

Abstract
Pericytes are cells in the blood–brain barrier (BBB) that degenerate in Alzheimer's disease (AD), a neurodegenerative disorder characterized by early neurovascular dysfunction, elevation of amyloid β‐peptide (Aβ), tau pathology and neuronal loss, leading to progressive cognitive decline and dementia. Pericytes are uniquely positioned within the neurovascular unit between endothelial cells of brain capillaries, astrocytes and neurons. Recent studies have shown that pericytes regulate key neurovascular functions including BBB formation and maintenance, vascular stability and angioarchitecture, regulation of capillary blood flow, and clearance of toxic cellular by‐products necessary for normal functioning of the central nervous system (CNS). Here, we review the concept of the neurovascular unit and neurovascular functions of CNS pericytes. Next, we discuss vascular contributions to AD and review new roles of pericytes in the pathogenesis of AD such as vascular‐mediated Aβ‐independent neurodegeneration, regulation of Aβ clearance and contributions to tau pathology, neuronal loss and cognitive decline. We conclude that future studies should focus on molecular mechanisms and pathways underlying aberrant signal transduction between pericytes and its neighboring cells within the neurovascular unit, that is, endothelial cells, astrocytes and neurons, which could represent potential therapeutic targets to control pericyte degeneration in AD and the resulting secondary vascular and neuronal degeneration.
Keywords: Alzheimer's disease, amyloid beta, blood–brain barrier, hypoperfusion, neurodegeneration, pericytes
Introduction
Cerebrovascular and neuronal functions are intimately entwined in the central nervous system (CNS) 75, 76, 194, 195, 196. Neurons have comparatively high metabolic rates with limited cellular reserves and require a nearly continuous supply of energy metabolites to maintain and/or replenish the ionic gradients and other metabolic functions required for rapid synaptic transmission 66. To meet these needs, neurons require continuous cerebral blood flow (CBF) for delivery of oxygen. Additionally, transport systems in brain endothelium of the blood–brain barrier (BBB) mediate the delivery of glucose and other nutrients from blood to brain, as well as the clearance of toxic metabolic by‐products from the CNS back to the circulation 195, 196. To ensure adequate vascular access, the mammalian brain has evolved to become a densely vascular structure with the diffusion distance between neurons and an adjacent capillary rarely exceeding 15 μm 166. In humans, the calculated total perfused vascular length of the cerebrovascular tree is 600–700 km, with major contribution coming from dense capillary networks 194. When CBF is disrupted and/or diffusion distance for transport exchange of metabolites between neurons and circulating blood is increased, neuronal damage may occur rapidly 109.
The coordinated matching of vascular supply to neuronal demand is not simply the result of a network of passive vascular conduits. Rather, individual vascular segments adjust both CBF and transport processes at the BBB in response to systemic, neuronal and glial signals 75, 195. Appropriate vascular responses require tight and coordinated cross‐talk between multiple cell types in the CNS—as reflected by the concept of the “neurovascular unit” (NVU) 177, 195, 196. At a cellular level, the NVU is composed of vascular cells [endothelial cells, vascular smooth muscle cells (vSMCs) and pericytes], glial cells (microglia and astrocytes) and neurons (Figure 1). One of these cell types—a perivascular cell known as the “pericyte”—is uniquely positioned within the NVU between vascular, neuronal and glial cells 4, 177. Pericytes are now believed to control key neurovascular functions necessary for neuronal homeostasis 14, 177. Only recently, a link between pericyte loss and/or malfunction and neurologic disease has begun to be elucidated.
Figure 1.
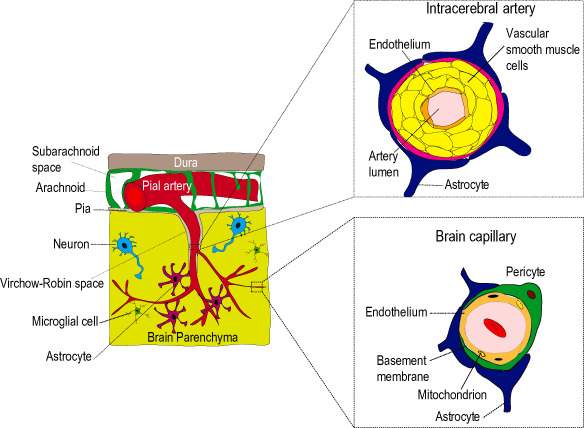
Cerebrovascular structure and the neurovascular unit (NVU). In the brain, pial arteries travel along the cerebrospinal fluid filled subarachnoid space and give rise to penetrating intracerebral arteries, which enter the brain parenchyma but are separated from neurons and glia by the perivascular Virchow‐Robin spaces defined by an outer wall of pia and astrocyte‐derived glial limitans membrane. These arteries branch into smaller arteries, arterioles and brain capillaries distally. At a cellular level, vascular function requires coordinated cross‐talk between multiple cell types of the NVU. The NVU is composed of endothelial cells, vascular mural cells (vascular smooth muscle and pericytes), glial cells (astrocytes and microglia) and neurons. The identity of mural cells changes along the arterial–venous axis. In intracerebral arteries, vascular smooth muscle cells occupy most of the vascular wall. At the level of brain capillaries, pericytes replace smooth muscle cells and are attached to the vascular basement membrane. Pericytes extend multiple cytoplasmic processes that encircle endothelial cells. The point of transition from smooth muscle to pericyte remains poorly defined. At each level, mural cells are further surrounded by astrocyte end‐feet and are in close proximity to neurons and microglia. Figure modified from Zlokovic 196.
Human neurodegenerative disorders, such as Alzheimer's disease (AD), have marked vascular pathophysiology, including endothelial and pericyte degeneration, instability and rupture of the vascular wall, disruption of the BBB and/or dysfunctional BBB transport systems 21, 50, 75, 196. The role of the pericyte in contributing to AD‐related neurovascular dysfunction has recently been suggested 14, 15, 139, 146, 177, 196. In the present review, we first introduce the pericyte and describe evidence supporting its role in regulating the BBB, microvascular structure, blood flow regulation and phagocytosis, and clearance of extracellular molecules from brain interstitial fluid (ISF). We then continue with the description of the vascular contributions to AD pathogenesis with an emphasis on pericyte‐derived sources of injury that are both independent and dependent on the AD neurotoxin amyloid β‐peptide (Aβ).
The Pericyte
Pericytes are a perivascular cell population embedded on the capillary wall in a shared basement membrane with the endothelium (Figure 1) 4, 41, 177. Along the arterial–venous axis, pericytes are predominately found in capillaries, post‐capillary venules and rarely terminal arterioles. According to the prevailing view, pericytes share certain characteristics with the vSMCs 4. However, the point of transition from vSMC to pericyte remains still poorly understood. Pericytes are composed of a cell body and elongated, multiple finger‐like cytoplasmic processes which elongate and cover the abluminal side of brain capillaries in a manner that is very different from vSMC 41, 177. At discrete points, lacking a basement membrane, pericytes and endothelial cells form direct cell‐to‐cell contacts, known as “peg‐and‐socket” contacts. At points of contact, connexin‐43 hemichannels form gap junctions permitting direct endothelial–pericyte communication 18. Fibronectin‐rich adhesion plaques connect the basement membrane to the plasma membrane and underlying actin cytoskeleton in both cell types 41. Pericyte–astrocyte and pericyte–neuron contacts are much less understood. However, it appears that pericytes may play a role in guiding the association of astrocyte end‐feet with the vessel wall 5.
The CNS has higher pericyte‐to‐endothelial cell ratios than peripheral vascular beds, that is, estimated 1:1–1:3 in the CNS vs. 1:10–100 in striated muscle, according to some early pioneering studies 148. Within the CNS, as much as 70%–80% of the capillary tube is covered with pericyte cell processes 5, 14, 15, 35, 139, 146, 178. Although coverage values are relatively consistent within cortical regions across independent reports 14, 176, 178, regional heterogeneity in more permeable regions, such as the motor neuron dense regions of the spinal cord and fetal germinal matrix, has also been described 20, 178. Whether a similar relationship exists in the brain's circumventricular organs which lack a BBB has yet to be determined.
Origin of CNS pericytes
CNS pericytes arise from several progenitor cells. During early embryonic vasculogenesis, chimerization experiments have established that neuroectoderm–mesodermal‐derived progenitor cells give rise to pericytes in the forebrain and midbrain, brainstem, spinal cord and peripheral organs 49, 89, 90. Later in the embryonic period and into the early postnatal period, intraparenchymal proliferation of existing pericytes—a process termed “longitudinal expansion”—contributes to the further expansion of the brain pericyte population 1, 68, 117, 118, 155, 157. Whether longitudinal recruitment continues throughout adulthood is not well understood at present. Recent studies suggest that the cerebral microvasculature is not quiescent and undergoes continued remodeling throughout life 64. However, the turnover of pericytes within the NVU under physiological conditions has yet to be determined. In models of ischemic injury, circulating bone marrow progenitor cells have been demonstrated to contribute to brain pericyte populations 88, 92, 131. However, it has yet to be shown whether circulating progenitors may also contribute to pericyte populations in the absence of CNS injury. Additionally, the cellular source for replenishing the CNS pericyte populations during postnatal CNS development and in the adult and aging CNS is not presently known.
Endothelial–pericyte molecular cross‐talk
During vasculogenesis and/or angiogenesis, endothelial cells secrete and/or present cell‐bound molecular cues that stimulate pericytes to proliferate, migrate and attach to nascent capillary tubes. This cross‐talk occurs through several well‐defined signal transduction cascades, including platelet‐derived growth factor B (PDGF‐B), transforming growth factor‐β (TGF‐β), NOTCH, sphingosine‐1 phosphate and angiopoietin cell signaling [reviewed in 4, 54, 177]. Each signaling cascade makes differential contributions to the process of “pericyte recruitment.” In simplified form, endothelial cells secrete PDGF‐BB, a homodimer of PDGF‐B, which binds to and activates the platelet‐derived growth factor receptor β (PDGFRβ) located on the pericyte plasma membrane, resulting in pericyte proliferation and migration 1, 54, 68, 97, 159. TGF‐β and NOTCH signaling then mediate pericyte attachment via endothelial upregulation of the adhesion molecule N‐cadherin 95. Bidirectional TGF‐β signaling between pericytes and endothelium regulates quiescence, maturation and differentiation in both cell types 42, 150, 156, 169. Maintenance of PDGF‐B/PDGFRβ and pericyte NOTCH3 signaling are required for maintenance of pericyte survival 14, 60. Recent evidence suggests that pericyte NOTCH3 may also facilitate pericyte proliferation during development 174.
Animal models of pericyte deficiency
Genetic manipulation of endothelial–pericyte signaling pathways results in animal models of pericyte deficiency. For example, deletion and/or genetic manipulation of Pdgfrβ and/or Pdgfb result in widely utilized pericyte deficient mouse models 5, 14, 35, 159, 176. Pericyte‐deficient transgenic mice have proved to be valuable tools in elucidating the functional roles of CNS pericytes in vivo and the effects of a chronic BBB disruption caused by pericyte degeneration on neuronal structure and function.
Neurovascular Functions of CNS Pericytes
Initially, pericytes were described as contractile cells around endothelial cells in small blood vessels by the French physiologist Rouget in 1873 136. Over a century later, the pericyte's functional role in the CNS has been expanded through organotypic slice preparations, in vitro BBB models and the in vivo characterization of cerebrovascular and neuronal phenotypes in pericyte‐deficient transgenic mice 177. Recent in vivo reports have established that CNS pericytes play pivotal regulatory roles in the induction and maintenance of the BBB, microvascular stability, capillary density and angiogenesis, capillary diameter, and blood flow regulation and the clearance of macromolecules from brain ISF.
Induction and maintenance of the BBB
The BBB tightly regulates the molecular exchange between circulating peripheral plasma and the CNS. The anatomic BBB is organized at the level of a continuous endothelial layer that lines the lumen of the cerebrovascular tree. Each endothelial cell is connected to adjacent endothelial cells via tight junctional complexes and adherens junctions, which limit unregulated paracellular transport of circulating molecules. Low levels of transendothelial vesicular transport further limits non‐specific transport of polar solutes and large macromolecules into the CNS 75, 195, 196. A series of tightly regulated transport systems for nutrients and energy metabolites and for brain clearance of metabolic waste products have evolved in the CNS endothelium to meet neuronal metabolic needs and maintain a microenvironment supportive of neuronal function. Large molecules, such as peptides and proteins, are in general transported slowly across the BBB via specialized transport systems 198, 200, 201 or are excluded from the brain in the absence of a specific transport system 193, 202.
Independent studies utilizing pericyte‐deficient mice have demonstrated that pericytes play a pivotal role in establishing and maintaining endothelial BBB properties 5, 14, 35. Early in embryogenesis, pericytes accompany endothelial cells as they invade neural tissue from the adjacent perineural vascular plexus 35, 68. Recent work in pericyte‐deficient Pdgfrβ+/− mice has demonstrated that pericytes induce formation of a functional BBB prior to the appearance of perivascular astrocytes 35. Similarly, embryonic pericyte detachment leads to BBB disruption, vascular leakage and overt hemorrhage in the immediate postnatal period 95. At a molecular level, initial BBB formation is achieved through pericyte‐driven downregulation of endothelial gene products which promotes permeability through transcytosis, for example, caveolin‐1 (Cav1) and plasmalemma vesicle‐associated protein (Plvap), and/or leukocyte infiltration, for example, intercellular adhesion molecule 1 (Icam1) 35, 67. Therefore, pericytes suppress a leaky and pro‐inflammatory phenotype of endothelial cells during the embryonic period.
Following birth, pericytes maintain endothelial BBB and blood–spinal cord barrier properties throughout life 5, 14, 95, 139, 178. Within the adult CNS, there is an inverse relationship between regional pericyte coverage and vascular permeability under physiologic conditions 178. In adult and aged pericyte‐deficient Pdgfrβ+/− mice, a loss of brain pericytes increases non‐specific paracellular endothelial transport through disrupted tight junctions 14, 178. For example, the levels of the essential tight junction transmembrane proteins occludin and claudin‐5, the adaptor protein zonula occludens‐1 and the adherens junction protein vascular endothelial cadherin are progressively reduced with aging in pericyte‐deficient mutants. This leads to brain accumulation of exogenous vascular tracers and circulating plasma proteins with vasculotoxic and neurotoxic properties, including thrombin, fibrinogen/fibrin, plasminogen/plasmin and different non‐immune and immune immunoglobulins 14. Other works in young pericyte‐deficient Pdgfbret/ret mutants has also demonstrated increased transendothelial vesicular transport. Therefore, in the adult brain, pericytes maintain BBB properties through promotion of endothelial tight junctional complexes and suppression of non‐specific vesicular transport 5. Unlike the embryonic period, brain pericyte loss does not lead to upregulation of inflammatory cytokines, chemokines or enhanced immune cell infiltration in early to mid‐adulthood, but only with advanced aging 14.
Microvascular stability
Vessels that lack pericytes are dilated and tortuous with frequent rupture prone out‐pouchings of the vascular wall—called “microaneurysms” 4, 48, 67, 95, 97. Evidence of rupture and/or vascular fragility is frequently evident in pericyte‐deficient mutants 14, 95. In humans, brain or spinal cord pericyte reduction is associated with hemorrhage in prematurity, amyotrophic lateral sclerosis (ALS) and AD 20, 146, 179. Recent works have suggested that pericyte–endothelial cell interactions stabilize the vascular wall through multiple mechanisms. Pericytes secrete the stabilizing molecule angiopoeitin 1, which activates endothelial receptor Tie2 receptors 79. Pericytes secrete transforming growth TGF‐β, which activates the TGFβR2‐Alk5‐Smad2/3 pathway within endothelium 3. This promotes endothelial maturation including formation of both stabilizing barrier properties and the vascular basement membrane (BM) 42, 156—a protein‐rich peri‐endothelial scaffolding of extracellular matrix necessary for vascular cell attachment 195. Pericyte‐endothelial signaling stimulates endothelial production of multiple BM proteins, including fibronectin, nidogen‐1, perlacan and laminin 156. Pericytes also directly synthesize and secrete fibronectin contributing to BM structure and downregulate and/or inhibit the activity of destabilizing matrix metalloproteinases (MMP), including MMP‐2 and MMP‐9, in the latter stages of angiogenesis 144, 156. In the setting of pericyte loss, disruption of the BM contributes to vascular instability, dilatation and rupture 14.
Angiogenesis and microvascular density
Pericytes play dynamic and at times opposing roles in angiogenesis. Early in angiogenesis, pericytes secrete angiogenic factors, that is, vascular endothelial growth factor‐A (VEGF‐A), which stimulates sprout formation, endothelial proliferation and/or survival 36, 118. Pericytes also express multiple MMPs, including MMP 2, 3 and 9, and urokinase plasminogen activator receptor to degrade BM proteins to facilitate endothelial migration 23, 45, 172. Some works have also suggested that pericyte migration precedes and guides endothelial tube formation, but this remains controversial 56, 117, 118, 172. Later in angiogenesis, pericytes inhibit endothelial proliferation and deposit BM proteins as described earlier. Pericytes also secrete the tissue inhibitor of metalloproteinase 3 (TIMP3) capable of inhibiting multiple MMPs, including MMP 1, 10 and 14 as well as a disintegrin and metallopeptidase domain 15 (ADAM15) 144, 156; this stabilizes the BM and concludes the angiogenic process. In pathologic states, pericyte angiogenic functions may become dysregulated with profound implications for CNS vascular structure. For example, overactivation of pericyte MMP‐9 and/or loss of pericyte‐derived trophic support contribute to capillary loss and ultimately hypoperfusion in adult mice 14, 15.
Continued brain angiogenesis is counterbalanced by vascular pruning throughout life 64. The net balance between these two processes regulates capillary density. Studies in pericyte‐deficient mutants have revealed context‐dependent effects of pericytes on regulation of capillary density. In the developing CNS, pericyte loss or detachment leads to endothelial hyperproliferation without changes in capillary branching and/or vascular density 67, 96. In the adult brain, however, variable levels of brain pericyte loss result in endothelial apoptosis, microvascular regression and reductions in capillary density 5, 14. Generation of a wide spectrum of pericyte deficient mutants through endothelial specific deletion of Pdgfb results in either vascular regression and/or vascular hyperproliferation depending on the magnitude of pericyte loss in the retina 48. This suggests that pericytes may have dose‐dependent as well as context‐dependent effects on vascular networks. However, inducible models of pericyte deficiency are required to better delineate the roles of pericytes during postnatal CNS development, growth, adulthood and aging.
Neurovascular coupling and blood flow regulation
Blood flow is carefully matched to neuronal metabolic need in the CNS—a process called “neurovascular coupling” 7, 129. It was previously thought that blood flow modulation was predominately regulated through alterations in vSMC tone in penetrating cerebral arteries and/or arterioles in response to synaptic transmission and release of vasoactive mediators [reviewed in 75]. This view, however, has more recently been complemented by multiple reports suggesting that pericytes may also play a role in blood flow modulation at the capillary level.
Pericytes express both contractile proteins 44, 62 and receptors for multiple vasoactive mediators including catecholamines, vasopression, angiotensin II, endothelin‐1, adenosine and VIP 41, 62. Exposure of pericytes to vasoactive substance results in elevation of intracellular calcium, which initiates the contractile apparatus 69, 83, 115. In organotypic cerebellar slice and retinal preparations, pericytes were shown to alter capillary diameter by constricting or dilating in response to neurotransmitters and/or electrical stimulation 126. It was also demonstrated that stimulation of the contractile response propagated among adjacent pericytes 126; therefore, pericytes may help coordinate responses of an entire microvascular segment. The absence and/or reductions in brain or retinal pericytes are associated with capillary dilatation in vivo, confirming that pericytes regulate capillary diameter in vivo 5, 67, 96, 97. Conversely, pericytes have been shown to assume a hypercontractile phenotype and obstruct capillary blood flow in response to pathologic injury, such as ischemia 187 and/or traumatic brain injury 46.
Despite possessing contractile properties, the relationship between pericyte‐mediated capillary dilatation and/or constriction and blood flow regulation remains less definitive in vivo. A loss of brain pericytes in the young mouse brain results in impairment of blow flow responses to brain activation in the presence of unaltered electrophysiological neuronal responses, suggesting that pericytes fulfill an important regulatory role in functional hyperemia 14. However, an independent study confirmed pericyte‐mediated constriction of cortical capillaries, but suggested that this did not alter functional hyperemic responses in vivo 52. This raises questions as to the significance of capillary diameter changes and its relevance to modulating blood flow responses to brain activation. More recently, carefully designed in vivo multiphoton experiments have suggested that pericytes dilate before arterioles in response to neuronal stimulation and may contribute up to ∼80% of the functional hyperemia response 6. Thus, the emerging view suggests that pericytes also contribute to blood flow regulation under physiologic and pathologic conditions, which should be investigated in greater detail by future studies.
Phagocytosis and clearance of extracellular molecules
Pericytes take up multiple soluble small molecules, for example, horseradish peroxidase, india ink and dextran, via non‐specific pinocytosis irrespective of route of administration—including peripheral injection, intraventricular injection and direct introduction in brain extracellular fluid [reviewed in 41, 162]. Engulfed molecules are transported to lysosomes for enzymatic degradation 41, 162. In chronic BBB disruption models, pericyte phagocytic properties help clear toxic circulating plasma proteins that are normally excluded from the brain, including immunoglobulins, fibrin and albumin 5, 14. In models of acute brain injury, pericytes phagocytose cellular debris 24, 103. Recently, neuroinflammation has been shown to accelerate pericyte phagocytic activity 130. In addition to non‐specific uptake, pericytes may also help specifically regulate the neuronal microenvironment by handling clearance of certain macromolecules in both physiologic and pathologic conditions. For example, pericytes express both the Aβ clearance receptor low‐density lipoprotein receptor‐related protein‐1 (LRP1) and the ATP‐binding cassette protein ABCA1 and may therefore contribute to brain Aβ and cholesterol homeostasis, respectively 139, 142, 175. Thus, pericyte degeneration may result in accumulation of different metabolites in the CNS that are normally cleared by pericytes.
Vascular Mechanisms in AD
AD is the most common cause of dementia worldwide and is characterized by three histopathologic hallmarks—accumulation of brain and vascular Aβ 133, tau hyperphosphorylation and formation of neurofibrillary tangles 10, 77, and neuronal loss 133, 194. Epidemiologic studies have identified considerable overlap between risk factors for cerebrovascular disease and AD 76, 81, 165. The presence of cerebral hypoperfusion 137, subclinical infarcts 170 and/or the presence of one or more cerebral infarcts increase risk for AD 152. Conversely, control of vascular risk factors reduces vascular lesions in AD and may delay disease progression 40, 93, 135. Most AD pathologic specimens have evidence of mixed vascular pathology and small vessel disease with vascular changes being found in >40% of AD pathologic specimens 80, 98, 145, 163. Pathologic changes include pericyte loss and degeneration 11, 139, 146, atrophy and degeneration of vSMC, endothelial loss and reduction in capillary density, formation of collapsed and acellular capillary tubes (so‐called string vessels), mitochondrial alterations, a thickened vascular basement membrane, loss of tight junctional and adherens junction proteins, increased endothelial pinocytosis and vesicular transport and BBB compromise leading to accumulation of plasma proteins in brain parenchyma, ISF and cerebrospinal fluid (CSF) [reviewed in 21, 50, 75, 196].
Although classically defined by the amyloid hypothesis 65, these findings have raised questions as to the role of Aβ in AD and have led to the development of a vascular two‐hit hypothesis of AD pathogenesis (Figure 2) 140, 141, 194, 196. This hypothesis maintains that reduced CBF gives rise to hypoxia and chronic perfusion stress, from one end, and BBB disruption gives rise to brain accumulation of plasma‐derived neurotoxins, from the other, which may converge and initiate neuronal dysfunction and degeneration independently and/or prior to Aβ deposition and the development of tau pathology 81, 141, 196. Vascular dysfunction and injury also impairs clearance of Aβ from the brain 39, 199, leads to increased influx of circulating Aβ into the brain 37, 47 and elevates expression and/or processing of the Aβ precursor protein (APP) 2, 189. This results in Aβ accumulation and deposition within brain parenchyma and surrounding cerebral blood vessels. Aβ may then accelerate both vascular 12, 139, 196 and neuronal injury 173, 186 and promote self‐propagation of cerebral and vascular β‐amyloidosis 47, 105. The following subsections describe some mechanisms through which microvascular pathology contributes to AD‐like neurodegeneration. The integral role of the pericyte in mediating vascular and neuronal changes in AD is described at length in a separate subsection (Figure 3) (please see the section Pericytes in AD).
Figure 2.

The vascular two hit hypothesis of Alzheimer's neurodegeneration. Vascular injury as result of long‐standing vascular risk factors, for example, hypertension, dyslipidemia, diabetes, smoking, or obesity, genetic risk, for example, apolipoprotein ε4, and/or other unidentified environmental or toxic injury leads to early vascular dysfunction characterized by abnormalities in endothelial cells, pericytes and vascular smooth muscle cells. Vascular cell dysfunction and/or degeneration results in hypoperfusion (oligemia) and blood–brain barrier (BBB) dysfunction (hit 1) leading to hypoxia and accumulation of multiple plasma‐derived neurotoxins, respectively, and contributes to neuronal dysfunction, degeneration and development of cognitive decline (solid lines). BBB dysfunction, mural cell loss and hypoperfusion/hypoxia reduce vascular amyloid β‐peptide (Aβ) clearance across the BBB and in vascular mural cells and increases production of Aβ from Aβ‐precursor protein (APP), causing Aβ accumulation in brain (hit 2, dashed lines). Pathologic elevations in soluble Aβ lead to the formation of neurotoxic Aβ oligomers and accelerates Aβ deposition around neurons and on blood vessels. Aβ species then amplify both vascular and neuronal injury. Tau phosphorylation and/or pathology, for example, caspase‐cleavage and/or formation of neurofibrillary tangles, within neurons results from convergence of vascular injury (hypoperfusion and BBB disruption) and direct Aβ neurotoxicity. Figure modified from Zlokovic 196.
Figure 3.

Pericyte degeneration leads to a chronic BBB disruption and vascular‐mediated secondary neuronal injury and degeneration. Pericyte loss and/or degeneration represent an important cellular source of hit 1 vascular injury in Alzheimer's disease. Pericyte degeneration leads to BBB disruption and unrestricted entry and accumulation of blood‐derived products in brain including erythrocyte‐derived hemoglobin and plasma‐derived proteins such as albumin, plasmin, thrombin, fibrin, immunoglobulin and others. Plasmin and thrombin have direct neurotoxic properties, whereas fibrin accelerates neurovascular injury. Brain degradation of hemoglobin liberates free iron, which catalyzes formation of reactive oxygen species (ROS) leading to further injury. Albumin increases oncotic pressure resulting in edema, microvascular compression and reduced blood flow. Pericyte loss also leads to endothelial cell death and microvascular regression leading to additional simultaneous reductions in blood flow. In mouse models, vascular injury in absence of Aβ as a result of pericyte loss is sufficient for neurodegeneration. Figure modified from Zlokovic 196.
BBB disruption in AD
In human subjects, post‐mortem brain tissue studies have shown that disruption of the BBB is associated with AD neuropathology and cognitive impairment 31, 33, 34, 51, 53, 57, 63, 74, 124, 138, 143, 146, 195, 196. The time point at which AD disruption occurs during disease pathogenesis remains unclear. Recent studies in human subjects at genetic risk for AD, such as carriers of the apolipoprotein ε4 allele (ApoE4), suggest that vascular permeability changes occur prior to cognitive decline 61. In transgenic mice overexpressing human APP, BBB permeability changes precede neuronal injury and AD pathology 139, 168. The mechanisms of BBB disruption remain poorly defined but may result from vasculotoxic effects of pericyte loss and/or endothelial injury as a result of vascular risk factors, that is, dyslipidemia and/or Aβ 19, 139, 146, 168. Heightened vascular permeability may arise from reduced expression or accelerated degradation of tight and adherens junctional proteins, increased bulk‐flow transcytosis, overt vascular rupture and/or combination thereof 195, 196. In transgenic mice expressing human ApoE4, increased activation of matrix metalloproteinase‐9 degrades tight and adherens junctional proteins leading to vascular leakage 15. A similar cascade has been identified in human ApoE4 carriers 61. Whether a similar pathogenic cascade is erroneously activated in sporadic AD in the absence of ApoE4 remains to be experimentally determined.
Irrespective of the mechanisms involved, the end by‐product of vascular disruption is accumulation of circulating plasma proteins and erythrocyte‐derived hemoglobin in brain ISF and parenchyma 177, 196. Extravasated proteins may remain in the extracellular compartment or become internalized by different cell types of the NVU, including microglia, pericytes and neurons, and frequently co‐localize with markers of cell death and/or injury 5, 14, 15, 101, 178. In multiple animal models, BBB disruption is an important contributor to neuronal injury and dysfunction 14, 15, 139, 178, 190. The mechanisms of injury are numerous. For example, brain accumulation of thrombin has been demonstrated in AD 192, and elevated thrombin concentrations lead to neuronal and vascular injury as well as cognitive impairment 26, 106. Plasmin degrades neuronal laminin, thereby disrupting neuron‐extracellular matrix and sensitizing neurons to secondary injury 27. Fibrin and/or its precursor fibrinogen propagate vascular injury and neuroinflammation through accelerated vascular regression and barrier disruption 124. Aβ interacts with and stabilizes fibrin clots 32, and greater fibrin deposition is seen in vessels inflicted with cerebral amyloid angiopathy (CAA) 74. Depletion of fibrinogen reduces CAA and cognitive decline in transgenic AD mice 32 and may therefore represent an important therapeutic target. In addition to plasma proteins, extravasation of erythrocytes due to microhemorrhage causes deposition of hemoglobin‐derived products, such as free iron, which generates reactive oxygen species and non‐specific oxidant injury 182, 190, 191. The presence of these proteins as well as albumin and immunoglobulin G contributes to brain edema and may alter perfusion dynamics through compression of brain capillaries 195, 196. Finally, disruption of the BBB may increase brain influx of circulating Aβ 37, 146.
Endothelial dysfunction
Abnormal endothelial function leads to excess generation of potentially neurotoxic and pro‐inflammatory molecules 58. For example, AD microvessels secrete neurotoxic factors, such as thrombin, which injure and kill neurons 59, 188. Isolated microvessels from human AD subjects also secrete and/or express a number of pro‐inflammatory mediators, such as nitric oxide, cytokines (tumor necrosis factor, interleukin‐1β and interleukin‐6), chemokines (monocyte chemoattractant protein 1 and interleukin‐8), prostaglandins and leukocyte adhesion molecules 58. In addition to neurotoxic and/or pro‐inflammatory properties, AD endothelial cells demonstrate abnormal metabolic functions 99, 100. The BBB prevents unregulated influx of circulating polar solutes, including glucose, from blood into brain. Therefore, expression of endothelial GLUT1, which is also known as the solute carrier family 2, facilitated glucose transporter membrane 1 (SLC2A1), is required for facilitated diffusion of glucose into the brain. In human AD, endothelial GLUT1 protein levels are reduced 70, 82, 107. Abnormalities in glucose transport and/or utilization are visualized early with 2‐[18F] fluoro‐2‐deoxy‐d‐glucose positron emission tomography imaging (FDG‐PET) in the AD hippocampus, parietotemporal cortex and/or posterior cingulate cortex 127. In those at genetic risk for AD, abnormal glucose handling precedes brain atrophy and neuronal dysfunction 108, 132. However, the significance of these findings remains controversial, and whether reduced glucose transport contributes to or, conversely, is the by‐product of ongoing neurodegeneration and resulting reductions in metabolic demand still awaits the final answer.
Altered vascular Aβ clearance
Pathologic accumulation of Aβ oligomers and insoluble fibrils may theoretically result from overproduction and/or diminished brain clearance of β‐amyloid. In sporadic AD, however, impaired Aβ clearance, but not overproduction, is the key factor leading to Aβ accumulation 102. Aβ may be cleared through transendothelial trafficking to circulating blood, enzymatic degradation such as neprilysin, insulin‐degrading enzyme, tissue plasminogen activator and MMPs, in astrocytes, vSMC, pericytes and neurons within parenchyma, and clearance through perivascular ISF‐CSF bulk flow 16, 196, 199. The vascular pathway is the major route of removal of Aβ from the brain 25, 149, 199. The predominate clearance protein—low‐density lipoprotein receptor‐related protein‐1 (LRP‐1)—binds Aβ on its abluminal plasma membrane and initiates its clearance to blood, as shown in multiple animal models 13, 39, 78, 149 and in vitro BBB models 39, 110, 184. ApoE4, but not ApoE2 or ApoE3, significantly inhibits LRP1‐mediated Aβ brain clearance 38. Apolipoprotein J, also known as clusterin, contributes to Aβ clearance via a related receptor—the low‐density lipoprotein receptor‐related protein‐2 (LRP‐2) 13. The efflux transporter P‐glycoprotein also makes contributions to vascular β‐amyloid clearance 29. In addition to transendothelial transport, LRP1 expression in both vSMC and pericytes mediate Aβ uptake and degradation in vascular mural cells 12, 84, 139, 175. Reductions in LRP1 levels and/or alterations in receptor function as a result of oxidation are associated with brain Aβ accumulation in rodents, non‐human primates, transgenic AD mice and human subjects with AD 9, 12, 39, 43, 149.
Brain hypoperfusion in AD
Neurons are exquisitely sensitive to reductions in CBF—called “hypoperfusion”. In human subjects and animal models, hypoperfusion is sufficient to induce cognitive decline 17, 76, 196. At a cellular level, moderate (∼20%) blood flow reductions may result in altered protein synthesis impairing processes, such as long‐term potentiation and synaptic plastic, required for learning and memory 71, 75. At greater reductions (>30%), the synthesis of ATP becomes impaired and anaerobic metabolism prevails. This results in reduced ATPase activity limiting generation of action potentials and cellular transport, thus contributing to neuronal dysfunction and/or alterations in the neuronal microenvironment, that is, lower cellular pH, electrolyte imbalances and cytotoxic edema 140, 196. Acute reductions in CBF (>80%) result in ischemic neuronal death 109.
Reductions in CBF have not only been observed in human AD subjects 8, 75, 104, 196, but in some studies precede neurodegenerative changes 75, 85, 86, 137, 147, 151. In APOE4 carriers, CBF reductions and reduced CBF responses to brain activation may occur prior to brain atrophy and Aβ accumulation and worsen as the disease progresses 85, 147. A similar relationship between blood flow changes and course of disease has been demonstrated in transgenic mouse models expressing both human APOE4 and APP 15, 112, 139. A number of studies in animal models suggest that reduced CBF accelerates multiple stages of AD neurodegeneration. In transgenic AD mouse models, bilateral occlusion of the common carotid artery increases neuronal Aβ levels and tau phosphorylation and accelerates cognitive decline and neuronal loss 87, 185. In human AD and transgenic models, hypoperfusion appears to accelerate vascular amyloid deposition and CAA, which may, in turn, lead to further ischemia and microinfarctions 116. Therefore, hypoperfusion and subsequent Aβ accumulation may constitute a feed‐forward loop.
AD‐related impairment in blood flow may result from at least two processes: aberrant vascular reactivity and microvascular degeneration. In human subjects, CBF changes occur prior to changes in vascular volume as evidenced by cerebral blood volume magnetic resonance imaging (MRI) 91. This suggests that abnormal vascular function may precede vascular degeneration. The mechanism(s) through which vascular reactivity becomes disrupted in AD have been shown to involve pathologic contractile properties in vSMC in small penetrating arteries. For example, studies in both human AD subjects and transgenic models have demonstrated upregulation of two transcription factors, myocardin (MYOCD) and serum response factor (SRF), important for controlling vSMC differentiation and leads to upregulation of vSMC contractile proteins, a hypercontractile vSMC phenotype, and, ultimately, impaired blood flow 12, 28. In human AD, reduced microvascular diameter suggestive of a hypercontractile phenotype has been observed in the hippocampus 22. MYOCD and SRF overexpression in microvascular pericytes has also been suggested (R.D. Bell, E.A. Winkler and B.V. Zlokovic, unpub. data) and may contribute to these changes. However, further investigation is needed to define the role of contractile protein expression in AD pericytes.
Aβ may also lead to vessel contraction and impairment in CBF as evidenced by the vaconstrictory effect of exogenous Aβ 111, 113, 114, 119 and aberrant vascular reactivity in transgenic models overexpressing APP 112, 139, 164, 183. Aβ impairs CBF on both luminal and abluminal targets 123. On the luminal membrane, Aβ binding to the receptor for advanced glycation end products (RAGE) leads to production of endothelin, a potent a potent vasoconstrictor 37, 195. Meanwhile, Aβ binding to the scavenging receptor CD36 on vascular and perivascular cells leads to oxidative stress and associated vascular dysfunction 121, 122. Deletion of CD36 protects against CBF reductions, vascular Aβ accumulation and slows cognitive decline in aged mice overexpressing APP 122. Therefore, improvement in CBF may delay AD histopathologic and behavior changes.
In addition to dysfunctional vasomotor responses, histologic studies have demonstrated focal vascular regression and reduced focal vascular density in human AD and transgenic models 11, 21, 58, 120, 139, 183. Neuroimaging studies demonstrating reductions in cerebral blood volume corroborate findings of microvascular degeneration 91, 167, 181. Genome‐wide transcriptional profiling in AD human endothelium has identified downregulation in the mesenchyme homeobox gene‐2 (MEOX2), a transcription factor important for vascular differentiation and patterning, as a key contributor to aberrant angiogenic responses to angiogenic stimuli, endothelial apoptosis and chronic hypoperfusion 183. In mouse models, heterozygosity for Meox2 is sufficient to induce neurodegenerative changes 14. Contributions of additional sources of vascular injury have been documented, including pericyte loss 11, 139, 146, Aβ vasculotoxicity 161 and/or Aβ‐mediated inhibition of angiogenesis 120. Whether pericyte loss precedes and/or leads to downregulation of endothelial Meox2 remains to be experimentally determined.
Pericytes in AD
Multiple independent reports have demonstrated pericyte loss and/or degeneration in both the hippocampus and cortex in human AD subjects 11, 50, 146. At an ultrastructural level, pericytes demonstrate large numbers of intracellular inclusions, pinocytotic vesicles, large lipid granules and mitochondrial abnormalities, suggesting cellular dysfunction and/or degeneration 11, 50. Pericyte degenerative changes are associated with capillary reductions and gross dilatation and tortuosity of surviving vessels 11. In individual AD subjects, reductions in pericyte coverage inversely correlate with evidence of BBB disruption, such as leakage of the plasma proteins including immunoglobulin G and fibrin 146. Although these observations are limited to post‐mortem tissue, pericyte dysfunction and/or loss are associated with key attributes of AD vascular pathology—vascular regression and disrupted vascular permeability.
Factors contributing to pericyte loss in AD
The mechanism(s) of pericyte loss have yet to be completely defined. Some studies have suggested that brain pericytes do not decrease with normal aging in rodents during adulthood 14, 125, 128. Additional studies in normally aged animals are needed, however, to confirm these findings. Preliminary data have suggested that vascular factors, for example, hypertension and dyslipidemia, may lead to pericyte injury and/or death 153, 158. Future works are still needed to establish a contributory link between vascular injury and early pericyte loss in AD. In later disease stages, Aβ accumulates on and around brain capillaries and pericytes 171, 180. Pericytes express the Aβ clearance receptor LRP‐1 which binds and internalizes different Aβ species for lysosomal degradation within brain pericytes 139, 175. At high concentrations and prolonged exposure, Aβ species overwhelm the clearance capacity and lead to pericyte cell death in vitro 139, 175. Reductions in pericyte cell number and/or coverage have been similarly observed in transgenic mice with β‐amyloidosis in vivo as a result of overexpression of human APP and accumulation of Aβ in pericytes 122, 139. Other works have also implicated the scavenging receptor CD36 in AD‐related pericyte loss 122.
A loss of brain pericytes, in turn, leads to reduced Aβ clearance through the LRP‐1 degradative pathway promoting Aβ accumulation and/or deposition in the brain 139. Therefore, pericytes are important for brain Aβ clearance but are susceptible to Aβ toxicity, a potential feed‐forward mechanism. In human AD subjects, extravascular Aβ deposits inversely correlate with pericyte coverage in the AD hippocampus—meaning those with greatest Aβ load have fewest pericytes 146. This finding does not permit directionality of this relationship to be established but may reflect the by‐product of both Aβ‐driven pericyte toxicity and a loss of pericyte‐dependent Aβ clearance.
Pericytes and Aβ‐independent neurodegeneration
Recent works have suggested that a loss and/or dysfunction of brain pericytes may accelerate AD‐like neurodegeneration cascade in vivo. In mouse models, a loss of brain pericytes is sufficient to induce neurodegenerative changes in the absence of Aβ through two major pathways—BBB disruption and hypoperfusion (Figure 3) 14, 177, 178. On the one hand, a loss of pericytes leads to heightened vascular permeability through a disrupted BBB. This, in turn, leads to an accumulation of multiple blood‐derived neurotoxic and vasculotoxic molecules seen in human AD, including fibrin 124, thrombin 26, 106, plasmin 27 and hemoglobin‐derived iron and reactive oxygen species 73, 134, 182. Concomitantly, a loss of pericytes also leads to regression of brain microvessels, mainly capillaries. This gives rise to a chronic perfusion stress and hypoxia. Toxin accumulation and hypoxia may then simultaneously converge at the neuronal interface resulting in injury, dysfunction and ultimately cell death 14.
The importance of both pathways to the neurodegenerative phenotype was demonstrated in comparative studies between two different mouse lines: a mouse deficient in PDGFRβ signaling (PdgfrβF7/F7 mice) and a mouse heterozygous for mesenchyme homeobox gene‐2 involved in vascular patterning (Meox2+/−mice). As previously mentioned, Meox2 is transcriptionally downregulated in AD and leads to reduced angiogenesis, microvascular reductions and diminished CBF 183. Meox2+/− mice have reduced microvascular density, but intact pericyte populations and normal BBB integrity 14, 183. In contrast, PdgfrβF7/F7 mice have a deficiency in brain pericytes, which leads to both a microvascular reduction and a disruption. In both mouse lines, neurodegeneration was observed, suggesting that chronic hypoperfusion is sufficient to induce neuronal injury. However, the magnitude of such changes was much larger in pericyte deficient mutants highlighting the synergy between blood‐derived toxin accumulation and hypoxia.
Pericytes and APOE genotype
Further evidence for the importance of Aβ‐independent vascular injury in neurodegenerative changes comes from works in two transgenic mouse lines overexpressing the human APOE4 gene 15, 197. The three major apoE isoforms (E2, E3 and E4) differ in single amino acid substitutions at two distinct residues and alter the structure and function of apoE at molecular and cellular levels 197. APOE4 is the strongest genetic risk factor for sporadic AD 160. Individuals homozygous for APOE4 have a 30% and 60% lifetime risk to develop disease at 75 and 85 years of age, respectively 55. The APOE4 gene may contribute to significant neurovascular dysfunction prior to cognitive decline and Aβ‐accumulation 194, 197 and is associated with greater deficits in BBB permeability in AD 74, 143, 192.
The mechanism of these BBB permeability changes was not established until recently. In mice, overexpression of human apoE4, but not apoE2 or apoE3, leads to upregulation of a pro‐inflammatory cyclophilin A (CypA)‐nuclear factor κB (NF‐κB)‐matrix metalloproteinase 9 (MMP‐9) pathway within brain pericytes (Figure 4). MMP‐9 is then secreted from pericytes and becomes activated. This, in turn, leads to enzymatic degradation of endothelial cell tight junctional complexes resulting in BBB breakdown and unregulated influx of the blood‐derived neurotoxins, including thrombin, fibrin and hemoglobin‐derived iron. Chronic barrier disruption in addition to caspase‐independent effects of apoE4 led to pericyte reductions, microvascular regression and CBF reductions. Importantly, apoE4‐driven vascular injury was sufficient to induce neuronal dysfunction, injury and ultimately cell death. Pharmacologic interventions inhibiting each step of the CypA‐NFκB‐MMP‐9 pathway with specific inhibitors of CypA and NF‐κB or inhibition of MMP‐9, and genetic deletion of CypA reduced both vascular and neuronal dysfunction in transgenic APOE4 mice, suggesting that dysfunction of pericytes can be therapeutically controlled and that pericytes may represent a novel cellular therapeutic target 15.
Figure 4.
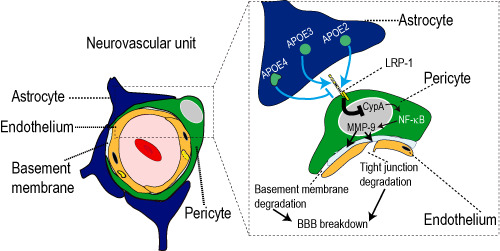
Apolipoprotein E4 triggers early pericyte dysfunction and blood–brain barrier (BBB) breakdown. Apolipoproteins are secreted by astrocytes. ApoE2 and apoE3, but not apoE4, bind to the low density lipoprotein receptor‐related protein‐1 (LRP‐1) on pericytes and suppress the levels of a pro‐inflammatory cytokine cyclophilin A (CypA). ApoE4 exhibits weak binding to LRP‐1 resulting in pathologic elevations in CypA protein levels, which, in turn, increases nuclear translocation of the pro‐inflammatory transcription factor nuclear factor‐κB (NF‐κB), and upregulation of pericyte matrix metalloproteinase‐9 (MMP‐9). Secretion and activation of MMP‐9 degrades endothelial tight and adherens junction proteins leading to disruption of the BBB. Pathologic elevations of MMP‐9 and a disrupted BBB lead to capillary loss and hypoperfusion. Vascular injury may then lead to neuronal dysfunction and degeneration. Figure modified from Bell et al 15.
This work has recently been extended to cognitively normal subjects including APOE4 carriers and APOE4 non‐carriers 61. This study has shown that individuals harboring the APOE4 allele develop an age‐dependent BBB breakdown as evidenced by elevated CSF : plasma albumin quotient, which correlated with increased CypA levels and activated MMP‐9 levels in the CSF, suggesting that activation of the pro‐inflammatory CypA pathway occur before cognitive decline 61. Additionally, a recent study reported that CypA mRNA levels are reduced in APOE2 carriers further supporting the role of CypA in regulating neurovascular function 30. Collectively, these data suggest that the BBB damage likely involving pericyte dysfunction might contribute to early neurovascular dysfunction seen in animal models and humans with the APOE4 allele.
Pericytes and Aβ accumulation
To determine whether a loss of brain pericytes also contributes to Aβ‐dependent toxicity in AD‐like neurodegeneration, our group recently published a study in which pericyte deficient mutants (Pdgfrβ+/− mice) were crossed with mice which overexpressing the Swedish mutation of human APP (APPsw/0) 139 (Figure 5). APPsw/0 mice accumulate parenchymal and vascular Aβ leading to amyloid plaques and memory deficits, but do not develop tau pathology and/or neuronal loss 72, 154. Enhanced pericyte loss (APPsw/0;Pdgfrβ+/− mice) leads to greater accumulation of soluble Aβ40 and Aβ42 species as a result of reduced pericyte‐dependent clearance of soluble Aβ species from brain ISF, as shown by in vivo microdialysis in the hippocampus. Impaired clearance of soluble Aβ then contributes to accelerated deposition of insoluble Aβ resulting in CAA and parenchymal β‐amyloid plaques.
Figure 5.

Pericyte degeneration and loss contributes to Alzheimer's type neurodegeneration through amyloid β‐peptide (Aβ) independent and dependent injury mechanisms. Brain pericyte loss as a result of disruption of the platelet derived growth factor receptor β (PDGFRβ) signaling triggers early Aβ‐independent vascular injury leading to microvascular loss and disruption, which results in hypoperfusion/hypoxia and brain accumulation of toxic plasma proteins, respectively (black). Loss of pericytes also leads to diminished clearance of soluble Aβ species from brain interstitial fluid (ISF) and further elevations in brain Aβ levels (red). Aβ may then overwhelm degradative pathway in surviving pericytes, resulting in further pericyte degeneration (dashed line). Both pericyte loss and Aβ acting simultaneously result in the early development of all facets of Alzheimer's disease neuropathology including Aβ plaques and neuronal tau pathology, degeneration and loss, which are not observed at an early disease stage when Aβ accumulation or pericyte‐driven vascular injury occurs in isolation. Figure modified from Sagare et al 139.
Pericytes and tau pathology
APPsw/0;Pdgfrβ+/− mice also developed tau pathology at the early age of 9 months including neuronal accumulation of hyperphosphoryled tau species, caspase‐cleaved tau and tau aggregates 139. Importantly, tau pathology at this relatively early disease stage was not observed in either APPsw/0 mice or Pdgfrβ+/− mice, suggesting that both pericyte‐driven vascular injury and Aβ elevations must be present to induce early appearance of tau pathology. With respect to neuronal loss, Pdgfrβ+/− mice did show a more moderate neuronal loss as a result of direct vascular injury as previously described 14. However, the magnitude of neuronal dysfunction and/or loss, and behavioral impairment on hippocampal‐dependent tasks was much more severe in APPsw/0;Pdgfrβ+/− mice 139. Consistent with the two‐hit vascular hypothesis, pericyte‐driven direct vascular injury and Aβ working together can induce tau pathology and neuronal loss in vivo amplifying cognitive decline. Aβ accumulation in pericytes in turn leads to pericyte loss, worsening AD‐related neurovascular and neuronal dysfunction (Figure 5).
Summary and Future Perspectives
The contributions of pericytes to neurodegenerative disease have only recently been recognized. Among chronic neurodegenerative diseases, pericyte loss and/or dysfunction have been implicated in AD pathogenesis. However, pericyte degeneration seems not to be unique to AD as pericyte loss and dysfunction have been recently reported in other neurodegenerative disorders including ALS 179 and cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) 60, as well as in transgenic models and individuals carrying APOE4, a major genetic risk factor for AD 15, 61.
As shown in transgenic models of pericyte deficiency and AD, pericytes may contribute to disease pathogenesis through both Aβ‐independent pathway and by altering Aβ metabolism and clearance. Both pathways may act synergistically after disease onset to amplify microvascular degeneration, causing neuronal dysfunction and degeneration. Whether pericyte injury and degeneration represents a common downstream pathway for neurodegenerative changes in AD has yet to be explored by using models with vascular risk factors alone (eg, hypertension, diabetes, dyslipidemia, homocysteinemia and pro‐coagulant profile) and/or in combination with models of Aβ and tau pathology. It would also be interesting to investigate whether pericytes play a role in the metabolism and clearance of other proteinaceous material implicated in the pathogenesis of other neurodegenerative diseases including Parkinson's disease (eg, α‐synuclein), prion disease and/or ALS (eg, superoxide dismutase).
Preliminary studies have supported that pericytes may be therapeutically targeted to stabilize peripheral vascular lesions 94. Therapies focused on CNS pericytes have yet to be developed experimentally and tested for chronic neurodegenerative disease and/or brain vascular lesions. Initial attempts have been made by showing successfully that either genetic or pharmacologic inhibition of the pro‐inflammatory CypA pathway in brain pericytes in rescues both the vascular and the neuronal phenotype in APOE4 transgenic mice 15. Future studies are needed, however, to extend these findings to humans with APOE4 genetic risk. Similarly, more work is needed to identify other key molecular players and pathways mediating aberrant signal transduction between pericytes, endothelial cells, astrocytes and neurons within the NVU in sporadic and inherited AD that could possibly represent new therapeutic targets to control pericyte degeneration in dementia and slow vascular and neuronal degeneration. Only then may pericyte‐targeted therapies find broader applications for neurological disorders and lead to the development of new neurovascular medicine.
References
- 1. Abramsson A, Kurup S, Busse M, Yamada S, Lindblom P, Schallmeiner E et al (2007) Defective N‐sulfation of heparan sulfate proteoglycans limits PDGF‐BB binding and pericyte recruitment in vascular development. Genes Dev 21:316–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ai J, Sun LH, Che H, Zhang R, Zhang TZ, Wu WC et al (2013) MicroRNA‐195 protects against dementia induced by chronic brain hypoperfusion via its anti‐amyloidogenic effect in rats. J Neurosci 33:3989–4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Allinson KR, Lee HS, Fruttiger M, McCarty JH, Arthur HM (2012) Endothelial expression of TGFbeta type II receptor is required to maintain vascular integrity during postnatal development of the central nervous system. PLoS ONE 7:e39336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Armulik A, Genove G, Betsholtz C (2011) Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell 21:193–215. [DOI] [PubMed] [Google Scholar]
- 5. Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C et al (2010) Pericytes regulate the blood‐brain barrier. Nature 468:557–561. [DOI] [PubMed] [Google Scholar]
- 6. Attwell D (2013) Cerebral pericytes are critical regulators of cerebral blood flow in health and disease [Abstract]. Aarhus CTTH workshop, Aarhus, Denmark.
- 7. Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA (2010) Glial and neuronal control of brain blood flow. Nature 468:232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Austin BP, Nair VA, Meier TB, Xu G, Rowley HA, Carlsson CM et al (2011) Effects of hypoperfusion in Alzheimer's disease. J Alzheimers Dis 26(Suppl. 3):123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bading JR, Yamada S, Mackic JB, Kirkman L, Miller C, Calero M et al (2002) Brain clearance of Alzheimer's amyloid‐beta40 in the squirrel monkey: a SPECT study in a primate model of cerebral amyloid angiopathy. J Drug Target 10:359–368. [DOI] [PubMed] [Google Scholar]
- 10. Ballatore C, Lee VM, Trojanowski JQ (2007) Tau‐mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci 8:663–672. [DOI] [PubMed] [Google Scholar]
- 11. Baloyannis SJ, Baloyannis IS (2012) The vascular factor in Alzheimer's disease: a study in Golgi technique and electron microscopy. J Neurol Sci 322:117–121. [DOI] [PubMed] [Google Scholar]
- 12. Bell RD, Deane R, Chow N, Long X, Sagare A, Singh I et al (2009) SRF and myocardin regulate LRP‐mediated amyloid‐beta clearance in brain vascular cells. Nat Cell Biol 11:143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bell RD, Sagare AP, Friedman AE, Bedi GS, Holtzman DM, Deane R, Zlokovic BV (2007) Transport pathways for clearance of human Alzheimer's amyloid beta‐peptide and apolipoproteins E and J in the mouse central nervous system. J Cereb Blood Flow Metab 27:909–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, Zlokovic BV (2010) Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 68:409–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z et al (2012) Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 485:512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bell RD, Zlokovic BV (2009) Neurovascular mechanisms and blood‐brain barrier disorder in Alzheimer's disease. Acta Neuropathol 118:103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bink DI, Ritz K, Aronica E, van der Weerd L, Daemen MJ (2013) Mouse models to study the effect of cardiovascular risk factors on brain structure and cognition. J Cereb Blood Flow Metab 33:1666–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bobbie MW, Roy S, Trudeau K, Munger SJ, Simon AM (2010) Reduced connexin 43 expression and its effect on the development of vascular lesions in retinas of diabetic mice. Invest Ophthalmol Vis Sci 51:3758–3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bowman GL, Kaye JA, Quinn JF (2012) Dyslipidemia and blood‐brain barrier integrity in Alzheimer's disease. Curr Gerontol Geriatr Res 2012:184042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Braun A, Xu H, Hu F, Kocherlakota P, Siegel D, Chander P et al (2007) Paucity of pericytes in germinal matrix vasculature of premature infants. J Neurosci 27:12012–12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brown WR, Thore CR (2011) Review: cerebral microvascular pathology in ageing and neurodegeneration. Neuropathol Appl Neurobiol 37:56–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Burke MC, Nelson L, Slade JY, Oakley AE, Khundakar AA, Kalaria RN (2014) Morphometry of the hippocampal microvasculature in post‐stroke and age‐related dementias. Neuropathol Appl Neurobiol 40:284–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Candelario‐Jalil E, Yang Y, Rosenberg GA (2009) Diverse roles of matrix metalloproteinases and tissue inhibitors of metalloproteinases in neuroinflammation and cerebral ischemia. Neuroscience 158:983–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Castejon OJ, Castellano A, Arismendi GJ, Medina Z (2005) The inflammatory reaction in human traumatic oedematous cerebral cortex. J Submicrosc Cytol Pathol 37:43–52. [PubMed] [Google Scholar]
- 25. Castellano JM, Deane R, Gottesdiener AJ, Verghese PB, Stewart FR, West T et al (2012) Low‐density lipoprotein receptor overexpression enhances the rate of brain‐to‐blood Abeta clearance in a mouse model of beta‐amyloidosis. Proc Natl Acad Sci U S A 109:15502–15507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen B, Cheng Q, Yang K, Lyden PD (2010) Thrombin mediates severe neurovascular injury during ischemia. Stroke 41:2348–2352. [DOI] [PubMed] [Google Scholar]
- 27. Chen ZL, Strickland S (1997) Neuronal death in the hippocampus is promoted by plasmin‐catalyzed degradation of laminin. Cell 91:917–925. [DOI] [PubMed] [Google Scholar]
- 28. Chow N, Bell RD, Deane R, Streb JW, Chen J, Brooks A et al (2007) Serum response factor and myocardin mediate arterial hypercontractility and cerebral blood flow dysregulation in Alzheimer's phenotype. Proc Natl Acad Sci U S A 104:823–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cirrito JR, Deane R, Fagan AM, Spinner ML, Parsadanian M, Finn MB et al (2005) P‐glycoprotein deficiency at the blood‐brain barrier increases amyloid‐beta deposition in an Alzheimer disease mouse model. J Clin Invest 115:3285–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Conejero‐Goldberg C, Gomar JJ, Bobes‐Bascaran T, Hyde TM, Kleinman JE, Herman MM et al (2014) APOE2 enhances neuroprotection against Alzheimer's disease through multiple molecular mechanisms. Mol Psychiatry [Epub ahead of print; doi: 10.1038/mp.2013.194]. [DOI] [PubMed] [Google Scholar]
- 31. Cordonnier C, van der Flier WM (2011) Brain microbleeds and Alzheimer's disease: innocent observation or key player? Brain 134(Pt 2):335–344. [DOI] [PubMed] [Google Scholar]
- 32. Cortes‐Canteli M, Paul J, Norris EH, Bronstein R, Ahn HJ, Zamolodchikov D et al (2010) Fibrinogen and beta‐amyloid association alters thrombosis and fibrinolysis: a possible contributing factor to Alzheimer's disease. Neuron 66:695–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cortes‐Canteli M, Zamolodchikov D, Ahn HJ, Strickland S, Norris EH (2012) Fibrinogen and altered hemostasis in Alzheimer's disease. J Alzheimers Dis 32:599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cullen KM, Kocsi Z, Stone J (2005) Pericapillary haem‐rich deposits: evidence for microhaemorrhages in aging human cerebral cortex. J Cereb Blood Flow Metab 25:1656–1667. [DOI] [PubMed] [Google Scholar]
- 35. Daneman R, Zhou L, Kebede AA, Barres BA (2010) Pericytes are required for blood‐brain barrier integrity during embryogenesis. Nature 468:562–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Darland DC, Massingham LJ, Smith SR, Piek E, Saint‐Geniez M, D'Amore PA (2003) Pericyte production of cell‐associated VEGF is differentiation‐dependent and is associated with endothelial survival. Dev Biol 264:275–288. [DOI] [PubMed] [Google Scholar]
- 37. Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E et al (2003) RAGE mediates amyloid‐beta peptide transport across the blood‐brain barrier and accumulation in brain. Nat Med 9:907–913. [DOI] [PubMed] [Google Scholar]
- 38. Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB et al (2008) apoE isoform‐specific disruption of amyloid beta peptide clearance from mouse brain. J Clin Invest 118:4002–4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K et al (2004) LRP/amyloid beta‐peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 43:333–344. [DOI] [PubMed] [Google Scholar]
- 40. Deschaintre Y, Richard F, Leys D, Pasquier F (2009) Treatment of vascular risk factors is associated with slower decline in Alzheimer disease. Neurology 73:674–680. [DOI] [PubMed] [Google Scholar]
- 41. Diaz‐Flores L, Gutierrez R, Madrid JF, Varela H, Valladares F, Acosta E et al (2009) Pericytes. Morphofunction, interactions and pathology in a quiescent and activated mesenchymal cell niche. Histol Histopathol 24:909–969. [DOI] [PubMed] [Google Scholar]
- 42. Dohgu S, Takata F, Yamauchi A, Nakagawa S, Egawa T, Naito M et al (2005) Brain pericytes contribute to the induction and up‐regulation of blood‐brain barrier functions through transforming growth factor‐beta production. Brain Res 1038:208–215. [DOI] [PubMed] [Google Scholar]
- 43. Donahue JE, Flaherty SL, Johanson CE, Duncan JA 3rd, Silverberg GD, Miller MC et al (2006) RAGE, LRP‐1, and amyloid‐beta protein in Alzheimer's disease. Acta Neuropathol 112:405–415. [DOI] [PubMed] [Google Scholar]
- 44. Dore‐Duffy P (2008) Pericytes: pluripotent cells of the blood brain barrier. Curr Pharm Des 14:1581–1593. [DOI] [PubMed] [Google Scholar]
- 45. Dore‐Duffy P, Owen C, Balabanov R, Murphy S, Beaumont T, Rafols JA (2000) Pericyte migration from the vascular wall in response to traumatic brain injury. Microvasc Res 60:55–69. [DOI] [PubMed] [Google Scholar]
- 46. Dore‐Duffy P, Wang S, Mehedi A, Katyshev V, Cleary K, Tapper A et al (2011) Pericyte‐mediated vasoconstriction underlies TBI‐induced hypoperfusion. Neurol Res 33:176–186. [DOI] [PubMed] [Google Scholar]
- 47. Eisele YS, Obermuller U, Heilbronner G, Baumann F, Kaeser SA, Wolburg H et al (2010) Peripherally applied Abeta‐containing inoculates induce cerebral beta‐amyloidosis. Science 330:980–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Enge M, Bjarnegard M, Gerhardt H, Gustafsson E, Kalen M, Asker N et al (2002) Endothelium‐specific platelet‐derived growth factor‐B ablation mimics diabetic retinopathy. EMBO J 21:4307–4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Etchevers HC, Vincent C, Le Douarin NM, Couly GF (2001) The cephalic neural crest provides pericytes and smooth muscle cells to all blood vessels of the face and forebrain. Development 128:1059–1068. [DOI] [PubMed] [Google Scholar]
- 50. Farkas E, Luiten PG (2001) Cerebral microvascular pathology in aging and Alzheimer's disease. Prog Neurobiol 64:575–611. [DOI] [PubMed] [Google Scholar]
- 51. Farrall AJ, Wardlaw JM (2009) Blood‐brain barrier: ageing and microvascular disease—systematic review and meta‐analysis. Neurobiol Aging 30:337–352. [DOI] [PubMed] [Google Scholar]
- 52. Fernandez‐Klett F, Offenhauser N, Dirnagl U, Priller J, Lindauer U (2010) Pericytes in capillaries are contractile in vivo, but arterioles mediate functional hyperemia in the mouse brain. Proc Natl Acad Sci U S A 107:22290–22295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fiala M, Liu QN, Sayre J, Pop V, Brahmandam V, Graves MC, Vinters HV (2002) Cyclooxygenase‐2‐positive macrophages infiltrate the Alzheimer's disease brain and damage the blood‐brain barrier. Eur J Clin Invest 32:360–371. [DOI] [PubMed] [Google Scholar]
- 54. Gaengel K, Genove G, Armulik A, Betsholtz C (2009) Endothelial‐mural cell signaling in vascular development and angiogenesis. Arterioscler Thromb Vasc Biol 29:630–638. [DOI] [PubMed] [Google Scholar]
- 55. Genin E, Hannequin D, Wallon D, Sleegers K, Hiltunen M, Combarros O et al (2011) APOE and Alzheimer disease: a major gene with semi‐dominant inheritance. Mol Psychiatry 16:903–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gerhardt H, Betsholtz C (2003) Endothelial‐pericyte interactions in angiogenesis. Cell Tissue Res 314:15–23. [DOI] [PubMed] [Google Scholar]
- 57. Goos JD, Kester MI, Barkhof F, Klein M, Blankenstein MA, Scheltens P, van der Flier WM (2009) Patients with Alzheimer disease with multiple microbleeds: relation with cerebrospinal fluid biomarkers and cognition. Stroke 40:3455–3460. [DOI] [PubMed] [Google Scholar]
- 58. Grammas P (2011) Neurovascular dysfunction, inflammation and endothelial activation: implications for the pathogenesis of Alzheimer's disease. J Neuroinflammation 8:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Grammas P, Moore P, Weigel PH (1999) Microvessels from Alzheimer's disease brains kill neurons in vitro . Am J Pathol 154:337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gu X, Liu XY, Fagan A, Gonzalez‐Toledo ME, Zhao LR (2012) Ultrastructural changes in cerebral capillary pericytes in aged Notch3 mutant transgenic mice. Ultrastruct Pathol 36:48–55. [DOI] [PubMed] [Google Scholar]
- 61. Halliday MR, Pomara N, Sagare AP, Mack WJ, Frangione B, Zlokovic BV (2013) Relationship between cyclophilin a levels and matrix metalloproteinase 9 activity in cerebrospinal fluid of cognitively normal apolipoprotein e4 carriers and blood‐brain barrier breakdown. JAMA Neurol 70:1198–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hamilton NB, Attwell D, Hall CN (2010) Pericyte‐mediated regulation of capillary diameter: a component of neurovascular coupling in health and disease. Front Neuroenergetics 2. pii: 5. doi: 10.3389/fnene.2010.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hanyu H, Tanaka Y, Shimizu S, Takasaki M, Abe K (2003) Cerebral microbleeds in Alzheimer's disease. J Neurol 250:1496–1497. [DOI] [PubMed] [Google Scholar]
- 64. Harb R, Whiteus C, Freitas C, Grutzendler J (2013) In vivo imaging of cerebral microvascular plasticity from birth to death. J Cereb Blood Flow Metab 33:146–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297:353–356. [DOI] [PubMed] [Google Scholar]
- 66. Harris JJ, Jolivet R, Attwell D (2012) Synaptic energy use and supply. Neuron 75:762–777. [DOI] [PubMed] [Google Scholar]
- 67. Hellstrom M, Gerhardt H, Kalen M, Li X, Eriksson U, Wolburg H, Betsholtz C (2001) Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J Cell Biol 153:543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hellstrom M, Kalen M, Lindahl P, Abramsson A, Betsholtz C (1999) Role of PDGF‐B and PDGFR‐beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development 126:3047–3055. [DOI] [PubMed] [Google Scholar]
- 69. Hirase H, Creso J, Singleton M, Bartho P, Buzsaki G (2004) Two‐photon imaging of brain pericytes in vivo using dextran‐conjugated dyes. Glia 46:95–100. [DOI] [PubMed] [Google Scholar]
- 70. Horwood N, Davies DC (1994) Immunolabelling of hippocampal microvessel glucose transporter protein is reduced in Alzheimer's disease. Virchows Arch 425:69–72. [DOI] [PubMed] [Google Scholar]
- 71. Hossmann KA (1994) Viability thresholds and the penumbra of focal ischemia. Ann Neurol 36:557–565. [DOI] [PubMed] [Google Scholar]
- 72. Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S et al (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274:99–102. [DOI] [PubMed] [Google Scholar]
- 73. Hua Y, Keep RF, Hoff JT, Xi G (2007) Brain injury after intracerebral hemorrhage: the role of thrombin and iron. Stroke 38(2 Suppl.):759–762. [DOI] [PubMed] [Google Scholar]
- 74. Hultman K, Strickland S, Norris EH (2013) The APOE varepsilon4/varepsilon4 genotype potentiates vascular fibrin(ogen) deposition in amyloid‐laden vessels in the brains of Alzheimer's disease patients. J Cereb Blood Flow Metab 33:1251–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Iadecola C (2004) Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat Rev Neurosci 5:347–360. [DOI] [PubMed] [Google Scholar]
- 76. Iadecola C (2013) The pathobiology of vascular dementia. Neuron 80:844–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ittner LM, Gotz J (2011) Amyloid‐beta and tau—a toxic pas de deux in Alzheimer's disease. Nat Rev Neurosci 12:65–72. [DOI] [PubMed] [Google Scholar]
- 78. Jaeger LB, Dohgu S, Hwang MC, Farr SA, Murphy MP, Fleegal‐DeMotta MA et al (2009) Testing the neurovascular hypothesis of Alzheimer's disease: LRP‐1 antisense reduces blood‐brain barrier clearance, increases brain levels of amyloid‐beta protein, and impairs cognition. J Alzheimers Dis 17:553–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Jeansson M, Gawlik A, Anderson G, Li C, Kerjaschki D, Henkelman M, Quaggin SE (2011) Angiopoietin‐1 is essential in mouse vasculature during development and in response to injury. J Clin Invest 121:2278–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Jellinger KA (2010) Prevalence and impact of cerebrovascular lesions in Alzheimer and Lewy body diseases. Neurodegener Dis 7:112–115. [DOI] [PubMed] [Google Scholar]
- 81. Kalaria RN, Akinyemi R, Ihara M (2012) Does vascular pathology contribute to Alzheimer changes? J Neurol Sci 322:141–147. [DOI] [PubMed] [Google Scholar]
- 82. Kalaria RN, Harik SI (1989) Reduced glucose transporter at the blood‐brain barrier and in cerebral cortex in Alzheimer disease. J Neurochem 53:1083–1088. [DOI] [PubMed] [Google Scholar]
- 83. Kamouchi M, Kitazono T, Ago T, Wakisaka M, Ooboshi H, Ibayashi S, Iida M (2004) Calcium influx pathways in rat CNS pericytes. Brain Res Mol Brain Res 126:114–120. [DOI] [PubMed] [Google Scholar]
- 84. Kanekiyo T, Liu CC, Shinohara M, Li J, Bu G (2012) LRP1 in brain vascular smooth muscle cells mediates local clearance of Alzheimer's amyloid‐beta. J Neurosci 32:16458–16465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kim SM, Kim MJ, Rhee HY, Ryu CW, Kim EJ, Petersen ET, Jahng GH (2013) Regional cerebral perfusion in patients with Alzheimer's disease and mild cognitive impairment: effect of APOE epsilon4 allele. Neuroradiology 55:25–34. [DOI] [PubMed] [Google Scholar]
- 86. Knopman DS, Roberts R (2010) Vascular risk factors: imaging and neuropathologic correlates. J Alzheimers Dis 20:699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Koike MA, Green KN, Blurton‐Jones M, Laferla FM (2010) Oligemic hypoperfusion differentially affects tau and amyloid‐{beta}. Am J Pathol 177:300–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kokovay E, Li L, Cunningham LA (2006) Angiogenic recruitment of pericytes from bone marrow after stroke. J Cereb Blood Flow Metab 26:545–555. [DOI] [PubMed] [Google Scholar]
- 89. Korn J, Christ B, Kurz H (2002) Neuroectodermal origin of brain pericytes and vascular smooth muscle cells. J Comp Neurol 442:78–88. [DOI] [PubMed] [Google Scholar]
- 90. Kurz H (2009) Cell lineages and early patterns of embryonic CNS vascularization. Cell Adh Migr 3:205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lacalle‐Aurioles M, Mateos‐Perez JM, Guzman‐De‐Villoria JA, Olazaran J, Cruz‐Orduna I, Aleman‐Gomez Y et al (2014) Cerebral blood flow is an earlier indicator of perfusion abnormalities than cerebral blood volume in Alzheimer's disease. J Cereb Blood Flow Metab 34:654–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Lamagna C, Bergers G (2006) The bone marrow constitutes a reservoir of pericyte progenitors. J Leukoc Biol 80:677–681. [DOI] [PubMed] [Google Scholar]
- 93. Larson EB, Yaffe K, Langa KM (2013) New insights into the dementia epidemic. N Engl J Med 369:2275–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Lebrin F, Srun S, Raymond K, Martin S, van den Brink S, Freitas C et al (2010) Thalidomide stimulates vessel maturation and reduces epistaxis in individuals with hereditary hemorrhagic telangiectasia. Nat Med 16:420–428. [DOI] [PubMed] [Google Scholar]
- 95. Li F, Lan Y, Wang Y, Wang J, Yang G, Meng F et al (2011) Endothelial Smad4 maintains cerebrovascular integrity by activating N‐cadherin through cooperation with Notch. Dev Cell 20:291–302. [DOI] [PubMed] [Google Scholar]
- 96. Li J, Huang X, Xu X, Mayo J, Bringas P Jr, Jiang R et al (2011) SMAD4‐mediated WNT signaling controls the fate of cranial neural crest cells during tooth morphogenesis. Development 138:1977–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Lindahl P, Johansson BR, Leveen P, Betsholtz C (1997) Pericyte loss and microaneurysm formation in PDGF‐B‐deficient mice. Science 277:242–245. [DOI] [PubMed] [Google Scholar]
- 98. Marchesi VT (2011) Alzheimer's dementia begins as a disease of small blood vessels, damaged by oxidative‐induced inflammation and dysregulated amyloid metabolism: implications for early detection and therapy. FASEB J 25:5–13. [DOI] [PubMed] [Google Scholar]
- 99. Marcus DL, Freedman ML (1997) Decreased brain glucose metabolism in microvessels from patients with Alzheimer's disease. Ann N Y Acad Sci 826:248–253. [DOI] [PubMed] [Google Scholar]
- 100. Marcus DL, de Leon MJ, Goldman J, Logan J, Christman DR, Wolf AP et al (1989) Altered glucose metabolism in microvessels from patients with Alzheimer's disease. Ann Neurol 26:91–94. [DOI] [PubMed] [Google Scholar]
- 101. Matz PG, Lewen A, Chan PH (2001) Neuronal, but not microglial, accumulation of extravasated serum proteins after intracerebral hemolysate exposure is accompanied by cytochrome c release and DNA fragmentation. J Cereb Blood Flow Metab 21:921–928. [DOI] [PubMed] [Google Scholar]
- 102. Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC et al (2010) Decreased clearance of CNS beta‐amyloid in Alzheimer's disease. Science 330:1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Mazlo M, Gasz B, Szigeti A, Zsombok A, Gallyas F (2004) Debris of “dark” (compacted) neurones are removed from an otherwise undamaged environment mainly by astrocytes via blood vessels. J Neurocytol 33:557–567. [DOI] [PubMed] [Google Scholar]
- 104. Mazza M, Marano G, Traversi G, Bria P, Mazza S (2011) Primary cerebral blood flow deficiency and Alzheimer's disease: shadows and lights. J Alzheimers Dis 23:375–389. [DOI] [PubMed] [Google Scholar]
- 105. Meyer‐Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E et al (2006) Exogenous induction of cerebral beta‐amyloidogenesis is governed by agent and host. Science 313:1781–1784. [DOI] [PubMed] [Google Scholar]
- 106. Mhatre M, Nguyen A, Kashani S, Pham T, Adesina A, Grammas P (2004) Thrombin, a mediator of neurotoxicity and memory impairment. Neurobiol Aging 25:783–793. [DOI] [PubMed] [Google Scholar]
- 107. Mooradian AD, Chung HC, Shah GN (1997) GLUT‐1 expression in the cerebra of patients with Alzheimer's disease. Neurobiol Aging 18:469–474. [DOI] [PubMed] [Google Scholar]
- 108. Mosconi L, Sorbi S, de Leon MJ, Li Y, Nacmias B, Myoung PS et al (2006) Hypometabolism exceeds atrophy in presymptomatic early‐onset familial Alzheimer's disease. J Nucl Med 47:1778–1786. [PubMed] [Google Scholar]
- 109. Moskowitz MA, Lo EH, Iadecola C (2010) The science of stroke: mechanisms in search of treatments. Neuron 67:181–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Nazer B, Hong S, Selkoe DJ (2008) LRP promotes endocytosis and degradation, but not transcytosis, of the amyloid‐beta peptide in a blood‐brain barrier in vitro model. Neurobiol Dis 30:94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Niwa K, Carlson GA, Iadecola C (2000) Exogenous A beta1‐40 reproduces cerebrovascular alterations resulting from amyloid precursor protein overexpression in mice. J Cereb Blood Flow Metab 20:1659–1668. [DOI] [PubMed] [Google Scholar]
- 112. Niwa K, Kazama K, Younkin SG, Carlson GA, Iadecola C (2002) Alterations in cerebral blood flow and glucose utilization in mice overexpressing the amyloid precursor protein. Neurobiol Dis 9:61–68. [DOI] [PubMed] [Google Scholar]
- 113. Niwa K, Porter VA, Kazama K, Cornfield D, Carlson GA, Iadecola C (2001) A beta‐peptides enhance vasoconstriction in cerebral circulation. Am J Physiol Heart Circ Physiol 281:H2417–H2424. [DOI] [PubMed] [Google Scholar]
- 114. Niwa K, Younkin L, Ebeling C, Turner SK, Westaway D, Younkin S et al (2000) Abeta 1‐40‐related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proc Natl Acad Sci U S A 97:9735–9740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Oishi K, Kamiyashiki T, Ito Y (2007) Isometric contraction of microvascular pericytes from mouse brain parenchyma. Microvasc Res 73:20–28. [DOI] [PubMed] [Google Scholar]
- 116. Okamoto Y, Yamamoto T, Kalaria RN, Senzaki H, Maki T, Hase Y et al (2012) Cerebral hypoperfusion accelerates cerebral amyloid angiopathy and promotes cortical microinfarcts. Acta Neuropathol 123:381–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Ozerdem U, Stallcup WB (2003) Early contribution of pericytes to angiogenic sprouting and tube formation. Angiogenesis 6:241–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Ozerdem U, Stallcup WB (2004) Pathological angiogenesis is reduced by targeting pericytes via the NG2 proteoglycan. Angiogenesis 7:269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Paris D, Humphrey J, Quadros A, Patel N, Crescentini R, Crawford F, Mullan M (2003) Vasoactive effects of A beta in isolated human cerebrovessels and in a transgenic mouse model of Alzheimer's disease: role of inflammation. Neurol Res 25:642–651. [DOI] [PubMed] [Google Scholar]
- 120. Paris D, Patel N, DelleDonne A, Quadros A, Smeed R, Mullan M (2004) Impaired angiogenesis in a transgenic mouse model of cerebral amyloidosis. Neurosci Lett 366:80–85. [DOI] [PubMed] [Google Scholar]
- 121. Park L, Wang G, Zhou P, Zhou J, Pitstick R, Previti ML et al (2011) Scavenger receptor CD36 is essential for the cerebrovascular oxidative stress and neurovascular dysfunction induced by amyloid‐beta. Proc Natl Acad Sci U S A 108:5063–5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Park L, Zhou J, Zhou P, Pistick R, El Jamal S, Younkin L et al (2013) Innate immunity receptor CD36 promotes cerebral amyloid angiopathy. Proc Natl Acad Sci U S A 110:3089–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Park L, Zhou P, Koizumi K, El Jamal S, Previti ML, Van Nostrand WE et al (2013) Brain and circulating levels of Abeta1‐40 differentially contribute to vasomotor dysfunction in the mouse brain. Stroke 44:198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Paul J, Strickland S, Melchor JP (2007) Fibrin deposition accelerates neurovascular damage and neuroinflammation in mouse models of Alzheimer's disease. J Exp Med 204:1999–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Peinado MA, Quesada A, Pedrosa JA, Torres MI, Martinez M, Esteban FJ et al (1998) Quantitative and ultrastructural changes in glia and pericytes in the parietal cortex of the aging rat. Microsc Res Tech 43:34–42. [DOI] [PubMed] [Google Scholar]
- 126. Peppiatt CM, Howarth C, Mobbs P, Attwell D (2006) Bidirectional control of CNS capillary diameter by pericytes. Nature 443:700–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Perrin RJ, Fagan AM, Holtzman DM (2009) Multimodal techniques for diagnosis and prognosis of Alzheimer's disease. Nature 461:916–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Peters A, Sethares C (2012) Age‐related changes in the morphology of cerebral capillaries do not correlate with cognitive decline. J Comp Neurol 520:1339–1347. [DOI] [PubMed] [Google Scholar]
- 129. Petzold GC, Murthy VN (2011) Role of astrocytes in neurovascular coupling. Neuron 71:782–797. [DOI] [PubMed] [Google Scholar]
- 130. Pieper C, Marek JJ, Unterberg M, Schwerdtle T, Galla HJ (2014) Brain capillary pericytes contribute to the immune defense in response to cytokines or LPS in vitro . Brain Res 1550:1–8. [DOI] [PubMed] [Google Scholar]
- 131. Piquer‐Gil M, Garcia‐Verdugo JM, Zipancic I, Sanchez MJ, Alvarez‐Dolado M (2009) Cell fusion contributes to pericyte formation after stroke. J Cereb Blood Flow Metab 29:480–485. [DOI] [PubMed] [Google Scholar]
- 132. Protas HD, Chen K, Langbaum JB, Fleisher AS, Alexander GE, Lee W et al (2013) Posterior cingulate glucose metabolism, hippocampal glucose metabolism, and hippocampal volume in cognitively normal, late‐middle‐aged persons at 3 levels of genetic risk for Alzheimer disease. JAMA Neurol 70:320–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Querfurth HW, LaFerla FM (2010) Alzheimer's disease. N Engl J Med 362:329–344. [DOI] [PubMed] [Google Scholar]
- 134. Regan RF, Guo Y (1998) Toxic effect of hemoglobin on spinal cord neurons in culture. J Neurotrauma 15:645–653. [DOI] [PubMed] [Google Scholar]
- 135. Richard E, Gouw AA, Scheltens P, van Gool WA (2010) Vascular care in patients with Alzheimer disease with cerebrovascular lesions slows progression of white matter lesions on MRI: the evaluation of vascular care in Alzheimer's disease (EVA) study. Stroke 41:554–556. [DOI] [PubMed] [Google Scholar]
- 136. Rouget C (1873) Memoire sur le developpement, la structures et les proprietes des capillaries sanguins et lymphatiques. Arch Physiol Norm Pathol 5:603–633. [Google Scholar]
- 137. Ruitenberg A, den Heijer T, Bakker SL, van Swieten JC, Koudstaal PJ, Hofman A, Breteler MM (2005) Cerebral hypoperfusion and clinical onset of dementia: the Rotterdam Study. Ann Neurol 57:789–794. [DOI] [PubMed] [Google Scholar]
- 138. Ryu JK, McLarnon JG (2009) A leaky blood‐brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer's disease brain. J Cell Mol Med 13(9A):2911–2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Sagare AP, Bell RD, Zhao Z, Ma Q, Winkler EA, Ramanathan A, Zlokovic BV (2013) Pericyte loss influences Alzheimer‐like neurodegeneration in mice. Nat Commun 4:2932. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 140. Sagare AP, Bell RD, Zlokovic BV (2012) Neurovascular dysfunction and faulty amyloid beta‐peptide clearance in Alzheimer disease. Cold Spring Harb Perspect Med 2:a011452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Sagare AP, Bell RD, Zlokovic BV (2013) Neurovascular defects and faulty amyloid‐beta vascular clearance in Alzheimer's disease. J Alzheimers Dis 33(Suppl. 1):S87–S100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Saint‐Pol J, Vandenhaute E, Boucau MC, Candela P, Dehouck L, Cecchelli R et al (2012) Brain pericytes ABCA1 expression mediates cholesterol efflux but not cellular amyloid‐beta peptide accumulation. J Alzheimers Dis 30:489–503. [DOI] [PubMed] [Google Scholar]
- 143. Salloway S, Gur T, Berzin T, Tavares R, Zipser B, Correia S et al (2002) Effect of APOE genotype on microvascular basement membrane in Alzheimer's disease. J Neurol Sci 203–204:183–187. [DOI] [PubMed] [Google Scholar]
- 144. Saunders WB, Bohnsack BL, Faske JB, Anthis NJ, Bayless KJ, Hirschi KK, Davis GE (2006) Coregulation of vascular tube stabilization by endothelial cell TIMP‐2 and pericyte TIMP‐3. J Cell Biol 175:179–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Schneider JA, Bennett DA (2010) Where vascular meets neurodegenerative disease. Stroke 41(10 Suppl.):S144–S146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Sengillo JD, Winkler EA, Walker CT, Sullivan JS, Johnson M, Zlokovic BV (2013) Deficiency in mural vascular cells coincides with blood‐brain barrier disruption in Alzheimer's disease. Brain Pathol 23:303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Sheline YI, Morris JC, Snyder AZ, Price JL, Yan Z, D'Angelo G et al (2010) APOE4 allele disrupts resting state fMRI connectivity in the absence of amyloid plaques or decreased CSF Abeta42. J Neurosci 30:17035–17040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Shepro D, Morel NM (1993) Pericyte physiology. FASEB J 7:1031–1038. [DOI] [PubMed] [Google Scholar]
- 149. Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B et al (2000) Clearance of Alzheimer's amyloid‐ss(1‐40) peptide from brain by LDL receptor‐related protein‐1 at the blood‐brain barrier. J Clin Invest 106:1489–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Sieczkiewicz GJ, Herman IM (2003) TGF‐beta 1 signaling controls retinal pericyte contractile protein expression. Microvasc Res 66:190–196. [DOI] [PubMed] [Google Scholar]
- 151. Smith CD, Andersen AH, Kryscio RJ, Schmitt FA, Kindy MS, Blonder LX, Avison MJ (1999) Altered brain activation in cognitively intact individuals at high risk for Alzheimer's disease. Neurology 53:1391–1396. [DOI] [PubMed] [Google Scholar]
- 152. Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR (1997) Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA 277:813–817. [PubMed] [Google Scholar]
- 153. Song W, Barth JL, Yu Y, Lu K, Dashti A, Huang Y et al (2005) Effects of oxidized and glycated LDL on gene expression in human retinal capillary pericytes. Invest Ophthalmol Vis Sci 46:2974–2982. [DOI] [PubMed] [Google Scholar]
- 154. Spires TL, Hyman BT (2005) Transgenic models of Alzheimer's disease: learning from animals. NeuroRx 2:423–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Stenzel D, Nye E, Nisancioglu M, Adams RH, Yamaguchi Y, Gerhardt H (2009) Peripheral mural cell recruitment requires cell‐autonomous heparan sulfate. Blood 114:915–924. [DOI] [PubMed] [Google Scholar]
- 156. Stratman AN, Malotte KM, Mahan RD, Davis MJ, Davis GE (2009) Pericyte recruitment during vasculogenic tube assembly stimulates endothelial basement membrane matrix formation. Blood 114:5091–5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Stratman AN, Schwindt AE, Malotte KM, Davis GE (2010) Endothelial‐derived PDGF‐BB and HB‐EGF coordinately regulate pericyte recruitment during vasculogenic tube assembly and stabilization. Blood 116:4720–4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Tagami M, Nara Y, Kubota A, Fujino H, Yamori Y (1990) Ultrastructural changes in cerebral pericytes and astrocytes of stroke‐prone spontaneously hypertensive rats. Stroke 21:1064–1071. [DOI] [PubMed] [Google Scholar]
- 159. Tallquist MD, French WJ, Soriano P (2003) Additive effects of PDGF receptor beta signaling pathways in vascular smooth muscle cell development. PLoS Biol 1:E52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Tanzi RE (2012) The genetics of Alzheimer disease. Cold Spring Harb Perspect Med 2:a006296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Thomas T, Thomas G, McLendon C, Sutton T, Mullan M (1996) beta‐Amyloid‐mediated vasoactivity and vascular endothelial damage. Nature 380:168–171. [DOI] [PubMed] [Google Scholar]
- 162. Thomas WE (1999) Brain macrophages: on the role of pericytes and perivascular cells. Brain Res Brain Res Rev 31:42–57. [DOI] [PubMed] [Google Scholar]
- 163. Toledo JB, Arnold SE, Raible K, Brettschneider J, Xie SX, Grossman M et al (2013) Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer's Coordinating Centre. Brain 136(Pt 9):2697–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Tong XK, Lecrux C, Rosa‐Neto P, Hamel E (2012) Age‐dependent rescue by simvastatin of Alzheimer's disease cerebrovascular and memory deficits. J Neurosci 32:4705–4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. de la Torre JC (2012) Cerebral hemodynamics and vascular risk factors: setting the stage for Alzheimer's disease. J Alzheimers Dis 32:553–567. [DOI] [PubMed] [Google Scholar]
- 166. Tsai PS, Kaufhold JP, Blinder P, Friedman B, Drew PJ, Karten HJ et al (2009) Correlations of neuronal and microvascular densities in murine cortex revealed by direct counting and colocalization of nuclei and vessels. J Neurosci 29:14553–14570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Uh J, Lewis‐Amezcua K, Martin‐Cook K, Cheng Y, Weiner M, Diaz‐Arrastia R et al (2010) Cerebral blood volume in Alzheimer's disease and correlation with tissue structural integrity. Neurobiol Aging 31:2038–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Ujiie M, Dickstein DL, Carlow DA, Jefferies WA (2003) Blood‐brain barrier permeability precedes senile plaque formation in an Alzheimer disease model. Microcirculation 10:463–470. [DOI] [PubMed] [Google Scholar]
- 169. Van Geest RJ, Klaassen I, Vogels IM, Van Noorden CJ, Schlingemann RO (2010) Differential TGF‐{beta} signaling in retinal vascular cells: a role in diabetic retinopathy? Invest Ophthalmol Vis Sci 51:1857–1865. [DOI] [PubMed] [Google Scholar]
- 170. Vermeer SE, Prins ND, den Heijer T, Hofman A, Koudstaal PJ, Breteler MM (2003) Silent brain infarcts and the risk of dementia and cognitive decline. N Engl J Med 348:1215–1222. [DOI] [PubMed] [Google Scholar]
- 171. Vinters HV, Secor DL, Read SL, Frazee JG, Tomiyasu U, Stanley TM et al (1994) Microvasculature in brain biopsy specimens from patients with Alzheimer's disease: an immunohistochemical and ultrastructural study. Ultrastruct Pathol 18:333–348. [DOI] [PubMed] [Google Scholar]
- 172. Virgintino D, Girolamo F, Errede M, Capobianco C, Robertson D, Stallcup WB et al (2007) An intimate interplay between precocious, migrating pericytes and endothelial cells governs human fetal brain angiogenesis. Angiogenesis 10:35–45. [DOI] [PubMed] [Google Scholar]
- 173. Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS et al (2002) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long‐term potentiation in vivo . Nature 416:535–539. [DOI] [PubMed] [Google Scholar]
- 174. Wang Y, Pan L, Moens CB, Appel B (2014) Notch3 establishes brain vascular integrity by regulating pericyte number. Development 141:307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Wilhelmus MM, Otte‐Holler I, van Triel JJ, Veerhuis R, Maat‐Schieman ML, Bu G et al (2007) Lipoprotein receptor‐related protein‐1 mediates amyloid‐beta‐mediated cell death of cerebrovascular cells. Am J Pathol 171:1989–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Winkler EA, Bell RD, Zlokovic BV (2010) Pericyte‐specific expression of PDGF beta receptor in mouse models with normal and deficient PDGF beta receptor signaling. Mol Neurodegener 5:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Winkler EA, Bell RD, Zlokovic BV (2011) Central nervous system pericytes in health and disease. Nat Neurosci 14:1398–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Winkler EA, Sengillo JD, Bell RD, Wang J, Zlokovic BV (2012) Blood‐spinal cord barrier pericyte reductions contribute to increased capillary permeability. J Cereb Blood Flow Metab 32:1841–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Winkler EA, Sengillo JD, Sullivan JS, Henkel JS, Appel SH, Zlokovic BV (2013) Blood‐spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol 125:111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Wisniewski HM, Wegiel J, Wang KC, Lach B (1992) Ultrastructural studies of the cells forming amyloid in the cortical vessel wall in Alzheimer's disease. Acta Neuropathol 84:117–127. [DOI] [PubMed] [Google Scholar]
- 181. Wu EX, Tang H, Asai T, Yan SD (2004) Regional cerebral blood volume reduction in transgenic mutant APP (V717F, K670N/M671L) mice. Neurosci Lett 365:223–227. [DOI] [PubMed] [Google Scholar]
- 182. Wu J, Hua Y, Keep RF, Schallert T, Hoff JT, Xi G (2002) Oxidative brain injury from extravasated erythrocytes after intracerebral hemorrhage. Brain Res 953:45–52. [DOI] [PubMed] [Google Scholar]
- 183. Wu Z, Guo H, Chow N, Sallstrom J, Bell RD, Deane R et al (2005) Role of the MEOX2 homeobox gene in neurovascular dysfunction in Alzheimer disease. Nat Med 11:959–965. [DOI] [PubMed] [Google Scholar]
- 184. Yamada K, Hashimoto T, Yabuki C, Nagae Y, Tachikawa M, Strickland DK et al (2008) The low density lipoprotein receptor‐related protein 1 mediates uptake of amyloid beta peptides in an in vitro model of the blood‐brain barrier cells. J Biol Chem 283:34554–34562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Yamada M, Ihara M, Okamoto Y, Maki T, Washida K, Kitamura A et al (2011) The influence of chronic cerebral hypoperfusion on cognitive function and amyloid beta metabolism in APP overexpressing mice. PLoS ONE 6:e16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A et al (1996) RAGE and amyloid‐beta peptide neurotoxicity in Alzheimer's disease. Nature 382:685–691. [DOI] [PubMed] [Google Scholar]
- 187. Yemisci M, Gursoy‐Ozdemir Y, Vural A, Can A, Topalkara K, Dalkara T (2009) Pericyte contraction induced by oxidative‐nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat Med 15:1031–1037. [DOI] [PubMed] [Google Scholar]
- 188. Yin X, Wright J, Wall T, Grammas P (2010) Brain endothelial cells synthesize neurotoxic thrombin in Alzheimer's disease. Am J Pathol 176:1600–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Zhiyou C, Yong Y, Shanquan S, Jun Z, Liangguo H, Ling Y, Jieying L (2009) Upregulation of BACE1 and beta‐amyloid protein mediated by chronic cerebral hypoperfusion contributes to cognitive impairment and pathogenesis of Alzheimer's disease. Neurochem Res 34:1226–1235. [DOI] [PubMed] [Google Scholar]
- 190. Zhong Z, Deane R, Ali Z, Parisi M, Shapovalov Y, O'Banion MK et al (2008) ALS‐causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat Neurosci 11:420–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Zhong Z, Ilieva H, Hallagan L, Bell R, Singh I, Paquette N et al (2009) Activated protein C therapy slows ALS‐like disease in mice by transcriptionally inhibiting SOD1 in motor neurons and microglia cells. J Clin Invest 119:3437–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Zipser BD, Johanson CE, Gonzalez L, Berzin TM, Tavares R, Hulette CM et al (2007) Microvascular injury and blood‐brain barrier leakage in Alzheimer's disease. Neurobiol Aging 28:977–986. [DOI] [PubMed] [Google Scholar]
- 193. Zlokovic BV (1995) Cerebrovascular permeability to peptides: manipulations of transport systems at the blood‐brain barrier. Pharm Res 12:1395–1406. [DOI] [PubMed] [Google Scholar]
- 194. Zlokovic BV (2005) Neurovascular mechanisms of Alzheimer's neurodegeneration. Trends Neurosci 28:202–208. [DOI] [PubMed] [Google Scholar]
- 195. Zlokovic BV (2008) The blood‐brain barrier in health and chronic neurodegenerative disorders. Neuron 57:178–201. [DOI] [PubMed] [Google Scholar]
- 196. Zlokovic BV (2011) Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci 12:723–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Zlokovic BV (2013) Cerebrovascular effects of apolipoprotein E: implications for Alzheimer disease. JAMA Neurol 70:440–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Zlokovic BV, Begley DJ, Chain‐Eliash DG (1985) Blood‐brain barrier permeability to leucine‐enkephalin, D‐alanine2‐D‐leucine5‐enkephalin and their N‐terminal amino acid (tyrosine). Brain Res 336:125–132. [DOI] [PubMed] [Google Scholar]
- 199. Zlokovic BV, Deane R, Sagare AP, Bell RD, Winkler EA (2010) Low‐density lipoprotein receptor‐related protein‐1: a serial clearance homeostatic mechanism controlling Alzheimer's amyloid beta‐peptide elimination from the brain. J Neurochem 115:1077–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Zlokovic BV, Hyman S, McComb JG, Lipovac MN, Tang G, Davson H (1990) Kinetics of arginine‐vasopressin uptake at the blood‐brain barrier. Biochim Biophys Acta 1025:191–198. [DOI] [PubMed] [Google Scholar]
- 201. Zlokovic BV, Mackic JB, Djuricic B, Davson H (1989) Kinetic analysis of leucine‐enkephalin cellular uptake at the luminal side of the blood‐brain barrier of an in situ perfused guinea‐pig brain. J Neurochem 53:1333–1340. [DOI] [PubMed] [Google Scholar]
- 202. Zlokovic BV, Segal MB, Begley DJ, Davson H, Rakic L (1985) Permeability of the blood‐cerebrospinal fluid and blood‐brain barriers to thyrotropin‐releasing hormone. Brain Res 358:191–199. [DOI] [PubMed] [Google Scholar]