

Background: Cancer cells evade death caused by extracellular matrix (ECM)-detachment to facilitate metastasis.
Results: ErbB2-expressing cancer cells form aggregates during ECM-detachment that promote survival signaling through EGFR.
Conclusion: Multicellular aggregation in ErbB2 positive cancer cells promotes survival by preventing EGFR degradation.
Significance: Disrupting aggregation or inhibiting EGFR may be effective strategies to eliminate ErbB2-expressing cancer cells during ECM-detachment.
Keywords: aggregation, anoikis, breast cancer, epidermal growth factor receptor (EGFR), extracellular matrix, metastasis
Abstract
The metastasis of cancer cells from the site of the primary tumor to distant sites in the body represents the most deadly manifestation of cancer. In order for metastasis to occur, cancer cells need to evade anoikis, which is defined as apoptosis caused by loss of attachment to extracellular matrix (ECM). Signaling from ErbB2 has previously been linked to the evasion of anoikis in breast cancer cells but the precise molecular mechanisms by which ErbB2 blocks anoikis have yet to be unveiled. In this study, we have identified a novel mechanism by which anoikis is inhibited in ErbB2-expressing cells: multicellular aggregation during ECM-detachment. Our data demonstrate that disruption of aggregation in ErbB2-positive cells is sufficient to induce anoikis and that this anoikis inhibition is a result of aggregation-induced stabilization of EGFR and consequent ERK/MAPK survival signaling. Furthermore, these data suggest that ECM-detached ErbB2-expressing cells may be uniquely susceptible to targeted therapy against EGFR and that this sensitivity could be exploited for specific elimination of ECM-detached cancer cells.
Introduction
The overwhelming majority of cancer-related deaths (90%) are a direct result of the metastasis of cancer cells from the primary tumor to distant sites (1–3). Metastasis is an inherently inefficient process as a significant percentage of cells that escape the primary tumor are not successful in colonizing secondary sites. It is currently understood that a significant contributor to this inefficiency is the induction of cell death, particularly in cells that lack attachment to the extracellular matrix (ECM)4 (4). Caspase-dependent programmed cell death that is caused by ECM-detachment is known as anoikis, and anoikis resistance is an important factor in determining the success of cancer cells in navigating the metastatic cascade (5, 6). In addition to anoikis, recent studies have discovered multiple, distinct cellular alterations that can impact the survival of ECM-detached cancer cells in an anoikis-independent fashion, suggesting that cancer cells may need to utilize a multifaceted approach to survive in the absence of ECM-attachment (7–14).
The activation of oncogenic signaling in cancer cells is of paramount importance to developing anoikis resistance and to rectifying other cellular alterations that compromise cell viability in absence of ECM-attachment (9). In particular, overexpression of the ErbB2 oncogene has been linked to the survival of ECM-detached cells in a number of different contexts (12, 13, 15–17). While these studies have unveiled distinct mechanisms by which ErbB2 can promote anchorage-independent survival, it remains unclear if ErbB2 can promote survival through additional molecular mechanisms. Given the tremendous biological heterogeneity in ErbB2-positive breast tumors, it seems likely that the ability of ErbB2 to promote the survival of ECM-detached cells is not solely limited to the aforementioned studies (18).
Among the signaling pathways associated with ErbB2 that have yet to be investigated during the survival of ECM-detached cancer cells are those regulating cell-cell adhesion. Signaling from the ErbB2 receptor has been shown to impinge upon key molecules that determine the nature and efficacy of cell-cell contacts in a number of settings (19). In addition, circulating tumor cells (which lack normal attachment to ECM) have often been discovered as multicellular aggregates. This is particularly true in malignancies like inflammatory breast cancer and epithelial ovarian cancer (20, 21), which are cancers that are oftentimes driven by ErbB2-mediated signaling. Furthermore, the oncogene TrkB, which has been shown to stimulate survival signaling pathways (including those also downstream of ErbB2) in a fashion that blocks anoikis and promotes metastasis (22), can enhance multicellular aggregation during ECM-detachment (23).
These data have motivated us to examine the relationship between ErbB2 and multicellular aggregation during ECM-detached conditions. In this study, we have discovered that ErbB2-induced multicellular aggregation is critical to the inhibition of anoikis. Interestingly, we have found that this multicellular aggregation during ECM-detachment promotes ErbB2/EGFR-mediated activation of ERK/MAPK by preventing EGFR from being internalized and trafficked to the lysosome. Furthermore, our data suggest that ErbB2 and E-cadherin-expressing cancer cells may be uniquely susceptible to therapies antagonizing EGFR activity during ECM-detachment.
EXPERIMENTAL PROCEDURES
Cell Culture
MCF-10A cells (ATCC) and derivatives were cultured in Dulbecco's Modified Eagle Medium/F12 supplemented with 5% horse serum (Invitrogen), 20 ng/ml epidermal growth factor (EGF), 10 μg/ml insulin, 500 μg/ml hydrocortisone, 100 ng/ml cholera toxin, and 1% penicillin/streptomycin. BT474 cells (ATCC), and derivatives were cultured in RPMI-1460 medium supplemented with 10% fetal bovine serum (Invitrogen) and 1% penicillin/streptomycin. SKBR3 cells (ATCC) were cultured in McCoy's 5A medium supplemented with 10% fetal bovine serum (Invitrogen) and 1% penicillin/streptomycin.
Reagents
1% weight to volume (w/v) methylcellulose was made by dissolving methylcellulose in line-specific medium. The following reagents were used at the doses indicated in the figure legends: U0126 (EMD Millipore), chloroquine diphosphate (Sigma-Aldrich), and Z-VAD-FMK (Apex Bio). Plates for detachment assays were made by coating with 6 mg/ml poly(2-hydroxyethyl methacrylate) (poly-HEMA) as previously described (12).
Caspase Activity Assays
Caspase activity was measured after 48 h by the Caspase-Glo 3/7 Assay System according to the manufacturer's instructions (Promega). Cells were plated at a density of 13,333 cells per well in 96-well poly-HEMA-coated plates. Representative data from at least three biological replicates are shown.
Cell Viability Assays
Cellular viability was measured after 48 h using the Cell Titer Glo Assay according to the manufacturer's instructions (Promega). Cells were plated at a density of 13,333 cells per well in 96-well poly-HEMA coated plates. Representative data from at least three biological replicates are shown.
Ethidium Homodimer Assay
Ethidium homodimer (Life Technologies) assays were measured after 72 h with an excitation of 528 nm and an emission of 671 nm. Cells were plated at a density of 13,333 cells per well in 96-well poly-HEMA-coated plates. Representative data from at least three biological replicates are shown.
Western Blot Analysis
Cells were plated at a density of 400,000 cells per well in 6-well poly-HEMA-coated plates. After 48 h, images were acquired, and cells were harvested, washed twice with ice-cold PBS, and then lysed in 1% Nonidet P-40 supplemented with protease inhibitors (leupeptin (5 μg/ml), aprotinin (1 μg/ml), and PMSF (1 mm), and the Halt® Phosphatase Inhibitor Mixture (Thermo Scientific)). Lysates were collected after spinning at 14,000 rpm and normalized by BCA Assay (Pierce Biotechnology). Samples were subjected to polyacrylamide gel electrophoresis (SDS-PAGE) as previously described (10). Representative data from at least three biological replicates are shown.
Measurement of Aggregate Size
Cells were plated at a density of 400,000 cells per well in 6-well poly-HEMA coated plates. After 48 h, images were acquired using a light box and camera and measured using the ImageJ analysis suite Fiji.
Antibodies
The following antibodies were used for Western blotting: Myc (Cell Signaling 2272), α-tubulin (Cell Signaling 2144), β-tubulin (Cell Signaling 2146) Total EGFR (Cell Signaling 4267), pEGFR (Y1173) (Cell Signaling 4407), Total Bad (Cell Signaling 9239), pBad S112 (Cell Signaling 5284), Bim (Cell Signaling 2933), Bcl-2 (Millipore 05–729), Mcl-1 (Millipore MABC43), Bcl-XL (Cell Signaling 2762), β-actin (Sigma A1978), pErk1&2 (pTpY185/187) (Invitrogen 368800), Total Erk1&2 (Cell Signaling 9102), cytochrome c (BD Pharmigen 556433), E-cadherin (AbCam ab40772), and ErbB2 (Dako A0485). The following antibodies were used for immunofluorescence: Total EGFR (Cell Signaling 4267) and LAMP1 (BD Pharmigen 555798). E-cadherin (Invitrogen 135700) was used for E-cadherin engagement and reconstituted according to manufacturer's instructions.
Utilization of Retrovirus to Generate Stable Cell Lines
VSV-psuedotyped retroviruses were produced as previously described (12). MCF-10A cells were plated at 4 × 105 cells and infected with retrovirus. Stable populations of MCF-10A:ErbB2, MCF-10A:MEKDD, and MCF-10A:Bcl-2 were obtained by selection with 2 μg/ml puromycin (Invivogen). Stable populations of MCF-10A:DNECAD cells were obtained by selection with 10 μg/ml blasticidin (24).
Immunoprecipitation
Cells were plated at a density of 400,000 cells per well in 6-well poly-HEMA-coated plates. After 48 h, cells were harvested, washed twice with ice-cold PBS, and lysed in lysis buffer (1% Triton X-100, 50 mm NaCl, 1 mm EDTA, 20 mm HEPES) supplemented with leupeptin (5 μg/ml), aprotinin (1 μg/ml), PMSF (1 mm), and the Halt® Phosphatase Inhibitor Mixture (Thermo Scientific). Lysates were collected following a spin at 14,000 rpm and normalized by BCA Assay (Pierce Biotechnology). Samples were precleared with Protein A-Sepharose Fast Flow beads (GE Healthcare) for 1 h and treated with 1:50 ErbB2 antibody (Dako) for 48 h at 4 °C. Proteins were captured with Protein A-Sepharose Fast Flow beads blocked with 2% BSA (Millipore). Proteins were washed three times with wash buffer (50 mm Tris-HCl pH 7.4, 150 mm NaCl, 1% Nonidet P-40, leupeptin (5 μg/ml), aprotinin (1 μg/ml), PMSF (1 mm), Halt Phosphatase Inhibitor Mixture)), eluted with SDS sample buffer, and analyzed by immunoblot. Representative data from at least three biological replicates are shown.
Cytochrome c Release Assay
Cytosolic cell extracts free of mitochondria were prepared as described previously (25). Briefly, cells were harvested, washed twice in ice-cold PBS, then lysed in lysis buffer (250 mm sucrose, 20 mm HEPES- KOH (pH 7.4), 10 mm KCl, 1.5 mm Na-EGTA, 1.5 mm Na-EDTA, 1 mm MgCl2, 1 mm DTT, the protease inhibitors leupeptin (5 μg/ml), aprotinin (1 μg/ml), Halt® Phosphatase Inhibitor Mixture (Thermo Scientific), and PMSF (1 mm)) by 25 strokes of a glass Dounce homogenizer and tight pestle. Lysates were normalized using a BCA Assay (Pierce Biotechnology) and analyzed as described above by immunoblot. Representative data from at least three biological replicates are shown.
shRNA Transduction
Mission (Sigma-Aldrich) shRNA for E-cadherin cdh1 (NM_004360; TRCN0000039665) was used. The pLKO.4 shRNA viruses were generated by cotransfection of HEK293T cells with the pCMV-D8.9 (0.5 μg), p-CMV-VSV-G (60 ng), and pLKO.4 (0.5 μg) with PLUS™ reagent (Invitrogen). Transfections were carried out using Lipofectamine® 2000 (Invitrogen). Virus was harvested, and cells were infected in the presence of 8 μg/ml of polybrene (Sigma-Aldrich). Cells were subsequently selected with 2 μg/ml puromycin (Invivogen), and knockdown was confirmed by Western blot.
siRNA Transfection
Cells were plated at a density of 400,000 cells per well in 6-well and allowed to grown overnight. A Dharmacon siRNA Smartpool (GE Healthcare) for Bad and ErbB2 was obtained and transfected according to manufacturer's instructions with Oligofectamine® 2000 (Invitrogen). Cells were incubated for 48 h for siErbB2 and 24 h for siBad, collected, and utilized in various assays. Representative data from at least three biological replicates are shown.
Immunofluorescence
Cells were plated at a density of 50,000 cells per well in 6-well poly-HEMA-coated plates in indicated conditions. After 48 h, cells were harvested, washed twice with ice-cold PBS, and deposited onto slides with a Shandon Cytospin3 (Thermo Scientific) at 800 RPM for 5 min. Cells were fixed in 4% paraformaldehyde and permeabilized with 0.5% Triton-X 100 in PBS. Cells were washed with 100 mm glycine in PBS three times and blocked with 10% goat serum (Invitrogen) in IF buffer (130 mm NaCL, 7 mm Na2HPO4, 3.5 mm NaH2PO4, 7.7 mm NaH3, 0.1% BSA (Millipore), 1.2% Triton-X 100, 0.5% Tween-20). Slides were stained with Total EGFR (Cell Signaling 4267) and LAMP1 (BD Pharmingen 555798) diluted 1:200 in IF block buffer. For secondary visualization, slides were treated with AlexaFluor® 488 (Invitrogen A11034) and AlexaFluor® 568 (Invitrogen A11031) at 1:200 in IF block buffer. Nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI) (Invitrogen) and mounted with ProLong® Gold Antifade Reagent. Images were collected on an Applied Precision DeltaVision OMX and colocalization was measured by Pearson's coefficient of correlation using Applied Precision softWoRx® software. Error bars represent S.E. Representative data from at least three biological replicates are shown.
RESULTS
Aggregate Formation upon ECM-detachment in ErbB2-overexpressing Cells Suppresses Anoikis
To assess the contribution of ErbB2 to the formation of cellular aggregates and the induction of anoikis, we utilized MCF-10A cells, a non-tumorigenic mammary epithelial cell line that has been widely used to study the biology of anoikis (9, 12, 26, 27). As has been observed previously (28), ErbB2 expression in MCF-10A cells (10A:ErbB2) is sufficient to abrogate the induction of anoikis when cells are grown in the absence of ECM-attachment for 48 h (Fig. 1A). When examining the morphology of these cells in the absence of ECM-attachment, we discovered that ErbB2-overexpressing cells formed multicellular aggregates that were considerably larger in size than those formed by control cells (Fig. 1B). Furthermore, apoptosis-deficient MCF-10A cells overexpressing Bcl-2 (10A:Bcl-2) did not form large multicellular aggregates suggesting that the striking phenotype observed in Fig. 1B cannot be simply explained by the inhibition of classical cell death mechanisms. Given the aforementioned link between circulating tumor cells and multicellular aggregates (29), we postulated that these aggregates may facilitate the survival of cells that lack proper ECM-attachment. Additionally, ErbB2 overexpression in MCF-10A cells did not result in any discernible changes in EMT markers suggesting that alterations in the EMT program are not involved in the observed changes in multicellular aggregation (Fig. 1C). To address whether multicellular aggregation is critical for survival, we treated ErbB2 overexpressing, ECM-detached cells with methylcellulose, which has been previously demonstrated to interfere with aggregation during ECM-detachment (30) (Fig. 1D). Upon methylcellulose treatment of ECM-detached 10A:ErbB2 cells, we observed an induction of caspase activity (Fig. 1D). In attached 10A:ErbB2 or 10A control cells, we did not observe any methylcellulose-induced caspase activation (Fig. 1D), suggesting that the caspase activation observed in 10A:ErbB2 cells was not the result of general toxicity associated with methylcellulose. To confirm our caspase data, we utilized a Cell Titer Glo viability assay (Fig. 1E) and an ethidium homodimer assay (Fig. 1F) to measure cell death following methylcellulose treatment in ECM-detached 10A:ErbB2 cells. As expected, these data also suggest that methylcellulose treatment induces anoikis in 10A:ErbB2 cells.
FIGURE 1.

Overexpression of ErbB2 results in anoikis suppression through the formation of multicellular aggregates. A, 10A parental (10A:Ctrl) or 10A cells overexpressing ErbB2 (10A:ErbB2) were grown in ECM-attached or -detached conditions for 48 h, and caspase activation was measured. a.u. is defined as fold change over attached. B, 10A:Ctrl, 10A:ErbB2, and 10A cells overexpressing the pro-survival protein Bcl-2 (10A:Bcl-2) were grown in attached and detached conditions for 48 h. Representative images are shown (10×). Average aggregate sizes for 10A:Ctrl and 10A:ErbB2 cells were quantified. C, expression of the indicated EMT markers was assessed via immunoblotting. BT549 cells were used as a positive control for N-cadherin and vimentin. D, 10A:Ctrl and 10A:ErbB2 cells were plated in 1% weight/volume (w/v) MC to disrupt cell-cell contact, and caspase activation was measured after 48 h in ECM-attachment or detachment. E, 10A:ErbB2 cells were plated in MC, and cellular viability (Cell Titer Glo) was measured after 48 h in ECM-detachment. F, 10A:ErbB2 cells were plated in MC, and relative cell death was measured after 48 h in ECM-detachment using an ethidium homodimer assay. a.u. is defined as fold change over control. All error bars represent S.D., and p values were determined using a two-tailed Student's t test.
E-cadherin-mediated Adhesion Underlies Aggregation-induced Anoikis Protection in ErbB2-expressing Cells
To further investigate how cellular aggregation facilitates anoikis evasion in ErbB2-expressing MCF-10A cells, we examined the role of E-cadherin-mediated cell-cell contacts. E-cadherin is typically expressed in epithelial cells where it is involved in cell-cell adhesion, and intracellular signaling pathways that are frequently mediated by mechanotransduction (31). Previous studies have demonstrated that the expression of an E-cadherin protein missing the extracellular domain behaves in a dominant-negative fashion to block cell-cell contacts (32). Therefore, we engineered 10A:ErbB2 cells to stably overexpress Myc-tagged dominant-negative E-cadherin (DNECAD) (Fig. 2A). As expected, the expression of DNECAD in these cells substantially impaired their ability to form large multicellular aggregates when detached from ECM (Fig. 2B). In addition, the aggregate disruption by DNECAD was sufficient to eliminate suppression of anoikis and results in a loss of cellular viability (Fig. 2C), suggesting that E-cadherin is necessary for aggregate-mediated protection from anoikis. The ability of DNECAD to induce anoikis was specific to 10A:ErbB2 cells as DNECAD expression in 10A:Ctrl cells did not reduce viability (Fig. 2D). To further assess the role of E-cadherin in promoting anoikis suppression, we disrupted cellular aggregation while simultaneously maintaining the interactions between E-cadherin molecules that mediate the inhibition of anoikis. To do this, we utilized an E-cadherin antibody that disrupts cell-cell contacts but mimics the engagement of E-cadherin (presumably through antibody-mediated lateral dimerization and clustering of E-cadherin) (33). Upon treatment of ErbB2-expressing cells with an E-cadherin engagement antibody, we did observe a stark inhibition in the formation of multicellular aggregates during ECM-detachment (similar to what we observed with DNECAD expression). This disruption of aggregation, however, does not eliminate ErbB2-mediated anoikis suppression or compromise cellular viability (Fig. 2E). These data further support a role for E-cadherin-mediated adhesion in the inhibition of caspase activation during ECM-detachment. Furthermore, treatment of 10A:CTRL cells with engagement antibody did not result in any changes in cellular viability (Fig. 2F).
FIGURE 2.

E-cadherin engagement is necessary for aggregate formation and caspase suppression. A, 10A:ErbB2 cells were engineered using retroviral transduction to express a Myc-tagged dominant-negative E-cadherin (DNECAD) and confirmed by immunoblotting for Myc. α-Tubulin was used as a loading control. a.u. is defined as fold change over control. B, representative images (10×, left) demonstrating that expression of DNECAD resulted in aggregate disruption upon ECM-detachment. C, caspase activation and cellular viability were measured upon 48 h of ECM-detachment in DNECAD cells. D, cellular viability was measured upon 48 h of ECM-detachment in 10A:Ctrl cells expressing DNECAD. E, 10A:ErbB2 cells were grown in detached conditions and treated with 1 μg/ml E-cadherin engagement antibody (Eng. Ab.) or vehicle (Veh) for 48 h to disrupt aggregation and caspase activity, and cellular viability was measured. Representative image (left) is shown (10×). F, 10A:Ctrl cells were grown in detached conditions and treated with 1 μg/ml E-cadherin engagement antibody (Eng. Ab.) or vehicle (Veh) for 48 h to disrupt aggregation, and cellular viability was measured. a.u. is defined as fold change over control. All error bars represent S.D., and p values were determined using a two-tailed Student's t test.
Aggregate Formation Facilitates Anoikis Evasion in E-cadherin-positive, ErbB2-positive Breast Cancer Cells
To extend our studies into breast cancer cells, we used two common ErbB2-positive cell lines: BT474 and SKBR3. We observed that upon ECM-detachment, BT474 cells form large multicellular aggregates that are subsequently reduced in size upon siRNA-mediated down-regulation of ErbB2 (Fig. 3A). As was observed with 10A:ErbB2 cells, the disruption of aggregation with methylcellulose resulted in a significant loss of anoikis protection in BT474 cells (Fig. 3B). In stark contrast, methylcellulose treatment did not appreciably alter anoikis resistance in SKBR3 cells (Fig. 3C). Interestingly, one significant difference between these two cell lines is that BT474 cells express E-cadherin while SKBR3 cells lack discernible E-cadherin due to a homozygous deletion in the CDH1 gene (34). Thus, perhaps the lack of E-cadherin in SKBR3 cells renders these cells insensitive to agents that disrupt E-cadherin-mediated cell-cell adhesion (e.g. methylcellulose), and these cells rely on alternative mechanisms for anoikis resistance. To examine this hypothesis, we engineered BT474 cells to be deficient in E-cadherin through lentiviral delivery of shRNA (Fig. 3D). This loss of E-cadherin substantially compromised the ability of methylcellulose to induce caspase activation in BT474 cells (Fig. 3E) suggesting that E-cadherin must be present for aggregate disruption to induce anoikis. Furthermore, we observed no perceptible changes in EMT markers following shRNA-mediated loss of E-cadherin in BT474 cells suggesting that alterations in the EMT program do not account for the observed changes sensitivity to methylcellulose-induced anoikis (Fig. 3F).
FIGURE 3.

Aggregate formation promotes anoikis evasion in ErbB2-positive, E-cadherin-positive breast cancer cells. A, siRNA-mediated reduction of ErbB2 in BT474 cells was confirmed by immunoblotting for ErbB2. β-Tubulin was used as a loading control. Aggregate size was quantified in nontargeting control and siErbB2 cells by ImageJ. B, E-cadherin positive, ErbB2-overexpressing BT474 cells were grown in detached conditions and treated with 1% w/v MC, and caspase activation was measured at 48 h. a.u. is defined as fold change over control. C, E-cadherin null, ErbB2-overexpressing SKBR3 cells were grown in detached conditions and treated with MC, and caspase activation was measured at 48 h. a.u. is defined as fold change over control. D and E, BT474 cells were transduced with lentiviral shRNA targeting either pLKO (empty vector) or E-cadherin (sh E-cadherin) and successful knockdown was confirmed by immunoblot for E-cadherin (D). β-Actin is used as a loading control. E, indicated cells were grown in MC, and caspase activation was measured. a.u. is defined as fold change over sh pLKO MC. F, expression of the indicated EMT factors was assessed via immunoblotting. BT549 cells were used as a positive control for N-cadherin and vimentin. All error bars represent S.D., and p values were determined using a two-tailed Student's t test.
Aggregate Formation Promotes ERK-mediated Survival Signaling in ErbB2-positive Cells during ECM-detachment
To examine the molecular mechanism underlying E-cadherin-mediated anoikis evasion, we investigated survival signaling pathways known to operate downstream of ErbB2. Previous studies have revealed that ERK/MAPK signaling can operate downstream of ErbB2 to block anoikis in MCF-10A cells (28). To investigate the activation of ERK following aggregate disruption in ECM-detached cells, we assessed the phosphorylation of ERK by immunoblotting. When multicellular aggregation was disrupted by either methylcellulose treatment or DNECAD expression, there is a significant reduction in phospho-ERK (Fig. 4A). In addition, treatment with the E-cadherin engagement antibody (which disrupts aggregation but mimics E-cadherin engagement, see Fig. 2C) does not alter the phosphorylation of ERK (Fig. 4B). These data suggest that ErbB2-induced aggregation during ECM-detachment can facilitate the activation of ERK to block anoikis. We were able to extend these findings to the aforementioned breast cancer cell lines. Methylcellulose treatment diminished phospho-ERK levels in E-cadherin-positive BT474 cells but did not affect ERK phosphorylation in E-cadherin-negative SKBR3 cells (Fig. 4C). To further ascertain the role of MAPK signaling in anoikis suppression, we expressed a mutant of MEK (MEKDD), which results in constitutive activation of ERK in the absence of any upstream (e.g. ErbB2-mediated) signaling (15). We hypothesized that 10A cells expressing MEKDD would not be sensitive to methylcellulose-mediated induction of anoikis and indeed, methylcellulose treatment of these cells did not promote anoikis (Fig. 4D). Taken together, these data suggest that ERK activation by E-cadherin-mediated aggregation is an important mechanism to block anoikis in ErbB2-expressing cells.
FIGURE 4.
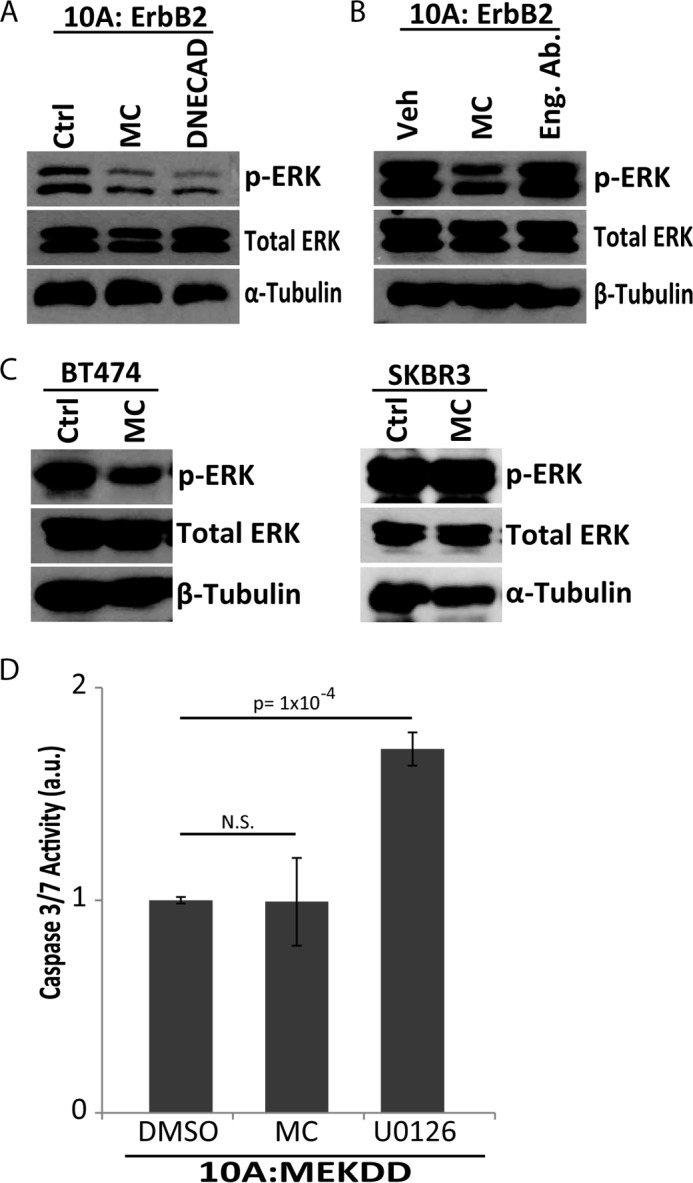
Aggregate formation by E-cadherin engagement promotes ERK/MAPK activation in ECM-detached ErbB2-overexpressing cells. A, phospho-ERK levels were measured in in 10A:ErbB2 Ctrl, 1% w/v MC, and DNECAD cells upon 48 h of ECM-detachment. Total Erk and α-tubulin were used as loading controls. B, 10A:ErbB2 cells were grown in ECM-detachment and treated with 1 μg/ml of E-cadherin engagement antibody (Eng. Ab.) or vehicle control. Activated MAPK levels were measured by immunoblot for phospho-ERK. Total ERK and β-tubulin were used as loading controls. C, activated MAPK levels were measured in BT474 and SKBR3 cells in MC upon 48 h of ECM-detachment by immunoblotting for phospho-ERK. Total ERK, α-tubulin, and β-tubulin were used as loading controls. D, 10A cells overexpressing constitutively active MAPK (10A:MEKDD) were plated in MC or treated with 3 μm U0126 in 48 h of ECM-detachment. a.u. is defined as fold change over DMSO. Caspase activation was measured as described above. All error bars represent S.D., and p values were determined using a two-tailed Student's t test.
Aggregate Formation Stabilizes EGFR, which Maintains ERK Signaling in ErbB2-expressing Cells
To better understand the molecular mechanism underlying the aggregation-induced activation of ERK in ErbB2-expressing cells, we investigated the role of EGFR in ERK activation. It is well known that when ErbB2 is overexpressed, it can heterodimerize with EGFR to stimulate the activation of ERK/MAPK (18). Interestingly, when multicellular aggregates of ECM-detached ErbB2-expressing cells were disrupted with methylcellulose, we observed a substantial reduction in the levels of EGFR protein (Fig. 5A). This reduction in EGFR protein was not limited to ErbB2-expressing MCF-10A cells as total EGFR levels were also diminished upon aggregate disruption in BT474 cells (Fig. 5B). This loss was not observed in SKBR3 cells (which lack endogenous E-cadherin) indicating that E-cadherin is required for aggregation-induced activation of ERK and for protection from anoikis (Fig. 5C). To expand upon these studies, we utilized SKBR3 cells that had been engineered to express E-cadherin and assessed the ability of aggregate disruption to regulate EGFR. Indeed, upon the introduction of E-cadherin into SKBR3 cells, methylcellulose treatment is sufficient to reduce total EGFR levels (Fig. 5D). These data further demonstrate that E-cadherin is necessary for aggregate disruption to induce reduced EGFR levels.
FIGURE 5.

EGFR stabilization by aggregate formation drives MAPK activation in ECM-detached ErbB2-overexpressing cells. A–C, total EGFR levels were measured by immunoblot in 10A:ErbB2 (A), BT-474 (B), or SKBR3 (C) cells grown in 1% w/v MC in 48 h of ECM-detachment. α-Tubulin was used as a loading control. D, SKBR3 cells with either empty vector or E-cadherin expression were treated with methylcellulose, and total EGFR was measured via immunoblot. E-cadherin expression was also confirmed by immunoblot, and α-tubulin was used as a control. E, EGFR targeting to the lysosome was observed using immunofluorescence in 10A:ErbB2 cells in control (n = 31) and MC (n = 32). At 48 h, cells were deposited by cytospin, fixed, and stained for LAMP1 (red) and EGFR (green). Nuclei were stained with DAPI (blue), and all cells were treated with 20 μm ZVAD-FMK to block caspase activation. Images were collected on an Applied Precision DeltaVision OMX, and colocalization was measured by Pearson's coefficient of correlation using Applied Precision softWoRx® software. Error bars represent S.E. F, 10A:ErbB2 cells were grown in MC in the presence of vehicle (Veh) or 12 μm chloroquine diphosphate (CQ), and caspase activation was measured at 48 h in ECM-detachment. a.u. is defined as fold change over control. Total EGFR and phospho-ERK levels were measured by immunoblot. Total ERK and α-tubulin were used as loading controls at 48 h. G, interaction between ErbB2 and E-cadherin was investigated in MC upon 48 h of ECM-detachment by immunoprecipitation of ErbB2 followed by immunoblot of either ErbB2 or E-cadherin. Rabbit IgG was used as a control for the immunoprecipitation. β-Tubulin was used as a loading control for input lysate. A two-tailed Student's t test was used to determine p values.
To further understand the mechanism by which EGFR levels were diminished upon aggregate disruption, we investigated whether aggregate formation could prevent targeting of EGFR for degradation. It is well established that degradation of EGFR is typically controlled by ligand-induced endocytosis, trafficking to the lysosome, and subsequent degradation (35). We found that upon aggregate disruption in ErbB2 overexpressing cells, EGFR localization to the lysosome is dramatically enhanced suggesting that aggregation inhibits EGFR targeting to the lysosome during ECM-detachment (Fig. 5E). To directly address the significance of EGFR targeting to the lysosome in the induction of anoikis by aggregate disruption, we assessed whether methylcellulose-induced anoikis in ErbB2-expressing cells could be rescued through treatment with the lysosomal inhibitor chloroquine. Indeed, chloroquine treatment protected cells from anoikis and rescued total EGFR and phospho-ERK levels in the presence of methylcellulose (Fig. 5F). These data strongly suggest that the formation of E-cadherin based multicellular aggregates prevents EGFR targeting to the lysosome, promotes downstream ERK activation, and inhibits the induction of anoikis. Previous research has demonstrated that EGFR family members (e.g. EGFR and ErbB2) can be regulated by a physical interaction with E-cadherin (36), and thus we hypothesized that E-cadherin binding to EGFR/ErbB2 during ECM-detachment could prevent its internalization and targeting to the lysosome. Interestingly, using immunoprecipitation of endogenous protein, we detected an interaction between ErbB2 and E-cadherin that was lost upon aggregate disruption (Fig. 5G). These data further support a model where E-cadherin-mediated cell-cell contacts promote the stabilization of EGFR at the plasma membrane and the subsequent induction of survival signaling.
Aggregate Disruption during ECM-detachment Results in Bad-mediated Anoikis
To investigate the effector molecules that block anoikis downstream of ErbB2, E-cadherin, and ERK, we assessed the release of cytochrome c from the mitochondrial intermembrane space following aggregate disruption in ECM-detached cells. As expected, aggregate disruption resulted in a significant increase in cytosolic cytochrome c (Fig. 6A), suggesting a role for the Bcl-2 family of proteins in aggregation-induced anoikis protection. Previous studies have linked multiple, distinct members of the Bcl-2 family to ERK-mediated inhibition of anoikis (13, 27, 28, 30, 37–39). Therefore, we looked for differences in the expression of either pro-apoptotic (e.g. Bim) or anti-apoptotic (e.g. Bcl-2, Bcl-XL, Mcl-1) proteins previously linked to anoikis. Interestingly, despite robust cytochrome c release upon aggregate disruption, we did not uncover any changes in the protein levels of these key regulators (Fig. 6B). Other investigations focusing on the regulation of Bcl-2 family members by ERK discovered that ERK can block mitochondrial cytochrome c release through phosphorylation of the pro-apoptotic protein Bad at serine 75 (murine 112) (S112) and subsequent sequestration from the mitochondria on 14-3-3 (26, 27, 40). Upon aggregate disruption, we observed a considerable loss of S112 phosphorylation on Bad and no change in total Bad levels (Fig. 6C). Additionally, aggregate disruption in 10A:MEKDD cells did not diminish Ser-112 phosphorylation on Bad (in line with data presented in Fig. 4D showing that aggregate disruption in these cells does not induce anoikis), while ERK inhibition by U0126 was effective in diminishing Ser-112 phosphorylation (Fig. 6D). Taken together, these data suggest that the disruption of aggregation in ECM-detached ErbB2-expressing cells alters Ser-112 phosphorylation on Bad in a manner that prevents anoikis induction. To confirm the role of Bad in anoikis induced by aggregate disruption, we eliminated Bad expression by siRNA and evaluated the ability of methylcellulose to induce anoikis. The siRNA-mediated reduction of Bad completely abrogated the ability of aggregate disruption to induce anoikis (Fig. 6E), further suggesting that Bad phosphorylation at Ser-112 by ERK is necessary for anoikis protection due to multicellular aggregation and that the reduction of Bad results in a loss of sensitivity to aggregate disruption.
FIGURE 6.
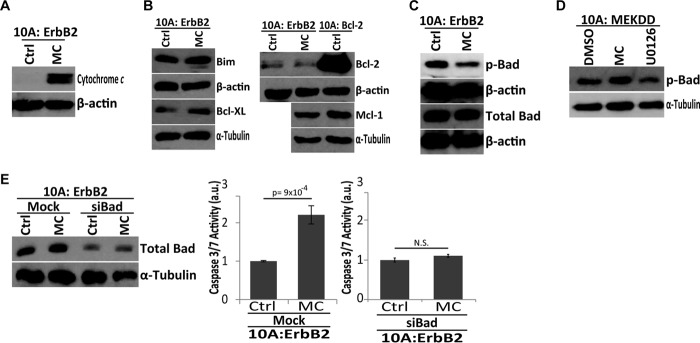
Aggregate disruption in ECM-detachment results in BAD-mediated cytochrome c release. A, cytosolic extracts of 10A:ErbB2 cells in 1% w/v MC were analyzed for cytochrome c and β-actin (load control) by immunoblotting at 48 h in ECM-detachment. B, pro-apoptotic (e.g. Bim) and pro-survival (e.g. Bcl-2, Bcl-XL, and Mcl-1) levels were measured by immunoblot in the presence or absence of MC after 48 h in ECM-detachment. α-Tubulin and β-actin were used as loading controls. C, serine-phosphorylated Bad (Ser-112) and total Bad levels were measured by immunoblot in the presence or absence of MC after 48 h of ECM-detachment. β-Actin was used as a loading control. D, immunoblotting of phospho-Bad Ser-112 was conducted in 10A:MEKDD cells in the presence or absence of MC or treated with DMSO or 3 μm U0126 after 48 h of ECM-detachment. α-Tubulin was used as a load control. E, bad siRNAs were transfected into 10A:ErbB2 cells, and success of transfection was confirmed using immunoblotting for total Bad and α-tubulin (loading control). Caspase activation was measured for the indicated cells plated in MC at 48 h of ECM-detachment. a.u. is defined as fold change over control. All error bars represent S.D., and p values were determined using a two-tailed Student's t test.
ECM-detachment Sensitizes ErbB2-overexpressing Cells to EGFR Therapy
Given that our data demonstrate a role for EGFR in ERK activation and anoikis inhibition (due to Ser-112 phosphorylation on Bad), we hypothesized that ECM-detached, ErbB2-expressing cells may be uniquely susceptible to targeted cancer therapies designed to inhibit EGFR. Interestingly, previous studies of anti-EGFR based therapies have found these therapies to be poorly effective at eliminating ErbB2-positive cancer cells; primarily due to sustenance of ERK signaling (41). However, many of these studies were completed in ECM-attached cells and thus we reasoned that targeted therapies aimed at EGFR may be specifically detrimental to ECM-detached cells that rely on aggregation-induced stabilization of EGFR for anoikis inhibition. Indeed, upon treatment of ErbB2-overexpressing cells with AG1478 (Fig. 7A) or gefitinib (Fig. 7B) (both EGFR inhibitors), caspase activation is induced in a dose-dependent fashion specifically in ECM-detached cells while caspase activation is not observed in attached cells. Immunoblotting for phospho-EGFR confirms the efficacy of these inhibitors in both conditions. These data suggest that aggregation-induced protection from anoikis in ErbB2-positive cancer cells may be a viable target for anti-EGFR-targeted therapies.
FIGURE 7.

ECM-detachment sensitizes ErbB2-overexpressing cells to EGFR therapy. A, 10:ErbB2 cells were treated with indicated concentrations of AG1478, and caspase activation was measured at 48 h of ECM-attachment or detachment (top). EGFR inhibition was confirmed by immunoblot for phospho-EGFR (Y1173) and β-actin (loading control) (bottom). a.u. is defined as fold change over DMSO. B, 10:ErbB2 cells were treated with indicated concentrations of gefitinib, and caspase activation was measured at 48 h in ECM-attachment or detachment (top). EGFR inhibition was confirmed by immunoblot for phospho-EGFR (Y1173) and β-actin (loading control) (bottom). a.u. is defined as fold change over DMSO. All error bars represent S.D., and p values were determined using a two-tailed Student's t test.
DISCUSSION
The evasion of anoikis by cancer cells is of paramount importance to their ability to successfully metastasize to distant sites. A better understanding of the molecular mechanisms and signal transduction pathways underlying anoikis evasion in cancer cells could potentially lead to the development of novel therapeutics that eliminate ECM-detached cancer cells. Here, we demonstrate that ErbB2-expressing breast cancer cells form large, multicellular aggregates when detached from the ECM and that these aggregates play a critical role in anoikis evasion (see model in Fig. 8). While this study focuses specifically on aggregate formation during ECM-detachment in ErbB2-positive cells, our data raise the possibility that signaling that resonates from other oncogenes or in other cancers may also promote aggregate formation and anoikis evasion during ECM-detachment. Indeed, previous research in squamous cell carcinoma cells characterized a process known as “synoikis” whereby cell-cell contacts facilitate survival through the elevation of Bcl-2 protein (42). That being said, evidence seems to suggest that the mechanism described in our study is specific to ErbB2-expressing cells. For example, in Fig. 4D, we show that cells engineered to have constitutive activation of ERK (via the expression of MEKDD) do not induce anoikis upon disruption of aggregation, suggesting that when ERK signaling is sustained by means other than ErbB2 overexpression, multicellular aggregation is not protective against anoikis. Given that our study suggests that stabilization of EGFR (potentially due to complex formation with ErbB2 and E-cadherin) is necessary for aggregation-induced anoikis evasion, it might be the case that ErbB2 expression is necessary for aggregation to be cytoprotective during ECM-detachment in breast cancer cells. Alternatively, other types of oncogenic signaling may stabilize EGFR through other mechanisms.
FIGURE 8.

Model for aggregate-mediated EGFR stabilization and anoikis suppression in ECM-detached ErbB2-overexpressing cells. A, model for aggregate-mediated ECM-detached survival in ErbB2-overexpressing cells. B, model for anoikis induction upon aggregate disruption in ECM-detached ErbB2-overexpressing cells.
The results presented here in our study also clearly delineate a role for E-cadherin-mediated cell-cell adhesion in protection from anoikis in ErbB2-expressing cells. One aspect of these data that we find particularly intriguing is that multiple types of cancer cells are well known to lose E-cadherin during an epithelial-mesenchymal transition (EMT), which facilitates migration and metastasis (43). In fact, EMT has itself been directly linked to the evasion of anoikis through eloquent studies involving the protein NRAGE (44). Therefore, how do we reconcile the data from this study implicating E-cadherin in anoikis protection with the fact that E-cadherin is often lost during tumor progression? Recent studies have revealed that while EMT does often occur during the early stages of metastasis, a re-acquisition of E-cadherin through a mesenchymal to epithelial transition (MET) can be critical for successful metastasis (45–47). These data raise the possibility that an MET (following an EMT) could facilitate aggregation-induced anoikis evasion at later stages of the metastatic cascade when disseminated cancer cells need to survive in foreign matrix environments. Furthermore, other studies have unequivocally demonstrated the presence of E-cadherin in a percentage of both primary breast tumors and distant metastases (48) and a correlation between cellular aggregation and successful metastasis was first published over 50 years ago (49, 50). More recent studies examining circulating tumor cells (CTCs) have provided evidence that these cells often exist in the bloodstream as multicellular aggregates (29, 51, 52). Our data may help explain the significance of these findings in that aggregation may occur in CTCs and may facilitate metastasis through the inhibition of anoikis.
In addition to our data demonstrating a role for multicellular aggregation in blocking anoikis, our study has also uncovered a unique vulnerability of ErbB2-expressing cells: the inhibition of EGFR during ECM-detachment (see Fig. 7). Numerous studies have demonstrated that EGFR inhibition is not an effective strategy for patients with ErbB2-positive breast cancer (41, 53). Our data suggest that while EGFR inhibition is not effective in ECM-attached cells, ECM-detached cells are sensitive to EGFR inhibitors. We postulate that this sensitivity is due to the reliance of ECM-detached cells on aggregation/E-cadherin-induced activation of EGFR for sustained ERK signaling. Indeed, in other tumors that rely heavily on EGFR signaling (e.g. NSCLC), E-cadherin expression can enhance the sensitivity of cancer cells to EGFR inhibitors (54). Therefore, it seems possible that EGFR inhibition may be helpful in eliminating ECM-detached, ErbB2-positive breast cancer cells, while alternative approaches to antagonize ERK signaling may be necessary to eliminate ErbB2-positive, ECM-attached cells.
Acknowledgments
We thank Cassandra Buchheit, Kelsey Weigel, Joshua Mason, Amy Leliaert, Kelsea Hosoda, Veronica Schafer, and the rest of the Schafer laboratory for helpful comments and discussion. We thank Lisa Checkley and Michael Ferdig at Notre Dame for providing chloroquine. Additionally, we thank Mary Ann McDowell at Notre Dame for the use of the Shandon cytospin3 and Michael Overholtzer at Memorial Sloan-Kettering Cancer Center for the E-cadherin-positive SKBR3 cells. We thank Kimbra Turner at the University of Notre Dame for help with figure design, layout, and formatting.
This work was supported by the Glynn Family Honors Program and the Notre Dame NSF-REU program in biology (to N. P.).
- ECM
- extracellular matrix
- DAPI
- 4,6-diamidino-2-phenylindole
- DNECAD
- dominant-negative E-cadherin
- EMT
- epithelial-mesenchymal transition
- MET
- mesenchymal to epithelial transition
- CTC
- circulating tumor cell
- MC
- methylcellulose
- EGFR
- epidermal growth factor receptor.
REFERENCES
- 1. Nguyen D. X., Massagué J. (2007) Genetic determinants of cancer metastasis. Nat. Rev. Genet 8, 341–352 [DOI] [PubMed] [Google Scholar]
- 2. Hanahan D., Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 3. Wan L., Pantel K., Kang Y. (2013) Tumor metastasis: moving new biological insights into the clinic. Nat. Med. 19, 1450–1464 [DOI] [PubMed] [Google Scholar]
- 4. Mehlen P., Puisieux A. (2006) Metastasis: a question of life or death. Nat. Rev. Cancer 6, 449–458 [DOI] [PubMed] [Google Scholar]
- 5. Frisch S. M., Francis H. (1994) Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell Biol. 124, 619–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Frisch S. M., Screaton R. A. (2001) Anoikis mechanisms. Curr. Opin. Cell Biol. 13, 555–562 [DOI] [PubMed] [Google Scholar]
- 7. Avivar-Valderas A., Bobrovnikova-Marjon E., Alan Diehl J., Bardeesy N., Debnath J., Aguirre-Ghiso J. A. (2013) Regulation of autophagy during ECM detachment is linked to a selective inhibition of mTORC1 by PERK. Oncogene. 32, 4932–4940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Avivar-Valderas A., Salas E., Bobrovnikova-Marjon E., Diehl J. A., Nagi C., Debnath J., Aguirre-Ghiso J. A. (2011) PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Mol. Cell Biol. 31, 3616–3629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Buchheit C. L., Rayavarapu R. R., Schafer Z. T. (2012) The regulation of cancer cell death and metabolism by extracellular matrix attachment. Semin Cell Dev. Biol. 23, 402–411 [DOI] [PubMed] [Google Scholar]
- 10. Davison C. A., Durbin S. M., Thau M. R., Zellmer V. R., Chapman S. E., Diener J., Wathen C., Leevy W. M., Schafer Z. T. (2013) Antioxidant enzymes mediate survival of breast cancer cells deprived of extracellular matrix. Cancer Res. 73, 3704–3715 [DOI] [PubMed] [Google Scholar]
- 11. Fung C., Lock R., Gao S., Salas E., Debnath J. (2008) Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol. Biol. Cell 19, 797–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schafer Z. T., Grassian A. R., Song L., Jiang Z., Gerhart-Hines Z., Irie H. Y., Gao S., Puigserver P., Brugge J. S. (2009) Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 461, 109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Whelan K. A., Caldwell S. A., Shahriari K. S., Jackson S. R., Franchetti L. D., Johannes G. J., Reginato M. J. (2010) Hypoxia suppression of Bim and Bmf blocks anoikis and luminal clearing during mammary morphogenesis. Mol. Biol. Cell 21, 3829–3837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Buchheit C. L., Weigel K. J., Schafer Z. T. (2014) Cancer cell survival during detachment from the ECM: multiple barriers to tumour progression. Nat. Rev. Cancer 14, 632–641 [DOI] [PubMed] [Google Scholar]
- 15. Grassian A. R., Schafer Z. T., Brugge J. S. (2011) ErbB2 stabilizes epidermal growth factor receptor (EGFR) expression via Erk and Sprouty2 in extracellular matrix-detached cells. J. Biol. Chem. 286, 79–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Haenssen K. K., Caldwell S. A., Shahriari K. S., Jackson S. R., Whelan K. A., Klein-Szanto A. J., Reginato M. J. (2010) ErbB2 requires integrin α5 for anoikis resistance via Src regulation of receptor activity in human mammary epithelial cells. J. Cell Sci. 123, 1373–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Buchheit C. L., Angarola B. L., Steiner A., Weigel K. J., Schafer Z. T. (2014) Anoikis evasion in inflammatory breast cancer cells is mediated by Bim-EL sequestration. Cell Death Differ December 19 doi: 10.1038/cdd.2014.1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Arteaga C. L., Engelman J. A. (2014) ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 25, 282–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Higgins M. J., Baselga J. (2011) Targeted therapies for breast cancer. J. Clin. Invest. 121, 3797–3803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Moss N. M., Barbolina M. V., Liu Y., Sun L., Munshi H. G., Stack M. S. (2009) Ovarian cancer cell detachment and multicellular aggregate formation are regulated by membrane type 1 matrix metalloproteinase: a potential role in I.p. metastatic dissemination. Cancer Res. 69, 7121–7129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Robertson F. M., Bondy M., Yang W., Yamauchi H., Wiggins S., Kamrudin S., Krishnamurthy S., Le-Petross H., Bidaut L., Player A. N., Barsky S. H., Woodward W. A., Buchholz T., Lucci A., Ueno N. T., Cristofanilli M. (2010) Inflammatory breast cancer: the disease, the biology, the treatment. CA Cancer J. Clin. 60, 351–375 [DOI] [PubMed] [Google Scholar]
- 22. Douma S., Van Laar T., Zevenhoven J., Meuwissen R., Van Garderen E., Peeper D. S. (2004) Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB. Nature 430, 1034–1039 [DOI] [PubMed] [Google Scholar]
- 23. Geiger T. R., Peeper D. S. (2007) Critical role for TrkB kinase function in anoikis suppression, tumorigenesis, and metastasis. Cancer Res. 67, 6221–6229 [DOI] [PubMed] [Google Scholar]
- 24. Onder T. T., Gupta P. B., Mani S. A., Yang J., Lander E. S., Weinberg R. A. (2008) Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 68, 3645–3654 [DOI] [PubMed] [Google Scholar]
- 25. Bossy-Wetzel E., Green D. R. (2000) Assays for cytochrome c release from mitochondria during apoptosis. Methods Enzymol. 322, 235–242 [DOI] [PubMed] [Google Scholar]
- 26. Adachi M., Zhang Y. B., Imai K. (2003) Mutation of BAD within the BH3 domain impairs its phosphorylation-mediated regulation. FEBS Lett. 551, 147–152 [DOI] [PubMed] [Google Scholar]
- 27. Wang H. G., Pathan N., Ethell I. M., Krajewski S., Yamaguchi Y., Shibasaki F., McKeon F., Bobo T., Franke T. F., Reed J. C. (1999) Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science 284, 339–343 [DOI] [PubMed] [Google Scholar]
- 28. Reginato M. J., Mills K. R., Becker E. B., Lynch D. K., Bonni A., Muthuswamy S. K., Brugge J. S. (2005) Bim regulation of lumen formation in cultured mammary epithelial acini is targeted by oncogenes. Mol. Cell Biol. 25, 4591–4601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stott S. L., Hsu C. H., Tsukrov D. I., Yu M., Miyamoto D. T., Waltman B. A., Rothenberg S. M., Shah A. M., Smas M. E., Korir G. K., Floyd F. P., Jr., Gilman A. J., Lord J. B., Winokur D., Springer S., Irimia D., Nagrath S., Sequist L. V., Lee R. J., Isselbacher K. J., Maheswaran S., Haber D. A., Toner M. (2010) Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc. Natl. Acad. Sci. U.S.A. 107, 18392–18397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Reginato M. J., Mills K. R., Paulus J. K., Lynch D. K., Sgroi D. C., Debnath J., Muthuswamy S. K., Brugge J. S. (2003) Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nat. Cell Biol. 5, 733–740 [DOI] [PubMed] [Google Scholar]
- 31. van Roy F. (2014) Beyond E-cadherin: roles of other cadherin superfamily members in cancer. Nat. Rev. Cancer 14, 121–134 [DOI] [PubMed] [Google Scholar]
- 32. Perl A. K., Wilgenbus P., Dahl U., Semb H., Christofori G. (1998) A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature 392, 190–193 [DOI] [PubMed] [Google Scholar]
- 33. Pece S., Chiariello M., Murga C., Gutkind J. S. (1999) Activation of the protein kinase Akt/PKB by the formation of E-cadherin-mediated cell-cell junctions. Evidence for the association of phosphatidylinositol 3-kinase with the E-cadherin adhesion complex. J. Biol. Chem. 274, 19347–19351 [DOI] [PubMed] [Google Scholar]
- 34. Pierceall W. E., Woodard A. S., Morrow J. S., Rimm D., Fearon E. R. (1995) Frequent alterations in E-cadherin and α- and β-catenin expression in human breast cancer cell lines. Oncogene. 11, 1319–1326 [PubMed] [Google Scholar]
- 35. Goh L. K., Sorkin A. (2013) Endocytosis of receptor tyrosine kinases. Cold Spring Harb. Perspect Biol. 5, a017459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bremm A., Walch A., Fuchs M., Mages J., Duyster J., Keller G., Hermannstädter C., Becker K. F., Rauser S., Langer R., von Weyhern C. H., Höfler H., Luber B. (2008) Enhanced activation of epidermal growth factor receptor caused by tumor-derived E-cadherin mutations. Cancer Res. 68, 707–714 [DOI] [PubMed] [Google Scholar]
- 37. Schmelzle T., Mailleux A. A., Overholtzer M., Carroll J. S., Solimini N. L., Lightcap E. S., Veiby O. P., Brugge J. S. (2007) Functional role and oncogene-regulated expression of the BH3-only factor Bmf in mammary epithelial anoikis and morphogenesis. Proc. Natl. Acad. Sci. U.S.A. 104, 3787–3792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Whelan K. A., Schwab L. P., Karakashev S. V., Franchetti L., Johannes G. J., Seagroves T. N., Reginato M. J. (2013) The oncogene HER2/neu (ERBB2) requires the hypoxia-inducible factor HIF-1 for mammary tumor growth and anoikis resistance. J. Biol. Chem. 288, 15865–15877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Weigel K. J., Jakimenko A., Conti B. A., Chapman S. E., Kaliney W. J., Leevy W. M., Champion M. M., Schafer Z. T. (2014) CAF-Secreted IGFBPs Regulate Breast Cancer Cell Anoikis. Mol. Cancer Res. 12, 855–866 [DOI] [PubMed] [Google Scholar]
- 40. She Q. B., Solit D. B., Ye Q., O'Reilly K. E., Lobo J., Rosen N. (2005) The BAD protein integrates survival signaling by EGFR/MAPK and PI3K/Akt kinase pathways in PTEN-deficient tumor cells. Cancer Cell 8, 287–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yonesaka K., Zejnullahu K., Okamoto I., Satoh T., Cappuzzo F., Souglakos J., Ercan D., Rogers A., Roncalli M., Takeda M., Fujisaka Y., Philips J., Shimizu T., Maenishi O., Cho Y., Sun J., Destro A., Taira K., Takeda K., Okabe T., Swanson J., Itoh H., Takada M., Lifshits E., Okuno K., Engelman J. A., Shivdasani R. A., Nishio K., Fukuoka M., Varella-Garcia M., Nakagawa K., Jänne P. A. (2011) Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci. Transl. Med. 3, 99ra86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shen X., Kramer R. H. (2004) Adhesion-mediated squamous cell carcinoma survival through ligand-independent activation of epidermal growth factor receptor. Am. J. Pathol. 165, 1315–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lamouille S., Xu J., Derynck R. (2014) Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 15, 178–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kumar S., Park S. H., Cieply B., Schupp J., Killiam E., Zhang F., Rimm D. L., Frisch S. M. (2011) A pathway for the control of anoikis sensitivity by E-cadherin and epithelial-to-mesenchymal transition. Mol. Cell Biol. 31, 4036–4051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ocaña O. H., Córcoles R., Fabra A., Moreno-Bueno G., Acloque H., Vega S., Barrallo-Gimeno A., Cano A., Nieto M. A. (2012) Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell 22, 709–724 [DOI] [PubMed] [Google Scholar]
- 46. Tsai J. H., Donaher J. L., Murphy D. A., Chau S., Yang J. (2012) Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 22, 725–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chao Y. L., Shepard C. R., Wells A. (2010) Breast carcinoma cells re-express E-cadherin during mesenchymal to epithelial reverting transition. Mol. Cancer 9, 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kowalski P. J., Rubin M. A., Kleer C. G. (2003) E-cadherin expression in primary carcinomas of the breast and its distant metastases. Breast Cancer Res. 5, R217–R222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fidler I. J. (1973) The relationship of embolic homogeneity, number, size and viability to the incidence of experimental metastasis. Eur. J. Cancer 9, 223–227 [DOI] [PubMed] [Google Scholar]
- 50. Watanabe S. (1954) The metastasizability of tumor cells. Cancer 7, 215–223 [DOI] [PubMed] [Google Scholar]
- 51. Fernandez S. V., Robertson F. M., Pei J., Aburto-Chumpitaz L., Mu Z., Chu K., Alpaugh R. K., Huang Y., Cao Y., Ye Z., Cai K. Q., Boley K. M., Klein-Szanto A. J., Devarajan K., Addya S., Cristofanilli M. (2013) Inflammatory breast cancer (IBC): clues for targeted therapies. Breast Cancer Res. Treat 140, 23–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yu M., Stott S., Toner M., Maheswaran S., Haber D. A. (2011) Circulating tumor cells: approaches to isolation and characterization. J. Cell Biol. 192, 373–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang S. E., Narasanna A., Perez-Torres M., Xiang B., Wu F. Y., Yang S., Carpenter G., Gazdar A. F., Muthuswamy S. K., Arteaga C. L. (2006) HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell 10, 25–38 [DOI] [PubMed] [Google Scholar]
- 54. Witta S. E., Gemmill R. M., Hirsch F. R., Coldren C. D., Hedman K., Ravdel L., Helfrich B., Dziadziuszko R., Chan D. C., Sugita M., Chan Z., Baron A., Franklin W., Drabkin H. A., Girard L., Gazdar A. F., Minna J. D., Bunn P. A., Jr. (2006) Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res. 66, 944–950 [DOI] [PubMed] [Google Scholar]