosis. Neurosci Lett. 2014;578:80–84. doi: 10.1016/j.neulet.2014.06.030. [DOI] [PubMed] [Google Scholar]
- 61.Balasubramanian R., Maruoka H., Jayasekara P.S., Gao Z.G., Jacobson K.A. AMP-activated protein kinase as regulator of P2Y6 receptor-induced secretion in MIN6 mouse pancreatic β cells. Biochem Pharmacol. 2013;85:991–998. doi: 10.1016/j.bcp.2012.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Parandeh F., Abaraviciene S.M., Amisten S., Erlinge D., Salehi A. Uridine diphosphate (UDP) stimulates insulin secretion by activation of P2Y6 receptors. Biochem Biophys Res Commun. 2008;370:499–503. doi: 10.1016/j.bbrc.2008.03.119. [DOI] [PubMed] [Google Scholar]
- 63.Sassmann A., Gier B., Gröne H.J., Drews G., Offermanns S., Wettschureck N. The Gq/G11-mediated signaling pathway is critical for autocrine potentiation of insulin secretion in mice. J Clin Invest. 2010;120:2184–2193. doi: 10.1172/JCI41541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vieira R.P., Müller T., Grimm M., von Gernler V., Vetter B., Dürk T. Purinergic receptor type 6 contributes to airway inflammation and remodeling in experimental allergic airway inflammation. Am J Respir Crit Care Med. 2011;184:215–223. doi: 10.1164/rccm.201011-1762OC. [DOI] [PubMed] [Google Scholar]
- 65.Malmsjö M., Hou M., Pendergast W., Erlinge D., Edvinsson L. Potent P2Y6 receptor mediated contractions in human cerebral arteries. BMC Pharmacol. 2003;3:4. doi: 10.1186/1471-2210-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Orriss I.R., Wang N., Burnstock G., Arnett T.R., Gartland A., Robaye B. The P2Y6 receptor stimulates bone resorption by osteoclasts. Endocrinology. 2011;152:3706–3716. doi: 10.1210/en.2011-1073. [DOI] [PubMed] [Google Scholar]
- 67.Boucher RC, Shaver SR, Pendergast W, Yerxa B, Rideout JL, Dougherty R, Croom D (2000) Uridine 5′-diphosphate and analogs useful for treating lung diseases. US 6,143,279
- 68.Carter R.L., Fricks I.P., Barrett M.O., Burianek L.E., Zhou Y., Ko H. Quantification of Gi-mediated inhibition of adenylyl cyclase activity reveals that UDP is a potent agonist of the human P2Y14 receptor. Mol Pharmacol. 2009;76:1341–1348. doi: 10.1124/mol.109.058578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gao Z.G., Wei Q., Jayasekara M.P.S., Jacobson K.A. The role of P2Y14 and other P2Y receptors in degranulation of human LAD2 mast cells. Purinergic Signal. 2013;9:31–40. doi: 10.1007/s11302-012-9325-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kiselev E., Barrett M.O., Katritch V., Paoletta S., Weitzer C.D., Hammes E. Exploring a 2-naphthoic acid template for the structure-based design of P2Y14 receptor antagonist molecular probes. ACS Chem Biol. 2014 doi: 10.1021/cb500614p. http://dx.doi.org/10.1021/cb500614p, (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tchilibon S., Kim S.K., Gao Z.G., Harris B.A., Blaustein J., Gross A.S. Exploring distal regions of the A3 adenosine receptor binding site: sterically-constrained N6-(2-phenylethyl)adenosine derivatives as potent ligands. Bioorg Med Chem. 2004;12:2021–2034. doi: 10.1016/j.bmc.2004.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim H.S., Ravi R.G., Marquez V.E., Maddileti S., Wihlborg A.-K., Erlinge D. Methanocarba modification of uracil and adenine nucleotides: high potency of Northern ring conformation at P2Y1, P2Y2, P2Y4 and P2Y11, but not P2Y6 receptors. J Med Chem. 2002;45:208–218. doi: 10.1021/jm010369e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jacobson K.A., X-d Ji, Li A.H., Melman N., Siddiqui M.A., Shin K.J. Methanocarba analogues of purine nucleosides as potent and selective adenosine receptor agonists. J Med Chem. 2000;43:2196–2203. doi: 10.1021/jm9905965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kiesewetter D.O., Lang L., Ma Y., Bhattacharjee A.K., Gao Z.G., Joshi B.V. Synthesis and characterization of [76Br]-labeled high affinity A3 adenosine receptor ligands for positron emission tomography. Nucl Med Biol. 2009;36:3–10. doi: 10.1016/j.nucmedbio.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]