

Background: L-type Cav1.2 Ca2+ channel undergoes extensive alternative splicing, generating functionally different channels.
Results: Neonatal rat heart expresses a higher level of a truncated Cav1.2 Ca2+ channel from alternative splicing.
Conclusion: The truncated channel can alter electrophysiological properties of a wild type channel.
Significance: Although aberrantly spliced Cav1.2 channels may not conduct Ca2+ ions, they can affect functional channels.
Keywords: Alternative Splicing, Calcium Channel, Cardiac Development, Electrophysiology, Ion Channel
Abstract
L-type Cav1.2 Ca2+ channel undergoes extensive alternative splicing, generating functionally different channels. Alternatively spliced Cav1.2 Ca2+ channels have been found to be expressed in a tissue-specific manner or under pathological conditions. To provide a more comprehensive understanding of alternative splicing in Cav1.2 channel, we systematically investigated the splicing patterns in the neonatal and adult rat hearts. The neonatal heart expresses a novel 104-bp exon 33L at the IVS3-4 linker that is generated by the use of an alternative acceptor site. Inclusion of exon 33L causes frameshift and C-terminal truncation. Whole-cell electrophysiological recordings of Cav1.233L channels expressed in HEK 293 cells did not detect any current. However, when co-expressed with wild type Cav1.2 channels, Cav1.233L channels reduced the current density and altered the electrophysiological properties of the wild type Cav1.2 channels. Interestingly, the truncated 3.5-domain Cav1.233L channels also yielded a dominant negative effect on Cav1.3 channels, but not on Cav3.2 channels, suggesting that Cavβ subunits is required for Cav1.233L regulation. A biochemical study provided evidence that Cav1.233L channels enhanced protein degradation of wild type channels via the ubiquitin-proteasome system. Although the physiological significance of the Cav1.233L channels in neonatal heart is still unknown, our report demonstrates the ability of this novel truncated channel to modulate the activity of the functional Cav1.2 channels. Moreover, the human Cav1.2 channel also contains exon 33L that is developmentally regulated in heart. Unexpectedly, human exon 33L has a one-nucleotide insertion that allowed in-frame translation of a full Cav1.2 channel. An electrophysiological study showed that human Cav1.233L channel is a functional channel but conducts Ca2+ ions at a much lower level.
Introduction
Voltage-gated calcium channels govern the depolarization-induced Ca2+ entry into cardiac muscles. The channel consists of a pore-forming α1 subunit associated with β, α2δ, and/or γ auxiliary subunits to form an oligomeric complex. In mammalian myocardium, excitation-contraction coupling is characterized by a transient increase in cytosolic Ca2+. The influx of Ca2+ through voltage-gated calcium channels subsequently induces Ca2+ release from sarcoplasmic reticulum in a process known as Ca2+-induced Ca2+ release. This process is prominent in the adult heart. The contraction of neonatal hearts relies more directly on the influx of Ca2+ through L-type voltage-gated calcium channels, in particular the Cav1.2 channels (1, 2).
L-type Cav1.2 channels are high voltage-activated channels and are expressed widely in cardiovascular and nervous systems (3). The human Cav1.2 gene, CACNA1C, contains 55 exons, of which >19 exons are subjected to alternative splicing (4). Novel splice variants of Cav1.2 channels were uncovered (5, 6) as of the first systemic screening of cardiac Cav1.2 channels (7). Alternative splicing of the Cav1.2 channels is coupled to the generation of splice variants with altered electrophysiological and pharmacological properties (4, 8–11). They also show tissue-specific expressions (8, 9, 12) and can be altered under pathological conditions (13, 14).
In this report we screened the major alternatively spliced loci of Cav1.2 channels and identified a novel exon 33L in neonatal rat hearts that has not been reported previously. Inclusion of exon 33L generates a truncated channel Cav1.233L that does not conduct Ca2+. However, Cav1.233L produced a dominant-negative effect when co-expressed with functional Cav1.2 channels.
MATERIALS AND METHODS
RT-PCR and Single-cell RT-PCR
Young adult male Wistar rats (2 months, 150–200 g) were sacrificed by CO2 and subsequent cervical dislocation. Neonatal hearts were harvested from postnatal day 1 rats of either sex. This study was approved and performed in accordance with the guidelines of the Institutional Animal Care and Use Committee of the University. Fetal human heart cDNA was purchased from Clontech. Adult human heart tissues were obtained from donors for heart transplantation in the National Heart Center of Singapore with the approval of the institutional review board. The work was approved by the Institutional Review Board committees of the National Heart Center of Singapore, Singapore General Hospital. Total RNA was isolated from the left ventricles. cDNA was generated from total RNA by incubating with reverse transcriptase and 18-mer oligo(dT). The PCR protocol includes: a denaturation step at 95 °C for 5 min; 35 cycles of 95 °C for 30 s, 52 °C or 55 °C for 45 s, and 72 °C for 1 min; a final extension step at 72 °C for 10 min. The primers used for the rat Cav1.2 exon 33L were: 5′-GCCTCTTCACGGTGGAG-3′ (forward) and 5′-TCCCAATCACTGCATAGATAA-3′ (reverse). The primers used for human Cav1.2 exon 33L are: 5′-CCAAGACCTAGAATACCGGG-3′ (forward) and 5′-CTACCACAGGGTGTTTCACC-3′ (reverse). The primers used for amplifying human Cav1.2 exons 18 to 24 as a control were: 5′-ATGAGGATAAGAGCCCCTACCC-3′ (forward) and 5′-ACTCGCAAGATCTTCACGACATTG-3′ (reverse). For single-cell RT-PCR, after electrophysiological characterization by the patch clamp method, the cells were collected into a vial containing reverse transcription reaction mix for first strand cDNA synthesis using the reverse primer. Single-cell PCR protocol was similar to RT-PCR, except for a 40-cycle amplification.
Colony Screening and Restriction Enzyme Digestion
This method was described previously to identify novel splice variations (7, 14). In brief, the PCR products were cloned into pGEM-T Easy vector (Promega). After being transformed into DH10B Escherichia coli cells, each transformant was selected and grown in a single well in a 96-well plate. Colony PCR was performed with the same set of primers and conditions to identify the component of exons in each colony. Usually 96 colonies were selected for each sample. The genotype of each PCR product amplified from a colony was predicted by size and randomly confirmed by DNA sequencing. To differentiate mutually exclusive exons that are of same size, restriction enzyme digestions were used to identify the expression of individual exons. The digestion was carried out in a 30-μl reaction volume at 37 °C for 5 h. To control for complete digestion, the same amount of PCR product of a positive control DNA was subjected to similar digestion conditions.
Sequencing, Analysis, and Cloning of 33L Channels
Rat exon 33L was verified by DNA sequencing. The sequences were compared with rat genome sequence from the Human Genome Sequencing Center, Baylor College of Medicine. To characterize the functions of exon 33L containing channels, a fragment containing exon 33L was cloned into a wild type rat Cav1.2 channel with AvrII and NheI sites. This wild type Cav1.2 channel contains the exons predominantly expressed in rat cardiac tissues.
Generation of Polyclonal Antibody against Exon 33L Polypeptide
Rat exon 33L was cloned in-frame into pGEX-4T-1. Amino acid sequences from adjacent exons were not included. GST-fused protein was purified with glutathione-agarose (Sigma). Purified protein was used to immunize female New Zealand White rabbit once a month. Complete Freund's adjuvant was first mixed with GST-33L for immunization, and incomplete Freund's adjuvant was used in subsequent injections once a month. Serum collected after immunization was preabsorbed with GST protein first to remove GST antibodies, and 33L polyclonal antibody was affinity-purified from immobilized 33L protein with an IgG elution buffer (Pierce). The antibody concentration is 1 μg/μl. Serum from rabbit before immunization was used as preimmune control.
Western Blot
For protein isolation, heart tissues were homogenized in HEPES lysis buffer: 20 mmol/liter HEPES, 137 mmol/liter NaCl, 1% Triton X-100, 10% glycerol, 1.5 mmol/liter MgCl2, and 1 mmol/liter EGTA supplemented with protease inhibitors (1:50 dilution, Roche Diagnostics). Homogenized samples were then centrifuged at 14,000 rpm for 15 min, and supernatants containing protein samples were collected. Protein concentration was determined using Bradford assay. 200 μg of lysates were separated by 8% SDS-PAGE and transferred overnight at 30 V onto PVDF membranes at 4 °C. Subsequently, the membrane was blocked with 1% BSA in 1× PBS + 0.1% Tween 20 for 1 h and probed with primary antibodies overnight at 4 °C. The primary antibodies used in the study include rabbit anti-exon 33L polyclonal (1:5000), rabbit anti-Cav1.2 or CaV1.3 (1:1000, Alomone), mouse anti-ubiquitin (1:1000, Invitrogen), and mouse anti-β-actin (1:5000, Sigma). The next day, the membrane was washed 3 times with 0.1% Tween 20 in 1× PBS. The membrane was then probed with HRP-conjugated secondary antibody for 1 h. After washing, the band was detected using Amersham Biosciences ECL Western blotting Analysis System (RPN2109, GE Healthcare).
Immunofluorescent Staining
Animals were sacrificed and perfused with saline and subsequently 4% paraformaldehyde. The heats were then collected and post-fixed with 4% paraformaldehyde for 2 h. Dehydration was subsequently carried out by immersing the hearts into 15% sucrose followed by 30% sucrose. Next, the rat heart was sectioned at 20 μm of thickness. After washing with 0.2% Triton X-100 phosphate-buffered saline (PBST), 100 μl of blocking serum (10% goat serum and 1% bovine serum albumin in 0.2% PBST) was added onto the sections for 1 h. The sections were then incubated with primary antibodies overnight at 4 °C. Monoclonal antibodies anticonnexin 43 (MAB 3068) was purchased from Chemicon International. On the following day, tissue sections were washed 3 times with TNT wash buffer (0.1 m Tris-HCl buffer, pH 7.5, containing 0.15 m NaCl and 0.05% Tween 20). The slides were incubated with FITC-conjugated or Texas red-labeled secondary antibodies for 1 h at room temperature. After washing three times of wash buffer, the slides were mounted with FluorSaveTM reagent (Merck). The results were visualized using laser scanning confocal microscope system (Fluoview BX61, Olympus). The negative control was done in an identical procedure except for primary antibody incubation, and no positive signal was identified.
Whole-cell Patch Clamp Electrophysiology
HEK293 cells were transiently transfected with rat α1 (1.25 μg), β2a (1.25 μg), and α2δ (1.25 μg) subunits with the calcium phosphate transfection method. The GFP-tagged β2a and α2δ clones were provided by Dr. Terrance Snutch (University of British Columbia). The rat Cav3.2 channel and Cav1.3 channel were described earlier (15, 16). ICa was recorded with a whole-cell patch clamp technique at room temperature (∼25 °C) 48–72 h post-transfection. Patch electrodes were pulled using a Flaming/Brown micropipette puller (Sutter Instrument) and polished with a microforge. The external solution contained 140 mmol/liter tetraethylammonium methanesulfonate, 10 mmol/liter HEPES, and 1.8 mmol/liter CaCl2 (the pH was adjusted to 7.4 with CsOH and osmolarity to 300–310 mosm with glucose). For IBa recording, 1.8 mm Ca2+ was replaced by 5 mm Ba2+. The internal solution (pipette solution) contained 138 mmol/liter Cs-MeSO3, 5 mmol/liter CsCl2, 0.5 mmol/liter ethylene glycol tetraacetic acid, 10 mmol/liter HEPES, 1 mmol/liter MgCl2, and 2 mg/ml MgATP, pH 7.3 (adjusted with CsOH). The osmolarity was adjusted to between 290 and 300 mosm with glucose. Under voltage clamp and using an Axopatch 200B amplifier (Axon Instruments), whole-cell currents were filtered at 1–5 kHz and sampled at 5–50 kHz. The series resistance was normally <5 megaohms after compensation. The capacity transient was compensated using an online P/4 protocol. The steady-state inactivation curves were obtained from experiments by stepping from a holding potential of −90 mV to a 30-ms normalizing pulse to 10 mV followed by a family of 15-s-long prepulses from −80 to +20 mV. A 104-ms test pulse to +10 mV was recorded finally. Each test pulse was normalized to the maximal current amplitude of the normalizing pulse. The steady-state inactivation (SSI)3 data were fitted with a single Boltzmann equation, Irelative = Imin + (Imax − Imin)/(1 + exp((V½ − V)/k), where Irelative is the normalized tail current, V½ is the potential for half-inactivation, and k is the slope value. G-V curves were obtained from a tail activation protocol. The cells were activated by a 20-ms test pulse of variable voltage family from −60 to 100 mV, and tail currents were measured after repolarization to −50 mV for 10 ms. The tail currents were normalized to the peak currents before fitting with a dual Boltzmann equation. G/Gmax = Flow/{1 + exp((V½,low −V)/klow)} + (1 − Flow)/{1 + exp((V½,high − V)/khigh)}, where G is the tail current, and Gmax is the peak tail current, Flow is the fraction of low threshold component, and V½,low, V½,high, klow, and khigh are the half-activation potentials and slope factors for the low and high threshold components, and V½act was calculated when G = 0.5Gmax.
RESULTS
Exon 31 and 32 Are Developmentally Regulated
Cav1.2 channel undergoes extensive alternative splicing, which affects its tissue distribution, pharmacology, and electrophysiological properties (4, 10). Although up to 20 exons are alternatively spliced (17), only exons 31 and 32 have been reported to be developmentally regulated in the heart (18). Mutually exclusive exons 31 and 32 encode the IVS3 transmembrane segment and part of the IVS2-S3 intracellular linker, whereas another alternatively spliced exon 33 encodes the IVS3-S4 extracellular linker. There exist extensive alternative splicings within this region (7) involved in many cardiovascular diseases (6, 13, 14, 19).
RT-PCR across exons 30–35 will generate a 382-bp amplicon containing exon 33 and one of the mutually exclusive exons 31 or 32. Enzyme digestion on exon 32 by NsiI produces two smaller fragments with the sizes of 68 and 314 bp, respectively (Fig. 1A). In the presence of exon 31, only one band exists (382 bp), as no digestion occurs. Deletion of the cassette exon 33 (33 nt in length) has rarely been found in rat hearts (8). When we digested RT-PCR products from the neonatal and adult rat hearts, partial digestions were observed in both samples, indicating that neonatal and adult hearts contain a mixture of exons 31 and 32. However, the extent of digestion by NsiI was different, as more PCR products from the adult hearts were digested than that from the neonatal hearts. Therefore, exon 32 expression is higher in adult rat hearts than in neonatal rat hearts (Fig. 1B). Such developmental changes of exon 31 and 32 have been reported by Diebold et al. (18), and our experiment supports the previous finding.
FIGURE 1.
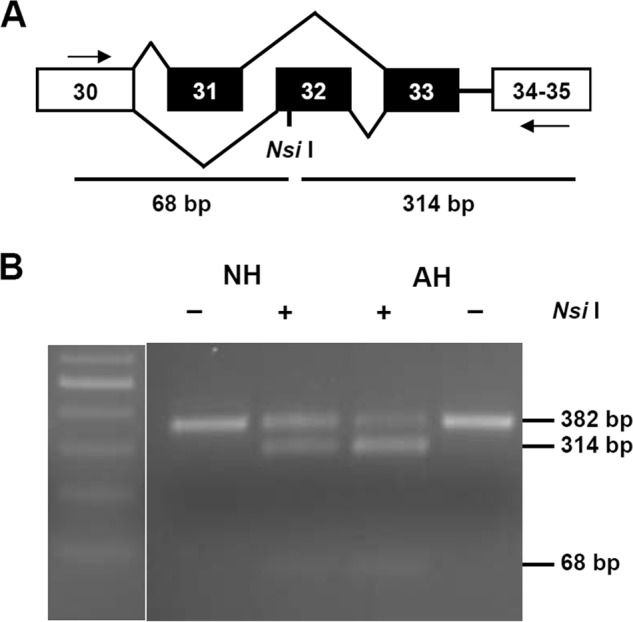
A, RT-PCR across exons 30–35 could produce a fragment of 382 bp when exon 33 (33 bp) is present. Exons 31 and 32 are mutually spliced exons. Digestion at exon 32 with NsiI will generate two fragments of 68 and 314 bp. B, partial digestion with NsiI was observed in both NH and AH. Exon 32 is expressed higher in AH as more PCR produce was digested. Equal amounts of DNA from both AH and NH were digested under same conditions in parallel.
Identification of Exon 33L from Neonatal Rat Heart
Besides mutually exclusive exons 31, 32, and cassette exon 33, there exists an additional 66-nt extension at the 5′ end of exon 34 with different acceptor sites, producing exon 34a, an isoform of exon 34 (6). To characterize the detailed expression of exon 33 and 34a, we used the colony screening method that is able to detect exons of lower expression (8). The inclusion or exclusion of exons 33 and/or 34a could be easily identified by differences in size of the PCR products, whereas mutually exclusive exons 31 and 32 cannot, as they are of the same length. Colony PCR screening showed that the size of the majority of colonies was ∼382 bp, indicating that the splice variants with exon 33 deletion or exon 34a inclusion are not predominant in neonatal and adult rat hearts (Fig. 2, A and B). DNA sequencing confirmed that the colonies with smaller size contained only exon 31 or 32 but with the exon 33 deletion. However, some colonies of a larger size have been found to appear more frequently in the neonatal heart than in the adult heart (indicated by arrows in Fig. 2, A and B). Subsequent DNA sequencing confirmed that these colonies did not contain exon 34a, but expressed a novel exon, which is named exon 33L here. We repeated RT-PCR-based colony screening experiments and confirmed that neonatal hearts (NH) express 9.7% of exon 33L, which represents a >2-fold increase to the 4.3% in adult hearts (AH) (Fig. 2C, p = 0.0127, NH (n = 5), AH (n = 5), Student's t test).
FIGURE 2.
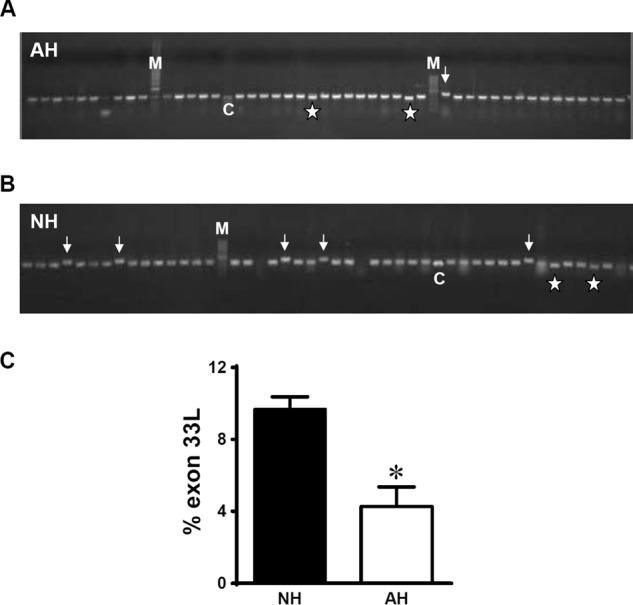
Identification of exon 33L from rat neonatal hearts. A and B, PCR-based colony screening on rat AH and NH using primers across exons 30–35. M, marker; C, control DNA with exon 33 (33 bp) deletion. Asterisk, colonies with exon 33 deletions. Arrows, colonies with novel exon 33L inclusion. C, summary of the expression of exon 33L in NH (n = 5) and AH (n = 5). *, p = 0.0127, unpaired Student's t test.
Exon 33L contains an additional 71-nt 5′ to exon 33 (Fig. 3A). To understand the nature of this 71-nt fragment, we searched the rat genome sequence from the Human Genome Sequencing Center from Baylor College of Medicine. The mechanism for the generation of the additional 71-nt 5′ of exon 33 can be rationalized by the inspection of the genomic sequence of the rat Cav1.2 channel gene. These 71 nt locate at chromosome 4 and 5′ to exon 33 of the rat Cav1.2 gene. No genomic sequence was found between the 71 nt and exon 33. More importantly, the canonical -ag- acceptor dinucleotides were found 5′ to the 71 nt. Therefore, the inclusion of the 71 nt could be explained by the use of an alternate acceptor site of exon 33. However, the inclusion of 71 nt will result in a frameshift of the coding sequences and produce a premature stop codon (Fig. 3, A and B). As the premature stop codon appeared in the IVS3-4 linker, it will generate a truncated channel lacking the last three transmembrane segments of domain IV and the entire C terminus (Fig. 3C). Thus, there exists two isoforms of exon 33. One is the original 33-nt exon 33 and the other is the new 104-nt exon 33L (71 nt + 33 nt). We further compared the DNA sequencing results and found that exon 33L can be linked either with exon 31 or exon 32.
FIGURE 3.
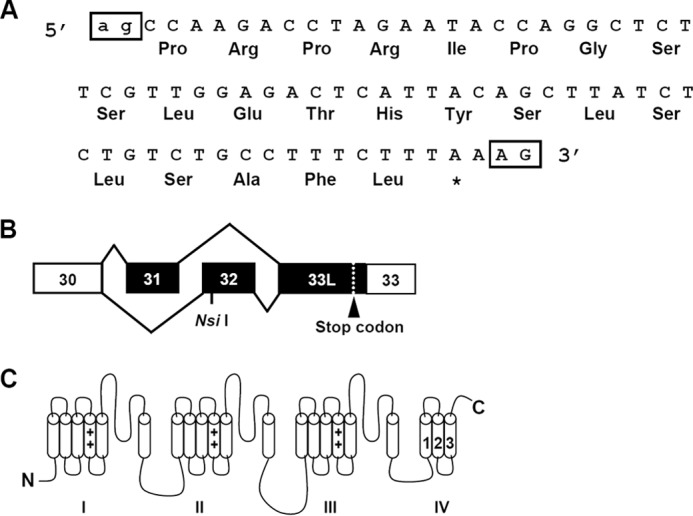
A, exon 33L contains an additional 71 nt that are 5′ to exon 33, encoding 22 amino acids with a frameshift. The canonical acceptors -ag- for exon 33L and -AG- for exon 33 are labeled in boxes. B, with the presence of either mutually exclusive exons 31 or 32, a stop codon is generated in exon 33L. C, exon 33L containing the Cav1.2 channel is truncated with the deletion of IVS4 and downstream entire C terminus.
Translational Expression of Exon 33L in Heart
To determine whether the aberrant splice variants are translated into proteins, a polyclonal antibody was generated against the 22-amino acid polypeptides encoded by exon 33L (Fig. 3A). The 22-amino acid polypeptides were expressed as a fusion protein with GST (Fig. 4A) and were injected into rabbits to produce exon 33L specific antibody α-33L. No adjacent amino acids were included for antibody production. Using α-33L, we detected by Western blot a band of 160 kDa in neonatal and adult hearts, having the same molecular mass of the truncated 33L containing Cav1.2 channel expressed in HEK 293 cells (Fig. 4B).
FIGURE 4.

Generation and characterization of a polyclonal antibody against exon 33L. A, The 22-amino acid polypeptides encoded by exon 33L were fused with GST (GST-33L) and purified for rabbit immunization. BSA standards of 1, 2, and 5 μg are shown. M, protein marker. C, crude lysate. B, representative Western blot using the exon 33L-specific antibody (α-33L) showed a band of 160 kDa in both NH and AH. Full-length Cav1.2 channel (∼270 kDa) and actin (42 kDa) were probed as controls. Lane 1, HEK cells transfected with pcDNA3 vector; lane 2, HEK cells transfected with Cav1.233L; lane 3, HEK cells transfected with wild type Cav1.2; lane 4, AH; lane 5, NH. C, immunofluorescent staining of Cav1.233L- or Cav1.2WT-transfected HEK cells with polyclonal antibody α-33L (red). GFP-tagged β2a was co-transfected together with the α1 subunits. D, representative immunofluorescent staining showed exon 33L expression in NH and AH. Connexin 43 (Cx43) was co-stained, and nuclei were labeled with DAPI. Scale bar: 50 μm.
Therefore, neonatal rat hearts expressed higher levels of Cav1.233L channel than adult hearts. To prove the specificity of α-33L antibody, HEK cells were transfected with Cav1.233L channel or the reference channel Cav1.2WT, which does not contain exon 33L. α-33L antibody stained Cav1.233-transfected cells but not Cav1.2WT-transfected cells (Fig. 4C). Next, we used an immunohistological method to examine the cellular expression of the Cav1.233L channel in rat hearts during development. Connexin 43 was labeled for the gap junctions between adjacent cardiac muscle cells. Again, α-33L staining showed a stronger fluorescent signal in neonatal heart than in adult heart, supporting a higher expression of Cav1.233L channel in neonatal rat heart (Fig. 4D). It should be noted that Cav1.233L did not colocalize with Connexin 43 in both neonatal and adult hearts, suggesting that the majority of Cav1.233L channels may not traffic to the plasma membrane.
Presence of Cav1.233L Channels Reduces Currents of Functional Cav1.2 Channels
The functional significance of the inclusion of exon 33L in Cav1.2 channels has not been reported. We sought to assess exon 33L functions by whole-cell patch clamp recording. Exon 33L was cloned into a reference rat cardiac-form Cav1.2 channel that contained exon 1a and 8a, with the absence of exon 9* (8). To control for transfection efficiency and the conditions of the HEK 293 cells, we performed the experiments in parallel by transfecting the HEK 293 cells on the same day and did the electrophysiological experiments alternately using the same solutions on the same day. The presence of successful transfection of Cav1.233L channels was evidenced by the single cell RT-PCR. All the cells patched with green fluorescence (β2a is tagged with GFP) contained Cav1.233L channels. The cells were maintained at a −90 mV holding potential before a test pulse of 0 mV for 900 ms was applied. No currents were recorded (Fig. 5A, n = 18) for Cav1.233L channel, indicating that the presence of exon 33L generates a novel Cav1.2 channel that is impermeable to Ca2+ ions. To investigate whether the presence of the 33L channels affects the reference cardiac channels, we co-transfected the 33L channels and the reference channels into HEK cells. The exon 33L expression in neonatal heart and adult heart is 9.7% and 4.3%, respectively (Fig. 2C). Therefore, we transfected Cav1.233L channels into HEK 293 cells at 5 and 10% to the reference wild type channels. Tail currents were recorded in 5 mm Ba2+ to exclude Ca2+-related effects. A slightly higher current density was observed in 5% Cav1.233L channels. However, there was no statistical significance between 5 and 10% Cav1.233L channels (Fig. 5B).
FIGURE 5.
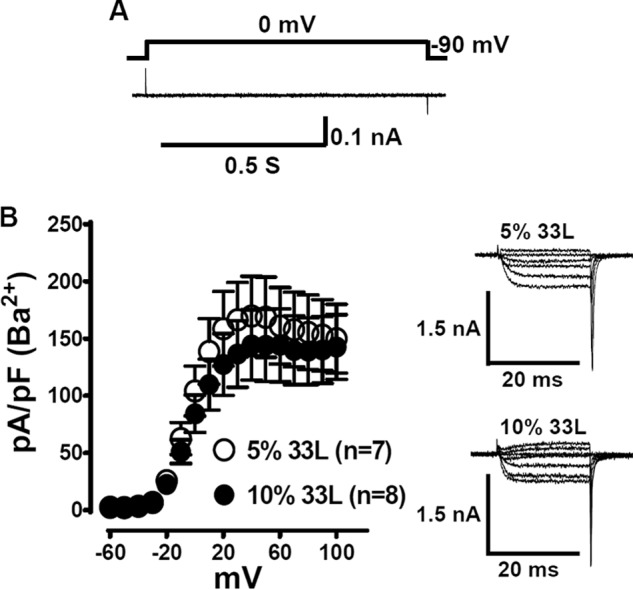
Electrophysiological characterization of exon 33L containing Cav1.233L channel. A, Cav1.233L channels were co-expressed with β2a and α2δ subunits in HEK cells. No Ca2+ or Ba2+ current was recorded when the cells were depolarized to 0 mV from a holding potential of −90 mV (n = 18). B, Cav1.233L channels were transfected into HEK cells at 5% or 10% to the reference channels. Tail current densities were measured in 5 mm Ba2+-containing bath solutions. No statistical difference was identified.
It is possible that the effect of Cav1.233L channels on the reference channels is masked by the variability of the data recorded. We decided to increase the 33L channel ratio by transfecting equal amounts of both Cav1.233L channels and the reference channels into HEK 293 cells for electrophysiological recordings. The control experiments were carried out by transfecting the same amount of empty pcDNA3 vector together with the reference channels. Single cell RT-PCR was performed to confirm that both the 33L channels and the reference channels were equally expressed (Fig. 6A). The data collected from cells with unequal expression were discarded. Similar to the dominant-negative effect observed by Ebihara et al. (20) in the three-domain Cav1.2 channels, the presence of Cav1.233L channels reduced the tail current density of the reference Cav1.2 channels (Fig. 6B) in 1.8 mm Ca2+ bath solution, mimicking a physiological condition. The tail current density at +100 mV was 41.6 ± 13.2 pA/pF (n = 7) when control pcDNA3 vectors were co-transfected with the reference channels, whereas the tail current density was 11.3 ± 2.3 pA/pF (n = 9) when Cav1.233L channels were co-transfected, representing a 73% decrease in the presence of Cav1.233L channels (p = 0.025, Student's t test). Measuring the ON-gating charge (QON) at the reversal potential (typically +50 mV, Fig. 6C) provides a convenient measure of the number of channels at the membrane (21). Therefore, we measured the QON of the Cav1.233L- and pcDNA3-transfected cells. Interestingly, 33L transfection greatly reduced the QON as compared with pcDNA3-transfected cells: 33L QON, 0.8374 ± 0.2296 femtocoulombs/pF, n = 7; pcDNA3 QON, 2.499 ± 0.4639 femtocoulombs/pF, n = 6; p = 0.0063, Student's t test. The reduction of 66.5% in QON was similar to the 73% reduction in current density.
FIGURE 6.
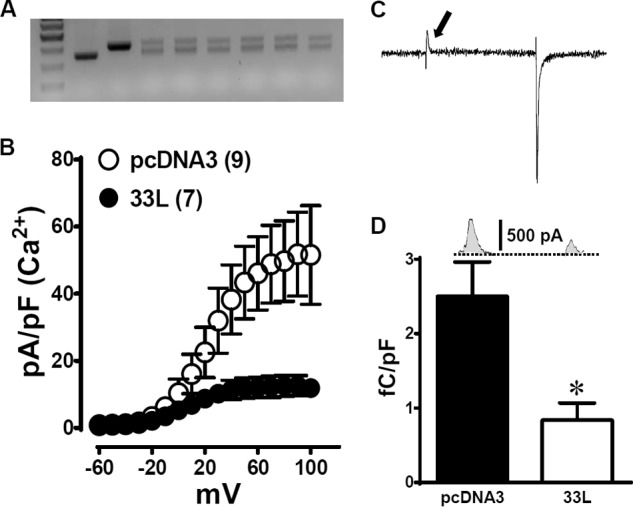
At a 1:1 ratio, Cav1.233L channels yield a dominant negative effect on the reference cardiac Cav1.2 channels. A, single-cell RT-PCR was performed after each patching experiment. The upper bands represent exon 33L, whereas lower bands represent the reference channel. Data from cells with unequal transfection were discarded. B, tail current density (pA/pF) was calculated in 1.8 mm Ca2+ from cells expressing the reference cardiac Cav1.2 channels together with the Cav1.233L channels (●, n = 7) or pcDNA3 vector (○, n = 9). C, a sample trace at +50-mV test pulse illustrates the measurement of gating current, indicated by an arrow. D, summary of the gating currents (femtocoulombs/pF (fC/pF)) measured from 33L channel or pcDNA3 transfected cells. *, p = 0.0063, Student's t test.
Cav1.233L Channels Exert a Dominant Negative Effect on Cav1.3 Channels but Not on Cav3.2 Channels
To examine whether Cav1.233L channels down-regulate other voltage-gated calcium channels, we cotransfected Cav1.233L channels with the paralogous L-type Cav1.3 channels or with the T-type Cav3.2 channels at a ratio of 1:1. Again, pcDNA3 vector was used as a control. One difference between Cav1.3 channels and Cav3.2 channels is that Cav1.3 channels require the β subunit for functional expression, whereas Cav3.2 channels do not need the β subunit. Current densities from I-V curves were measured in 1.8 mm Ca2+ solution. Interestingly, the dominant negative effect of Cav1.233L channels is prominent in Cav1.3-expressed cells but not in Cav3.2-expressed cells (Fig. 7). Compared to the pcDNA3 control, the presence of Cav1.233L channels reduced Cav1.3 current densities at −10 mV from −16.16 ± 2.205 pA/pF (n = 8) to −7.952 ± 1.250 pA/pF (n = 15); p = 0.0021, Student's t test (Fig. 7A). However, in Cav3.2-expressed cells, the current densities were similar in pcDNA3 and Cav1.233L-transfected cells (Fig. 7B). At −20 mV, the current densities of Cav1.233L-expressed cells were −16.93 ± 4.158 pA/pF, n = 9, whereas those in pcDNA3-expressed cells were −17.13 ± 3.257 pA/pF, n = 7 (p = 0.9698, Student's t test).
FIGURE 7.
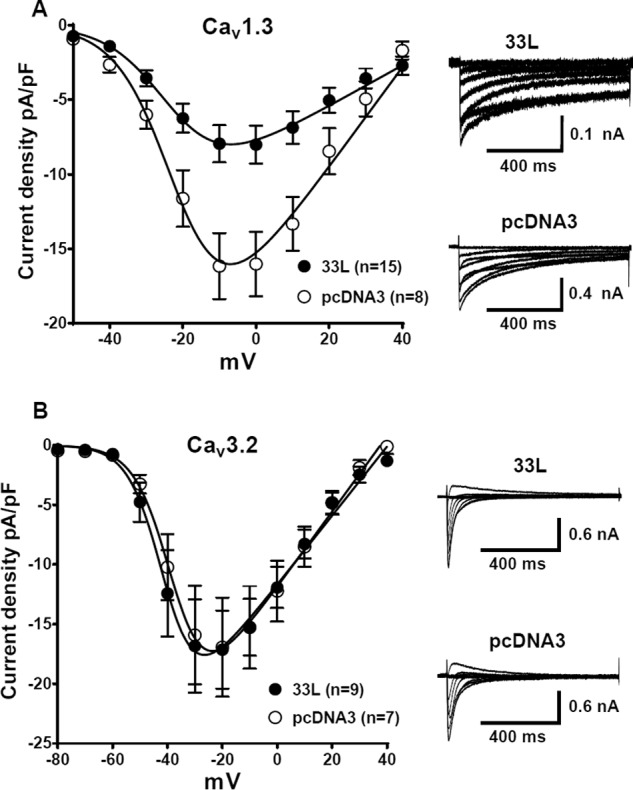
Cav1.233L channels yield dominant negative effect on Cav1.3 channels but not on Cav3.2 channels. Cav1.233L channels were cotransfected with Cav1.3 or Cav3.2 channels at a ratio of 1:1. I-V curves were obtained in a bath solution containing 1.8 mm Ca2+. As a control, pcDNA3 vectors were transfected at the same molar ratio. A, in Cav1.3 channels, which require β subunits, Cav1.233L channels greatly inhibited Cav1.3 currents. At −10 mV, the reduction of current density from −16.16 ± 2.205 pA/pF (n = 8) to −7.952 ± 1.250 pA/pF (n = 15), p = 0.0021, Student's t test, represents an almost 50% decrease. B, Cav1.233L cotransfection had no effect on Cav3.2 channels, which do not require β subunits.
Presence of Cav1.233L Channels Changes Electrophysiological Properties of Functional Cav1.2 Channels
To investigate whether the electrophysiological properties of the reference cardiac channel is altered with the presence of Cav1.233L channels, we used a recording protocol for the SSI and a tail protocol for the activation properties, which were described previously (8, 9). Interestingly, the presence of Cav1.233L channels negatively shifted both the SSI and the activation properties of the reference channels (Fig. 8). The V½ of the SSI was hyperpolarize-shifted for −3.54 mV (p = 0.0174, Student's t test), V½SSIpcDNA (−22.96 ± 0.86 mV (n = 6)) versus V½SSI33L (−26.5 ± 0.9 mV (n = 6)), whereas the V½ of the activation curve was hyperpolarize-shifted for −5.73 mV (p = 0.0036, Student's t test), V½ActpcDNA (18.43 ± 1.05 mV (n = 9)) versus V½Act33L (12.7 ± 1.28 mV (n = 7)). Therefore, the calcium window current of the reference cardiac channel was shifted to a more negative potential in the presence of Cav1.233L channels.
FIGURE 8.
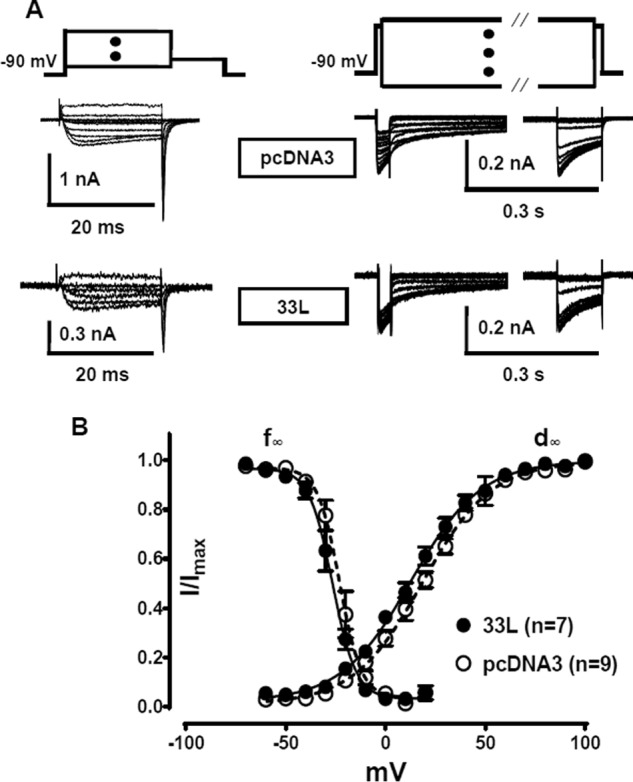
Effect of Cav1.233L channels on the gating properties of the reference Cav1.2 channels when transfected at 1:1 ratio. A, sample traces from the recordings for the tail currents and steady-state inactivation. B, plots of steady-state inactivation (f∞) and activation (d∞) curves of the reference cardiac Cav1.2 channels co-expressed with Cav1.233L (●) and pcDNA3 vector (○). Cav1.233L channels slightly shifts activation and SSI curves of the reference channels toward negative potential. All recordings were performed in 1.5 mm Ca2+ bath solution. The protocols and fittings were described under “Materials and Methods.”
Cav1.233L Channels Down-regulate Functional Cav1.2 Channels by Enhancing Protein Degradation via the Ubiquitin-Proteasome System
To investigate the mechanism for the dominant negative effect of Cav1.233L, the channels were first co-transfected with the reference channels at 1:1 ratio. A Western blot showed that the total protein level (membrane and cytosol) of the reference Cav1.2 channels was reduced by the presence of Cav1.233L (Fig. 9A). Treatment with 3 μm MG132, a proteasome inhibitor, successfully increased the expression of the reference Cav1.2 channels, suggesting that Cav1.233L might enhance proteasomal degradation. A similar down-regulation effect of Cav1.233L was observed on Cav1.3 channels (Fig. 9B) but with a smaller reduction. Next, we examined protein ubiquitination using immunoprecipitation and subsequent Western blot. Interestingly, transfection with Cav1.233L greatly enhanced wild type Cav1.2 channel ubiquitination. The ubiquitinated Cav1.2 channels were further increased with the treatment of 3 μm MG132 (Fig. 9C). Again, Cav1.3 channel ubiquitination was also increased with Cav1.233L co-expression (Fig. 9D).
FIGURE 9.
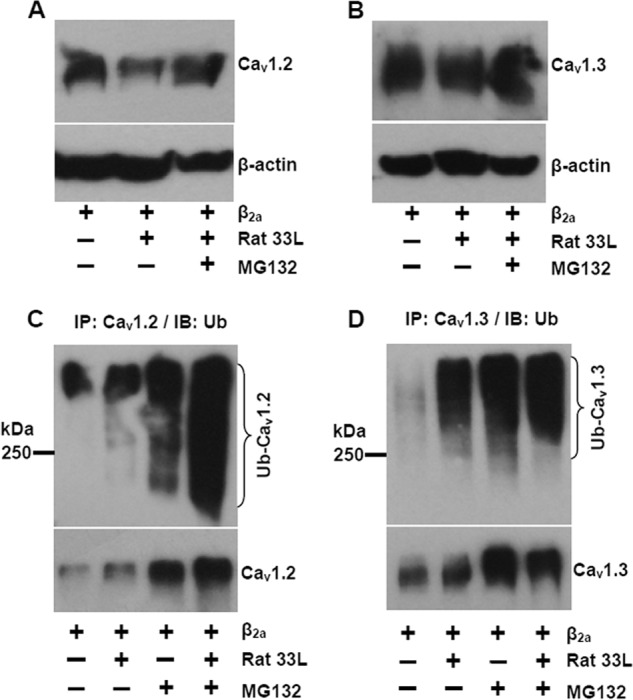
Cav1.233L channels down-regulates the reference Cav1.2 channels by enhancing protein degradation via ubiquitin-proteasome system. A, Western blot for the expression of the reference Cav1.2 channels with or without Cav1.233L channels and proteasome inhibitor MG132 (3 μm). B, Western blot for the expression of Cav1.3 channels with or without Cav1.233L channels and MG132. C, immunoprecipitation (IP) using anti-Cav1.2 antibody to detect ubiquitinated (Ub) Cav1.2 channels with or without Cav1.233L channels and MG132 treatments. Ubiquitinated Cav1.2 channels were labeled by immunoblotting (IB) using a mouse anti-ubiquitin antibody. D, ubiquitination of Cav1.3 channels with or without Cav1.233L channels and MG132 treatments. The results are representative of three independent experiments.
Human Exon 33L-containing Channel Conducts Ca2+ Ions
When we compared the exon 33L sequences of mouse, rat, and human (Fig. 10A), we found that the acceptor for exon 33L is preserved across the three species, indicating that exon 33L may exist in all these species. The mouse and the rat shared exactly the same sequences of exon 33L, whereas the human owns a different sequence. Interestingly, the human genomic sequence had an additional base within exon 33L compared to mouse or rat. Therefore, no frameshift was generated (Fig. 10 B), and the human Cav1.233L channel may conduct Ca2+ ions. To prove this, we first performed RT-PCR on human heart samples using primers specific for exon 33L. The sequences of the primers are described under “Materials and Methods.” A band with a size of 502 bp was found in the left and right ventricles of a human adult heart and in the left ventricle of a fetal human heart (Fig. 10C). DNA sequencing confirmed that the bands contained human exon 33L and exon 33 sequences, as predicted in Fig. 10A. The human fetal heart expressed a higher level of exon 33L as compared with adult hearts, which is similar to the rat exon 33L expression during cardiac development. Next, we cloned the human exon 33L into a reference Cav1.2 channel and performed patch clamp experiments. Fig. 10D shows that the human Cav1.233L channel conducted Ca2+ ions when it was cotransfected with β2a and α2δ subunits. Compared to the wild type reference Cav1.2 channel, the Cav1.233L channel had a lower current density. For the Cav1.233L channel, current density at 0 mV was −4.095 ± 0.8472, n = 15, whereas in wild type channel the current density was −10.85 ± 1.668; n = 15; p = 0.0012, Student's t test. Conductance was further calculated using the equation G = I/(Vm − ECa) and normalized by the maximum conductance value and fit with the Boltzmann equation (Fig. 10E), where ECa is reverse potential. The V½ of human Cav1.233L channel was −9.17 ± 1.24 mV, which is significantly higher than the V½ of the wild type channel (−13.3 ± 0.6 mV, p = 0.0056).
FIGURE 10.

Human Cav1.233L channels conduct Ca2+ ions and are developmentally regulated in heart. A, alignment of the exon 33L DNA sequences from Rattus norvegicus, Mus musculus, and Homo sapiens. The canonical -ag- acceptors (indicated in the gray box) is preserved in all three species. The mouse and the rat share the same exon 33L sequence. Human exon 33L is predicted to contain an additional nucleotides. All the differences in DNA sequences are highlighted by boxes. B, alignment of the amino acid sequences of exon 33L predicted from R. norvegicus, M. musculus, and H. sapiens. The differences in amino acid sequences are labeled by boxes. No stop codon is generated by the inclusion of human exon 33L. C, using human exon 33L-specific primers, a band of 502 bp was found in human adult left ventricle (LV), adult right ventricle (RV), and fetal left ventricle (FH). M, marker. DNA sequencing confirmed the sequence to be human exon 33L. PCR using primers across exon 18–exon 24 was illustrated as the loading control. As a water control (–), cDNA was replaced by water to exclude possible contaminations. D, human exon 33L was cloned into a Cav1.2 reference channel. I-V curves were obtained in a bath solution containing 1.8 mm Ca2+. In HEK cells, human Cav1.233L channels indeed conduct Ca2+ ions with a smaller current density. E, conductance was calculated as G = I/(Vm − ECa) and normalized by the maximum conductance value and fit with Boltzmann equation, where ECa is reverse potential.
DISCUSSION
Truncated Na+ and Ca2+ channels have been found in brain and muscles (22, 23). In rabbit heart aberrant splicing in the II-III loop of Cav1.2 channels produces two truncated channels that contain only domains I and II (23). The truncation is caused by the deletion of exons 17 and 18 or the deletion of exon 19, which results in a frameshift leading to premature termination of translation. In contrary, alternative splicing of exon 33L of the Cav1.2 channels we report here yielded 2 significant findings. 1) The functional modification is species-specific in that rat exon 33L results in a truncated channel that likely competes for β subunit binding to wild type channels. However, the human exon 33L has a single nucleotide insertion resulting in a longer functional full-length channel that, however, produced reduced current density. 2) The truncated rat Cav1.233L variant not only competes for β subunit binding to expose the wild type Cav1.2 channel to presumably ERAD degradation (24), but it also similarly down-regulated the expression of another β subunit requiring L-type Cav1.3 channel but not the T-type Cav3.2 channel.
The presence of rat exon 33L shifts the coding frame and leads to a deletion of the last three transmembrane segments of the domain IV and the downstream C terminus. Therefore, the predicted topography of this rat Cav1.233L channels is larger than the two domain channels reported previously (23).
Rat Cav1.233L channels do not conduct Ca2+ ions, indicating that the last three transmembrane segments within domain IV are critical in forming a complete functional channel. The pore of the channel is likely to be disrupted in particular with the deletion of IVS4 containing positively charged amino acids. Another result from this study is that the presence of Cav1.233L channels could down-regulate the current flux through the reference wild type channels. The inhibitory effect is not prominent when Cav1.233L channels are expressed at 5% or 10%, possibly due to data variability when recording small changes in current density. However, when Cav1.233L was expressed in equal amount to the reference channels, the dominant negative effect is clear. We found a 73% current density reduction, largely in correlation with a 66.5% decrease in gating current, indicating that the current density reduction is mainly due to a decrease of the functional Cav1.2 channels on the cell membrane. The slight difference between the changes in current density and gating current (∼6.5%) suggests that a small number of channels on the surface membrane may not conduct Ca2+ ions. This small population of channels is likely the truncated Cav1.233L channels. Immunostaining of Cav1.233L channels in rat neonatal and adult hearts suggests that the majority of Cav1.233L channels were not expressed on the plasma membrane. Therefore, only very few Cav1.233L channels may traffic to the membrane, contributing to a small portion of the gating currents.
The mechanism for the inhibitory effect of Cav1.233L channels could be explained in that Cav1.233L channels compete with the reference channels, presumably for β subunit for expression on the plasma membrane. It is well known that the β subunit is required to target the functional L-type Ca2+ channels to the cell surface (25), and the binding requires a conserved motif in the domain I-II cytoplasmic loop of the Cav1.2 channels (26). As the I-II loop is well preserved in Cav1.233L channels, the expression of this truncated channel may scavenge β subunits and, therefore, down-regulate the surface expression of the reference channels. This mechanism is supported by a drastic reduction of gating currents caused by Cav1.233L channels. Another possible mechanism is that the truncated channels may prevent the correct folding of wild type channels and inhibit channel synthesis or stability. This hypothesis was shown by Dolphin and co-workers (27) in two domain truncated Cav2.2 channels, in which the dominant negative effect is independent on β subunit. Therefore, such modulation is generally channel-specific, seldom affecting other channels with a different protein structure. Our results from experiments performed on Cav1.3 and Cav3.2 channels suggest that the second mechanism may not explain the effect Cav1.233L has on wild type Cav1.2 and Cav1.3 channels. The dominant negative effect of Cav1.233L channels was exerted as well on Cav1.3 channels, which require β subunits, but not on Cav3.2 channels, which do not need β subunits, suggesting that β subunits are critical for Cav1.233L suppression. The binding of β subunits has been demonstrated by the Zamponi and co-workers (24) to increase membrane expression of Cav1.2 channels by preventing endoplasmic reticulum-associated protein degradation. Without β subunits, Cav1.2 channels undergo robust ubiquitination. Therefore, if Cav1.233L channels compete with wild type Cav1.2 channels for β subunits, wild type channels without β subunit association will be degraded. Our biochemical results support this hypothesis. In the presence of Cav1.233L channels, wild type Cav1.2 channel ubiquitination was enhanced for degradation, and thus total protein level was reduced.
However, we cannot exclude the mechanism proposed by Dolphin completely. If the number of intracellular β subunits is fixed, the expression of Cav1.233L channels should correlate with the inhibitory effect on the reference channels, which is an ∼50% decrease when the Cav1.233L channel and the reference channel are expressed at a 1:1 ratio. However, the current density is reduced by 73%, more than the predicted 50%, indicating the presence of the second mechanism. Furthermore, the Cav1.233L channel suppressed the current density in Cav1.3 channels from −16.16 pA/pF to −7.952 pA/pF, representing an ∼50% reduction, strongly correlating with the molar ratio of Cav1.233L channels to Cav1.3 channels, which is 1:1. As Cav1.3 channels have a different structure from Cav1.2 channels, the second mechanism is unlikely to apply to Cav1.3 channels. Therefore, we would hypothesize that the inhibitory effect of Cav1.233L channels on Cav1.3 channels is solely via competing for β subunits.
Cav1.233L channels are also different from another three-domain Cav1.2 channel that was reported earlier (20). In that truncated channel, the N terminus and the first domain as well as the entire I-II loop are deleted. Thus, there will be no competition for β subunits from this three-domain channel, which could explain why the inhibitory effect was not found on other channels. Interestingly, the three-domain Cav1.2 channel exhibits a more prominent dominant-negative effect on wild type channels compared with the Cav1.233L channel. As the C terminus is well preserved in the three-domain Cav1.2 channel and the rest of the channel is the same as in the Cav1.233L channel, the potent inhibitory effect is likely due to the intact C terminus. Zamponi and co-workers (24) showed that the endoplasmic reticulum retention motif of Cav1.2 channel locates at the C terminus and likely interferes with the β subunits and the I-II linker sterically or allosterically in a properly folded channel. The three-domain Cav1.2 channel with a complete C terminus may interrupt such interactions, yielding a dominant negative effect. Additional experiments are, therefore, needed.
Our results further showed that the presence of Cav1.233L channels could alter the electrophysiological properties of the reference cardiac Cav1.2 channels. The negative shifting of SSI and activation curves produced a calcium window current that peaks at more hyperpolarized membrane potential. The exact mechanism of such alterations is not fully understood. In a previous study on Cav1.1 channel (α1S), the two-domain (I and II)-containing hemichannels were expressed on the cell membrane, and the gating characteristics were modified significantly, presumably by interactions with domains III and IV (28). Thus, it is possible that the interaction between the intact I and II domains of 33L channels with the reference Cav1.2 channels may change the electrophysiological properties of those reference channels. The deletion of the C terminus in 33L channels may also cause the electrophysiological property changes in reference channels. The C terminus of Cav1.2 channels is known to regulate Ca2+-dependent and voltage-dependent channel inactivation (29). Therefore, the interaction with the truncated 33L channels may alter the regulatory functions of C terminus in the reference channel. Additional experimentations are needed to elucidate the regulatory role of the 33L channels on functional Cav1.2 channels.
The truncated Cav1.233L channel may not have a major physiological impact on neonatal heart function, as the expression level is only at 9.7% and even lower in adult hearts. However, during the search for more altered exonal expression in neonatal hearts, at least one more site was identified in the IIIS2 region involving exons 21 and 22 in forming an aberrant channel (data not shown). This aberrant channel again loses the ability for Ca2+ influx but exhibits a dominant-negative effect on functional channels. Therefore, we believe that aberrant channels in neonatal hearts may have some functions during cardiac development. More experiments with gene manipulation can help to identify the physiology of these aberrant channels.
When we searched human genomic sequence of Cav1.2 channel, we identified a similar acceptor site for human exon 33L. Unexpectedly, human exon 33L sequence contained an additional nucleotide and was unlikely to generate a stop codon that is found in rat exon 33L. We successfully cloned human exon 33L into a reference Cav1.2channel, and electrophysiology studies on these channels eventually showed that they are functional in conducting Ca2+ ions, albeit with a smaller current density. At 0 mV, the current density of human Cav1.233L channels is only ∼40% that in wild type channels. The mechanism for the smaller current remains unknown. Exon 33L codes for the extracellular loop between IVS3 and IVS4, which has been shown to affect channel activation property (7). It was proposed that a longer IVS3-S4 loop shifts the activation potential to a more positive potential. This trend was previously shown for alternatively spliced exon 33, which is of 33 bp in length. Deletion of exon 33 in smooth muscles shifts the activation and inactivation properties of Cav1.2 channels to a more negative potential. Such structural variations are believed to contribute to the electrophysiological differences between cardiac and smooth muscle Cav1.2 channels (8). With the inclusion of human exon 33L, which encodes an additional 24 amino acids, the loop between IVS3 and IVS4 is much longer and may change the channel properties as well. Indeed, when we compared the activation properties of the human Cav1.233L channel with the wild type channel, the V½ of the human Cav1.233L channel is significantly more positive than that of wild type channel.
Interestingly, human exon 33L is also developmentally regulated in heart, with a similar trend as seen in rat exon 33L, suggesting that the mechanism for regulating exon 33L expression during cardiac development is conserved in human and rodents. In the brain, two exons, 9* and 33, of Cav1.2 channel are developmentally regulated (30). Fox proteins 1 and 2 regulate the expression of the two exons during embryonic mouse brain development. In this study, the longer exon 33L is formed by adopting an alternate acceptor site. As there is no intron between exon 33L and the downstream exon 33, the splicing mechanism of 33L is likely regulated by Fox proteins as well. The increase of Fox proteins during embryonic development causes more inclusion of exon 33 in cerebral Cav1.2 channel (30). Therefore, the lower level of Fox proteins in fetal rats may affect the selection of the acceptor sites for exon 33 or exon 33L. Further experiments in changing the Fox binding elements locating within the intron after exon 33 will help to elucidate the underlying mechanism.
To the best of our knowledge, only one report showed an alteration of the structure of IVS3 segment (encoded by alternatively spliced exons 31/32) of Cav1.2 channels during cardiac development (18). Interestingly, subsequent studies revealed a reappearance of the fetal Cav1.2 channel in hearts of rat myocardial infarction and human heart failure (13, 19), indicating an alteration of Cav1.2 channel in response to cardiac malfunctions.
In conclusion, we report here for the first time that the neonatal rat heart expresses a higher level of an aberrant truncated Cav1.233L channel. Albeit not permeable to Ca2+ ions, Cav1.233L channels affect the functional reference channels, mainly via competing for β subunits. Due to a genomic difference, human Cav1.233L channels are complete and functional channels that conduct Ca2+ ions. Importantly, human exon 33L is also developmentally regulated in heart. Neonatal Cav1.2 splice variants have been shown to re-emerge in chronic heart diseases (13, 19). Therefore, it is important to understand whether these neonatal channels appear and function in cardiac diseases.
This work was supported by a grant from Singapore Ministry of Health's National Medical Research Council (to T. W. S.) and a grant from Singapore Ministry of Health's National Medical Research Council (to P. L.).
- SSI
- steady-state inactivation
- nt
- nucleotide(s)
- NH
- neonatal heart
- AH
- adult heart
- pF
- picofarad.
REFERENCES
- 1. Vornanen M. (1996) Excitation-contraction coupling of the developing rat heart. Mol. Cell Biochem. 163, 5–11 [DOI] [PubMed] [Google Scholar]
- 2. Vornanen M. (1996) Contribution of sarcolemmal calcium current to total cellular calcium in postnatally developing rat heart. Cardiovasc. Res. 32, 400–410 [DOI] [PubMed] [Google Scholar]
- 3. Catterall W. A. (2000) Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev. Biol. 16, 521–555 [DOI] [PubMed] [Google Scholar]
- 4. Liao P., Yong T. F., Liang M. C., Yue D. T., Soong T. W. (2005) Splicing for alternative structures of Cav1.2 Ca2+ channels in cardiac and smooth muscles. Cardiovasc. Res. 68, 197–203 [DOI] [PubMed] [Google Scholar]
- 5. Cheng X., Liu J., Asuncion-Chin M., Blaskova E., Bannister J. P., Dopico A. M., Jaggar J. H. (2007) A novel Cav1.2 N terminus expressed in smooth muscle cells of resistance size arteries modifies channel regulation by auxiliary subunits. J. Biol. Chem. 282, 29211–29221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tiwari S., Zhang Y., Heller J., Abernethy D. R., Soldatov N. M. (2006) Atherosclerosis-related molecular alteration of the human Cav1.2 calcium channel α1C subunit. Proc. Natl. Acad. Sci. U.S.A. 103, 17024–17029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tang Z. Z., Liang M. C., Lu S., Yu D., Yu C. Y., Yue D. T., Soong T. W. (2004) Transcript scanning reveals novel and extensive splice variations in human l-type voltage-gated calcium channel, Cav1.2 α1 subunit. J. Biol. Chem. 279, 44335–44343 [DOI] [PubMed] [Google Scholar]
- 8. Liao P., Yu D., Li G., Yong T. F., Soon J. L., Chua Y. L., Soong T. W. (2007) A smooth muscle Cav1.2 calcium channel splice variant underlies hyperpolarized window current and enhanced state-dependent inhibition by nifedipine. J. Biol. Chem. 282, 35133–35142 [DOI] [PubMed] [Google Scholar]
- 9. Liao P., Yu D., Lu S., Tang Z., Liang M. C., Zeng S., Lin W., Soong T. W. (2004) Smooth muscle-selective alternatively spliced exon generates functional variation in Cav1.2 calcium channels. J. Biol. Chem. 279, 50329–50335 [DOI] [PubMed] [Google Scholar]
- 10. Abernethy D. R., Soldatov N. M. (2002) Structure-functional diversity of human L-type Ca2+ channel: perspectives for new pharmacological targets. J. Pharmacol. Exp. Ther. 300, 724–728 [DOI] [PubMed] [Google Scholar]
- 11. Zühlke R. D., Bouron A., Soldatov N. M., Reuter H. (1998) Ca2+ channel sensitivity towards the blocker isradipine is affected by alternative splicing of the human α1C subunit gene. FEBS Lett. 427, 220–224 [DOI] [PubMed] [Google Scholar]
- 12. Welling A., Ludwig A., Zimmer S., Klugbauer N., Flockerzi V., Hofmann F. (1997) Alternatively spliced IS6 segments of the α1C gene determine the tissue-specific dihydropyridine sensitivity of cardiac and vascular smooth muscle L-type Ca2+ channels. Circ. Res. 81, 526–532 [DOI] [PubMed] [Google Scholar]
- 13. Yang Y., Chen X., Margulies K., Jeevanandam V., Pollack P., Bailey B. A., Houser S. R. (2000) L-type Ca2+ channel α 1c subunit isoform switching in failing human ventricular myocardium. J. Mol. Cell Cardiol. 32, 973–984 [DOI] [PubMed] [Google Scholar]
- 14. Liao P., Li G., Yu de J., Yong T. F., Wang J. J., Wang J., Soong T. W. (2009) Molecular alteration of Cav1.2 calcium channel in chronic myocardial infarction. Pflugers Arch. 458, 701–711 [DOI] [PubMed] [Google Scholar]
- 15. Tao J., Hildebrand M. E., Liao P., Liang M. C., Tan G., Li S., Snutch T. P., Soong T. W. (2008) Activation of corticotropin-releasing factor receptor 1 selectively inhibits CaV3.2 T-type calcium channels. Mol. Pharmacol. 73, 1596–1609 [DOI] [PubMed] [Google Scholar]
- 16. Huang H., Yu D., Soong T. W. (2013) C-terminal alternative splicing of Cav1.3 channels distinctively modulates their dihydropyridine sensitivity. Mol. Pharmacol. 84, 643–653 [DOI] [PubMed] [Google Scholar]
- 17. Liao P., Zhang H. Y., Soong T. W. (2009) Alternative splicing of voltage-gated calcium channels: from molecular biology to disease. Pflugers Arch. 458, 481–487 [DOI] [PubMed] [Google Scholar]
- 18. Diebold R. J., Koch W. J., Ellinor P. T., Wang J. J., Muthuchamy M., Wieczorek D. F., Schwartz A. (1992) Mutually exclusive exon splicing of the cardiac calcium channel α1 subunit gene generates developmentally regulated isoforms in the rat heart. Proc. Natl. Acad. Sci. U.S.A. 89, 1497–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gidh-Jain M., Huang B., Jain P., Battula V., el-Sherif N. (1995) Reemergence of the fetal pattern of L-type calcium channel gene expression in non infarcted myocardium during left ventricular remodeling. Biochem. Biophys. Res. Commun. 216, 892–897 [DOI] [PubMed] [Google Scholar]
- 20. Ebihara T., Komiya Y., Izumi-Nakaseko H., Adachi-Akahane S., Okabe S., Okamura Y. (2002) Coexpression of a Cav1.2 protein lacking an N terminus and the first domain specifically suppresses L-type calcium channel activity. FEBS Lett. 529, 203–207 [DOI] [PubMed] [Google Scholar]
- 21. Takahashi S. X., Miriyala J., Colecraft H. M. (2004) Membrane-associated guanylate kinase-like properties of β-subunits required for modulation of voltage-dependent Ca2+ channels. Proc. Natl. Acad. Sci. U.S.A. 101, 7193–7198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Plummer N. W., McBurney M. W., Meisler M. H. (1997) Alternative splicing of the sodium channel SCN8A predicts a truncated two-domain protein in fetal brain and non-neuronal cells. J. Biol. Chem. 272, 24008–24015 [DOI] [PubMed] [Google Scholar]
- 23. Wielowieyski P. A., Wigle J. T., Salih M., Hum P., Tuana B. S. (2001) Alternative splicing in intracellular loop connecting domains II and III of the α1 subunit of Cav1.2 Ca2+ channels predicts two-domain polypeptides with unique C-terminal tails. J. Biol. Chem. 276, 1398–1406 [DOI] [PubMed] [Google Scholar]
- 24. Altier C., Garcia-Caballero A., Simms B., You H., Chen L., Walcher J., Tedford H. W., Hermosilla T., Zamponi G. W. (2011) The Cavβ subunit prevents RFP2-mediated ubiquitination and proteasomal degradation of L-type channels. Nat. Neurosci. 14, 173–180 [DOI] [PubMed] [Google Scholar]
- 25. Chien A. J., Zhao X., Shirokov R. E., Puri T. S., Chang C. F., Sun D., Rios E., Hosey M. M. (1995) Roles of a membrane-localized β subunit in the formation and targeting of functional L-type Ca2+ channels. J. Biol. Chem. 270, 30036–30044 [DOI] [PubMed] [Google Scholar]
- 26. Pragnell M., De Waard M., Mori Y., Tanabe T., Snutch T. P., Campbell K. P. (1994) Calcium channel β-subunit binds to a conserved motif in the I-II cytoplasmic linker of the α1-subunit. Nature 368, 67–70 [DOI] [PubMed] [Google Scholar]
- 27. Raghib A., Bertaso F., Davies A., Page K. M., Meir A., Bogdanov Y., Dolphin A. C. (2001) Dominant-negative synthesis suppression of voltage-gated calcium channel Cav2.2 induced by truncated constructs. J. Neurosci. 21, 8495–8504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ahern C. A., Arikkath J., Vallejo P., Gurnett C. A., Powers P. A., Campbell K. P., Coronado R. (2001) Intramembrane charge movements and excitation-contraction coupling expressed by two-domain fragments of the Ca2+ channel. Proc. Natl. Acad. Sci. U.S.A. 98, 6935–6940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kobrinsky E., Tiwari S., Maltsev V. A., Harry J. B., Lakatta E., Abernethy D. R., Soldatov N. M. (2005) Differential role of the α1C subunit tails in regulation of the Cav1.2 channel by membrane potential, β subunits, and Ca2+ ions. J. Biol. Chem. 280, 12474–12485 [DOI] [PubMed] [Google Scholar]
- 30. Tang Z. Z., Zheng S., Nikolic J., Black D. L. (2009) Developmental control of Cav1.2 L-type calcium channel splicing by Fox proteins. Mol. Cell. Biol. 29, 4757–4765 [DOI] [PMC free article] [PubMed] [Google Scholar]