

Abstract
Outcome Objectives
1.Investigate the role of reflux, specifically pepsin, in laryngopharyngeal carcinogenesis. 2.Evaluate effects of chronic pepsin exposure on cell migration, apoptosis, and colony forming ability in hypopharyngeal cells.
Study Design
Translation research.
Setting
Academic research laboratory.
Methods
Human hypopharyngeal squamous carcinoma FaDu cells were chronically exposed to nonacidic pepsin (exposed for 24 hours, 4 times over 2 weeks at the following concentrations: 0.01mg/ml, 0.1 mg/ml, or 1mg/ml). Precise wounds were created in confluent cell plates and rates of cell migration into wounds were quantified. Separately, cell viability of chronic pepsin exposed FaDu cells acutely treated with paclitaxel was measured. Finally, a clonogenic assay was performed on these cells to measure effects of chronic pepsin exposure on colony forming ability.
Results
An increased rate of relative wound density was observed in chronic pepsin treated (0.01mg/ml, 0.1mg/ml) cells compared to control (P<0.001), suggesting greater rates of cell migration. Pepsin treated (0.1 mg/ml) cells demonstrated on average, greater cell viability compared to control after exposure to paclitaxel suggesting possible apoptotic resistance, however this was not statistically significant. Chronic pepsin exposure (0.1mg/ml, 1mg/ml) was associated with dose dependent increase in colony forming ability relative to control (P<0.001).
Conclusion
Hypopharyngeal squamous cell line chronically exposed to pepsin demonstrated increased cell migration, and colony forming ability relative to control cells. These experiments indicate that chronic pepsin exposure acts as a promoter of tumorigenesis and metastasis of airway epithelium, suggesting a role for pepsin in laryngopharyngeal carcinogenesis attributed to gastric reflux.
Keywords: pepsin, laryngopharyngeal reflux, laryngeal cancer, reflux
Introduction
Laryngopharyngeal reflux (LPR), reflux of gastric contents into the upper airway, is becoming increasingly prevalent and more widely recognized, currently affecting an estimated 20-60% of the United States population.1,2 While LPR is implicated in the development of various inflammatory disorders and laryngeal cancers, the long term implications of this chronic disease have yet to be fully understood.3,4 In gastroesophageal reflux, chronic exposure of the esophageal epithelium to gastric refluxate creates chronic inflammation (reflux esophagitis) characterized by an expression profile of proinflammatory cytokines within esophageal mucosa that is highly correlative with grading of endoscopic findings. This chronic inflammation is thought to lead to metaplastic changes of the cells lining the lower esophagus as seen in Barrett's esophagus.5,6 Similarly, patients with LPR demonstrate laryngeal mucosal injury.4 The expression of laryngeal stress proteins and protective proteins such as carbonic anhydrase, which neutralizes refluxed acid, and mucins, which comprise the mucous barrier, is altered in the laryngeal mucosa of LPR patients. Hypopharyngeal cells exposed to nonacid pepsin in vitro demonstrate a similar proinflammatory cytokine expression profile as that observed in the esophageal response to reflux.7,8,9
Initial investigations to identify the causative agents of LPR-attributed disease evaluated the effects of gastric acid on the mucosal lining of the upper aerodigestive tract. However, injury to the upper aerodigestive tract was seen despite proton pump inhibitor therapy and esophageal and laryngeal injury was found to occur even in nonacidic conditions.10-13 Further studies found that pepsin played a significant role in laryngeal injury despite nonacidic conditions.14-16 Recent evidence has shown that, at neutral pH, pepsin is taken up by laryngeal and hypopharyngeal epithelial cells by receptor mediated endocytosis and retained in the low pH environment of intracellular vesicles such as endosomes and Golgi.14 This retention of pepsin in a low pH environment would allow pepsin to restore it's proteolytic activity and potentially cause further mucosal injury.15,16
Clinical studies have evaluated the incidence of LPR in patients with laryngopharyngeal cancer.17,18 Some have demonstrated a higher rate of LPR in nonsmokers and nondrinkers with laryngeal cancer.19 While a correlation between LPR and laryngeal cancer has been demonstrated in prior studies, causality has been difficult to prove.20 Specifically, identifying LPR as an independent risk factor for laryngeal cancer has been difficult, as these investigations are often confounded with patients with prior tobacco or alcohol dependence. Additionally, these research studies and clinicians use different methods to measure and diagnose LPR. Currently, a causal role of reflux in laryngeal cancer has not been adequately demonstrated.21-23 Understanding the molecular effects of chronic pepsin exposure in the laryngopharynx may provide further support for its’ role in carcinogenesis.
We previously looked at 84 genes implicated in carcinogenesis and evaluated the effects of acute pepsin exposure on expression of these genes in human hypopharyngeal squamous cell carcinoma (SCC) FaDu cells. Three genes were noted to have increased expression by more than 1.5 fold and 24 genes, including many tumor suppressors, demonstrated reduced expression by more than 1.5 fold.24 Additionally, growth curve assays showed that acute pepsin exposure increased the percentage of cells in the replicative S phase in both human hypopharyngeal SCC FaDu cells and primary laryngeal epithelial cells. The pepsin-induced increase in cell proliferation exhibited both a dose and time dependent response, suggesting that acute pepsin exposure increases cell proliferation and may cause aberrant cell growth.24 The effects of chronic pepsin exposure were also analyzed in a study conducted by Allen et al25 using a hamster buccal pouch model to demonstrate the role of pepsin as a cofactor in tumor growth. The study showed that co-treatment of the hamster buccal pouch with 7,12-dimethylbenzanthracene (DMBA), a known carcinogen, and acidified pepsin (three times daily for 14 weeks) generated significantly larger tumors than DMBA alone.25 Thereby demonstrating a potential role for pepsin in malignant transformation. Cumulatively, these data reveals that pepsin exposure increases cell proliferation and alters expression of genes involved in carcinogenesis suggesting that uncontrolled chronic reflux of pepsin could cause unregulated cell growth and malignancy.
We hypothesized that reflux of nonacidic pepsin contributes to oncogenic transformation, by acting as a tumor promoter. To investigate the role of reflux, specifically pepsin, in laryngopharyngeal cancer, the effects of chronic pepsin exposure on cell migration, apoptosis, and anchorage-independent growth were analyzed in a human hypopharyngeal SCC FaDu cell line. A FaDu cell line was utilized as these cells are technically optimal cells for this initial investigation and would provide information regarding the impact of pepsin on promoting tumor growth, which is one step in oncogenic transformation.
Materials and Methods
Cell Culture
Human hypopharyngeal SCC FaDu cells (ATCC, Manassas, VA) were cultured in Minimum Essential Medium (MEM; ATCC, Manassas, VA) supplemented with 10% Fetal Bovine Serum (FBS) (ATCC) and 1% Antibiotic/Antimycotic (Life Technologies, Grand Island, NY). When exposing these cells to pepsin, porcine pepsin was used, as it is commercially available and has similar activity as human pepsin. Pepsin is at a concentration of 1 mg/ml in the stomach. It presumably is diluted in aerodigestive secretions as the refluxate travels proximally to the laryngopharynx. Physiological concentrations were used ranging from 0.01mg/ml to 1 mg/ml.
Cell Migration Assay
Hypopharyngeal SCC FaDu cells were grown to 100% confluence on a 96-well Essen BioScience ImageLock Plate (Essen Bioscience, Ann Arbor, Michigan) at 37°C with 5% CO2. The cells were exposed to porcine pepsin (0.01 or 0.1mg/ml; Sigma-Aldrich, St. Louis, MO) at pH7 or with media at pH7 alone (control) for 24 hours in six technical replicates. Wells were washed with phosphate buffered solution (PBS) and fresh growth media was replaced. Precise wounds were made in each well using a WoundMaker (Essen Bioscience), a tool designed to make a wound with consistent size and decreased variability among each well. The plate was placed in an IncuCyte™ incubator, a live-cell imaging system (Essen Bioscience), and images were collected at 1 hour time intervals.
Images of cell migration within each well were obtained while in the Incucyte, an incubator that provides real time images of cells to document migration during the incubation process. The Incucyte software uses the parameter of Relative Wound Density (RWD) as the most robust metric to quantify cell migration. At each time point, the RWD, which corresponds to the spatial cell density in the wound area relative to the spatial cell density outside the wound area, was calculated by the Incucyte software RWD algorithm. The RWD and its’ rate of change was compared between control and pepsin-treated conditions. Additionally, the area under the curve was calculated for each condition and used to determine statistically significant differences.
Apoptosis Resistance Assay
This assay was performed to assess the effects of pepsin on disruption of the apoptotic pathway and cell viability. Human hypopharyngeal SCC FaDu cells were treated for 24 hours with porcine pepsin (0.01, 0.1, or 1 mg/ml) at pH7 or with media at pH7 alone (control) two times over one week time period. There were 3 technical replicates for each condition. Cells were maintained at <90% confluence and allowed to rest at least 24 hours between each treatment. Cells were then treated with 50uM paclitaxel (Sigma-Aldrich), an apoptosis promoting agent, or media alone (control) and incubated for 12 hours at 37°C with 5% CO2. Viable cells that were observed after treatment with paclitaxel were then counted. Poisson regression with over-dispersion adjustment was performed to determine statistical difference among the conditions.
Clonogenic Assay
The clonogenic assay was performed to assess the effect of pepsin on anchorage independent growth and colony forming ability. Often cells need a solid surface to attach for further proliferation; and normal cells fail to grow when suspended in viscous fluid or gel (agar). As this assay is performed using a previously transformed cell line, comparison of colony growth in each condition would provide ability of pepsin exposure to promote tumor growth. Prior to the assay hypopharyngeal SCC FaDu cells were treated for 24 hours with porcine pepsin (0.1, or 1 mg/ml; Sigma-Aldrich) or with media at pH7 (control) in replicates of six, four times over a two week time period. The concentration of 0.01 mg/ml pepsin was not used as prior investigation evaluating the impact of pepsin exposure on cell proliferation did not demonstrate a statistically significant difference at this concentration.24 Cells were maintained at <90% confluence and allowed to rest at least 24 hours between each treatment. Bottom agar was prepared with 0.5% agarose melted and added to 50% normal growth media. One mililiter aliquots of the bottom agar was placed into the six well plates. Twenty-five hundred cells of each of the replicates for the four conditions were mixed with 500 microliters of 0.3% agarose in 75% normal growth media and overlayed onto solidified bottom agar. The bottom agar served as a barrier from the solid plate and the cells suspended in the top agar. Therefore cells that only possessed anchorage independence would grow. Cells were placed in an incubator at 37°C with 5% CO2 for two weeks. One hundred microliters of growth media was added to each well as needed during the incubation period. After two weeks of incubation, colonies were noted in the agar plates. Wells were stained with 0.005% crystal violet and washed with sterile water. Images were obtained of each well and condition (Photo Doc-It Imaging System, UVP, Upland, CA). Colony number was assessed using Image J 1.47 software (imagej.nih.gov/ij/download/). The data obtained was the mean of six wells for each condition. Statistically significant differences were determined.
Statistical Methodology
Poisson regression with over-dispersion adjustment was performed on the apoptosis resistance assay and the clonogenic assay. For the data from the cell migration assay, area under the curve of the relative wound density (%) over 24 hours were calculated for the curve for each sample, then a linear model was fit to examine the differences of the three types of samples. P-values were adjusted for multiple comparisons by the Tukey-Kramer method. The significance level was set at 0.05. All data analyses were carried out using the Statistical Analysis System, version 9.2(SAS institute, Cary, NC, USA).
Results
Cell Migration Assay
The ability of non-acidic pepsin to alter cell migration was assessed by measuring the relative wound density (RWD) over time in the wound healing assay (Figures 1 and 2). The maximal difference in RWD was detected at 24 hours post-wounding; the control group had a relative wound density of 49% at 24 hours and cells exposed to 0.01mg/ml pepsin and 0.1mg/ml pepsin had a RWD of 62% and 82% at 24 hours, respectively. The area under the curve of RWD was calculated for each of the groups (control, 0.01 mg/ml pepsin, and 0.1 mg/ml pepsin). There was a significant difference comparing the area under the curve of RWD of the three groups, p<0.001. It was highest in the 0.1 mg/ml pepsin group and lowest in the control group. There were significant differences observed when comparing any of the two groups among the three (p<0.01). No significant differences were noted in cell morphology amongst the 3 different conditions.
Figure 1. Effect of Pepsin on Cell Migration over 24 hours.

Initial and 24-hour scratch wound masks generated by Incucyte for quantitation of relative wound density of pepsin-treated and control hypopharyngeal FaDu cells. Magnification: 10x. The black region represents the cell-covered area remaining after the initial wound (initial wound mask). The gray area represents the area of the wound covered by cells 24 hours after the initial wound. The white region represents the remaining cell-free wound area at 24 hours.
Figure 2. Cell migration as percent change in Relative Wound Density (RWD) per hour.
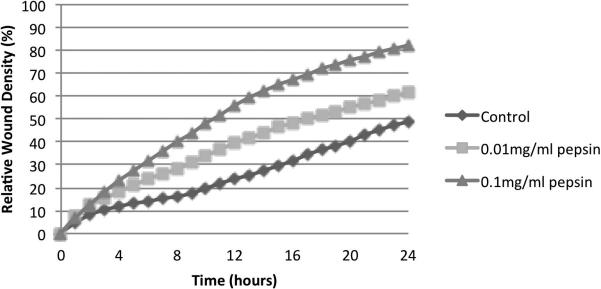
Graphical representation of the concentration–dependent effect of pepsin treatment on the migration of FaDu cells using the RWD index of cell migration.
Apoptotic Resistance Assay
FaDu cells were treated acutely with an apoptosis promoting agent, paclitaxel, after chronic exposure to pepsin at three different concentrations to assess the effects of pepsin on apoptosis. The mean number of viablecells counted following paclitaxel treatment of the 0.01mg/ml pepsin treatment condition was 1.1-fold higher than the mean number of viable cells counted in the control condition not treated with pepsin but treated with paclitaxel.The mean number of viable cells counted following paclitaxel of the 0.1mg/ml pepsin treatment condition was 1.75-fold higher than the mean number of viable cells counted in the control condition not treated with pepsin but treated with paclitaxel.. The mean number of viable cells of the 1 mg/ml pepsin treatment condition that were counted following paclitaxel was 1.48-fold higher than the mean number of viable cells counted in the control condition. Results are displayed in Figure 3. There was no statistically significant difference of the four conditions (control, 0.01 mg/ml pepsin, 0.1 mg/ml pepsin, 1 mg/ml pepsin), p=0.078. Moreover, it was not significantly different for the pairwise comparison of any of the two groups.
Figure 3. Effects of chronic pepsin exposure on apoptosis.

Number of viable cells counted following acute paclitaxel exposure of human hypopharyngeal FaDu cells chronically treated with pepsin (pH7) or pH7 media alone (control). Error bars represent standard deviation.
Clonogenic Assay
Colony formation was noted on pepsin treated plates after 2 weeks of incubation, with a unique morphology compared to controls (Figures 4 and 5). Soft agar clonogenic assay shows increased colony formation in those FaDu cells exposed to pepsin. The mean of six replicates demonstrated that 11 colonies formed in control wells, 293 colonies in those cells treated with 0.1 mg/ml pepsin and 305 colonies in those cells treated with 1 mg/ml pepsin. A greater than 25-fold increase in colony number was noted in those wells containing cells that were exposed to chronic pepsin-treatment compared with control/untreated wells as displayed in Figure 6. There was a significant difference among the three conditions (p<0.001). Among the three, control group was significantly different from the 0.1 mg/ml pepsin and 1 mg/ml pepsin groups, with p-value <0.001. However, 0.1mg/ml pepsin was not significantly different from 1 mg/ml pepsin group (p=0.972).
Figure 4. Effect of pepsin on anchorage-independent cell growth.

Images of crystal violet-stained colonies of human hypopharyngeal FaDu cells chronically treated with pepsin (pH7) or pH7 media alone (control) grown in soft agar plates for 2 weeks. Magnification:0.
Figure 5.

Images of Hypopharyngeal SCC FaDu cells in soft agar assay. Magnification: 100x. A. Demonstrates control cells. B. Demonstrates treated cells with a change in morphology relative to controls. C. Demonstrates treated cells.
Figure 6. Cell Clonogenic Assay Results.
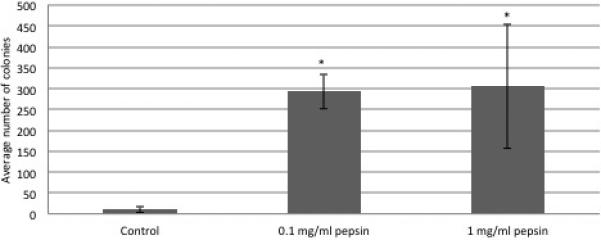
FaDu cells treated with porcine pepsin (0.1mg/ml and 1 mg/ml) had a statistically significant increase in colony forming ability compared to untreated cells. Error bars represent standard deviation.
Discussion
An association between laryngopharyngeal reflux and laryngeal cancer has been demonstrated in a number of previous studies.17,23 Demonstration of the ability of pepsin to cause oncogenic transformation would lend further support to the role of reflux in carcinogenesis. While our previous work focused on the effect of acute pepsin exposure on cell proliferation, the hamster buccal model utilized by Allen and colleagues25 suggested a role for chronic pepsin exposure in oncogenic transformation. The study reported herein corroborates previous findings of the tumorigenic potential of chronic pepsin exposure in the upper airway and suggests a role for pepsin in metastasis.
The results of the wound healing assay confirmed that chronic nonacidic pepsin exposure dose-dependently induced an increase in cell migration, a process that is imperative in a number of biologic pathways including chronic inflammation and tumor metastasis. Our previous work has shown that pepsin-treatment induces altered expression of metastasis-related genes including extracellular matrix genes and those involved in cell adhesion, cell cycle, growth, proliferation, and apoptosis.3,14,24,26 In vivo evidence similarly supports a role for LPR in promotion of metastasis. Several authors studied the effects of refluxate on cell adhesion proteins and identified a decline in E-cadherin expression in laryngeal biopsies of patients with documented LPR. E-cadherin provides vital structural role in in cells of tissue/organs and with loss of expression of this protein, may decrease the adhesive function of cells and increase possibility of tumor cell invasion and metastasis.27,28 The five-year survival rate for laryngeal cancer is poor for advanced stage disease. While trends in tobacco use have declined over the past few decades the survival rates have continued to decline, specifically in patients with regional and distant disease suggesting the influence of other factors in this disease process.29 Advances in our understanding of the factors that contribute to laryngeal SCC could be of fundamental importance in reducing the frequency of advanced stage disease.
Our prior investigation of the influence of an acute pepsin exposure on gene expression using the Cancer PathwayFinder SuperArray demonstrated a reduced expression of pro apoptotic genes.14 Accordingly, an anti-apoptotic effect of chronic pepsin exposure was evaluated in our apoptotic resistance assay. These results were obtained to evaluate the effect of pepsin on the balance between cell growth and apoptosis and potential for unregulated cell growth. There were more viable cells that were repeatedly exposed to pepsin after treatment with the apoptosis-promoting agent, paclitaxel, compared to controls, however this was not significant in our analysis.
Additionally, a clonogenic assay was performed to measure the capacity of pepsin to induce anchorage independence and loss of contact inhibition. A common feature of carcinoma development and growth is the ability of transformed cells to survive under anchorage independence. The soft agar assay for colony formation, an anchorage independent growth assay in soft agar, is considered the most stringent assay for detecting malignant transformation of cells. As expected, there were colonies noted on the control plates, as the FaDu cells used are a tumorogenic SCC derived cell line. However, FaDu cells demonstrated significantly greater colony forming ability after chronic pepsin exposure, suggesting that pepsin promotes anchorage independence. This role of chronic pepsin exposure as a tumor promoter in a hypopharyngeal squamous cell linefurther supports the role the pepsin in carcinogenesis of the laryngopharynx.
There were several limitations with this study. One limitation includes the use of an in vitro cell line. While in vitro experiments provide obvious benefits including simplicity and ease, they do not completely recreate the complex in vivo environment. Additionally, the cell line used was an immortal, tumor-derived cell line. Further investigation using primary laryngeal epithelial cells (currently ongoing in our laboratory) and animal models will be needed to validate the role of pepsin in laryngeal carcinogenesis attributed to LPR and the potential utility of pepsin inhibitors in the prevention of LPR-attributed laryngeal cancer.
Conclusion
Pepsin in laryngopharyngeal reflux continues to be investigated as a contributor to the development of laryngopharyngeal carcinogenesis. In the study described herein, chronic pepsin exposure elicited increased cell proliferation and migration, anchorage independent cell growth, and increased colony formation in oncogenetically transformed human hypopharyngeal cells, lending further support for the role of pepsin as an agent of oncogenic transformation.
Acknowledgements
We thank Aniko Szabo, PhD, and Shi Zhao, MS, for biostatistics consultation and data analysis. The statistical support and interpretation of the data is provided, in part, by grant 1UL1RR031973 from the clinical and translational Science Award program of the national center for research resources, National Institutes of Health.
Footnotes
The Department of Otolaryngology and Communication Sciences, Medical College of Wisconsin, Milwaukee, Wisconsin sponsored this research study.
Presented at the Annual Meeting of the American Academy of Otolaryngology-Head and Neck Surgery, Vancouver, BC, September 30, 2013
References
- 1.Koufman JA, Amin MR, Panetti M. Prevalence of reflux in 113 consecutive patients with laryngeal and voice disorders. Otolaryngol Head Neck Surg. 2000;123(4):385–388. doi: 10.1067/mhn.2000.109935. [DOI] [PubMed] [Google Scholar]
- 2.Reulbach TR, Belafsky PC, Blalock PD, Koufman JA, Postma GN. Occult laryngeal pathology in a community-based cohort. Otolaryngol Head Neck Surg. 2001;124(4):448–450. doi: 10.1067/mhn.2001.114256. [DOI] [PubMed] [Google Scholar]
- 3.Samuels TL, Johnston N. Pepsin as a causal agent of inflammation during nonacidic reflux. Otolaryngol Head Neck Surg. 2009;141(5):559–563. doi: 10.1016/j.otohns.2009.08.022. [DOI] [PubMed] [Google Scholar]
- 4.Koufman JA. The otolaryngologic manifestations of gastroesophageal reflux disease (GERD): a clinical investigation of 225 patients using ambulatory 24-hour pH monitoring and an experimental investigation of the role of acid and pepsin in the development of laryngeal injury. Laryngoscope. 1991;101(4 Pt 2 Suppl 53):1–78. doi: 10.1002/lary.1991.101.s53.1. [DOI] [PubMed] [Google Scholar]
- 5.Fitzgerald RC, Onwuegbusi BA, Bajaj-Elliott M, et al. Diversity in the oesophageal phenotypic response to gastro-oesophageal reflux: immunological determinants. Gut. 2002;50(4):451–459. doi: 10.1136/gut.50.4.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guillem PG. How to make a Barrett esophagus: pathophysiology of columnar metaplasia of the esophagus. Dig. Dis. Sci. 2005;50(3):415–424. doi: 10.1007/s10620-005-2451-x. [DOI] [PubMed] [Google Scholar]
- 7.Johnston N, Knight J, Dettmar PW, Lively MO, Koufman J. Pepsin and carbonic anhydrase isoenzyme III as diagnostic markers for laryngopharyngeal reflux disease. Laryngoscope. 2004;114(12):2129–2134. doi: 10.1097/01.mlg.0000149445.07146.03. [DOI] [PubMed] [Google Scholar]
- 8.Samuels TL, Handler E, Syring ML, et al. Mucin gene expression in human laryngeal epithelia: effect of laryngopharyngeal reflux. Ann. Otol. Rhinol. Laryngol. 2008;117(9):688–695. doi: 10.1177/000348940811700911. [DOI] [PubMed] [Google Scholar]
- 9.Johnston N, Dettmar PW, Lively MO, et al. Effect of pepsin on laryngeal stress protein (Sep70, Sep53, and Hsp70) response: role in laryngopharyngeal reflux disease. Ann. Otol. Rhinol. Laryngol. 2006;115(1):47–58. doi: 10.1177/000348940611500108. [DOI] [PubMed] [Google Scholar]
- 10.Mainie I, Tutuian R, Shay S, et al. Acid and non-acid reflux in patients with persistent symptoms despite acid suppressive therapy: a multicentre study using combined ambulatory impedance-pH monitoring. Gut. 2006;55(10):1398–1402. doi: 10.1136/gut.2005.087668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tutuian R, Vela MF, Hill EG, et al. Characteristics of symptomatic reflux episodes on Acid suppressive therapy. Am. J. Gastroenterol. 2008;103(5):1090–1096. doi: 10.1111/j.1572-0241.2008.01791.x. [DOI] [PubMed] [Google Scholar]
- 12.Sharma N, Agrawal A, Freeman J, Vela MF, Castell D. An analysis of persistent symptoms in acid-suppressed patients undergoing impedance-pH monitoring. Clin Gastroenterol Hepatol. 2008;6(5):521–524. doi: 10.1016/j.cgh.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 13.Oelschlager BK, Quiroga E, Isch JA, Cuenca-Abente F. Gastroesophageal and pharyngeal reflux detection using impedance and 24-hour pH monitoring in asymptomatic subjects: defining the normal environment. J. Gastrointest. Surg. 2006;10(1):54–62. doi: 10.1016/j.gassur.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 14.Johnston N, Wells CW, Samuels TL, Blumin JH. Pepsin in nonacidic refluxate can damage hypopharyngeal epithelial cells. Ann. Otol. Rhinol. Laryngol. 2009;118(9):677–685. doi: 10.1177/000348940911800913. [DOI] [PubMed] [Google Scholar]
- 15.Johnston N, Dettmar PW, Bishwokarma B, Lively MO, Koufman JA. Activity/stability of human pepsin: implications for reflux attributed laryngeal disease. Laryngoscope. 2007;117(6):1036–1039. doi: 10.1097/MLG.0b013e31804154c3. [DOI] [PubMed] [Google Scholar]
- 16.Johnston N, Wells CW, Blumin JH, Toohill RJ, Merati AL. Receptor-mediated uptake of pepsin by laryngeal epithelial cells. Ann. Otol. Rhinol. Laryngol. 2007;116(12):934–938. doi: 10.1177/000348940711601211. [DOI] [PubMed] [Google Scholar]
- 17.Tae K, Jin BJ, Ji YB, et al. The Role of Laryngopharyngeal Reflux as a Risk Factor in Laryngeal Cancer: A Preliminary Report. Clin Exp Otorhinolaryngol. 2011;4(2):101. doi: 10.3342/ceo.2011.4.2.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Copper MP, Smit CF, Stanojcic LD, et al. High incidence of laryngopharyngeal reflux in patients with head and neck cancer. Laryngoscope. 2000;110(6):1007–1011. doi: 10.1097/00005537-200006000-00023. [DOI] [PubMed] [Google Scholar]
- 19.Langevin SM, Michaud DS, Marsit CJ, et al. Gastric reflux is an independent risk factor for laryngopharyngeal carcinoma. Cancer Epidemiol. Biomarkers Prev. 2013;22(6):1061–1068. doi: 10.1158/1055-9965.EPI-13-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bacciu A, Mercante G, Ingegnoli A, et al. Effects of gastroesophageal reflux disease in laryngeal carcinoma. Clin Otolaryngol Allied Sci. 2004;29(5):545–548. doi: 10.1111/j.1365-2273.2004.00851.x. [DOI] [PubMed] [Google Scholar]
- 21.Coca-Pelaz A, Rodrigo JP, Takes RP, et al. Relationship between reflux and laryngeal cancer Eisele DW, ed. Head Neck. 2013 doi: 10.1002/hed.23208. n/a–n/a. [DOI] [PubMed] [Google Scholar]
- 22.Weaver EM. Association between gastroesophageal reflux and sinusitis, otitis media, and laryngeal malignancy: a systematic review of the evidence. Am. J. Med. 2003;115(3):81–89. doi: 10.1016/s0002-9343(03)00203-1. [DOI] [PubMed] [Google Scholar]
- 23.Qadeer MA, Colabianchi N, Strome M, Vaezi MF. Gastroesophageal reflux and laryngeal cancer: causation or association? A critical review. Am J Otolaryngol. 2006;27(2):119–128. doi: 10.1016/j.amjoto.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 24.Johnston N, Yan JC, Hoekzema CR, et al. Pepsin promotes proliferation of laryngeal and pharyngeal epithelial cells. Laryngoscope. 2012;122(6):1317–1325. doi: 10.1002/lary.23307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Allen J, Tinling SP, Johnston N, Belafsky P. Review article: effects of pepsin and alginate in an animal model of squamous cell carcinoma. Aliment. Pharmacol. Ther. 2011;33:21–28. [Google Scholar]
- 26.Johnston N, Wells CW, Samuels TL, Blumin JH. Rationale for targeting pepsin in the treatment of reflux disease. Ann. Otol. Rhinol. Laryngol. 2010;119(8):547–558. doi: 10.1177/000348941011900808. [DOI] [PubMed] [Google Scholar]
- 27.Reichel O, Mayr D, Durst F, Berghaus A. E-cadherin but not β-catenin expression is decreased in laryngeal biopsies from patients with laryngopharyngeal reflux. Eur Arch Otorhinolaryngol. 2008;265(8):937–942. doi: 10.1007/s00405-007-0568-6. [DOI] [PubMed] [Google Scholar]
- 28.Galera-Ruiz H, Ríos-Moreno MJ, González-Cámpora R, et al. The cadherin–catenin complex in laryngeal squamous cell carcinoma. Eur Arch Otorhinolaryngol. 2011;269(4):1183–1188. doi: 10.1007/s00405-011-1892-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cosetti M, Yu G-P, Schantz SP. Five-year survival rates and time trends of laryngeal cancer in the US population. Arch. Otolaryngol. Head Neck Surg. 2008;134(4):370–379. doi: 10.1001/archotol.134.4.370. [DOI] [PubMed] [Google Scholar]