

Background: TLR4 is an important receptor, promoting both protective immunity and inflammatory diseases.
Results: A revised model of TLR4 signaling is proposed, based upon new observations.
Conclusion: TLR4 forms an autoinhibitory complex in the absence of ligand; disrupting this complex induces downstream signaling.
Significance: This new mechanism may explain TLR4 responsiveness to diverse ligands and guide the design of novel therapeutics.
Keywords: Immunology, Receptor Regulation, Receptor Structure-Function, Signal Transduction, Toll-like Receptor 4 (TLR4)
Abstract
Toll-like receptors (TLRs) mediate immune recognition of both microbial infections and tissue damage. Aberrant TLR signaling promotes disease; thus, understanding the regulation of TLR signaling is of medical relevance. Although downstream mediators of TLR signaling have been identified, the detailed mechanism by which ligand binding-mediated dimerization induces downstream signaling remains poorly understood. Here, we investigate this question for TLR4, which mediates responsiveness to bacterial LPS and drives inflammatory disease. TLR4 exhibits structural and functional features that are unique among TLRs, including responsiveness to a wide variety of ligands. However, the connection between these structural features and the regulation of signaling is not clear. Here, we investigated how the unique intracellular structures of TLR4 contribute to receptor signaling. Key conclusions include the following. 1) The unique intracellular linker of TLR4 is important for achieving LPS-inducible signaling via Toll/IL-1 receptor (TIR) domain-containing adapter-inducing interferon-β (TRIF) but less so for signaling via myeloid differentiation primary response 88 (MyD88). 2) Membrane-bound TLR4 TIR domains were sufficient to induce signaling. However, introducing long, flexible intracellular linkers neither induced constitutive signaling nor ablated LPS-inducible signaling. Thus, the initiation of TLR4 signaling is regulated by a mechanism that does not require tight geometric constraints. Together, these observations necessitate refining the model of TLR4 signal initiation. We hypothesize that TLR4 may interact with an inhibitory partner in the absence of ligand, via both TIR and extracellular domains of TLR4. In this speculative model, ligand binding induces dissociation of the inhibitory partner, triggering spontaneous, switchlike TIR domain homodimerization to initiate downstream signaling.
Introduction
Toll-like receptors (TLRs)3 play a central role in immunological defense by activating the innate arm of the immune system in response to ligands, including both damage-associated and pathogen-associated molecular patterns (1–3). Humans express 10 distinct TLR proteins, each of which contains an extracellular ligand-binding domain, a single transmembrane domain, and an intracellular Toll/IL-1 receptor (TIR) domain. The canonical model of TLR biosensing is that ligand binding to the ectodomains (ECDs) induces receptor dimerization, which promotes dimerization of TIR domains to generate a scaffold that recruits downstream signaling mediators (4).
TLR signaling is tightly regulated, and aberrant TLR signaling promotes a range of pathologies, ranging from acute threats like septic shock to chronic disease, including autoimmunity and cancer (5–7). Consequently, modulation of TLR signaling is a promising therapeutic strategy (8, 9). However, despite substantial progress in the identification of intracellular mediators of TLR signaling (10) and the structural solution of several TLR-ligand complexes (11), the mechanisms by which the initiation of signaling is regulated in the absence of ligand and promoted upon formation of a signaling complex remain poorly understood (4). A better understanding of how this initial induction of TLR signaling is regulated could help to explain the roles of TLRs in pathological and beneficial processes and suggest novel strategies for therapeutically modulating TLR signaling.
This study focuses on elucidating the mechanisms by which TLR4 signal induction is regulated, because this TLR is of particular medical relevance and, in many ways, is unique among this protein family. TLR4 induces potent inflammation in response to lipid A, the core component of bacterial LPS. Although this response provides a first line of defense against bacterial incursion, excessive TLR4 signaling promotes harmful inflammation and, in its extreme, septic shock (3, 6–8). TLR4 is also the only TLR that requires an adaptor protein for ligand recognition, myeloid differentiation factor 2 (MD2) (12). MD2 binds to the ECD of TLR4 during protein biosynthesis and facilitates surface localization (although MD2 is not strictly required for surface localization (13)), such that this heterodimer comprises the ligand-free cell surface receptor (14). When the hydrophobic core of MD2 binds lipid A, MD2 exposes an interface that binds a second TLR4 ECD to promote the formation of a heterotetrameric TLR4·MD2 signaling complex, which is stabilized by protein-protein interactions between TLR4 ECDs (15). Although such structural characterizations provide insights into the mechanisms by which ligand binding induces the formation of a TLR4 signaling complex, how TLR4 signaling is regulated in the absence of lipid A remains much more poorly understood.
Both functional and structural evidence suggests that TLR4 signaling may be regulated via mechanisms that are unique among this receptor family. Unlike other TLRs, TLR4 responds to a wide range of non-canonical ligands, including nickel (16), cobalt (17), fibrinogen (18), high mobility group box 1 (19, 20), respiratory syncytial virus (21), and the envelope protein of the vesicular stomatitis virus (22). Why TLR4 seems particularly susceptible to activation by non-canonical ligands is not known. A potentially relevant structural insight is that TLR4 TIR domains exhibit a propensity to homodimerize, probably to a larger degree than do TIR domains from other TLRs (23). Although TLR4 TIR dimers have not been crystallized, a recent computational analysis predicted that an energetically favorable conformation of such a dimer would place membrane-proximal sequences on the same interface and enable adapter protein docking, suggesting that spontaneous TIR dimerization could induce signaling (24). The TLR4 ECD appears to play a role in preventing constitutive activation because replacing the ECD of TLR4 with a smaller monomeric protein, such as mCherry or the N-terminal fragment of Escherichia coli gyrase B, results in constitutive signaling (25). Replacing the TLR4 ECD with another large protein (i.e. ovalbumin) prevented constitutive activation, suggesting that steric occlusion is sufficient to prevent TLR4 signaling. However, it is not clear that steric occlusion is the dominant mechanism by which the TLR4 ECD prevents constitutive signaling because even TLR4 ECDs with substantial N-terminal truncations were sufficient to prevent constitutive signaling. A final structural idiosyncrasy is that TLR4 is the only TLR in which the linker between the transmembrane domain and TIR domain is hydrophobic. This hydrophobic linker has been hypothesized to contribute to oligomerization of TLR4 (observed via immunoblotting) because other TLRs do not appear to oligomerize in a similar fashion (26). Although deletion of this hydrophobic linker ablates TLR4 signaling, the role of this feature in regulating TLR4 signaling in the presence or absence of ligand is not clear. In sum, although features that contribute to the promotion or regulation of TLR4 signaling have been identified, we currently lack an overall model explaining how this ensemble of features collectively governs TLR4 signaling.
In this study, we sought to investigate how the structural features that distinguish TLR4 from other TLRs contribute to the signaling properties of this receptor. Together, our results indicate that the unique intracellular linker of TLR4 is not required to initiate LPS-inducible signaling, although it does contribute to signaling in a pathway-specific manner. Moreover, although membrane-bound TIR domains were sufficient to induce signaling, constitutive signaling was suppressed in the absence of ligand by a mechanism that did not require tight geometric constraints, which argues against a mechanism reliant upon steric occlusion. To integrate our observations and the existing literature, we hypothesize that TLR4 signaling may be regulated by a switchlike mechanism involving an inhibitory partner.
EXPERIMENTAL PROCEDURES
Plasmid Construction
mTLR4 was isolated from cDNA of RAW264.7 cell line using PCR and inserted into the pcDNA 3.1(−) backbone. mCherry fusion proteins used mCherry from pmCherry-C1 (Clontech) (27). The pMD2 expression plasmid was constructed from pDUO-MD2/CD14 (InvivoGen) by removing the CD14-coding sequence. The pIFN-βluc and pSV-βgal plasmids were gifts from David Segal (28). pHR-SFFV-dCas9-BFP-KRAB and pMLM3636 were acquired from Addgene (catalog nos. 46911 and 43860, respectively) (29). Expression constructs for small guide RNA (sgRNA) specific for TMED7 (TMED7-sgRNA2 and TMED7-sgRNA3) were constructed by inserting annealed, overlapping oligonucleotides into pMLM36363. A human TMED7 cDNA plasmid was acquired from the DNASU Plasmid Repository (pLX304-TMED7). pTMED7-Delta was generated by modifying TMED7 from pLX304-TMED7 by PCR as described (30) and inserting this construct into pcDNA 3.1(−). Primer sequences are listed in supplemental Table S1.
Cell Culture and Transfection
Cells were maintained at 37 °C in 5% CO2 in Dulbecco's modified growth medium supplemented with 10% fetal bovine serum (FBS), 1% penicillin-streptomycin, and 4 mm l-glutamine. HEK293FT cells (Life Technologies, Inc.) were used for all immunohistochemistry. Unless stated otherwise, transfections were performed in biological triplicate in 12-well plates seeded with 3 × 105 cells in 1.5 ml of medium 12 h before the transfection. 1.25 μg of DNA/well was transfected using the CaCl2-HEPES-buffered saline method.
TLR4 Signaling Assays
HEK293 cells stably transduced with an NF-κB promoter driving expression of GFP were used to measure NF-κB activity (31). Each well was transfected with 0.25 μg of pDsRed-Express2 transfection control (Clontech), 0.025 μg of pMD2, 0.875 μg of pcDNA 3.1(−), and 0.1 μg of pTLR4 (or variant) or 0.1 μg of pcDNA 3.1(−) for mock transfections. 16 h post-transfection, medium was changed, and 100 ng/ml LPS from E. coli K12 (InvivoGen) was added to culture medium, cells were incubated for 24 h, and GFP fluorescence was analyzed by flow cytometry.
For TMED7 overexpression and TMED7-Delta experiments, each well was transfected with 0.1 μg of pLX304-TMED7 or pTMED7-Delta, 0.25 μg of pDsRed-Express2 transfection control (Clontech), 0.025 μg of pMD2, 0.775 μg of pcDNA 3.1(−), and 0.1 μg of pTLR4 (or variant) or 0.1 μg of pcDNA 3.1(−) for mock transfections. For TMED7 knockdown experiments, each well was transfected with 0.35 μg of pHR-SFFV-dCas9-BFP-KRAB, 0.175 μg of TMED7-sgRNA2, 0.175 μg of TMED7-sgRNA3, 0.25 μg of pDsRed-Express2 transfection control (Clontech), 0.025 μg of pMD2, 0.175 μg of pcDNA 3.1(−), and 0.1 μg of pTLR4 (or variant) or 0.1 μg of pcDNA 3.1(−) for mock transfections. All other steps were performed as described above.
For LPS-induced TRIF signaling assays, each well was transfected with 0.25 μg of pDsRed-Express2 transfection control (Clontech), 0.025 μg of pMD2, 0.25 μg of pSV-βgal, 0.25 μg of pIFN-βluc, 0.875 μg of pcDNA 3.1(−), and 0.1 μg of pTLR4 (or variant) or 0.1 μg of pcDNA 3.1(−) for mock transfections. 24 h post-transfection, medium was changed, and 100 ng/ml LPS from E. coli K12 (InvivoGen) was added to the culture medium. Cells were incubated for 6 h, washed with PBS, and lysed using 200 μl of Glo lysis buffer (Promega). For luciferase assays, 25 μl of cell lysate was mixed with 25 μl of ONE-GloTM luciferase assay substrate (Promega). Luminescence was read using a BioTek Synergy H1 plate reader. For β-galactosidase assays, 50 μl of cell lysate was incubated with 50 μl of β-galactosidase 2× assay buffer (Promega) for 30 min. 150 μl of 1 m Tris base was added to stop the reaction, and absorbance measurements at 420 nm were recorded using a BioTek Synergy H1 plate reader.
Immunolabeling
HEK293FT cells were transfected with 0.25 μg of pcDNA-BFP as a transfection control, 0.5 μg of pMD2, and 0.5 μg of pTLR4 (variant) or mock-transfected with empty pcDNA3.1(−). Cells were harvested 36 h post-transfection using PBS with 2 mm EDTA. For intracellular staining, cells were then fixed in 1% paraformaldehyde at room temperature for 15 min and permeabilized with 0.5% saponin. 5 × 105 cells were blocked with 10 μg of human IgG and subsequently incubated with 0.1 μg of PE·Cy7 anti-mouse TLR4 (CD284)·MD2 complex (Biolegend), which cross-reacts with the human antigens. Cells were resuspended in PBS with 5% bovine serum albumin (BSA) and 2 mm EDTA prior to analysis by flow cytometry.
For labeling mCherry-TLR4 fusion constructs, 7.5 × 105 HEK293FT cells were transfected with 0.5 μg of pcDNA-BFP as a transfection control, 1 μg of pcDNA3.1(−), and 1 μg of mCherry-TLR4 (or variant; each with N-terminal FLAG tags) or pcDNA3.1(−) for mock transfections, in 6-well plates. 5 × 105 cells were incubated with 0.5 μg of SureLight® allophycocyanin anti-FLAG (AbCam).
Flow Cytometry
2 × 104 live cells from each transfected well were characterized using an LSRII flow cytometer (BD Biosciences) running FACSDiva software. Data were electronically compensated and analyzed using FlowJo software (Tree Star). Live single cells were gated based on scatter, and transfected cells were gated by expression of the appropriate fluorescent transfection control. For LPS-induced signaling experiments, reporter activity was quantified and normalized with respect to cells transfected with wild-type (WT) pTLR4 and treated with LPS.
RESULTS
Intracellular Linker Properties Required for LPS-inducible Signaling
We first investigated how the unique intracellular linker (ICL) of TLR4 may play a role in receptor function. In TLR4, the linker between the TMD and TIR domain is hydrophobic, which is a feature unique among TLR ICLs, and removal of this sequence ablates LPS-mediated induction of TLR4 signaling (26). However, the mechanism by which this linker enables TLR4 function remains unclear. We first investigated whether LPS-inducible signaling requires the native linker sequence, for example by mediating sequence-specific interactions between hydrophobic linkers on multiple receptors or between the hydrophobic linker and another component of the receptor-adaptor protein complex. The hydrophobic linker sequence was reversed (TLR4-H1) or scrambled in two different ways (TLR4-H2 and TLR4-H3) (Fig. 1A), such that the overall composition of the linker was maintained. TLR4-H1 and TLR4-H2 were both expressed on the cell surface and signaled inducibly (Fig. 1, B and C). Whereas the maximal magnitude of NF-κB reporter activation was lower than that conferred by the wild-type receptor (TLR4-WT), the -fold increase upon the addition of LPS was similar to that observed for WT TLR4 (Fig. 1C). The lower magnitude of NF-κB reporter activation could be due to reduced localization of the modified TLRs to the cell surface. LPS-induced TLR4 signaling via myeloid differentiation primary response 88 (MyD88), which eventually drives translocation of NF-κB into the nucleus, must initiate at the cell surface (32). In contrast, TLR4-H3 was not LPS-inducible, and its surface expression was weaker than that of either the -H1 or -H2 variants (Fig. 1, B and C). The observed LPS-inducible signaling of TLR4-H1 and TLR4-H2 indicates that the necessary function mediated by the ICL is not highly specific to linker sequence.
FIGURE 1.

Contributions of TLR4 hydrophobic linker sequence to receptor signaling. A, above is a schematic of TLR4 modifications. Light gray, WT TLR4; dark gray, portion of TLR4 being modified. Below is a comparison of the amino acid sequence of the ICL of WT TLR4 compared with a reversed (TLR4-H1) or scrambled (TLR4-H2 and TLR4-H3) linker. B, surface and intracellular expression of modified TLR4s (gray-filled histogram, mock-transfected). PE-Cy7 was evaluated after gating on transfected cells. C, NF-κB reporter activation by TLR4 and variants following activation by LPS. Mean fluorescence intensity (MFI) of GFP was measured in biological triplicate for each sample after gating on transfected cells. Measurements were normalized relative to WT TLR4 + LPS. Error bars, S.D.
To examine whether the ICL must be hydrophobic to confer LPS-inducible signaling, the WT linker was replaced with linkers of other compositions. Linkers characterized by flexible glycines (TLR4-G), positively charged residues (TLR4-P), negatively charged residues (TLR4-N), or polar uncharged residues (TLR4-U) were each evaluated (Fig. 2A). TLR4-G and TLR4-P were expressed on the cell surface and exhibited LPS-inducible signaling (Fig. 2, B and C). TLR4-U was not inducible, despite being expressed on the surface of the cell (Fig. 2, B and C). TLR4-N was expressed intracellularly but not on the cell surface and did not demonstrate LPS-inducible signaling (Fig. 2, B and C). Given that only some of our hydrophobic ICL variants were competent for LPS-inducible signaling (Fig. 1B), we cannot exclude the possibility that other TLR4 variants with polar uncharged or negatively charged ICL sequences would also be competent for LPS-inducible signaling. Most importantly, the inducibility of TLR4-G and TLR4-P demonstrates that the TLR4 ICL need not be highly hydrophobic to confer LPS-inducible signaling.
FIGURE 2.

Contributions of TLR4 ICL composition to receptor signaling. A, above is a schematic of TLR4 modifications. Light gray, WT TLR4; dark gray, portion of TLR4 being modified. Below is a comparison of the amino acid sequence of the ICL of WT TLR4 with a flexible glycine (TLR4-G), positively changed (TLR4-P), polar uncharged (TLR4-U), or negatively charged (TLR4-N) linker. B, surface and intracellular expression of modified TLR4s (gray-filled histogram, mock-transfected). PE-Cy7 was evaluated after gating on transfected cells. C, NF-κB reporter activation by TLR4 and variants following activation by LPS. MFI of GFP was measured in biological triplicate for each sample after gating on transfected cells, and measurements were normalized relative to WT TLR4 + LPS. Error bars, S.D.
We next investigated how linker length, in addition to linker composition, may affect TLR4 signaling. TLR4 constructs were engineered in which the hydrophobic linker was removed (TLR4-L0) or replaced by flexible linkers with lengths of 6 amino acids (TLR4-L6), 11 amino acids (TLR4-L11), 23 amino acids (TLR4-L23), and 48 amino acids (TLR4-L48) (Fig. 3A). Both ligand-inducible and ligand-independent signaling were evaluated (Fig. 3, B and C). Consistent with the results of Nishiya et al. (26), TLR4-L0 did not exhibit LPS-inducible signaling despite being expressed on the surface of the cell (Fig. 3, B and D). However, TLR4-L6, TLR4-L11, TLR4-L23, and TLR4-L48 all exhibited LPS-inducible signaling (Fig. 3, B and C). TLR4-L6 signaled as well as variants with longer linkers, and it is possible that the minimal linker length required for signaling competency is shorter than the shortest (6-amino acid) linker length characterized here. Each of these TLR4 constructs was also expressed on the cell surface (Fig. 3D). Again, the magnitude of LPS-induced NF-κB reporter activation was lower for the modified TLR4s compared with wild-type TLR4, but the -fold induction of NF-κB was equal to or higher than that conferred by WT TLR4 (Fig. 3, B and C). Thus, even very long linkers between the transmembrane and TIR domains of TLR4 maintain inducibility and do not result in constitutive activation, despite the reported propensity of TLR4 TIR domains to homodimerize (23).
FIGURE 3.

Contributions of TLR4 ICL length to receptor signaling. A, at the left is a schematic of TLR4 modifications. Light gray, WT TLR4; dark gray, portion of TLR4 being modified. At the right is a comparison of the amino acid sequence of the ICL of WT TLR4 with no ICL (TLR4-L0) or flexible linkers with lengths of 6 amino acids (TLR4-L6), 11 amino acids (TLR4-L11), 25 amino acids (TLR4-L25), and 48 amino acids (TLR4-L48). B and C, NF-κB reporter activation by TLR4 and variants following activation by LPS. MFI of GFP was measured in biological triplicate for each sample after gating on transfected cells, and measurements were normalized relative to WT TLR4 + LPS. Error bars, S.D. D, surface and intracellular expression of modified TLR4s (gray-filled histogram, mock-transfected). PE-Cy7 was evaluated after gating on transfected cells.
Replacement of the TLR4 Transmembrane Domain Ablates Ligand-inducible Signaling
There exists some evidence that TLR TMDs oligomerize even in the absence of other TLR sequences (33, 34). We hypothesized that TLR4 function may require a TLR4-derived (or TLR-derived) TMD with this propensity for oligomerization. To investigate this question, TLR4 constructs were generated in which the TLR4 TMD was replaced with that of two cell surface proteins, IL-10 receptor α-chain (IL-10Rα) (TLR4-TMa) or cluster of differentiation 28 (CD28) (TLR4-TM28) (Fig. 4A). Non-TLR-derived TMD sequences were selected to avoid potential substitution of TLR4 TMD functions by other TLR TMDs. We previously incorporated the CD28 TMD into engineered receptor proteins that were monomeric and expressed at the cell surface (35). Replacement of the TLR4 TMD in TLR4-TMa and TLR4-TM28 prevented trafficking of these constructs to the cell surface but did not ablate overall receptor expression (Fig. 4B). The TMDs of intracellular TLRs mediate receptor localization via interaction with proteins such as UNC93B1 (36), but to our knowledge, no specific TMD interaction partner has been identified as necessary for surface localization of TLR4 (13). Transmembrane Emp24 protein transport domain-containing protein 7 (TMED7) mediates transport of TLR4 from the endoplasmic reticulum to the Golgi (30, 37), and transport of TLR4 from the Golgi to the cell surface involves Rab10 (38). Thus, our TMD modifications could impair one or more of these interactions. TLR4-TMa exhibited a low level of ligand-independent signaling, which was dependent on overexpression of MD2 (Fig. 4C). Unlike TLR4-TMa, TLR4-TM28 did not exhibit ligand-independent signaling. To control for artifacts due to the manner in which fusion proteins were engineered, we also evaluated a construct in which the sequence derived from CD28 was lengthened to include 4–5 additional flanking residues derived from CD28 on each side of the TMD (TLR4-TM28L) (Fig. 4A). Whereas this modification neither modulated overall expression nor restored surface expression (Fig. 4B), MD2-dependent signaling was observed for TLR4-TM28L, but this signaling was not LPS-inducible (Fig. 4C). We note that in some experiments (including data presented in subsequent figures), TLR4-TM28L generated background signaling more comparable with that of WT TLR4, yet TLR4-TM28 signaling was consistently non-inducible by LPS. To further investigate the contributions of TMD-proximal sequences added in TLR4-TM28L, these CD28-derived flanking sequences were inserted into WT TLR4 next to the TMD on the extracellular side (TLR4-CD28flank_ex), intracellular side (TLR4-CD28flank_in), or both sides (TLR4-CD28flank_both) (Fig. 4D). Overall, these substitutions increased background signaling and reduced but did not eliminate LPS-mediated increases in signaling. Together, these data suggest a model wherein TMD substitutions shift TLR4 localization to subcellular compartments that are incompetent for signaling (TLR4-TM28, TLR4-TMa) or that are competent for signaling but preclude interactions with LPS (TLR4-TM28L).
FIGURE 4.

Contributions of TLR4 transmembrane domain to receptor signaling. A, at the left is a schematic of TLR4 modifications. Light gray, WT TLR4; dark gray, portion of TLR4 being modified. At the right is a comparison of the amino acid sequence of the TMD of WT TLR4 with the TMD of IL-10Rα (TLR4-TMa), CD28 (TLR4-TM28), or CD28 with the flanking charged residues (TLR4-TM28L). B, surface and intracellular expression of modified TLR4s (gray-filled histogram, mock-transfected). PE-Cy7 was evaluated after gating on transfected cells. C and D, NF-κB reporter activation by TLR4 and variants following activation by LPS. In D, all wells were transfected with MD2. MFI of GFP was measured in biological triplicate for each sample after gating on transfected cells, and measurements were normalized relative to WT TLR4 + LPS. Error bars, represent S.D. *, p < 0.05; **, p < 0.01 (two-tailed Student's t test).
The TIR Domain of TLR4 Is Sufficient to Induce Constitutive Signaling
Because TLR4-TMa, TLR4-TM28, and TLR4-TM28L did not localize to the cell surface, we could not use these constructs to evaluate whether the TMD of TLR4 is required for LPS-inducible signaling. However, because TLR4 signals constitutively when the ECD is replaced with mCherry (25), which is much smaller than the TLR4 ECD, we used this model to evaluate whether the TMD of TLR4 (and its potential to oligomerize (33, 34)) is required for constitutive signaling. Constructs were engineered, replacing the ECD of TLR4 with mCherry and including either the TMD of TLR4 (M-4) or the TMD of CD28 (M-28) (Fig. 5A). Consistent with the observations of Panter et al. (25), M-4 exhibited constitutive signaling (Fig. 5B). Replacement of the TMD to generate M-28 did not eliminate constitutive signaling (Fig. 5B), demonstrating that the TMD of TLR4 is not required for constitutive signaling.
FIGURE 5.

Contributions of the transmembrane domain and ICL to constitutive the signaling by TLR4 variants lacking an inhibitory ectodomain. A, schematic representation of mCherry-TLR4 fusion proteins. Gray, WT TLR4, black ECD, mCherry; light gray TMD with black outline, CD28 TMD; light gray ICL, an 11-amino acid glycine linker; dark gray ICL, a polar uncharged linker. B and C, NF-κB reporter activation by mCherry-TLR4 fusion proteins compared with WT TLR4 (WT TLR4-expressing cells were treated with LPS, but mCherry fusion protein-expression cells were not treated with LPS). MFI of GFP was measured in biological triplicate for each sample after gating on transfected cells, and measurements were normalized relative to WT TLR4 + LPS. Error bars, S.D. D, surface expression of mCherry-TLR4 fusion proteins (gray-filled histogram, mock-transfected). PE was evaluated after gating on transfected cells.
To determine whether the hydrophobic ICL and/or TIR domains are required for the constitutive activation observed when the ECD of TLR4 is replaced with mCherry, we constructed other mCherry-TLR4 fusions proteins in which the hydrophobic linker was removed (M-4-X and M-28-X) or, alternatively, replacing the hydrophobic linker with a flexible glycine linker of the same length (M-4-G and M-28-G) (Fig. 5A). When the hydrophobic linker was simply removed, constitutive signaling was ablated for both M-4-X and M-28-X (Fig. 5, B and C). Replacing the hydrophobic linker with a flexible glycine linker, however, maintained constitutive signaling for M-4-G and M-28-G. Although TLR4-U exhibited neither background nor LPS-inducible signaling (Fig. 2C), replacing the ICL of M-4 with the polar linker of TLR4-U did not prevent constitutive activation (Fig. 5, A and B). Each of these mCherry fusion proteins was robustly expressed at the cell surface (Fig. 5D), including TMD/ICL combinations whose surface localization was impaired when fused to the TLR4 ECD (Fig. 4B). Together, these results again suggest that a linker is required to initiate signaling (via recruitment of MyD88 and Mal (MyD88 adapter-like), which also include TIR domains). However, the specific sequence and hydrophobic nature of the WT linker may not be required. Thus, the only TLR4-derived domain required to initiate signaling from the plasma membrane is the TIR domain, and in the absence of a TLR4 ECD, this signaling is constitutive.
TLR4 Intracellular Domains Impact Signaling in a Pathway-specific Fashion
TLR4 signaling is unique among TLRs, in that TLR4 signals via both MyD88 and TRIF (TIR domain-containing adapter-inducing interferon-β) (4). Ligand binding first initiates signaling via MyD88 at or near the cell surface, and subsequently, TRIF signaling is initiated from early endosomal compartments (32, 37, 39). To investigate whether our TLR4 variants differentially impact MyD88- and TRIF-mediated signaling, we evaluated LPS-induced activation of an interferon-β reporter (28) for several key constructs (Fig. 6A). Whereas LPS-induced TRIF signaling via WT TLR4 was comparable with that in prior reports (30), TRIF signaling via all other constructs was reduced or ablated. Notably, only TLR4-H1 (whose hydrophobic ICL differs from that of WT TLR4 in sequence but not composition) retained LPS-inducible TRIF signaling. In contrast, receptors with glycine-rich ICL that had exhibited LPS-inducible MyD88 signaling (TLR4-G, -L6, and -L48) did not confer LPS-inducible TRIF signaling. Finally, constructs that failed to exhibit LPS-inducible MyD88 signaling (TLR4-L0, -TM28, -TM28L) also failed to exhibit LPS-inducible TRIF signaling.
FIGURE 6.
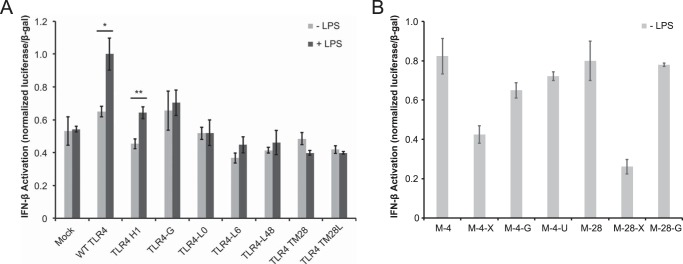
Pathway-specific signaling by TLR4 variants. A and B, TRIF reporter activation by TLR4 variants. Luminescence data were normalized by β-galactosidase signal, and these ratios were normalized to that of the wild-type (WT) TLR4 sample treated with LPS. Error bars, S.D.; *, p < 0.05; **, p < 0.01 (two-tailed Student's t test).
When constructs lacking the TLR4 ECD were similarly evaluated, we observed a striking similarity between the patterns of constitutive (LPS-independent) signaling via MyD88 (Fig. 5, B and C) and TRIF (Fig. 6, B and C). These data indicate that ablating the inhibition mediated by the TLR4 ECD (25) is sufficient to drive both MyD88- and TRIF-mediated signaling, the latter of which was not previously evaluated. Notably, replacement of the ICL with an equivalently sized glycine linker conferred constitutive signaling via both pathways when the ECD comprised mCherry (M-4-G), but when the TLR4 ECD was retained (TLR4-G), LPS induced signaling via MyD88 but not TRIF. Overall, these observations suggest that the hydrophobicity (but not strictly the sequence) of the ICL may be essential for conferring LPS-induced TRIF-mediated signaling. However, this ICL feature is not required for LPS-induced initiation of MyD88 signaling. Because mCherry constructs signaled comparably via both MyD88 and TRIF pathways, it is possible that these receptors localize (constitutively) to compartments competent for initiating both signaling pathways, potentially bypassing the requirement for interactions mediated by the hydrophobic ICL.
Negative Regulators of TLR4 Do Not Contribute to Differential Signaling of Intracellular Domain Variants
Given that our receptor variants exhibited differential LPS-inducible signaling efficiencies, we hypothesized that negative regulators of TLR4 might differentially act upon these variants (40). Given the sites of variation between our constructs, plausible negative regulators include RP105 and TMED7. RP105 (also called CD180) is a TLR4 homolog that is co-expressed with TLR4 and inhibits TLR4 signaling via direct physical interactions mediated by receptor ECDs (41, 42). TMED7 is a transmembrane adaptor protein that has been implicated in multiple aspects of TLR4 signaling (30, 43). TMED7 appears to be required for trafficking of TLR4 from the endoplasmic reticulum to the Golgi via COPII-coated vesicles, and release of TLR4 from TMED7 in the Golgi precedes trafficking of TLR4 to the cell surface (30, 37). TMED7 may also associate with TLR4 after stimulation with LPS to attenuate LPS-induced signaling (43). Thus, we hypothesized that TLR4 variants exhibiting impaired background and/or LPS-inducible signaling (i.e. TLR4-TMa, -TM28, and -TM28L) may be impaired in their association or disassociation from these regulators.
To investigate whether RP105 or TMED7 may differentially regulate our TLR4 variants, we first evaluated expression of regulator genes in our HEK293 cells (Fig. 7A). While basal TMED7 expression was robust, consistent with previous reports (30), RP105 mRNA was not detected. Thus, RP105 cannot explain our observations, and we focused on TMED7 in subsequent work. We perturbed TMED7 in three ways: overexpression, knockdown, and expression of a TMED7 truncation mutant (TMED7-Delta) (Fig. 7, B–D). TMED7-Delta lacks the cytoplasmic C-terminal domain and localizes diffusely throughout the cell, and co-expression of this mutant with TLR4 has been reported to increase ligand-independent signaling via both MyD88 and TRIF (30). Knockdown of TMED7 was implemented using a CRISPR-based system, in which a modified Cas9 protein directs transcriptional silencing of a specific locus targeted by a small guide RNA (29). Analysis of TMED7 mRNA in mock samples from Fig. 7, B and C, confirmed that overexpression and knockdown of TMED7 were both substantial (Fig. 7E); TMED7 overexpression and knockdown in transfected cells is expected to be of even greater magnitude than the population-averaged values depicted in Fig. 7E (see figure legend).
FIGURE 7.

Modulation of TLR4 variant signaling by potential negative regulators. A, evaluation of basal gene expression of RP105 and TMED7 in HEK293 cells. B–D, NF-κB reporter activation by TLR4 variants under overexpression of TMED7 (B), knockdown of TMED7 (C), or co-expression with TMED7-Delta truncation mutant (D). E, changes in TMED7 gene expression for mock samples in B and C. In these experiments, transfection efficiency was uniformly ∼10%, as measured by flow cytometric detection of the fluorescent protein transfection control; thus, TMED7 overexpression and knockdown in transfected cells is expected to be of greater magnitude than the population-averaged values depicted in E. Error bars, S.D.; ND, not detected (below the lowest value on the standard curve and not distinguishable from negative control); *, p < 0.05; **, p < 0.01 (two-tailed Student's t test).
Overall, TMED7 overexpression minimally altered signaling in TLR4 variants, except to slightly enhance background signaling and diminish LPS-induced signaling for WT TLR4 (Fig. 7B). This pattern is consistent with previous reports (note that our NF-κB assays integrate reporter output over 24 h) (30, 43). TMED7 knockdown substantially diminished LPS-induced signaling for WT TLR4 (Fig. 7C), which is consistent with a model in which TMED7 is required for TLR4 to traffic to LPS-inducible compartments. Surprisingly, expression of the TMED7-Delta mutant diminished LPS-independent signaling for both WT TLR4 and TLR4-TMa, and LPS-induced signaling was diminished for WT TLR4 as well (Fig. 7D). These results differ superficially from those reported previously (30), although we note that our NF-κB assay spanned 24 h, whereas Liaunardy-Jopeace et al. (30) examined NF-κB-mediated reporter expression 6 h after stimulation with LPS. Altogether, these data are most consistent with a model whereby TMED7 is required for trafficking of TLR4 to signaling-competent compartments, as expected, but regulation by TMED7 cannot explain why some TLR4 variants (e.g. TLR4-TM28L) were not induced by LPS. Thus, these data bolster the explanation posed above; the TMD substitutions investigated here impair trafficking of TLR4 to the cell surface, and thus LPS-inducible signaling is also ablated.
DISCUSSION
Altogether, this investigation helps to clarify the role played by the unique ICL and TIR domains of TLR4. Collectively, our results also indicate that the current model of TLR4 signaling requires refinement.
First, our analysis provides new insights into the role of the unique TLR4 ICL bridging the TMD and TIR domains. In TLR4, this linker is hydrophobic, whereas it is hydrophilic in all other TLRs, and the functional role of this property was not known (26). As previously reported by Nishiya et al. (26), deletion of the hydrophobic linker prevented LPS-inducible signaling (Fig. 3), probably by sterically occluding TIR dimerization or interactions with downstream signaling mediators, because all TLRs have a linker of some sort between the TMD and TIR domain. However, we found that by replacing the WT ICL with linkers of identical composition but altered sequence (Fig. 1) or with comparably sized linkers composed either entirely of glycine or of nonstructured residues, including positively charged amino acids (Fig. 2), LPS-inducible signaling via MyD88/NF-κB was largely retained. However, glycine ICL substitutions (of various lengths) that retained LPS-inducible MyD88 signaling proved to ablate LPS-inducible signaling via TRIF (Figs. 2, 3, and 6). Interestingly, one construct in which the ICL sequence was reversed (preserving composition) retained LPS-inducible signaling via TRIF, although the magnitude of reporter activation was diminished (Fig. 6).
Overall, these observations suggest that the hydrophobicity (but not strictly the sequence) of the ICL may be essential for conferring LPS-induced TRIF-mediated signaling, although hydrophobicity is not required for LPS-induced initiation of MyD88 signaling. One possible explanation is that the hydrophobic ICL facilitates the unique TLR4 mechanism by which LPS-mediated activation initiates at or near the cell surface, followed by translocation of the TLR4 signaling complex to endosomal compartments that are competent for initiation of signaling via TRIF (32, 37). Because TLR4 variants lacking the inhibitory ECD (25) signaled comparably via both MyD88 and TRIF pathways (Figs. 5 and 6), it is possible that these receptor variants localize (constitutively) to compartments competent for initiating both signaling pathways. Thus, differences in signaling rates between these variants might be ascribed to differential efficiencies with which TLR4 TIR domains dimerize and recruit adapter molecules Mal and TRAM (TRIF-related adapter molecule) to form complexes with MyD88 and TRIF, respectively.
Our analysis of TLR4 ICL variants also revealed particularly interesting insights into how constitutive signaling may be suppressed. First, we demonstrated that membrane-bound TLR4 TIR domains (lacking all other TLR4 sequences) are sufficient to initiate constitutive signaling via MyD88 (Fig. 5), as predicted via computational analysis (24) but to our knowledge not previously observed. Second, we observed that the TLR4 variant including a 48-amino acid-long ICL (TLR4-L48) did not exhibit elevated signaling in the absence of ligand, and indeed signaling was LPS-inducible (Fig. 3). This finding is important because it cannot be explained by the current model in which the TLR4 ECD suppresses constitutive signaling via steric occlusion (25). When fully extended (within the constraints of the peptide backbone), this 48-residue linker may span ∼168 Å, nearly twice the distance required to bridge the widest part of the TLR4 ECD (15). Therefore, if TIR dimerization is suppressed only by steric occlusion between TLR4 ECDs, receptors bearing such linkers would be expected to enable TIR domain dimerization and signaling in the absence of ligand. Moreover, Panter et al. (25) discovered that removal of up to 542 N-terminal amino acids (LRR1–LRR20), which results in an ECD of only 10 kDa (comprising LRR21, LRR22, and LRRCT), was sufficient to prevent constitutive activation of the TLR4 receptor. However, a slightly smaller construct in which the ECD comprised LRR22 and LRRCT (TLR4 Δ563) exhibited constitutive signaling. A final observation that challenges the steric occlusion model is that inserting linkers between the TMD and ECD domain of TLR4 decreased LPS-inducible signaling but did not lead to constitutive signaling (44).
Therefore, we hypothesize that the role of the TLR4 ECD in preventing signaling is more than steric occlusion mediated by its large size, and it is likely that the C-terminal region of the TLR4 ECD (potentially a region in or near LRR21) mediates the inhibition of constitutive signaling via a distinct mechanism. Moreover, our TLR4-L48 construct was LPS-inducible, which does not support a mechanism by which LPS-induced receptor dimerization or conformational rearrangement of the ECDs leads to an intracellular conformational change that simply alleviates steric inhibition of TIR dimerization. Thus, a distinct mechanism is necessary to explain how ligand binding is transduced into signal induction for this relatively “floppy” receptor variant. Altogether, these observations indicate the need for a revised model of TLR4 signaling.
To integrate both our observations and those previously reported, we propose a revised, albeit speculative, model of switchlike TLR4 signaling (Fig. 8). We hypothesize that in the absence of ligand, TLR4 is retained in a signaling-incompetent conformation via interactions between TLR4 and one or more additional, as yet unidentified species. We hypothesize that this TLR4-inhibitory complex (TIC) binds to TLR4 via interactions involving the C-terminal region of the TLR4 ECD (putatively within the region spanned by LRR21, LRR22, and LRRCT), the TIR domain, and one or more interaction partners, which may be proteins (potentially including known adapter proteins) or other molecules. In this model, TIC-bound TIR domains are held in a signaling-incompetent conformation, and upon LPS binding to the ECDs to induce dimerization and/or rearrangement, interactions with TIC are disrupted, enabling the TIR domains to spontaneously dimerize and initiate downstream signaling. For example, TIC may retain the TIR domains in a membrane-proximal location, which prevents signaling (Fig. 3), or TIC may otherwise preclude interactions between TIR domains and adapter proteins. Notably, the TLR4 TIR domain is predicted to possess a flat but slightly curved membrane proximal surface, which is common in proteins that interact with the membrane (24). The proposed inhibitory interactions could occur in cis, wherein the ECD and TIR domain of one TLR4 protein are engaged with a single TIC, or in trans, where the ECD and TIR domains of different TLR4 proteins interact via TIC (Fig. 8).
FIGURE 8.
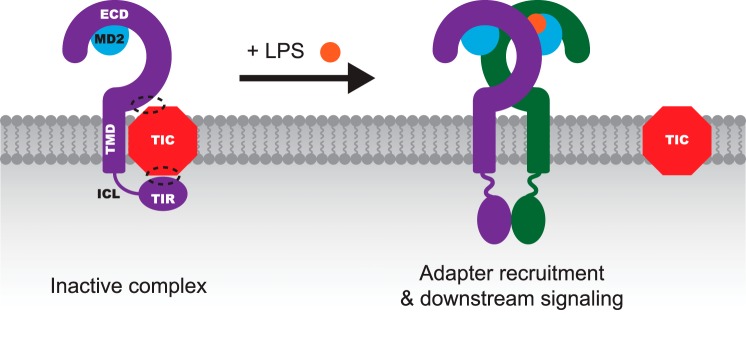
Proposed model of TLR4 switchlike signaling. This schematic summarizes the proposed model by which TLR4 signaling is regulated. In the absence of ligand, the TIC associates with TLR4 and maintains the receptor in an inactive conformation that prohibits constitutive signaling. Dashed circles, putative sites of interactions between TLR4 and TIC. Although this schematic depicts TLR4-TIC interactions occurring exclusively in cis (involving one TLR4 protein and one TIC), it is also possible that some interactions occur in trans and involve multiple TLR4 proteins, as described under “Discussion.” LPS (orange circle) binding to MD2 promotes a conformational change that induces TLR4 ectodomain dimerization, leading to dissociation of TIC from TLR4. In the absence of TIC, TLR4 TIR domains spontaneously dimerize and induce adapter recruitment and downstream signaling.
The proposed TIC hypothesis is consistent with the key observations reported here and discussed above as well as with other reported properties of TLR4 signaling. Most importantly, the TIC hypothesis could explain why 1) the C-terminal fragment of the TLR4 ECD (LRR21-LRRCT) is sufficient (and necessary) to prevent constitutive signaling, 2) TLR4-L48 exhibits both low background signaling and LPS-inducible signaling, 3) insertion of external linkers between the TMD and ECD does not confer constitutive signaling, and 4) LPS-mediated induction of TLR4 signaling does not require specific ICL sequences. According to the TIC hypothesis, ICL length and composition would not affect inducible signaling so long as the ICL does not interfere with TLR4 associating or dissociating with TIC. For example, the polar, uncharged linker evaluated here might stabilize interactions with TIC, such that ligand binding does not induce signaling, which could explain why TLR4-U was not inducible yet M-4-U (which lacks the TLR4 ECD hypothesized to be necessary for TIC binding) signaled constitutively (Figs. 2 and 5). Similarly, according to the TIC hypothesis, we would predict that inserting external linkers would not preclude association with TIC, although the linkers may diminish the extent to which LPS-induced ECD dimerization or rearrangement disrupts TIC binding. Indeed, the TIC hypothesis best explains why inserting linkers between the ECD and TMD has such a different impact on signaling than does inserting linkers between the TMD and TIR domain.
The TIC hypothesis could also explain why TLR4 signaling is induced by numerous non-canonical ligands (16–22). If induction of TLR4 signaling requires only disruption of TIC binding, rather than ligand-guided formation of a specific dimer conformation, then there may exist multiple mechanisms for inducing signaling. For example, nickel and cobalt have been shown to trigger activation of human TLR4 via interaction with non-conserved histidine residues, and these metals may mediate TLR4 ECD dimerization even in the absence of MD2 (16, 17). Under the TIC hypothesis, these and other ligands may also induce TLR4 signaling by disrupting TIC binding, even without inducing TLR4 ECD dimerization. Interestingly, human TLR4 contains a non-conserved histidine at position His-555 (16), and although this residue has not yet been investigated for conferring or enhancing responsiveness to metal this residue falls within LRR21, which was previously implicated as being crucial for suppressing constitutive signaling (25).
Further investigation is certainly required to confirm or refute the existence of the proposed TIC mechanism and to identify the components involved. Our experiments exclude RP105 and TMED7, two important negative regulators of TLR4, as mediating the TIC mechanism in our experiments. RP105 was not expressed in our cells, so although this protein may participate in TIC in other cell types, it cannot explain our observations. Thus, RP105 would not be a required component of TIC. In both our experiments and previous reports, TMED7 does not regulate TLR4 signaling in a manner consistent with TIC. For example, knockdown of TMED7 expression diminished LPS-inducible TLR4 signaling rather than substantially boosting constitutive signaling, as one would predict for knockdown of TIC (Fig. 7) (30). Similarly, overexpression of TMED7 failed to diminish ligand-independent background signaling, and LPS-induced signaling was suppressed modestly (Fig. 7) or not at all (30). Thus, although we cannot formally exclude TMED7 as a participant in TIC, it is clear that TMED7 does not mediate the main functions hypothesized to be meditated by TIC.
Finally, it is intriguing to consider the consequences of the hypothesized TIC mechanism. Although a TIC-like mechanism may also apply to other TLRs, suppression of ligand-independent signaling may be particularly important for TLR4 because it drives a wide range of inflammatory responses (4). The TIC mechanism may also confer switchlike signaling to TLR4; if the LPS concentration exceeds the threshold required to transiently disrupt TIC binding, then the propensity of the TIR domains to homodimerize may result in robust downstream signaling in response to even low concentrations of LPS. Finally, whether or not the TIC mechanism proves to be correct, the observations reported here necessitate revising the conceptual model of TLR4 signaling, which could ultimately inform the design of novel strategies for therapeutically manipulating this important receptor.
Supplementary Material
Acknowledgments
We acknowledge the use of the Northwestern University Flow Cytometry Facility. Traditional sequencing services were performed at the Northwestern University Genomics Core Facility.
This work was supported, in whole or in part, by National Institutes of Health, NCI, Cancer Center Support Grant CA060553. This work was also supported by Defense Advanced Research Projects Agency Award W911NF-11-2-0066 and by the National Academies Keck Futures Initiative (NAKFI-SB6).

This article contains supplemental Table S1.
- TLR
- Toll-like receptor
- CD28
- cluster of differentiation 28
- ECD
- ectodomain
- IL-10Rα
- IL-10 receptor α-chain
- ICL
- intracellular linker
- Mal
- MyD88-adapter-like
- MD2
- myeloid differentiation factor 2
- MyD88
- myeloid differentiation primary response 88
- sgRNA
- small guide RNA
- TIC
- TLR4-inhibitory complex
- TIR
- Toll/IL-1 receptor
- TMD
- transmembrane domain
- TMED7
- transmembrane Emp24 protein transport domain-containing protein 7
- TRIF
- TIR-domain-containing adapter-inducing interferon-β
- MFI
- mean fluorescence intensity
- PE
- phycoerythrin.
REFERENCES
- 1. Takeda K., Kaisho T., Akira S. (2003) Toll-like receptors. Annu. Rev. Immunol. 21, 335–376 [DOI] [PubMed] [Google Scholar]
- 2. Iwasaki A., Medzhitov R. (2004) Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 5, 987–995 [DOI] [PubMed] [Google Scholar]
- 3. Trinchieri G., Sher A. (2007) Cooperation of Toll-like receptor signals in innate immune defence. Nat. Rev. Immunol. 7, 179–190 [DOI] [PubMed] [Google Scholar]
- 4. Gay N. J., Symmons M. F., Gangloff M., Bryant C. E. (2014) Assembly and localization of Toll-like receptor signalling complexes. Nat. Rev. Immunol. 14, 546–558 [DOI] [PubMed] [Google Scholar]
- 5. Mills K. H. (2011) TLR-dependent T cell activation in autoimmunity. Nat. Rev. Immunol. 11, 807–822 [DOI] [PubMed] [Google Scholar]
- 6. Wang J. Q., Jeelall Y. S., Ferguson L. L., Horikawa K. (2014) Toll-like receptors and cancer: MYD88 mutation and inflammation. Front. Immunol. 5, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Savva A., Roger T. (2013) Targeting toll-like receptors: promising therapeutic strategies for the management of sepsis-associated pathology and infectious diseases. Front. Immunol. 4, 387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hennessy E. J., Parker A. E., O'Neill L. A. (2010) Targeting Toll-like receptors: emerging therapeutics? Nat. Rev. Drug Discov. 9, 293–307 [DOI] [PubMed] [Google Scholar]
- 9. Fekonja O., Avbelj M., Jerala R. (2012) Suppression of TLR signaling by targeting TIR domain-containing proteins. Curr. Protein Pept. Sci. 13, 776–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee C. C., Avalos A. M., Ploegh H. L. (2012) Accessory molecules for Toll-like receptors and their function. Nat. Rev. Immunol. 12, 168–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kang J. Y., Lee J. O. (2011) Structural biology of the Toll-like receptor family. Annu. Rev. Biochem. 80, 917–941 [DOI] [PubMed] [Google Scholar]
- 12. Gioannini T. L., Teghanemt A., Zhang D., Coussens N. P., Dockstader W., Ramaswamy S., Weiss J. P. (2004) Isolation of an endotoxin-MD-2 complex that produces Toll-like receptor 4-dependent cell activation at picomolar concentrations. Proc. Natl. Acad. Sci. U.S.A. 101, 4186–4191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Visintin A., Halmen K. A., Khan N., Monks B. G., Golenbock D. T., Lien E. (2006) MD-2 expression is not required for cell surface targeting of Toll-like receptor 4 (TLR4). J. Leukoc. Biol. 80, 1584–1592 [DOI] [PubMed] [Google Scholar]
- 14. Nagai Y., Akashi S., Nagafuku M., Ogata M., Iwakura Y., Akira S., Kitamura T., Kosugi A., Kimoto M., Miyake K. (2002) Essential role of MD-2 in LPS responsiveness and TLR4 distribution. Nat. Immunol. 3, 667–672 [DOI] [PubMed] [Google Scholar]
- 15. Park B. S., Song D. H., Kim H. M., Choi B. S., Lee H., Lee J. O. (2009) The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 458, 1191–1195 [DOI] [PubMed] [Google Scholar]
- 16. Schmidt M., Raghavan B., Müller V., Vogl T., Fejer G., Tchaptchet S., Keck S., Kalis C., Nielsen P. J., Galanos C., Roth J., Skerra A., Martin S. F., Freudenberg M. A., Goebeler M. (2010) Crucial role for human Toll-like receptor 4 in the development of contact allergy to nickel. Nat. Immunol. 11, 814–819 [DOI] [PubMed] [Google Scholar]
- 17. Raghavan B., Martin S. F., Esser P. R., Goebeler M., Schmidt M. (2012) Metal allergens nickel and cobalt facilitate TLR4 homodimerization independently of MD2. EMBO Rep. 13, 1109–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Smiley S. T., King J. A., Hancock W. W. (2001) Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J. Immunol. 167, 2887–2894 [DOI] [PubMed] [Google Scholar]
- 19. Park J. S., Svetkauskaite D., He Q., Kim J. Y., Strassheim D., Ishizaka A., Abraham E. (2004) Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J. Biol. Chem. 279, 7370–7377 [DOI] [PubMed] [Google Scholar]
- 20. Kim S., Kim S. Y., Pribis J. P., Lotze M., Mollen K. P., Shapiro R., Loughran P., Scott M. J., Billiar T. R. (2013) Signaling of high mobility group box 1 (HMGB1) through toll-like receptor 4 in macrophages requires CD14. Mol. Med. 19, 88–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Marr N., Turvey S. E. (2012) Role of human TLR4 in respiratory syncytial virus-induced NF-κB activation, viral entry and replication. Innate Immun. 18, 856–865 [DOI] [PubMed] [Google Scholar]
- 22. Georgel P., Jiang Z., Kunz S., Janssen E., Mols J., Hoebe K., Bahram S., Oldstone M. B., Beutler B. (2007) Vesicular stomatitis virus glycoprotein G activates a specific antiviral Toll-like receptor 4-dependent pathway. Virology 362, 304–313 [DOI] [PubMed] [Google Scholar]
- 23. Zhang H., Tay P. N., Cao W., Li W., Lu J. (2002) Integrin-nucleated Toll-like receptor (TLR) dimerization reveals subcellular targeting of TLRs and distinct mechanisms of TLR4 activation and signaling. FEBS Lett. 532, 171–176 [DOI] [PubMed] [Google Scholar]
- 24. Núñez Miguel R., Wong J., Westoll J. F., Brooks H. J., O'Neill L. A., Gay N. J., Bryant C. E., Monie T. P. (2007) A dimer of the Toll-like receptor 4 cytoplasmic domain provides a specific scaffold for the recruitment of signalling adaptor proteins. PLoS One 2, e788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Panter G., Jerala R. (2011) The ectodomain of the Toll-like receptor 4 prevents constitutive receptor activation. J. Biol. Chem. 286, 23334–23344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nishiya T., Kajita E., Miwa S. (2006) Ligand-independent oligomerization of TLR4 regulated by a short hydrophobic region adjacent to the transmembrane domain. Biochem. Biophys. Res. Commun. 341, 1128–1134 [DOI] [PubMed] [Google Scholar]
- 27. Shaner N. C., Campbell R. E., Steinbach P. A., Giepmans B. N., Palmer A. E., Tsien R. Y. (2004) Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22, 1567–1572 [DOI] [PubMed] [Google Scholar]
- 28. Bell J. K., Askins J., Hall P. R., Davies D. R., Segal D. M. (2006) The dsRNA binding site of human Toll-like receptor 3. Proc. Natl. Acad. Sci. U.S.A. 103, 8792–8797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gilbert L. A., Larson M. H., Morsut L., Liu Z., Brar G. A., Torres S. E., Stern-Ginossar N., Brandman O., Whitehead E. H., Doudna J. A., Lim W. A., Weissman J. S., Qi L. S. (2013) CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liaunardy-Jopeace A., Bryant C. E., Gay N. J. (2014) The COP II adaptor protein TMED7 is required to initiate and mediate the delivery of TLR4 to the plasma membrane. Sci. Signal. 7, ra70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Leonard J. N., Ghirlando R., Askins J., Bell J. K., Margulies D. H., Davies D. R., Segal D. M. (2008) The TLR3 signaling complex forms by cooperative receptor dimerization. Proc. Natl. Acad. Sci. U.S.A. 105, 258–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kagan J. C., Su T., Horng T., Chow A., Akira S., Medzhitov R. (2008) TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-β. Nat. Immunol. 9, 361–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Godfroy J. I., 3rd, Roostan M., Moroz Y. S., Korendovych I. V., Yin H. (2012) Isolated Toll-like receptor transmembrane domains are capable of oligomerization. PLoS One 7, e48875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Reuven E. M., Fink A., Shai Y. (2014) Regulation of innate immune responses by transmembrane interactions: lessons from the TLR family. Biochim. Biophys. Acta 1838, 1586–1593 [DOI] [PubMed] [Google Scholar]
- 35. Daringer N. M., Dudek R. M., Schwarz K. A., Leonard J. N. (2014) Modular extracellular sensor architecture for engineering mammalian cell-based devices. ACS Synth. Biol. 10.1021/sb400128g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim Y. M., Brinkmann M. M., Paquet M. E., Ploegh H. L. (2008) UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature 452, 234–238 [DOI] [PubMed] [Google Scholar]
- 37. Liaunardy-Jopeace A., Gay N. J. (2014) Molecular and cellular regulation of toll-like receptor-4 activity induced by lipopolysaccharide ligands. Front. Immunol. 5, 473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang D., Lou J., Ouyang C., Chen W., Liu Y., Liu X., Cao X., Wang J., Lu L. (2010) Ras-related protein Rab10 facilitates TLR4 signaling by promoting replenishment of TLR4 onto the plasma membrane. Proc. Natl. Acad. Sci. U.S.A. 107, 13806–13811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Husebye H., Halaas Ø., Stenmark H., Tunheim G., Sandanger Ø., Bogen B., Brech A., Latz E., Espevik T. (2006) Endocytic pathways regulate Toll-like receptor 4 signaling and link innate and adaptive immunity. EMBO J. 25, 683–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kondo T., Kawai T., Akira S. (2012) Dissecting negative regulation of Toll-like receptor signaling. Trends Immunol. 33, 449–458 [DOI] [PubMed] [Google Scholar]
- 41. Divanovic S., Trompette A., Atabani S. F., Madan R., Golenbock D. T., Visintin A., Finberg R. W., Tarakhovsky A., Vogel S. N., Belkaid Y., Kurt-Jones E. A., Karp C. L. (2005) Inhibition of TLR-4/MD-2 signaling by RP105/MD-1. J. Endotoxin Res. 11, 363–368 [DOI] [PubMed] [Google Scholar]
- 42. Divanovic S., Trompette A., Atabani S. F., Madan R., Golenbock D. T., Visintin A., Finberg R. W., Tarakhovsky A., Vogel S. N., Belkaid Y., Kurt-Jones E. A., Karp C. L. (2005) Negative regulation of Toll-like receptor 4 signaling by the Toll-like receptor homolog RP105. Nat. Immunol. 6, 571–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Doyle S. L., Husebye H., Connolly D. J., Espevik T., O'Neill L. A., McGettrick A. F. (2012) The GOLD domain-containing protein TMED7 inhibits TLR4 signalling from the endosome upon LPS stimulation. Nat. Commun. 3, 707. [DOI] [PubMed] [Google Scholar]
- 44. Treeby M., Vasl J., Ota P., Friedrich J., Jerala R. (2009) Different functional role of domain boundaries of Toll-like receptor 4. Biochem. Biophys. Res. Commun. 381, 65–69 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.