

Abstract
One of the less understood aspects of membrane transporters is the dynamic coupling between conformational change and substrate transport, because the dynamic uninhibited states of transporters have been largely inaccessible for structural studies. Here, we use NMR approaches to investigate conformational heterogeneity of the GTP/GDP carrier (GGC) from yeast mitochondria. NMR residual dipolar coupling (RDC) analysis of GGC in DNA-origami nanotube liquid crystal shows that several structured segments have different generalized degree of order (GDO), indicating the presence of conformational heterogeneity. Complete GDO mapping reveals asymmetry between domains of the transporter and even within certain transmembrane helices. Substrate titration measurements show that nucleotide binding partially reduces local structural heterogeneity, and that the substrate binds to multiple sites along the transport cavity. Our observations suggest that mitochondrial carriers in the uninhibited states are intrinsically plastic and structural plasticity is asymmetrically distributed among the three homologous domains.
Keywords: transporter mechanism, conformational heterogeneity, GTP/GDP carrier, NMR spectroscopy
Graphical Abstract
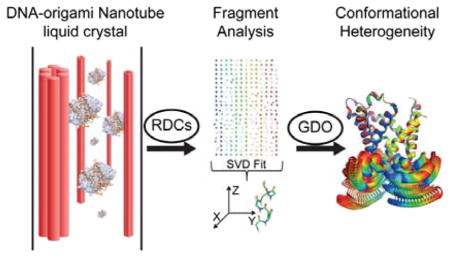
Liquid-state NMR spectroscopy has been used to study conformational heterogeneity of mitochondrial GTP/GDP transporter. The data reveal that the carrier is intrinsically plastic. Nevertheless of the threefold pseudo symmetry of the carrier, the plasticity is asymmetrically distributed among the domains.
Most solute transporters in membrane adopt the so-called alternating access mode of transport, in which access to the central binding site by substrate can be switched between two opposite sides of the membrane.[1] This mechanism involves at least two conformational states: the outward-facing open conformation where the substrates can enter the binding site from the extracellular space and the inward-facing conformation where the binding site is exposed to the intracellular compartment, for the release of the bound substrate and for receiving new substrates.[1–2]
A recurring theme in these transporters is the presence of conformationally homologous domains, which are related to each other by a distinct symmetry axis. Most of the transporter architectures have a two-fold pseudo symmetry[3] with the symmetry axis being parallel to the substrate translocation pathway. These transporters are generally known to switch between the “V” and inverted “V” types of structures during their transport cycles.[4] There are, however, also transporters that adopt three-fold pseudo symmetry and their mechanism of transport is much less understood.
A major transporter family with three-fold pseudo symmetry is the mitochondrial carrier family. This family of transporters selectively catalyze the trafficking of metabolites, nucleotides, ions and vitamins across the mitochondrial inner membrane;[2a, 5] they are driven mainly by substrate concentration gradient and have been proposed to adopt the alternating access mechanism.[2a, 6] To date, structures of only two mitochondrial carriers are available and they all have been determined under inhibited conditions; they are the crystal structures of AAC in complex with CATR[7] and the NMR-derived backbone structure of UCP2 in the presence of GDP.[8] The overall architecture of the carrier protein resembles an open-top container formed with three structurally similar domains, and each domain consists of two packed transmembrane (TM) helices (labelled ‘H’) separated in sequence by an amphipathic (AP) helix (labelled ‘h’) (Figure S1a).[7a] The domains contain several conserved sequence/structure motifs, among which the important ones are the proline kinks (P-kinks) of H1, H3, and H5, which act as the “pivots” of the transporter,[2a] and the glycine-linker (G-linker) that modulates the relative orientation between TM and AP helices in each domain (Figure S1b).[9] The P-kinks are approximately at the midpoint of the transporter along the membrane normal and divide the transporter into two sections, referred to as the cytosolic section (c-section) and matrix section (m-section) in this paper (Figure S1a).
NMR spectroscopy is emerging as a technology for investigating conformational dynamics of transporters and ion channels and 7-TM helices receptors, as exemplified by the studies on EmrE[4b], KcsA[10], rhodopsin[11] and β2A receptor.[12] In earlier work, we demonstrated the use of NMR in achieving full-scale characterization of the backbone conformation of a 34 kDa mitochondrial transporter known as the uncoupling protein 2 (UCP2) in the inhibited state.[8] Encouraged by this NMR study, we sought to use NMR to investigate the conformational dynamics of carrier proteins in the uninhibited form to facilitate the understanding of whether the conversion involves purely rigid body movements of the pseudo-symmetric protein domains, or whether it also requires intrinsic structural variability on both global and local levels.
The carrier protein we chose for the current study is the GTP/GDP carrier (GGC) from yeast mitochondria.[13] GGC is a 300 amino acid residues transporter that transports external GTP into the mitochondrial matrix while exporting internal GDP out of the matrix.[13] The structure of GGC is unknown. Here, we combine dipolar coupling tensor analysis and chemical shift perturbation to investigate the local and global conformational variability of GGC in the absence of substrate and how ligand bindings alter the conformational heterogeneity of GGC.
GGC with C-terminal His-tag was expressed in E. coli cells, purified, and solubilized in dodecylphosphocholine (DPC) detergent as described in the Supplementary Information. The 1H-15N transverse relaxation-optimized spectroscopy (TROSY) spectrum of the reconstituted GGC shows good spectral resolution and resonance intensity (Figure 1). Backbone assignments have been obtained for 94% of all non-proline residues (Figure S2, Table S1). We built a structural model of GGC (Figure S3) based on alignment of signature sequence motifs and secondary structures between GGC and AAC (PDB code: 1OKC). The secondary structures of GGC were determined using a combination of RDC-based molecular fragment replacement (MFR) method[8, 14] and NMR chemical shifts (TALOS+)[15] (See Figure S2, S4 and Methods in Supplementary Information).
Figure 1.

Backbone amide resonance assignments of GGC. The 2D [15N-1H] TROSY-HSQC spectrum of U-[2H, 15N, 13C]-GGC in DPC micelles shown here was collected on a Bruker 600 MHz spectrometer with cryogenic probe at 30 °C. Sequence specific resonance assignments are indicated by the residue labels. To resolve the peaks in the crowded region, the spectrum is over-processed with a sharp apodization function in the 1H dimension with a 63° shifted squared sine-bell window.
We then used NMR chemical shift perturbation to examine GGC binding with nucleotides including GDP, GTP, and AMP. We measured NMR resonance shifts using the 3D TROSY HNCO spectrum, which correlates the chemical shifts of backbone 1H, 15N, and 13C’ nuclides. GDP and GTP both induced substantial chemical shift changes whereas the effect of nucleoside monophosphate AMP is insignificant (Figure S5). This result is consistent with previous observations that GGC in liposomes catalyzed the transport of GTP and GDP with high efficiency, as well as the transport of the corresponding deoxynucleotides and the structurally related inosine di- and triphosphate.[13]
Addition of GDP and GTP to GGC induced similar pattern of chemical shift perturbation (Figure S6), indicating that the two nucleotides have common binding sites and/or same allosteric effect on the carrier. Despite the three-fold pseudo symmetry in conformation, the chemical perturbation is highly asymmetric. The m-section of the protein shows greater substrate-induced chemical perturbation than the c-section (Figure S6e,f). More specifically, the helical segments of the P-kinks and G-linkers in the m-section show pronounced chemical shift changes. Among the three similar domains, the N-terminus and C-terminus, H2 of domain I and h2 of domain II show distinctly larger chemical shift perturbation than the rest. The asymmetric and widespread changes in chemical environment induced by nucleotide binding suggest a rather global change in conformation and/or dynamics in GGC upon substrate binding.
To examine substrate affinity, we calculated residue-specific dissociation constant (KD) for GDP and GTP (examples of binding saturation curves are shown in Figure 2a, S7 and S8a). The different KD observed for different amino acids of the protein suggest multiple binding sites along the transport path (Table S2). Mapping the KD values onto the GGC model shows that regions with highest affinity (or lowest KD) are clustered around the “pivots” region above the P-kinks consisting of residues in H1 (24 – 28), H3 (132, 133), H5 (228) and middle regions of H2 (86 – 90), H4 (176 – 182), H6 (276, 281 – 282) (Figure 2b,c and S8b,c). These regions appear to constitute a well-defined, polar binding site for nucleotide at the centre of the transporter, and this is consistent with a binding mode proposed earlier based on sequence alignment and the pseudo-three fold symmetry of the mitochondrial carrier family.[9, 16–17] Overall, the affinity for GTP (mean KD of the central binding site (CBS) = 6.6 mM) is about 4-fold stronger than that of GDP (mean KD of the CBS = 23 mM). The 3–4 fold difference in substrate affinities is qualitatively consistent with earlier measurements of GGC transport activity that the apparent KM of GTP and GDP transport are 1.2 ± 0.1 μM and 4.5 ± 0.7 μM, respectively.[13]
Figure 2.

Residue-specific KD of GGC–GTP binding. (a) Sample chemical shift perturbation from 3D TROSY HNCO spectra of GGC upon incremental addition of GTP. The NMR peaks at different substrate/protein molar ratio are shown with the linear color spectrum scale from ratio = 0 (red) to ratio = 35 for GTP (magenta). For each of the perturbed resonances, the chemical shift changes (defined in Supplementary Information) vs. GTP/GGC molar ratio are plotted and fitted to the standard equation of binding equilibrium (right panels). (b) Mapping residue-specific KD onto the GGC model for GTP binding with color-code defined in the Figure. The common binding site (CBS) of nucleotide carrier proteins as proposed based on conservation of amino acid, comparative model and chemical properties,[16] is indicated by the green arrow. (c) The KD values in b are shown in the context of the schematic diagram of the tripartite topology and of the cytoplasmic and matrix views or providing a conceptual view of nucleotide binding sites.
In addition to the central binding site, the KD mapping also revealed two other regions of substrate affinity. One in the c-section consisting of the N-terminus of H1 and the C-terminus of H6 that may function to initially recruit the substrates to the transporter via charge-charge interaction (Figure 2c). Another region is in the m-section and composes of the G-linker of domain I, and the amphipathic helix, h2, of domain II. It is interesting to note that this region show not only large chemical shift perturbation (Figure S6), but also low KD values (Figure 2c, S8c), suggesting that these parts of the GGC play important roles in the binding and transport of nucleotides.
During RDC-based structural analysis of GGC, we found that different structured segments, as identified by RDC-based MFR, are subject to different alignment tensors, indicating that these segments do not align together as a rigid body in solution and that they move or reorient relative to each other. This observation presents a fundamental problem for structure determination: there is not a convergent conformation in the statistical ensemble to be determined, in contrast to the previous structure determination of UCP2[8], which allowed us to refine the entire structure against RDCs without introducing very high dipolar coupling energy. However, RDC can also provide dynamics information complementary to R1 and R2 as it covers motions faster than the timescale at which chemical shifts are measured, including the ns and μs-ms motions. We thus used a previously introduced parameter of alignment tensor, known as the generalized degree of order (GDO, ϑ),[18] to investigate local and global conformational heterogeneity. The parameter ϑi was determined for all seven-residue segments centred at i along the GGC sequence, except for those that did not have structurally convergent fragments (See Figure S9, S10 and details in Supplementary Information).
Mapping ϑi onto the GGC topology showed that in the absence of substrate, GGC has large global conformational heterogeneity, as the GDO values from the c- and m-sections of the protein are very different. Moreover, even within the m-section of the protein, the GDO values differ significantly between the amphipathic helices (h1 and h3) (Figure 3a) and between the helical segments next to the P-kinks. GTP binding overall reduces GDO dispersion (Figure 3b, 4b) though asymmetric differences persisted between domains, for example, domain I (large perturbation in H2) and domain III (Figure 4b). In contrast, the GDO analysis of the structured fragments of UCP2 bound to the inhibitor GDP shows much less GDO variations (Figure S11), indicating that the conformation of the inhibited UCP2 is more homogeneous. The large differences between the GDO maps of uninhibited GGC and inhibited UCP2 are consistent with the common notion that transporters are generally dynamic and structural homogeneity often requires their inactivation. The analysis of GGC and UCP2 data are also validation of the approach of using GDO as a probe for structural heterogeneity.
Figure 3.

Mapping conformational heterogeneity of the odd helices. a) GDO variation in GGC in the absence of GTP and b) in presence of 30mM of GTP, as shown by the plot of GDO values vs. sliding seven-residue fragments for H1, H3 and H5.
Figure 4.

Mapping conformational heterogeneity of the amphipathic and even helices. a) GDO variation in GGC in the absence of GTP and b) in presence of 30mM of GTP, as shown by the plot of GDO values vs. sliding seven-residue fragments for H2, H4 and H6.
To reveal potential local conformational heterogeneity within the secondary elements of GGC, we mapped Δϑi =|ϑi-ϑi+3|, which is the GDO differences between seven-residue segments that are shifted relative to each other by three residues, onto the GGC topology (Figure 5). In the substrate-free state, H1 and h1 of domain I show high local conformational heterogeneity. Although GDO values are not available for the H2 helix of domain I due to missing NMR resonances or lack of convergent fragments, it is likely that this TM helix also undergoes large conformational exchange, which could have broadened the NMR resonances. In addition, the regions of H4 and H5 in the m-section show large Δϑi. Overall, the m-section of the transporter has higher local conformational plasticity than the c-section. GTP binding significantly reduces the majority of the Δϑi values, with most pronounced effects localized to the P-kinks and the helical segments following the P-kinks in the m-section. This result suggests that GTP binding partially stabilizes the pivot region of the transporter. But local conformational heterogeneity persists for H2 and h1 of domain I and G-linker of domain II. Similar to the chemical shift perturbation caused by substrate binding, the overall distribution of local and global conformational heterogeneity as indicated by RDC tensor analysis is also highly asymmetric.
Figure 5.
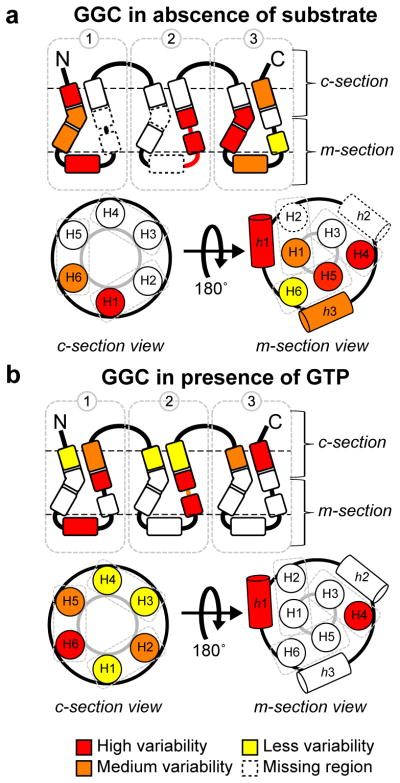
Mapping local GDO variation for GGC a) in the absence and b) in the presence of GTP. Local GDO variation is defined as the GDO difference between seven-residue segments that are shifted relative to each other by three residues.
The model in Figure 2b, based on the crystal structure of AAC locked in c-state by the inhibitor CATR, confirms the central binding site and shows that it is readily accessible by substrate from the c-side, and should thus represent the c-state conformation. In addition to the central pivot region, large perturbation in the N-terminus and C-terminus at the c-side mouth of the GGC cavity suggests the existence of a secondary binding site in the c-section of the protein that may actively recruit substrates before they enter the cavity. Moreover, a large chemical shift perturbation is observed on H2 (Figure S6e,f). This perturbation can be related to the translocation of the substrate inside the cavity and this is consistent with the mechanism of electrostatic funnelling of substrate proposed earlier for AAC.[19]
Substrate binding also causes substantial chemical shift change in the m-section of all three domains (Figure S6e,f). In particular, the G-linkers of domain I and the AP helix h2 show lower than average KD values (Figure 2c, S8c). These perturbed residues do not appear to constitute a continuous binding region in our structural model of GGC built based on the c-state of AAC. A plausible explanation is that NMR observes the time and ensemble average of the c- and m-states. Likewise these observed chemical shift perturbations are due to substrate binding to the m-state, which presumably differ significantly from the c-state. It is also possible that substrate binding induces conformation change in the m-section of the protein, which is also expected to give rise to large chemical shift perturbation. Finally, even in the c-state conformation, GGC can conceivably recruit substrate using the highly basic amphipathic helices, e.g., an ATP binding site between h1 and the loop before h2 on the m-side of AAC has been proposed based on MD simulation.[20]
GGC in the absence of substrate has large global conformational heterogeneity as indicated by the different GDO values throughout the three domains of the protein. The helical segments in the c- and m-sections of the transporter also have very different GDO values. These results are consistent with conformational equilibrium between the c- and m-states of GGC in the absence of ligand. GGC is capable of performing bidirectional transport, i.e., GGC reconstituted in liposome can catalyze GDP/GDP and GTP/GTP homoexchange,[13] and the intrinsic “molecular breathing” would allow the entrance of substrates into the transporter from either side of the membrane.
An unexpected finding, however, is that GGC in the absence of substrates also shows large local conformational heterogeneity, e.g., GDO values vary significantly even within the H1, H4, and H5 helices (Figure 3a, 4a and 5a). It has been proposed that TM helices of membrane proteins are often malleable, possibly due to the shifting of backbone hydrogen bond partners during functional cycles of the proteins.[21] Although the implication of the structural plasticity of TM helices remains to be investigated, we propose that it is important in facilitating the large-scale conformational switch. GTP binding reduces local GDO variation in the P-kink motifs of domains I and III, which indicates partial stabilization of the central substrate-binding region in the middle of the transporter. This result is consistent with GTP binding to the pivot region of GGC as confirmed by the residue-specific KD mapping in Figure 2c. GTP binding induces an asymmetry between the c-section and the m-section of the transporter.
In both apo and GTP-bound states of GGC, the local conformational heterogeneity is asymmetrically distributed with the largest dispersion observed for domain I of the protein. Moreover, GTP binding appears to significantly alter this distribution. This observation is coherent with previous MD simulation of AAC without inhibitor, which showed asymmetric behaviour of the three domains.[22] The finding implies that structural rearrangement that interconvert c- and m-states may not be symmetric as suggested by the threefold pseudo symmetry of the carrier architecture. Conformational heterogeneity is the consequence of conformational plasticity, and we believe such plasticity is important for substrate assess to the apo state and for substrate release from the GTP-bound state.
Supplementary Material
Footnotes
This work was supported by the NIH grant GM094608 (to J.J.C.).
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201xxxxxx.
Contributor Information
Dr. Remy Sounier, Department of Biological Chemistry and Molecular Pharmacology Harvard Medical School Boston, Massachusetts 02115.
Dr. Gaetan Bellot, Institut de Génomique Fonctionnelle, Centre National de la Recherche Scientifique (CNRS) Unité Mixte de Recherche (UMR) 5203, Institut National de la Santé et de la Recherche Médicale (INSERM) U661, Universités de Montpellier 1 and 2 F-34000 Montpellier, France
Dr. James J. Chou, Department of Biological Chemistry and Molecular Pharmacology Harvard Medical School Boston, Massachusetts 02115.
References
- 1.Jardetzky O. Nature. 1966;211:969–970. doi: 10.1038/211969a0. [DOI] [PubMed] [Google Scholar]
- 2.a) Klingenberg M. Biochim Biophys Acta. 2008;1778:1978–2021. doi: 10.1016/j.bbamem.2008.04.011. [DOI] [PubMed] [Google Scholar]; b) West IC. Biochim Biophys Acta. 1997;1331:213–234. doi: 10.1016/s0304-4157(97)00007-5. [DOI] [PubMed] [Google Scholar]
- 3.Forrest LR, Kramer R, Ziegler C. Biochim Biophys Acta. 2011;1807:167–188. doi: 10.1016/j.bbabio.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 4.a) Erkens GB, Hanelt I, Goudsmits JM, Slotboom DJ, van Oijen AM. Nature. 2013;502:119–123. doi: 10.1038/nature12538. [DOI] [PubMed] [Google Scholar]; b) Morrison EA, DeKoster GT, Dutta S, Vafabakhsh R, Clarkson MW, Bahl A, Kern D, Ha T, Henzler-Wildman KA. Nature. 2012;481:45–50. doi: 10.1038/nature10703. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Forrest LR, Zhang YW, Jacobs MT, Gesmonde J, Xie L, Honig BH, Rudnick G. Proc Natl Acad Sci USA. 2008;105:10338–10343. doi: 10.1073/pnas.0804659105. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zhao Y, Terry D, Shi L, Weinstein H, Blanchard SC, Javitch JA. Nature. 2010;465:188–193. doi: 10.1038/nature09057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palmieri F, Agrimi G, Blanco E, Castegna A, Di Noia MA, Iacobazzi V, Lasorsa FM, Marobbio CM, Palmieri L, Scarcia P, Todisco S, Vozza A, Walker J. Biochim Biophys Acta. 2006;1757:1249–1262. doi: 10.1016/j.bbabio.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 6.Kunji ER, Robinson AJ. Curr Opin Struct Biol. 2010;20:440–447. doi: 10.1016/j.sbi.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 7.a) Pebay-Peyroula E, Dahout-Gonzalez C, Kahn R, Trezeguet V, Lauquin GJ, Brandolin G. Nature. 2003;426:39–44. doi: 10.1038/nature02056. [DOI] [PubMed] [Google Scholar]; b) Ruprecht JJ, Hellawell AM, Harding M, Crichton PG, McCoy AJ, Kunji ER. Proc Natl Acad Sci USA. 2013;111:E426–434. doi: 10.1073/pnas.1320692111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berardi MJ, Shih WM, Harrison SC, Chou JJ. Nature. 2011;476:109–113. doi: 10.1038/nature10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palmieri F, Pierri CL. FEBS Lett. 2010;584:1931–1939. doi: 10.1016/j.febslet.2009.10.063. [DOI] [PubMed] [Google Scholar]
- 10.a) Imai S, Osawa M, Takeuchi K, Shimada I. Proc Natl Acad Sci USA. 2010;107:6216–6221. doi: 10.1073/pnas.0911270107. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Imai S, Osawa M, Mita K, Toyonaga S, Machiyama A, Ueda T, Takeuchi K, Oiki S, Shimada I. J Biol Chem. 2012;287:39634–39641. doi: 10.1074/jbc.M112.401265. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Chill JH, Louis JM, Baber JL, Bax A. J Biomol NMR. 2006;36:123–136. doi: 10.1007/s10858-006-9071-4. [DOI] [PubMed] [Google Scholar]
- 11.a) Gautier A, Kirkpatrick JP, Nietlispach D. Angew Chem Int Ed. 2008;47:7297–7300. doi: 10.1002/anie.200802783. [DOI] [PubMed] [Google Scholar]; b) Reckel S, Gottstein D, Stehle J, Lohr F, Verhoefen MK, Takeda M, Silvers R, Kainosho M, Glaubitz C, Wachtveitl J, Bernhard F, Schwalbe H, Guntert P, Dotsch V. Angew Chem Int Ed. 2011;50:11942–11946. doi: 10.1002/anie.201105648. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Stehle J, Silvers R, Werner K, Chatterjee D, Gande S, Scholz F, Dutta A, Wachtveitl J, Klein-Seetharaman J, Schwalbe H. Angewandte Chemie. 2014 doi: 10.1002/anie.201309581. [DOI] [PubMed] [Google Scholar]
- 12.a) Kofuku Y, Ueda T, Okude J, Shiraishi Y, Kondo K, Maeda M, Tsujishita H, Shimada I. Nature communications. 2012;3:1045. doi: 10.1038/ncomms2046. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nygaard R, Zou Y, Dror RO, Mildorf TJ, Arlow DH, Manglik A, Pan AC, Liu CW, Fung JJ, Bokoch MP, Thian FS, Kobilka TS, Shaw DE, Mueller L, Prosser RS, Kobilka BK. Cell. 2013;152:532–542. doi: 10.1016/j.cell.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kofuku Y, Ueda T, Okude J, Shiraishi Y, Kondo K, Mizumura T, Suzuki S, Shimada I. Angewandte Chemie. 2014 doi: 10.1002/anie.201406603. [DOI] [PubMed] [Google Scholar]
- 13.Vozza A, Blanco E, Palmieri L, Palmieri F. J Biol Chem. 2004;279:20850–20857. doi: 10.1074/jbc.M313610200. [DOI] [PubMed] [Google Scholar]
- 14.Delaglio F, Kontaxis G, Bax A. J Am Chem Soc. 2000;122:2142–2143. [Google Scholar]
- 15.Shen Y, Delaglio F, Cornilescu G, Bax A. J Biomol NMR. 2009;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robinson AJ, Kunji ER. Proc Natl Acad Sci USA. 2006;103:2617–2622. doi: 10.1073/pnas.0509994103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palmieri F, Pierri CL, De Grassi A, Nunes-Nesi A, Fernie AR. The Plant Journal. 2011;66:161–181. doi: 10.1111/j.1365-313X.2011.04516.x. [DOI] [PubMed] [Google Scholar]
- 18.Tolman JR, Al-Hashimi HM, Kay LE, Prestegard JH. J Am Chem Soc. 2001;123:1416–1424. doi: 10.1021/ja002500y. [DOI] [PubMed] [Google Scholar]
- 19.a) Wang Y, Tajkhorshid E. Proc Natl Acad Sci USA. 2008;105:9598–9603. doi: 10.1073/pnas.0801786105. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dehez F, Pebay-Peyroula E, Chipot C. J Am Chem Soc. 2008;130:12725–12733. doi: 10.1021/ja8033087. [DOI] [PubMed] [Google Scholar]
- 20.Di Marino D, Oteri F, Morozzo Della Rocca B, D'Annessa I, Falconi M. J Mol Model. 2011;18:2377–2386. doi: 10.1007/s00894-011-1255-5. [DOI] [PubMed] [Google Scholar]
- 21.Cao Z, Bowie JU. Proc Natl Acad Sci USA. 2012;109:8121–8126. doi: 10.1073/pnas.1201298109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnston JM, Khalid S, Sansom MS. Molecular Membrane Biology. 2008;25:506–517. doi: 10.1080/09687680802459271. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.