

Background: UL36 is a large multifunctional protein that is conserved across herpesviridae.
Results: The 970 central residues of UL36 have been analyzed by several biophysical and structural methods.
Conclusion: These UL36 residues constitute an elongated fiber that is able to form monomers and dimers.
Significance: This provides a framework for understanding how UL36 fulfills its functions.
Keywords: Crystal Structure, Herpesvirus, Structural Biology, Virus Assembly, Virus Structure, HSV1, Tegument, ul36, vp1/2
Abstract
The tegument of all herpesviruses contains a capsid-bound large protein that is essential for multiple viral processes, including capsid transport, decapsidation at the nuclear pore complex, particle assembly, and secondary envelopment, through mechanisms that are still incompletely understood. We report here a structural characterization of the central 970 residues of this protein for herpes simplex virus type 1 (HSV-1 UL36, 3164 residues). This large fragment is essentially a 34-nm-long monomeric fiber. The crystal structure of its C terminus shows an elongated domain-swapped dimer. Modeling and molecular dynamics simulations give a likely molecular organization for the monomeric form and extend our findings to alphaherpesvirinae. Hence, we propose that an essential feature of UL36 is the existence in its central region of a stalk capable of connecting capsid and membrane across the tegument and that the ability to switch between monomeric and dimeric forms may help UL36 fulfill its multiple functions.
Introduction
Herpesviruses, such as herpes simplex virus type 1 (HSV-1), share a common four-layer organization. Their double-stranded DNA genome is contained within a T = 16 icosahedral capsid, surrounded by an intermediate proteinaceous tegument and finally an external lipid envelope. The tegument is asymmetric with a minimal thickness of 15 nm and a maximal thickness of 50 nm in extracellular virions. It is subdivided in an “inner” tegument that is tightly bound to the capsid and an “outer” tegument that is connected to the viral envelope (1–4). The HSV-1 tegument is made of more than 20 proteins. Three of them, US3, UL36, and UL37, have been identified as inner tegument proteins according to their localization during fractionation of viral particles and to their association with capsids or membranes during particle assembly and disassembly (5–8). Two of them, UL36 and UL37, form a complex of unknown stoichiometry (9, 10).
The essential tegument protein UL36 is tightly bound to the capsid vertices through an interaction with the vertices' specific component UL25 (11–13). UL36 is involved in several processes during the virus cycle. After fusion of the viral membrane with the plasma membrane during herpesvirus entry, most of the viral tegument is detached, whereas UL36 is one of the few proteins that remain associated with the capsid (14). UL36 together with UL37 play an essential role during entry. It is necessary for capsid transport toward the nucleus during the entry phase and toward the final viral particle assembly sites during egress. Both transports are abolished in the absence of UL36, and they are strongly impaired in the absence of UL37(15–18). Viral capsids are transported along the microtubule network, and several cellular proteins are recruited to this end, including molecular motors kinesin 1 and 2, dynein, and its dynactin cofactor as well as the dystonin/BPAG1, a cross-linker of cytoskeleton elements (14, 18–20).
Besides capsid transport, UL36 is required for capsid routing at the nuclear pore complex (21) and subsequent uncoating (22–24). In contrast, UL37 is not required for this final step (24). The docking process also involves the capsid protein UL25 that is able to bind the cellular nucleoporins CAN/Nup214 and HCG1 (13). These nucleoporins are localized on the cytoplasmic side of the nuclear pore complex and could thus act as a nuclear receptor for the capsid. Finally, an antibody directed against Nup358, a component of the cytoplasmic fibrils of the nuclear pore complex, is also able to block this association. Although several proteins involved in this process have been identified, the detailed mechanisms triggering the DNA ejection and the precise role of UL36 are still unknown (22).
Finally, UL36 has been involved in the last stages of viral assembly. A null mutation in the UL36 gene leads to an accumulation of unenveloped DNA-filled capsids in the cytoplasm and a massive production of defective “light particles” lacking capsid, thus suggesting that UL36 is necessary for associating the preassembled outer viral layers to the capsids (25, 26). The outer tegument protein UL48 has been shown to interact with UL36 (10, 27). However, disrupting this interaction reduces the incorporation of UL48 in viral particles, but it is not sufficient for preventing the formation of viral particles, thus suggesting that other unidentified direct or indirect interactions between UL36 and outer tegument proteins might be involved (28). Recently, the disruption of interactions between UL37 and the envelope proteins gK and UL20 has been shown to impair secondary envelopment (29). The absence of UL36 would thus abolish the recruitment of UL37 and, as a consequence, of the viral envelope as well.
In addition to these roles in the virus cycle, UL36 has a deubiquitinase domain that is active on both Lys-48 and Lys-63 ubiquitinylations (30, 31). It is able to deubiquitinylate several substrates, including TRAF3 and UL36 itself (32, 33).
Despite the large amount of available functional data, the way by which UL36 fulfills its functions is still elusive. The large size of the protein (336 kDa) and the scarcity of structural data are hindering the study of these mechanisms. Fibrous structures extending radially from the capsid vertices are observed in “T36” capsids, i.e. capsids with the inner tegument still bound, but it is unclear whether these fibers are made of UL36, UL37, or both proteins (3). Except for the human cytomegalovirus N-terminal deubiquitinase domain (34), no high resolution data are available for any herpesvirus UL36 proteins, and the structural domains of the protein have not been identified. In silico analyses suggest the presence of several coiled coil motifs between amino acids 980 and 1740. Here, we were able to produce in a recombinant system a large fragment (residues 760–1733) of UL36 containing almost one-third of the protein, including these putative coiled coils. Biophysical analyses establish that this central part of UL36 forms a monomeric fiber sufficiently extended to bridge the capsid and membrane. We solved the atomic structure of the C-terminal part of this fiber that surprisingly crystallized as a domain-swapped antiparallel dimer. Molecular dynamics simulations explain how a transition from a monomeric to a dimeric state could occur. Homology modeling suggests that these properties are conserved among alphaherpesvirinae. We discuss what roles extended monomeric and antiparallel dimeric forms of UL36 could have in the virus cycle.
EXPERIMENTAL PROCEDURES
Sequence Analyses
The protein sequence alignments of HSV-1, HSV-2, PRV,3 VZV, GHV-2, and CHV-1 UL36 were performed using ClustalW and were displayed with ESPRIPT. The coiled coils were predicted with PCOILS. The secondary structure predictions were performed using SOPMA.
Cloning, Expression, and Purification of UL36 Fragments
Amino acids 1600–1733 and 760–1733 of strain 17 HSV-1 UL36 (accession number FJ593289.1) were cloned between the BamHI and EcoRI restriction sites of the pGEX-6P1 (GE-Healthcare) expression vector, resulting in fusions of these fragments with an N-terminal cleavable glutathione S-transferase. Both fragments were produced in Escherichia coli BL21 (DE3) cells (Stratagene). Freshly transformed cells were incubated overnight in 5 ml of LB supplemented by ampicillin at 37 °C. These cultures were diluted in 1 liter of LB supplemented by 50 μg/ml ampicillin and grown at 37 °C up to an absorbance of 0.6. The temperature was then lowered to 18°, and the protein expression was induced by the addition of 1 mm isopropyl β-d-thiogalactopyranoside. Cells were harvested 16 h after induction.
Cells expressing construct 760–1733 were pelleted by centrifugation at 5000 × g for 20 min in a JLA9.1,000 rotor (Beckman Coulter). The pellet was resuspended in 50 mm HEPES, pH 7.0, 500 mm NaCl, 10% glycerol, 1 mm DTT supplemented by a tablet of EDTA-free “Complete” protease inhibitor mixture tablet (Roche Diagnostics) and lysed by sonication at 4 °C. The lysate was then centrifuged at 4 °C for 5 min at 2700 × g. The supernatant was recovered and centrifuged at 4 °C for 30 min at 48,000 × g in a JA25.50 rotor (Beckman Coulter). The soluble lysate was loaded on Protino glutathione-agarose 4B beads (Macherey Nagel). The column was washed with 20 volumes of 20 mm HEPES, pH 7.0, 200 mm NaCl, 1 mm DTT and eluted with 20 mm HEPES, pH 7.0, 200 mm NaCl, 1 mm DTT, 20 mm glutathione. The GST tag was then removed by the addition of 120 μg of PreScission protease for 150 min at 20 °C. The fragment was then purified by size exclusion chromatography on a Superose 6 10/300 GL pre-equilibrated in 20 mm HEPES, pH 7.0, 200 mm NaCl, 1 mm DTT. Finally, it was loaded again on glutathione beads, and the flow-through containing the fragment of interest was concentrated to 1 mg/ml and stored at −80 °C.
Similarly, cells expressing construct 1600–1733 were lysed in 50 mm HEPES, pH 7.0, 500 mm NaCl, 1 mm DTT supplemented by a tablet of EDTA-free Complete protease inhibitor mixture tablet. After two successive centrifugations at 2700 × g for 5 min then 48,000 × g for 30 min, the soluble lysate was loaded on a GSTrap FF affinity column (GE Healthcare). The column was washed with 20 volumes of 20 mm HEPES, pH 7.0, 200 mm NaCl and eluted with 20 mm HEPES, pH 7.0, 200 mm NaCl, 20 mm glutathione. After removal of the GST tag by the addition of 50 μg of PreScission protease for 2 h at 20 °C, the protein was purified on a Superdex 200 10/300 GL column and finally loaded on a second GSTrap FF column. The flow-through containing the fragment of interest was concentrated to 1 mg/ml and flash-frozen in liquid nitrogen.
SEC-MALLS Measurements
The experiments were performed with a Wyatt Technology instrument (mini DAWN TREOS + QELS + OPTILab T-rEX) coupled to a Shimadzu HPLC system. For all samples, 30 μl of samples were used. Fragment 1600–1733 in a 20 mm HEPES, pH 7.0, 500 mm NaCl buffer was injected at an initial concentration of 2 mg/ml in a KW-803 column. Fragment 760–1733 in a 20 mm HEPES, pH 7.0, 200 mm NaCl, 1 mm DTT buffer was injected at an initial concentration of 2 mg/ml in a Biosec-3 column.
Analytical Ultracentrifugation
Sedimentation velocity and equilibrium analyses were performed at 20 °C using an An-60Ti rotor in an XLA70 Beckman Coulter centrifuge. For sedimentation velocity, 360 μl of a 5 μm or 9.4 μm solution of the fragment 760–1733 in 20 mm HEPES, pH 7.0, 200 mm NaCl, 1 mm DTT were introduced in a cuvette with a 12-mm optical pathway and centrifuged at 40,000 rpm (116,370 × g). A280 measurements were performed each 5 min. The results were analyzed by SEDFIT. For equilibrium analyses, 110 μl of a 3.3, 5.4, or 8.6 μm solution of the fragment 760–1733 in 20 mm HEPES, pH 7.0, 200 mm NaCl, 1 mm DTT was introduced in a cuvette with a 12-mm pathway and centrifuged successively at 8200, 9800, and 14,200 rpm (corresponding to 4890 × g, 6985 × g, and 14,665 × g). The results were analyzed using SEDPHAT.
Electron Microscopy
The purified 760–1733 fragment was diluted to 0.1 mg/ml in 2 mm HEPES, pH 7, 20 mm NaCl, 50 mm ammonium acetate. The sample was then adsorbed onto airglow discharge carbon-coated grids and stained with sodium phospho-tungstic acid adjusted to the sample, pH 7.5. The images were recorded in an electron microscope (model CM12; Philips) operated at 80 kV, with a nominal magnification of 35,000. The length and width of the objects were measured using ImageJ.
Crystallization, Structure Determination, and Analysis
The 1600–1733 fragment was concentrated to 5 mg/ml. Crystals were grown by vapor diffusion at 20 °C in hanging drops using 0.1 μl of protein and 0.1 μl of the well solution containing 0.2 m (NH4)2HPO4, 20% PEG 3350 (native dataset), or 100 mm HEPES, pH 7.5, and 2 m ammonium formate (KI soak dataset). In both cases, small hexagonal crystals grew within 1–4 days and were harvested 2 weeks later. The native crystals were flash-cooled in 0.2 m (NH4)2HPO4, 20% PEG 3350, 20% glycerol, although the derivative crystals were incubated for 10 min in HEPES, pH 7.5, 1 m ammonium formate, 0.8 m potassium iodide, 25% glycerol before flash-freezing.
A native dataset was collected at 100 K on a single crystal at the PROXIMA 1 beamline at the Soleil synchrotron. A highly redundant iodine SAD dataset was collected at 100 K on a single crystal at a wavelength of 1.90745 Å at the ID23-1 beamline of the European Synchrotron Radiation Facility. Both datasets were processed using the XDS package (35) and were found to belong to the same crystal form despite the different crystallization conditions. SIRAS phasing was not attempted due to nonisomorphism. The structure was solved by SAD phasing with the KI soak dataset. SHELXD (36) could localize eight iodine atoms per asymmetric unit. After density modification using SHELXE, interpretable density maps were obtained, initially to 2.8 Å resolution. The structure was refined and analyzed using the PHENIX (37) and CCP4 (38) suites and built using COOT (39). Phenix.autobuild was able to place about 50% of the amino acids in the initial map. Despite significant data anisotropy, we gradually extended refinement resolution for subsequent refinement and building, following the current best practice (40). Thus, we included data according to CC½ and validated this choice by comparing CC(work) and CC(free) to CC* (Table 1). Coordinates were refined for the two 1600–1733 molecules in the asymmetric unit with local NCS restraints. TLS and individual B-factors were refined with three TLS groups as follows: residues 1628–1723 of both molecules, residues 1595–1627 of molecule B and residues 1597–1627 of molecule A (residues N-terminal to 1600 represent the residual affinity tag). In later stages of refinement, all iodine ions were identified in log-likelihood gradient maps. The asymmetric unit contains 42 iodine atoms (23 with occupancy of at least 50%). The final refined protein model was used as the starting point for refinement against the native dataset keeping the same test set. After rigid body refinement, the same refinement protocol was used as for the KI soak. There is additional weak density in the KI soak that could be ascribed to residues 1724–1725 stabilized by an iodine ion site. We did not include these residues in the final models as no density is apparent in the native dataset. The KI soak (respectively native) models have no Ramachandran outliers, and 98% (respectively, 97%) of residues are in the favored region of the Ramachandran plot. CC* (40) is 0.86 (respectively 0.91) in the highest resolution shell. Crystal packing and interfaces were analyzed with PISA (41) and the NOXclass server (42). Structure visualization was performed and structural figures made with PyMOL. Atomic coordinates and structure factors have been deposited to the RCSB Protein Data Bank under accession number 4TT0 (potassium iodide derivative) and 4TT1 (native crystals).
TABLE 1.
Data collection and refinement statistics
Statistics for the highest resolution shell are shown in parentheses.
| Iodine derivative (SAD phasing) (4TT0) | Native (4TT1) | |
|---|---|---|
| Resolution range (Å) | 48.04–2.599 (2.692–2.599) | 38.32–2.75 (2.848–2.75) |
| Space group | P 61 2 2 | P 61 2 2 |
| Unit cell a, b, c | 110.939, 110.939, 154.93 | 110.217 110.217 159.939 90 90 120 |
| α, β, γ | 90, 90, 120 | |
| Total reflections | 208,952 (19,540) | 198,088 (17,771) |
| Unique reflections | 17,958 (1,728) | 15,528 (1,500) |
| Multiplicity | 11.6 (11.3) | 12.8 (11.8) |
| Completeness (%) | 99.97 (99.77) | 99.94 (99.60) |
| Mean I/σ(I) | 14.53 (1.80) | 21.00 (1.59) |
| Wilson B-factor | 48.76 | 76.69 |
| R-merge | 0.1792 (1.185) | 0.09051 (1.604) |
| R-pim | 0.053 (0.379) | 0.026 (0.484) |
| CC1/2 | 0.997 (0.577) | 1 (0.697) |
| R-work | 0.1853 (0.2858) | 0.1950 (0.3598) |
| R-free | 0.2452 (0.3598) | 0.2431 (0.4095) |
| CC* | 0.999 (0.855) | 1 (0.906) |
| CC(work) | 0.950 (0.749) | 0.973 (0.731) |
| CC(free) | 0.926 (0.466) | 0.945 (0.564) |
| No. of non-hydrogen atoms | 2014 | 1942 |
| Macromolecules | 1941 | 1923 |
| Ligands | 42 | 10 |
| Water | 31 | 9 |
| Protein residues | 256 | 253 |
| r.m.s. (bonds) | 0.008 | 0.009 |
| r.m.s. (angles) | 0.97 | 1.14 |
| Ramachandran favored (%) | 98 | 97 |
| Ramachandran allowed (%) | 2 | 3 |
| Ramachandran outliers (%) | 0 | 0 |
| Clashscore | 6.57 | 3.34 |
| Average B-factor | 59.90 | 98.70 |
| Macromolecules | 59.70 | 98.50 |
| Ligands | 81.60 | 142.80 |
| Solvent | 42.90 | 91.20 |
Tryptophan Fluorescence
The fragment 1600–1733 was diluted to 100 μg/ml in 20 mm HEPES, pH 7, 200 mm NaCl. Identical spectra obtained with other buffers ranging from pH 6 to pH 8 and from 20 mm to 1 m NaCl were also tested. Emission spectra were recorded at 25 °C on a PerkinElmer Life Sciences LS50B fluorimeter. Excitation was performed at 290 nm (slit width 2.5 nm), and the emission was measured between 310 and 400 nm (slit width 2.5 nm) at a speed of 60 nm per min. The displayed spectrum is the average of 10 spectra.
Modeling and Molecular Dynamics Simulations
A putative folded-back 1600–1723 monomer was modeled by including residues 1600–1675 from one crystallographic molecule (chain B) and residues 1680–1723 of the other (chain A). The four intervening residues 1676–1679 were added so as to be stereochemically reasonable. Homology modeling was performed with program MODELLER (43) release 9.14 with default parameters using the alignments shown in Fig. 5A. Molecular dynamics simulations of a crystallographic monomer (residues 1600–1722 of chain B) of the modeled folded-back monomer and of homology models corresponding to HSV-1 UL36 residues 1632–1723 were performed using the AMBER 9 program suite (44) with the Parm99SB force field. Hydrogen atoms were added, and proteins were neutralized with 2–4 Na+ cations and immersed in an explicit TIP3P water box with a solvation shell at least 12 Å-deep using the LEaP program. The systems were then minimized and used to initiate molecular dynamics. All simulations were performed in the isothermal isobaric ensemble (p = 1 atm, T = 300 K), regulated with the Berendsen barostat and thermostat (45), using periodic boundary conditions and Ewald sums for treating long range electrostatic interactions (46). The hydrogen atoms were constrained to the equilibrium bond length using the SHAKE algorithm (47). A 2-fs time step for the integration of Newton's equations was used. The nonbonded cutoff radius of 10 Å was used. For targeted molecular dynamics simulations, the starting and target systems were built so as to be chemically identical (same protein residues, same number of water molecules). Targeted molecular dynamics adds an additional term to the energy function as shown in Equation 1,
where kforce corresponds to the force constant applied during the simulation (kcal/mol·Å2); N is the number of atoms; r.m.s.d. is the mass weighted root mean square deviation of the positions of protein Cα atoms at time t from the corresponding positions in the target structure. r.m.s.d.0 is the target r.m.s.d. that was set to 0 here. All simulations were run with the SANDER module of the AMBER package.
FIGURE 5.

Stability of crystallographic and folded-back models of fragment 1600–1733 and interconversion between them. A, sequence conservation in alphaherpesviruses for helix C (residues 1659–1696 in the crystal structure of 1600–1733). B, molecular dynamics simulations for a crystallographic molecule and a putative monomer folded back at residues 1676–1679. Both systems were neutralized, hydrated in explicit solvent, and minimized. Protein structures are displayed as ribbons at the beginning (left) and the end (right) of the 10-ns production time. They are aligned on the stable central three-helix bundle (residues 1633–1675, colored black). Residues N-terminal to 1633–1675 are colored pale yellow, and the C-terminal residues are colored pale green, including Trp-1686, whose side chain is displayed as sticks. Middle panels, change over time for the crystallographic molecule (black lines) and the folded-back model (salmon lines) of the r.m.s.d. for all atoms in residues 1633–1722 (top) and of the distance between Trp-1686 Nϵ and Leu-1640 O (bottom). C, targeted molecular dynamics simulations between the observed and modeled conformers. Top, all atom r.m.s.d. between the simulated, initially extended molecule and the folded-back target for four values of the force constant a·.Å2). The time points are indicated below the structures.
RESULTS
Biophysical Characterization of UL36 Fragments
The boundaries of the structural domains of UL36 are so far unknown. Sequence analysis software predicts that several segments of UL36 (981–1035, 1292–1328, 1503–1532, 1586–1613, and 1710–1737) organize as coiled coils. All of them are localized within a 760-amino acid segment, suggesting it could correspond to a structural element. Consistent with this, we were able to express and purify a fragment of UL36 composed of residues 760–1733 in E. coli (Fig. 1A) containing these predicted coiled coil regions. The C-terminal part of this fragment 1600–1733 containing the last two repeats of a predicted helix-turn-helix motif could also be produced (Fig. 1A).
FIGURE 1.

Oligomeric status and shape of HSV-1 UL36 fragments in solution. A, all secondary structure prediction servers predict the presence of three regions with a high propensity to form α-helices and β-strands (rectangles) and two large segments with a higher probability to be random coils (thin stretches 230–490 and 1520–3087). Reported interaction regions and predicted coiled coils are indicated. Two fragments, 1600–1733 and 760–1733, have been used in this work. B, SEC-MALLS analyses of fragments 1600–1733 (top) and 760–1733 (bottom). Two independent experiments are presented for fragment 760–1733. The protein concentrations at the peak are indicated. Insets, 4 μg of the purified proteins used in SEC-MALLS were analyzed by 16% (1600–1733) or 14% (760–1733) SDS-PAGE with Coomassie staining. C, molecular weight of fragment 760–1733 derived from sedimentation equilibrium ultracentrifugation at 3.3, 5.4, and 8.6 μm. D, sedimentation velocity experiment of fragment 760–1733 at 5 and 9.4 μm. The experimental curves obtained during a centrifugation at 40,000 rpm in an An-60Ti rotor (top) and the fitted data obtained after analyses by Sedfit (bottom) are represented.
Coiled coils can be made of helices belonging to a single polypeptide, but they frequently constitute oligomerization regions. To check for a possible oligomerization, these fragments were analyzed by size exclusion chromatography associated with SEC-MALLS. Although 1600–1733 was less well behaved than 760–1733 and was not recovered quantitatively in SEC, it eluted as a single monomeric species (12.8 ± 0.2 kDa for an expected 14.8 kDa calculated from the fragment sequence) at an effective concentration of 30 μg/ml (Fig. 1B, top). At 70 μg/ml 760–1733 also eluted as a monomeric species (101.4 ± 0.2 kDa and 100.0 ± 0.1 kDa in two independent experiments for an expected 104.6 kDa) (Fig. 1B, bottom), but a minor form could also be detected, which in some preparations was in sufficient quantity for its mass to be accurately determined (Fig. 1B, bottom, purple curve). This mass of 217.3 ± 0.4 kDa is consistent with that of a dimer. The observation of distinct peaks shows that the conversion between monomeric and dimeric 760–1733 is not possible or is at least slower than the characteristic time scale of the experiment. Analytical ultracentrifugation (sedimentation equilibrium and sedimentation velocity) was performed at much higher protein concentrations for 760–1733. Sedimentation equilibrium experiments show that 760–1733 is still a monomer at least up to a concentration of 0.90 mg/ml (Fig. 1C).
Given that the inner tegument has been described as fibrous (3), there is a possibility that some or all of these fragments could be elongated. To characterize this, this fragment was analyzed by sedimentation velocity (Fig. 1D). A monomodal distribution corresponding to a 3.3 S species (s20,w = 3.5) was observed, and taking into account that 760–1733 is monomeric, it means that this fragment has a f/f0 ratio of 2.3. This very high value can only be explained by an extremely elongated shape and is typically observed with fibrous proteins.
Because of its large size, fragment 760–1733 could be directly observed by negative staining electron microscopy, which confirms that it is an elongated fiber (Fig. 2, A–D). The length distribution of fibers is bimodal with a main population at 33.7 ± 5.5 nm and a minor population at 59.7 ± 2.3 nm, whereas the width distribution is monomodal with a single population at 2.9 ± 0.4 nm (Fig. 2E). Additionally, the shorter fibers were sometimes associated via one of their ends, thus constituting “V”-shaped objects (Fig. 2D).
FIGURE 2.

Electron microscopy of fragment 760–1733. A–D, negative staining observation. The scale bar for A has a length of 100 nm. Several different morphologies are observed. Objects indicated by a black arrow are enlarged in B–D. E, histograms of the measured lengths (top) and widths (bottom) of the fibers.
The fibers are surprisingly straight for objects with such a large length to width ratio. Only a minority of fibers display one or two kinks separated with straight segments, suggesting that the fragment is made of several rods separated by a discrete number of articulations. At least in the conditions in which the observations were performed, the most extended conformation was dominant.
Crystal Structure of Fragment 1600–1733
To get more detailed information on this fiber, crystallization of the different fragments was undertaken. It was possible to determine the crystal structure of fragment 1600–1733 that crystallized in several different conditions. The two that allowed useful data collection yielded the same crystal form with almost superimposable asymmetric units despite a substantial difference in cell parameters (Table 1 and see under “Experimental Procedures”). The high resolution cutoffs of the two models were validated according to Diederichs and Karplus (40). The asymmetric unit contains two molecules, and residues 1600–1723 could be built for both. The two molecules are very similar, being almost identical over residues 1628–1723 and differing only in different bends of their N-terminal helices at residues 1624–1627. Additional amino acids left over from cleavage of the purification tag could also be observed at the N termini, extending the N-terminal helix of molecule B by five residues (i.e. all the residues left from the tag despite their sequence GPLGS having no helical propensity). In accordance to the biophysical analyses, each molecule appears as an elongated fiber about 120 Å long and with a maximal width between α carbons of ∼15 Å. Each molecule is constituted by two repeats of a long helix-turn-antiparallel short helix motif, A-B and C-D (Fig. 3A). The intrachain contacts are extremely scarce and limited to two short regions. The first region involves residues 1633–1672, including the C-terminal part of helix A, the entire helix B, and the N-terminal part of helix C. This region organizes as a short three-helix bundle. The second region is constituted by the C-terminal part of helix C and the totality of helix D as well as β-like contacts in loop CD. Surprisingly, there is a cluster of exposed hydrophobic residues in both regions (Fig. 3C). The first one involves Leu-1636, Leu-1639, Leu-1640, Val-1642, Val-1643, Pro-1646, Ile-1662, and Leu-1669, whereas the second one involves Val-1684, Trp-1686, Leu-1687, Phe-1694, Leu-1709, Leu-1710, and Leu-1720. These hydrophobic residues are strongly conserved through alphaherpesviruses, suggesting that these surfaces have a structural and/or functional role (Fig. 3B).
FIGURE 3.

Architecture of a single molecule of fragment 1600–1733. A, four helices termed A, B, C, and D are observed in the crystal structure of fragment 1600–1733. The N-terminal extremity is on the left end, and the C terminus is on the right end. B, surface representation of the A-B-C three-helix bundle using the same orientation as in C. Residues are colored according to their conservation according to server Consurf from blue (fully conserved residues) to red (least conserved residues). The sequence alignment used by Consurf contained 136 representative sequences of alphaherpesvirus UL36. C, trace representation of residues 1636–1720. Two clusters of conserved and exposed hydrophobic residues are displayed as sticks. Residues buried in the dimer interface are colored by atom type (red, oxygen; blue, nitrogen; carbons, green and cyan in either hydrophobic patch).
Extended contacts are observed between the two molecules in the asymmetric unit (Fig. 4A). The two molecules are wrapped around each other, and they adopt an overall boomerang shape with the N terminus at the extremity and the C terminus in the center. This makes up an extremely elongated dimer with a length of 158 Å and a maximal width of 24 Å. The dimerization involves the formation of a long antiparallel coiled coil between the two C helices. Moreover, the two exposed hydrophobic surfaces of one monomer are now interacting with those of the other, thus constituting two new five-helix bundles involving helices A, B, and C of one monomer and helices C and D of the other monomer. The highly conserved Trp-1686 that would be completely exposed in the isolated monomer is now buried in a pocket of the other molecule (visible on Fig. 3B). It is engaged in multiple interchain hydrophobic contacts with Leu-1640, Val-1643, Val-1645, Pro-1646, Val-1649, Val-1666, and Leu-1669. The NH of the indole group is also involved in an intermolecular hydrogen bond with the carbonyl of Leu-1640 (Fig. 4C).
FIGURE 4.

Dimeric structure of fragment 1600–1733. A, architecture of the crystallographic dimer. One monomer is colored in blue and the other one in gray. B, tryptophan fluorescence emission spectrum of fragment 1600–1733. The maximal emission was observed at a wavelength of 338 nm. The spectrum was recorded in a 50 mm Tris, pH 8, 200 mm NaCl buffer. Spectra with a maximum emission at 338 nm were recorded in all the tested buffers, including (50 mm Tris, pH 8, 20 mm to 1 m NaCl), (50 mm HEPES, pH 7, 200 mm NaCl), and (50 mm MES, pH 6, 200 mm NaCl). C, environment of tryptophan 1686 in the crystallographic monomer (left) and dimer (right). The residues with a side chain less than 4.5 Å away from the indole group of W1686_A (red) are represented as orange (chain A) or yellow (chain B) sticks. The interchain hydrogen bond between the nitrogen of the indole group of W1686_A and the carbonyl of L1640_B is displayed on the right.
The NOXclass server (42) reports 2140 Å2 of buried surface per molecule for this dimeric interface, accounting for 20% of the accessible surface. Taking into account the amino acid composition of the interface, the server assigns a 99.99% probability that the crystallographic dimer is biologically relevant. However, dimers can be either obligate or nonobligate, i.e. the dimer can be stable or it can be in equilibrium with monomers. Here, the calculated probability that it constitutes an obligate dimer is only 89.6%. Thus, the dimer might be a transient species, sensitive to external parameters.
In addition to this major interface, the PISA program (41) predicts no other contact in the crystals to be stable in solution. The second and third most prominent crystal contacts (burying 880 and 520 Å2 per molecule, respectively) involve antiparallel coiled coils between helices A of two molecules B and two molecules A, respectively. The contacts involving helices A are not identical in these two coiled coils, as there is one helical turn shift between the two. The larger buried surface for molecule B is due in part to additional contacts between helices A and C involving the N-terminal extremity of the fragment, including the residues stemming from the purification tag.
The crystallographic data strongly suggest that fragment 1600–1733 is dimeric, whereas the biophysical tools could only detect a monomer in solution. It could suggest that the crystallographic dimer is artifactual; however, it was crystallized under at least two highly different conditions, suggesting that this species is present in the preparation. The high concentration encountered during the crystallization process has then shifted the equilibrium toward this dimeric species. The structure of the main monomeric species is thus still unknown.
Many hydrophobic residues are exposed in the crystallographic monomer, although they are buried in the dimer where they constitute the hydrophobic core of an intermolecular structural domain. It is thus unlikely that the crystallographic monomer is the solution monomer without at least a major structural reorganization. To check for this hypothesis, we decided to take advantage of Trp-1686 that is the single tryptophan in the fragment. In the crystallographic monomer it is highly exposed, although it is completely buried in the dimer in a hydrophobic environment, except for the hydrogen bond between the NH of the side chain of Trp-1686 and the carbonyl of Leu-1640. A tryptophan fluorescence emission spectrum displays a maximum at a wavelength of 338 nm for fragment 1600–1733 (Fig. 4B) independently of the pH (between 6 and 8) and of the salt concentration (between 20 mm and 1 m NaCl). This is not consistent with a fully exposed tryptophan but is fully compatible with the environment observed in the dimer (48). Thus, the tryptophan in the solution monomer is similarly buried as in the crystallographic dimer.
The DALI server was used to detect whether the crystallographic and the model folded-back monomers were homologous to other proteins in the Protein Data Bank. The highest Z-score obtained for the crystallographic protomer corresponded to the copper-sensitive operon regulator (Z = 5.2). However, a detailed analysis reveals that the homology only extends to helix A, which suggests that it is not relevant. Higher Z-scores were obtained for the model folded-back bulge, including the histone-lysine N-methyltransferase MLL (3LQH, Z = 6.6), the general control of amino acid synthesis protein 5 GCN5 (3D7C, Z = 6.2), and the human transcription intermediary factor 1α (1YYN, Z = 5.8). All these proteins belong to the bromodomain family. This domain only contains four helices, whereas fragment 1600–1733 has five helices. However, MLL displays an additional segment at the expected position for helix D of fragment 1600–1733, suggesting that the 1600–1733 belongs to this family.
Modeling of the UL36 1600–1733 Monomer and Molecular Dynamics Simulations
The contacts between both monomers involve the two five-helix bundles, but there is a break in the contacts between helices C at their center close to the noncrystallographic 2-fold axis. The last interchain contact on either side of the break involves a salt bridge between Arg-1677 and Asp-1680. However, this region is not strictly conserved; Arg-1677 may be replaced by a lysine in gallid herpesvirus 2 (GHV-2), an alanine in cercopithecine herpesvirus 1 (CHV-1), and a proline in PRV. An even more severe mutation is observed in varicella zoster virus where a glycine is inserted (Fig. 5A). These mutations are hardly compatible with a continuous helix C extending from residue 1659 to residue 1696, but it is compatible with two smaller helices separated by a short linker around residue 1677. Taken together, these data suggest that these residues are not necessarily folded as a helix turn as it is the case in the crystal structure and could constitute a conformational switch.
A chimera constituted by residues 1600–1675 of molecule A and 1680–1733 of molecule B would be geometrically possible while explaining the otherwise discordant results. Indeed, residues 1675 of one chain and 1680 of the other chain are sufficiently close (11 Å between their Cα) for the intervening residues 1676 and 1679 to loop between them. The chimera would be monomeric in solution, given that there would not be any interchain contact anymore. Trp-1686 would still be buried as indicated by the tryptophan fluorescence analysis. Under this conformation, fragment 1600–1733 would have a lollipop shape with the N terminus of helix A constituting the stalk and a five-helix bundle made of the C terminus of helix A, the two parts of former helix C and helices B and D, constituting the bulge (Fig. 5B, bottom left).
To test this hypothesis, we performed all atom molecular dynamics simulations in explicit solvent of such a folded-back lollipop model. In parallel, we also simulated the dynamics of the isolated extended crystallographic molecule. The small central three-helix bundle 1633–1670 (in black on Fig. 5B) is quite stable whether in the extended or folded-back molecule. After 10 ns of simulation, it is little changed in both and thus still displays the hydrophobic patch that is prominent in the crystallographic dimer interaction, including the Trp-1686 pocket (Figs. 3B and 4C). In contrast, the N-terminal helix A upstream of Leu-1633 (in yellow on Fig. 5B) breaks up in both simulations. This is consistent with its observed static flexibility in the two crystallographic molecules (see above). The major difference between the two simulations lies in the C-terminal part downstream of residue 1673 (green on Fig. 5B). The starting helix C of the extended molecule breaks at residues 1673–1685, whereas the folded-back molecule remains stable over the whole C terminus. Thus, the r.m.s.d. of all atoms for residues 1633–1722 quickly stabilizes at a value below 2.5 Å for the latter molecule, although it rises above 10 Å and has not converged after 10 ns for the extended crystallographic molecule (Fig. 5B, top center). Similarly, Trp-1686 remains buried and in hydrogen bonding distance of Leu-1640 throughout the simulation for the folded-back molecule, whereas it goes from 38 Å to below 25 Å for the extended molecule and has not yet stabilized after 10 ns. These results confirm that the crystallographic molecule is not the 1600–1733 solution monomer, whereasthe folded-back molecule could be. They further highlight residues 1633–1670 and to a lesser extent 1686–1720 as folding units capable of forming together a stable five-helix bundle. In contrast, the middle of helix C (residues 1673–1685) and the N terminus of helix A (residues 1600–1633) are clearly not stable in solution in the absence of stabilizing contacts. To establish whether extended and folded-back monomers can spontaneously interconvert, we performed targeted molecular dynamics simulations between the two conformations, starting from one and taking the other as the target. Even a very weak force of 0.02 kcal/(mol·Å2) is sufficient to guide conversion toward the folded-back conformation (Fig. 5C). Conversion back from the folded-back conformation is also readily accomplished, although at slightly higher force constant values (data not shown). This indicates that there is no large energy barrier between the observed and hypothesized conformations. Finally the energies of the two chemically identical systems are −2.474,1 105 kcal/mol (extended) and −2.483,5 105 kcal/mol (folded back). These small differences are consistent with an equilibrium, and one favoring the folded-back conformation.
Finally, we further probed the interconversion hypothesis and sought to extend our results to other alphaherpesviruses by producing homology models taking HSV-1 UL36 residues 1632–1723 as template for HSV-2, PRV, VZV, and the avian alphaherpesvirus GHV-2 (Fig. 6A). The extended models (Fig. 6A, top) illustrate that a straight helix C is indeed not to be found in PRV and VZV and also not in HSV-2 (despite 84% sequence identity) due to the HSV-2 Q1682P substitution (Fig. 5A). Molecular dynamics simulations of these extended molecules show the same features as described for the crystallographic HSV-1 monomer, namely instability of helix C and stability of the two helix bundles on either side. In contrast, the folded-back monomer can be modeled with little deviation from the HSV-1 template (Fig. 6A, bottom). The largest difference is found in PRV, in which a two-residue deletion prevents straight modeling of helix B either for the extended or folded-back monomer (Fig. 6A). As exemplified by the case of GHV-2 (despite only 35% sequence identity to HSV-1 UL36 fragment 1632–1723), the modeled structures are stable over 10 ns of simulation in each case. The conserved counterpart of Trp-1686 is inserted into a hydrophobic pocket with its hydrogen bond to the counterpart of Leu-1640 maintained throughout the simulation (Fig. 6B). In the case of PRV, only the pocket as modeled is not complete, although it is completely closed and stable in the other three cases.
FIGURE 6.

Homology modeling taking residues 1632–1723 of HSV-1 UL36 as templates for other alphaherpesviruses. A, models for HSV-2, VZV, PRV, and GHV-2. Top, based on the crystallographic molecule, only the avian GHV-2 fragment can be modeled in a similar conformation. Bottom, in contrast, models are readily generated that closely match the folded-back monomer for all viruses. The single tryptophan finds a hydrophobic pocket and hydrogen bonds (displayed in magenta) to the same leucine as in the HSV-1 template. B, stability of this hydrogen bond and r.m.s.d. for all atoms in a 10-ns molecular dynamics simulation for GHV-2 folded-back monomer.
DISCUSSION
The structural data presented in this study reveal that a fragment of HSV-1 UL36 comprising residues 760–1733 adopts a very elongated conformation. The major species is a stalk conformation with a length of 34 nm and a width of 2.9 nm. A smaller subpopulation of 760–1733 appeared to be even longer with an average length of 60 nm. It could either reveal a conformational change between an extended 60-nm monomeric species and a 34-nm hairpin conformation or a rare dimerization of two 34-nm monomeric fragments via one of their extremities. No significant width difference could be measured between the 34- and 60-nm species, arguing in favor of the second hypothesis.
The measured lengths for UL36 residues 760–1733 have to be compared with the dimensions of the elongated tufts detected at the surface of T36 particles (3). The width of fragment 760–1733 observed by electron microscopy and the width of the five helix bundle in the folded-back model of fragment 1600–1733 are close to the main peak of the distribution (2.9 nm). Moreover, the 760–1733 fragment has a length of 34 nm, whereas the tuft distribution shows three main peaks at 28, 45, and 67.8 nm. Thus, the similar width is compatible with the 760–1733 fragment constituting a major part of the T36 tufts. However, the significantly larger lengths measured for the T36 tufts suggest that the 760–1733 cannot be their sole constituent and that it must also contain other parts of UL36 or UL37. Interestingly, secondary structure predictions suggest that the region between residues 520 and 1920 is a succession of helices separated by short loops, implying that the entire 520–1920 part of UL36 could constitute a very long stalk in the middle of the protein.
The presence of this stalk would divide the entire protein in three main regions. The N-terminal part of the protein contains the deubiquitinase domain, the binding sites to the outer tegument protein UL48 (27) and UL37 (49) and an essential nuclear localization signal (50). The C terminus of UL36 contains the two capsid-binding sites. The intermediate fiber would provide a long stalk between the two extremities. Although the amino acid conservation in the stalk of UL36 is significantly lower than in other parts of UL36 such as the deubiquitinase domain and the capsid-binding sites, it has to be noted that the length of sequence is conserved across all three families of herpesviruses, suggesting that a precise length for UL36 could be an essential property of the protein.
Unexpectedly, the 1600–1733 fragment exists under at least two different conformations, a main monomeric and a minority dimeric species that could be crystallized. Molecular dynamics simulations show that the folded-back monomer is stable at least at the 10-ns scale. In contrast, the intervening region is unstable in the crystallographic monomer, but the 1633–1720 five-helix bundle of the folded-back monomer does not undergo any large rearrangement. Moreover, the folded-back monomer is in agreement with the different analyses in solution, which strongly suggests that it is the main species in solution. Of note, this organization still makes a very extended molecule (80 Å long over 122 residues) while strengthening its local structure, in accordance with the observed stiffness of fragment 760–1733.
In addition to a possible role during assembly, these elongated fibers could be involved in the other main functions of UL36, including capsid transport. Molecular motors typically contain a long arm connecting the motor and cargo-binding domains, thus limiting hindrance between a possible large cargo and the cytoskeleton. Given that UL36 cross-links the viral capsids to kinesin motors, the presence of this stalk could further increase the distance between the capsid and the microtubule, depending on the location of the UL36 kinesin-binding motif. However, in the case of PRV, the complex dynein binding motif of UL36 is bipartite and involves both the first 1285 amino acids of UL36 (corresponding to residues 1–1610 of HSV-1 UL36) and the large C-terminal proline-rich domain, thus suggesting that capsid transport is possible if the molecular motor binding domains are in the C terminus of UL36 (20).
Finally, the thermosensitive HSV-1 UL36 Y1453H mutation (51, 52) is worth reconsidering in the light of our high resolution structural data. This single mutation is sufficient for decreasing the virus production by 5 logs in plaque formation and induces a complex phenotype. During the entry phases, an accumulation of capsid at the nuclear pore is observed, but the viral genome is not efficiently released, and the production of viral proteins is reduced, suggesting that the transport phase has not been altered but that the DNA ejection into the nucleus is impaired. An additional defect in the later stages of the virus cycle is also present (51, 52). At least one and possibly more essential functions of UL36 have thus been altered in a temperature-dependent manner by this single mutation. If the UL36 stalk indeed contains strengthening nodes along its length similar to the one we modeled around Trp-1686, a substitution such as Y1453H could lead to destabilization of one such node, thus inducing a temperature-dependent loss of stiffness. As a consequence, the relative positions of the two extremities of the multifunctional UL36 would be affected, thus inducing a complex thermosensitive phenotype.
The data presented here are probably relevant across all alphaherpesviruses. Sequence conservation is sufficient for accurate homology modeling even to avian alphaherpesviruses based on our crystal structure for residues 1632–1723. Such modeling shows that the two halves of the bulge are complementary across alphaherpesviruses, arguing for a functional interface underlying monomeric folding, dimeric interface, or both. Despite overall low amino acid conservation in other parts of the 760–1733 segment, it is striking that UL36 proteins from all alphaherpesviruses are all predicted to contain a long segment with a high α-helical content in their central part. Moreover, despite fairly low sequence identity, the sizes and order of the predicted helices seem to be strongly constrained, suggesting that the fiber-like structural element presented here could be an essential feature conserved among all alphaherpesviruses (Fig. 7). It is likely that this fiber is also present in other herpesviruses, but the amino acid conservation in this region is not sufficient for performing reliable sequence alignments. However, the presence of multiple amphipathic helices with a characteristic 4-3-4 spacing of hydrophobic residues is predicted in UL36 from all three families of herpesviruses over a 1000–1200-amino acid-long segment corresponding to residues 760–1733 of UL36 HSV-1. This suggests that this fiber is a strongly conserved element even if its amino acid sequence is much less constrained. Interestingly, it has been recently shown that the homologous protein in the human cytomegalovirus also has a fibrous shape (53). There could thus be a strong evolutionary pressure for maintaining a precise organization of the protein with a central fiber bridging capsid binding elements at the C-terminal end and membrane-interacting elements at the N-terminal end.
FIGURE 7.
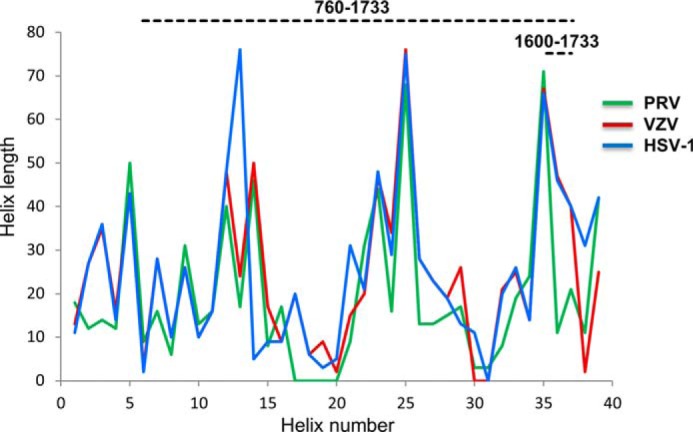
Secondary structure of the central stalk is strongly constrained across alphaherpesvirinae. The secondary structures of fragments 584–1836 of HSV-1 (blue), 522–1708 of VZV (red), and 398–1419 of PRV (green) UL36 were predicted using SOPMA. Plotted are the predicted lengths of successive helices (numbered 1–39 from N to C terminus) in these fragments. Three insertions are plotted as helices of length 0 in viruses where they are not present (helices 17–20 in PRV, helix 31 in HSV-1, and helices 30–31 in VZV). The HSV-1 and VZV curves are superimposed between helix numbers 4 and 12.
Acknowledgments
We are grateful to the ESRF and SOLEIL Synchrotron facilities for allocation of beamtime and especially to Beatriz Guimaraes for help in data collection. We thank Malika Ouldali for help in electron microscopy imaging. We thank Paloma Fernandez-Varela, Christophe Velours, and Karine Madiona of the Structural and Proteomic Biology pole of the IMAGIF integrated platform for the SEC-MALLS and analytical ultracentrifugation experiments. We acknowledge Ines Gallay of the core platform “Cristallisation et Determination de la Structure 3D” (IBBMC, Paris Sud) for crystallization of the 1600–1733 fragment. We also acknowledge Thibaut Leger and Camille Gracia of the “Plateforme Protéomique Structurale et Fonctionnelle/Spectrométrie de Masse de l'Institut Jacques-Monod” for the mass spectrometry analysis of fragments 760–1733 and 1600–1733.
The atomic coordinates and structure factors (codes 4TT0 and 4TT1) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PRV
- pseudorabies virus
- r.m.s.d.
- root mean square deviation
- SEC-MALLS
- multi-angle laser light scattering
- VZV
- varicella zoster virus.
REFERENCES
- 1. Chen D. H., Jiang H., Lee M., Liu F., Zhou Z. H. (1999) Three-dimensional visualization of tegument/capsid interactions in the intact human cytomegalovirus. Virology 260, 10–16 [DOI] [PubMed] [Google Scholar]
- 2. Grünewald K., Desai P., Winkler D. C., Heymann J. B., Belnap D. M., Baumeister W., Steven A. C. (2003) Three-dimensional structure of herpes simplex virus from cryo-electron tomography. Science 302, 1396–1398 [DOI] [PubMed] [Google Scholar]
- 3. Newcomb W. W., Brown J. C. (2010) Structure and capsid association of the herpesvirus large tegument protein UL36. J. Virol. 84, 9408–9414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhou Z. H., Chen D. H., Jakana J., Rixon F. J., Chiu W. (1999) Visualization of tegument-capsid interactions and DNA in intact herpes simplex virus type 1 Virions. J. Virol. 73, 3210–3218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aggarwal A., Miranda-Saksena M., Boadle R. A., Kelly B. J., Diefenbach R. J., Alam W., Cunningham A. L. (2012) Ultrastructural visualization of individual tegument protein dissociation during entry of herpes simplex virus 1 into human and rat dorsal root ganglion neurons. J. Virol. 86, 6123–6137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Antinone S. E., Zaichick S. V., Smith G. A. (2010) Resolving the assembly state of herpes simplex virus during axon transport by live-cell imaging. J. Virol. 84, 13019–13030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Granzow H., Klupp B. G., Mettenleiter T. C. (2005) Entry of pseudorabies virus: an immunogold-labeling study. J. Virol. 79, 3200–3205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luxton G. W., Haverlock S., Coller K. E., Antinone S. E., Pincetic A., Smith G. A. (2005) Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc. Natl. Acad. Sci. U.S.A. 102, 5832–5837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Klupp B. G., Fuchs W., Granzow H., Nixdorf R., Mettenleiter T. C. (2002) Pseudorabies virus UL36 tegument protein physically interacts with the UL37 protein. J. Virol. 76, 3065–3071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vittone V., Diefenbach E., Triffett D., Douglas M. W., Cunningham A. L., Diefenbach R. J. (2005) Determination of interactions between tegument proteins of herpes simplex virus type 1. J. Virol. 79, 9566–9571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Coller K. E., Lee J. I., Ueda A., Smith G. A. (2007) The capsid and tegument of the alphaherpesviruses are linked by an interaction between the UL25 and VP1/2 proteins. J. Virol. 81, 11790–11797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee J. I., Luxton G. W., Smith G. A. (2006) Identification of an essential domain in the herpesvirus VP1/2 tegument protein: the carboxy terminus directs incorporation into capsid assemblons. J. Virol. 80, 12086–12094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pasdeloup D., Blondel D., Isidro A. L., Rixon F. J. (2009) Herpesvirus capsid association with the nuclear pore complex and viral DNA release involve the nucleoporin CAN/Nup214 and the Capsid protein pUL25. J. Virol. 83, 6610–6623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Radtke K., Kieneke D., Wolfstein A., Michael K., Steffen W., Scholz T., Karger A., Sodeik B. (2010) Plus- and minus-end directed microtubule motors bind simultaneously to herpes simplex virus capsids using different inner tegument structures. PLoS Pathog. 6, e1000991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Luxton G. W., Lee J. I., Haverlock-Moyns S., Schober J. M., Smith G. A. (2006) The pseudorabies virus VP1/2 tegument protein is required for intracellular capsid transport. J. Virol. 80, 201–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pasdeloup D., Labetoulle M., Rixon F. J. (2013) Differing effects of herpes simplex virus 1 and pseudorabies virus infections on centrosomal function. J. Virol. 87, 7102–7112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sandbaumhüter M., Döhner K., Schipke J., Binz A., Pohlmann A., Sodeik B., Bauerfeind R. (2013) Cytosolic herpes simplex virus capsids not only require binding inner tegument protein pUL36 but also pUL37 for active transport prior to secondary envelopment. Cell. Microbiol. 15, 248–269 [DOI] [PubMed] [Google Scholar]
- 18. McElwee M., Beilstein F., Labetoulle M., Rixon F. J., Pasdeloup D. (2013) Dystonin/BPAG1 promotes plus-end-directed transport of herpes simplex virus 1 capsids on microtubules during entry. J. Virol. 87, 11008–11018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pasdeloup D., McElwee M., Beilstein F., Labetoulle M., Rixon F. J. (2013) Herpesvirus tegument protein pUL37 interacts with dystonin/BPAG1 to promote capsid transport on microtubules during egress. J. Virol. 87, 2857–2867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zaichick S. V., Bohannon K. P., Hughes A., Sollars P. J., Pickard G. E., Smith G. A. (2013) The herpesvirus VP1/2 protein is an effector of dynein-mediated capsid transport and neuroinvasion. Cell Host Microbe 13, 193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Abaitua F., Hollinshead M., Bolstad M., Crump C. M., O'Hare P. (2012) A nuclear localization signal in herpesvirus protein VP1–2 is essential for infection via capsid routing to the nuclear pore. J. Virol. 86, 8998–9014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Copeland A. M., Newcomb W. W., Brown J. C. (2009) Herpes simplex virus replication: roles of viral proteins and nucleoporins in capsid-nucleus attachment. J. Virol. 83, 1660–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jovasevic V., Liang L., Roizman B. (2008) Proteolytic cleavage of VP1–2 is required for release of herpes simplex virus 1 DNA into the nucleus. J. Virol. 82, 3311–3319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Roberts A. P., Abaitua F., O'Hare P., McNab D., Rixon F. J., Pasdeloup D. (2009) Differing roles of inner tegument proteins pUL36 and pUL37 during entry of herpes simplex virus type 1. J. Virol. 83, 105–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Desai P. J. (2000) A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA-filled capsids in the cytoplasm of infected cells. J. Virol. 74, 11608–11618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fuchs W., Klupp B. G., Granzow H., Mettenleiter T. C. (2004) Essential function of the pseudorabies virus UL36 gene product is independent of its interaction with the UL37 protein. J. Virol. 78, 11879–11889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ko D. H., Cunningham A. L., Diefenbach R. J. (2010) The major determinant for addition of tegument protein pUL48 (VP16) to capsids in herpes simplex virus type 1 is the presence of the major tegument protein pUL36 (VP1/2). J. Virol. 84, 1397–1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Svobodova S., Bell S., Crump C. M. (2012) Analysis of the interaction between the essential herpes simplex virus 1 tegument proteins VP16 and VP1/2. J. Virol. 86, 473–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jambunathan N., Chouljenko D., Desai P., Charles A.-S., Subramanian R., Chouljenko V. N., Kousoulas K. G. (2014) Herpes simplex virus 1 protein UL37 interacts with viral glycoprotein gK and membrane protein UL20 and functions in cytoplasmic virion envelopment. J. Virol. 88, 5927–5935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kattenhorn L. M., Korbel G. A., Kessler B. M., Spooner E., Ploegh H. L. (2005) A deubiquitinating enzyme encoded by HSV-1 belongs to a family of cysteine proteases that is conserved across the family herpesviridae. Mol. Cell 19, 547–557 [DOI] [PubMed] [Google Scholar]
- 31. Schlieker C., Korbel G. A., Kattenhorn L. M., Ploegh H. L. (2005) A deubiquitinating activity is conserved in the large tegument protein of the herpesviridae. J. Virol. 79, 15582–15585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bolstad M., Abaitua F., Crump C. M., O'Hare P. (2011) Autocatalytic activity of the ubiquitin-specific protease domain of herpes simplex virus 1 VP1–2. J. Virol. 85, 8738–8751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang S., Wang K., Li J., Zheng C. (2013) Herpes simplex virus 1 ubiquitin-specific protease UL36 inhibits β interferon production by deubiquitinating TRAF3. J. Virol. 87, 11851–11860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schlieker C., Weihofen W. A., Frijns E., Kattenhorn L. M., Gaudet R., Ploegh H. L. (2007) Structure of a herpesvirus-encoded cysteine protease reveals a unique class of deubiquitinating enzymes. Mol. Cell 25, 677–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sheldrick G. M. (2008) A short history of SHELX. Acta Crystallogr. A 64, 112–122 [DOI] [PubMed] [Google Scholar]
- 37. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Collaborative Computational Project No. 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 39. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Diederichs K., Karplus P. A. (2013) Better models by discarding data? Acta Crystallogr. D Biol. Crystallogr. 69, 1215–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 42. Zhu H., Domingues F. S., Sommer I., Lengauer T. (2006) NOXclass: prediction of protein-protein interaction types. BMC Bioinformatics 7, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sali A., Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 44. Case D. A., Cheatham T. E., 3rd, Darden T., Gohlke H., Luo R., Merz K. M., Jr., Onufriev A., Simmerling C., Wang B., Woods R. J. (2005) The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Berendsen H. J., Postma J. P., Gunsteren W. F., van DiNola A., Haak J. R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 [Google Scholar]
- 46. Darden T., York D., Pedersen L. (1993) Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 [Google Scholar]
- 47. Ryckaert J.-P., Ciccotti G., Berendsen H. J. (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 [Google Scholar]
- 48. Shen C., Menon R., Das D., Bansal N., Nahar N., Guduru N., Jaegle S., Peckham J., Reshetnyak Y. K. (2008) The protein fluorescence and structural toolkit: Database and programs for the analysis of protein fluorescence and structural data. Proteins 71, 1744–1754 [DOI] [PubMed] [Google Scholar]
- 49. Mijatov B., Cunningham A. L., Diefenbach R. J. (2007) Residues F593 and E596 of HSV-1 tegument protein pUL36 (VP1/2) mediate binding of tegument protein pUL37. Virology 368, 26–31 [DOI] [PubMed] [Google Scholar]
- 50. Abaitua F., O'Hare P. (2008) Identification of a highly conserved, functional nuclear localization signal within the N-terminal region of herpes simplex virus type 1 VP1–2 tegument protein. J. Virol. 82, 5234–5244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Abaitua F., Souto R. N., Browne H., Daikoku T., O'Hare P. (2009) Characterization of the herpes simplex virus (HSV)-1 tegument protein VP1–2 during infection with the HSV temperature-sensitive mutant tsB7. J. Gen. Virol. 90, 2353–2363 [DOI] [PubMed] [Google Scholar]
- 52. Abaitua F., Daikoku T., Crump C. M., Bolstad M., O'Hare P. (2011) A single mutation responsible for temperature-sensitive entry and assembly defects in the VP1–2 protein of herpes simplex virus. J. Virol. 85, 2024–2036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tullman J. A., Harmon M.-E., Delannoy M., Gibson W. (2014) Recovery of an HMWP/hmwBP (pUL48/pUL47) complex from virions of human cytomegalovirus: subunit interactions, oligomer composition, deubiquitylase activity. J. Virol. 88, 8256–8267 [DOI] [PMC free article] [PubMed] [Google Scholar]