

Background: Barth syndrome is an inherited cardiomyopathy due to mutations in the TAZ gene.
Results: A screen using taz1Δ yeast cells identified genes whose deletion aggravated its fitness.
Conclusion: The protease Yme1 is required for efficient mitophagy in the absence of TAZ1.
Significance: This is the first study linking mitochondrial quality control to mitophagy as important in cells lacking TAZ1 function.
Keywords: Cardiolipin, Cardiomyopathy, Inborn Error of Metabolism, Lipid, Lipid Metabolism, Membrane Lipid, Mitophagy, Phospholipid Metabolism, Saccharomyces cerevisiae, Yeast Genetics
Abstract
The Saccharomyces cerevisiae TAZ1 gene is an orthologue of human TAZ; both encode the protein tafazzin. Tafazzin is a transacylase that transfers acyl chains with unsaturated fatty acids from phospholipids to monolysocardiolipin to generate cardiolipin with unsaturated fatty acids. Mutations in human TAZ cause Barth syndrome, a fatal childhood cardiomyopathy biochemically characterized by reduced cardiolipin mass and increased monolysocardiolipin levels. To uncover cellular processes that require tafazzin to maintain cell health, we performed a synthetic genetic array screen using taz1Δ yeast cells to identify genes whose deletion aggravated its fitness. The synthetic genetic array screen uncovered several mitochondrial cellular processes that require tafazzin. Focusing on the i-AAA protease Yme1, a mitochondrial quality control protein that degrades misfolded proteins, we determined that in cells lacking both Yme1 and Taz1 function, there were substantive mitochondrial ultrastructural defects, ineffective superoxide scavenging, and a severe defect in mitophagy. We identify an important role for the mitochondrial protease Yme1 in the ability of cells that lack tafazzin function to maintain mitochondrial structural integrity and mitochondrial quality control and to undergo mitophagy.
Introduction
Barth syndrome is an X-linked inherited disorder due to mutations in the TAZ gene. Patients with a defective TAZ gene present primarily with dilated cardiomyopathy along with a combination of neutropenia, recurrent infections, mouth ulcers, myopathy, exercise intolerance, delayed motor milestones, mild learning disabilities, growth delay, delayed puberty, and aciduria (1). If Barth syndrome goes undiagnosed, sudden cardiac arrest due to ventricular arrhythmia or bacterial septicemia due to neutropenia leads to fatality for most infants within the first few years of birth. Although there is no known cure, the disease can be managed with standard heart failure medications or ventricular assist devices to prevent arrhythmia and with prophylactic antibiotic treatment and granulocyte colony-stimulating factor to treat recurrent infections (1, 2). Despite symptom management that has extended the life span for most Barth syndrome patients, quality of life and life span are still not near the norm. There is a need for a deeper understanding of the molecular basis of Barth syndrome to aid in moving toward more effective treatments.
The understanding of the molecular basis of Barth syndrome was increased substantially when it was determined that the protein encoded by the TAZ gene, referred to as tafazzin, is a monolysocardiolipin (MLCL)2 transacylase that transfers unsaturated fatty acyl chains from phosphatidylcholine or phosphatidylethanolamine to MLCL to produce cardiolipin (CL) (Fig. 1) (3, 4). CL is a phospholipid found exclusively in the mitochondria, where it accounts for 15% of mitochondrial membrane lipid, with the majority of CL localized to the mitochondrial inner membrane (MIM) (5–7). Tafazzin also localizes to the mitochondria, where it acts to remodel newly synthesized CL; newly synthesized CL primarily contains saturated fatty acyl chains and tafazzin remodels this CL such that it contains unsaturated fatty acyl chains. In the absence of tafazzin function there is an increase in its substrate, MLCL, and a concomitant decrease in its product, unsaturated CL. This observation is conserved across all models of tafazzin deficiency including human, mouse, zebrafish, fruit fly and yeast (8–12). It is clear that mitochondrial dysfunction(s) due to altered CL metabolism is front and center with respect to the pathogenesis of Barth syndrome; however, how this metabolic defect causes the pathology observed in Barth syndrome patients is not well understood.
FIGURE 1.
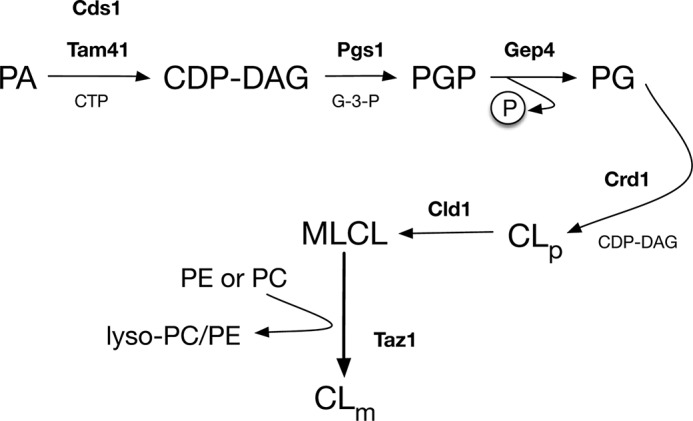
Cardiolipin synthesis and remodeling pathway. CL synthesis is initiated by the formation of cytidine diphosphate diacylglycerol (CDP-DAG) from PA by the transfer of CDP to PA from cytidine triphosphate (CTP). Both Cds1 and Tam41 can catalyze this reaction, but in yeast, the majority of CL is made from CDP-DAG supplied by Tam41. The rate-limiting production of phosphatidylglycerol phosphate (PGP) by the addition of glycerol 3-phosphate (G-3-P) to CDP-DAG is catalyzed by PGP synthase (Pgs1). After the removal of phosphate from PGP by Gep4, phosphatidylglycerol (PG) is then condensed with CDP-DAG by CL synthase (Crd1) to form premature CL (CLp). The de novo premature CL lacks the abundance of unsaturated fatty acyl species in mature CL (CLm), and it undergoes fatty acyl remodeling by the actions of CL deacylase 1 (Cld1) and Taz1. First, premature CL is deacylated by Cld1 to generate MLCL (which has only three acyl chains), which is then reacylated by Taz1 by the transfer of acyl chains from PC or PE to produce mature CL.
Electron micrographs of lymphoblasts from Barth syndrome patients showed abnormal clusters of giant and fragmented mitochondria with MIM adhesions and subsequent loss of intracristae space (13). The stability and assembly of ETC supercomplexes were partially compromised, and a decrease in mitochondrial respiration and ATP synthesis was observed (14, 15). This respiratory defect appears to be offset in Barth patient lymphoblasts by a compensatory increase in total mitochondrial mass (16). Mitochondria from Barth syndrome patient lymphoblasts also produce slightly more reactive oxygen species (ROS) than normal cells (16), although whether this increase in ROS contributes to the pathogenesis of Barth syndrome is not clear.
Studies on Saccharomyces cerevisiae lacking tafazzin function have recapitulated many of the observed defects seen in cells from Barth syndrome patients, including defective assembly of ETC supercomplexes, decreased mitochondrial respiration, and a slight increase in ROS (11, 15, 17, 18). Indeed, it is difficult to predict the severity of Barth syndrome due to a lack of known genotype-phenotype correlations. Clinical manifestation of the disease has been shown to vary within the same family containing the same TAZ mutation, implying that modifier genes exist that contribute to disease severity (19, 20). To further understand the molecular basis of Barth syndrome, we sought to identify modifier genes whose inactivation affected the fitness of yeast cells lacking tafazzin function. We discovered several genes that do so, and we reveal an important role for the mitochondrial quality control protein Yme1, a mitochondrial i-AAA protease. Yme1 is required to ensure effective and efficient mitophagy in cells lacking tafazzin function.
MATERIALS AND METHODS
Reagents
Custom oligonucleotides were acquired from Integrated DNA Technologies, HiFi TaqDNA polymerase from Invitrogen was used to perform PCR amplifications, and all of the restriction enzymes and T4 DNA ligase used in cloning procedures were purchased from New England Biolabs. Plasmids from bacteria were purified using the QIAprep spin miniprep kit from Qiagen. Genomic DNA was purified from yeast with the yeast DNA extraction reagent (Y-DER) kit from Pierce. DNA fragments and PCR amplicons used for ligation were cleaned with the GeneCleanII kit from Bio-101. For phospholipid labeling, [32P]orthophosphoric acid was purchased from PerkinElmer Life Sciences. Phospholipids were separated by thin layer chromatography (TLC) on LK5 silica gel 150 A TLC plates from Whatman. Complete protease inhibitor tablets with and without EDTA were from Roche Applied Science. Neurseothricin (NAT) or ClonNAT was from Werner BioAgents. The yeast strain CWY-152 (W3031-α OM45-GFP::TRP1) used to assess mitophagy was a kind gift from Dr. Kanki (Kyushu University School of Medical Sciences, Fukuoka, Japan). Native 3–12% precast BisTris polyacrylamide gels from Invitrogen were used to separate mitochondrial proteins.
Media
Yeast and bacterial media were purchased from Difco except for synthetic complete (SC) media (SC and SC-uracil), which were from Sunrise Science products. Yeasts were normally grown in rich medium containing 1% bacto-yeast extract, 2% bacto-peptone, 2% carbon source (2% agar was included to make solid medium). The fermentable carbon source dextrose or the non-fermentable carbon source lactate was added to the yeast extract bacto-peptone mixture (YP) to make YP with dextrose (YPD) or YP with lactate (YPL), respectively. Synthetic defined (SD) medium (minimal medium) typically contained 2% dextrose, 0.67% bacto-yeast nitrogen base, 20 mg/liter adenine sulfate, 20 mg/liter l-histidine HCl, l00 mg/liter l-leucine, 30 mg/liter l-lysine, 20 mg/liter l-methionine, 20 mg/liter l-tryptophan, and 20 mg/liter uracil to account for auxotrophies in our laboratory yeast strains. In certain experiments, such as in synthetic genetic array (SGA) analysis, some amino acid(s) were excluded to facilitate selection of yeast cells. When G418 was required in SD medium, the medium used 1% monopotassium glutamate (as nitrogen source), 0.17% yeast nitrogen base devoid of both ammonium sulfate and amino acids, 2% dextrose, and the required amino acid supplements. SD medium containing 2% dextrose and 0.17% yeast nitrogen base without ammonium sulfate and amino acids (SD−N) was used to induce mitophagy in yeast. SC medium usually contained 2% dextrose, 0.67% bacto-yeast nitrogen base, and 85.6 mg/liter of the following amino acids and supplements: l-alanine, l-arginine HCl, l-asparagine, l-aspartic acid, l-cysteine HCl, glutamine, l-glutamic acid, glycine, l-histidine HCl, myo-inositol, l-isoleucine, l-lysine HCl, l-methionine, l-phenylalanine, l-proline, l-serine, l-threonine, l-tryptophan, l-tyrosine, uracil, and l-valine. It also contained 21 mg/liter adenine, 173.4 mg/liter l-leucine, and 8.6 mg/liter para-aminobenzoic acid. However, in experiments that forced yeast to undergo mitochondrial respiration, dextrose in SC was substituted with 2% lactate (SC medium with lactate). SC medium lacking uracil (SC−uracil) was used to select for yeast transformed with pRS416-based plasmids. Bacteria used for cloning and propagation of yeast plasmids were grown in Luria-Bertani (LB; containing 10 g/liter tryptone, 5 g/liter yeast extract, 10 g/liter sodium chloride) broth or on LB plates (with 2% agar). Ampicillin (100 mg/liter) was included in LB medium to select for colonies with plasmids after transformation.
Yeast Strain Construction
For a complete list of all of the yeast strains used in this study, see Table 1. The TAZ1 gene in CWY-153 and CBY-303 cells was disrupted and replaced with a NAT resistance marker by homologous recombination using a PCR-generated taz1Δ::NATMX4 gene disruption cassette (21). The primer pair F-taz_M (TCT TTT AGG GAT GTC CTA GAA AGA GGA GAT TTT TTA GAA GCC TAA CAT GGA GGC CCA GAA TAC CC) and R-taz_M (ATC ATC CTT ACC CTT TGG TTT ACC CTC TGG AGG CAG AAA CTT TTG CAG TAT AGC GAC CAG CAT TCA C) was used to amplify the NATMX4 gene from the plasmid pAG25. Each primer contained 44 (F-taz_M; italicized above) or 47 (R-taz_M; italicized above) nucleotides identical to the regions adjacent to TAZ1 translational start and stop sites at their 5′ ends. At their 3′ ends they contained a 20-nucleotide-long stretch of NATMX4-specific sequence. This resulted in a PCR product that contained the NATMX4 cassette with overhangs that are homologous to the regions proximal to the translation start and stop site of the TAZ1 gene. The PCR amplicon was purified and transformed into strain W3031-a, and BY2454 yeast strains and transformants were plated on YPD medium with 2% agar. The plated cells were then grown overnight at 25 °C to allow for homologous recombination. To select for transformants with a disrupted TAZ1 coding sequence, the lawn of cells from the YPD plate was replica-plated to plates with YPD medium containing ClonNAT (100 mg/liter; Werner BioAgents). Genomic DNA from these transformants was purified and used for PCR amplification with primers TAZ5 flanking forward (CCATTGTCTCTCCAATTGGTG) and TAZ3 flanking reverse (TCATGCTTTAGGTCAGCCTGA) that bound (367 bp) upstream and downstream (408 bp) of the TAZ1 coding sequence, respectively. The generated PCR product comprised a NdeI site if it harbored the wild type TAZ1 coding sequence, and it was abolished and replaced by an XmaI site if disrupted by the taz1Δ::NATMX4 cassette. Therefore, PCR amplicons generated from the transformants were subjected to restriction enzyme digestion with XmaI (New England Biolabs) or NdeI (New England Biolabs) to confirm the successful disruption of the TAZ1 coding sequence.
TABLE 1.
Yeast strains used in this study
| Strain | Genotype | Source |
|---|---|---|
| BY4741 (CBY 300) | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | EUROSCARF |
| CBY 301 | BY4741 yme1Δ::KANMX4 | EUROSCARF |
| Y2454 (CBY 302) | MATα mfa1Δ::MFA1pr-HIS3 can1Δ0 his3Δ1 leu2Δ0 ura3Δ0 lys2Δ0 | Ref. 1 |
| CBY 303 | Y2454 taz1Δ::NATMX4 | This study |
| CBY 304 | BY4741 crd1Δ::KANMX4 | EUROSCARF |
| W3031-a (CWY 150) | MATa leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15 | Thermo Scientific |
| W3031-α (CWY 151) | MATα leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15 | Thermo Scientific |
| CWY 152 | W3031-α OM45-GFP::TRP | Ref. 2 |
| CWY 153 | W3031-a taz1Δ::NATMX4 | This study |
| CWY 154 | W3031-α yme1Δ::KANMX4 | This study |
| CWY 155 | W3031-a taz1Δ::NATMX4 yme1Δ::KANMX4 | This study |
| CWY 156 | W3031-a OM45-GFP::TRP | This study |
| CWY 157 | W3031-a OM45-GFP::TRP taz1Δ::NATMX4 | This study |
| CWY 158 | W3031-a OM45-GFP::TRP yme1Δ::KANMX4 | This study |
| CWY 159 | W3031-a OM45-GFP::TRP yme1Δ::KANMX4 taz1Δ::NATMX4 | This study |
The CWY-154 strain was generated in a manner similar to cells' inactivation of the TAZ1 gene with the NATMX4 cassette; however, the gene disruption cassette yme1Δ::KANMX4 was PCR-amplified from genomic DNA purified from CBY-301 (BY4741 yme1Δ::KANMX4) using primers Yme1F (AGCAAGCACAGCTTAAAGGA) and Yme1R (ATGAAGCAAAAGCGAAACCG). To enable homologous recombination between the disruption cassette and the wild type YME1 allele of W3031-α, the primers were designed such that they were homologous to the upstream and downstream flanking regions of the disrupted YME1 coding sequence. The PCR-generated yme1Δ::KANMX4 was also used to transform the CWY-151 strain, and transformants were selected on agar plates with G418 (200 mg/liter; Invitrogen). To confirm YME1 open reading frame (ORF) disruption, genomic DNA purified from transformants was used to amplify the flanking regions of YME1 coding sequence using primers cYme1F (GTTAAATCTCTACCTTGCGTTTTTGA) and cYme1R (TGCTGGTGTTATGAAGCAAAAGC) that bound (127 bp) upstream and (100 bp) downstream of the translation start and stop codons, respectively. Successful disruption of YME1 generated shorter PCR products (1.86 kbp) compared with amplicons (2.47 kbp) generated from transformants with wild type YME1.
Yeast strains carrying a chromosomal version of OM45-GFP were utilized to monitor mitophagy (22). The strain CWY-152 (W3031-α OM45-GFP::TRP1), a gift from Dr. Kanki (Kyushu University School of Medical Sciences), was used via matings and tetrad dissections to replace the OM45 with OM45-GFP::TRP1 allele in taz1Δ yme1Δ cells and in taz1Δ and yme1Δ single mutants. To generate a MATa version of wild type yeast with OM45-GFP::TRP1, CWY-152 was mated with wild type W3031-a. The diploids were selected on SD medium lacking tryptophan, (SD−trp), after which they were sporulated, and the resulting spores were dissected and allowed to germinate on YPD medium. Haploid colonies that grew on SD−trp agar plates were selected and confirmed for Om45-GFP tagging by observing mitochondrial GFP fluorescence using the GFP filter fitted to a Zeiss Axiovert 200M microscope. CWY-156 (W3031-a OM45-GFP::TRP1) was selected for mitophagy assays and for the generation of CWY-158 (W3031-a yme1Δ:KANMX4 OM45-GFP::TRP1) cells. Yeast strains CWY-157 (W3031-a taz1Δ:NATMX4 OM45-GFP::TRP1), CWY-159 (W3031-a taz1Δ:NATMX4 yme1Δ:KANMX4 OM45-GFP::TRP1) and CWY-158 (W3031-a yme1Δ:KANMX4 OM45-GFP::TRP1) were generated as described for CWY-156 cells. The CWY-152 strain was mated with CWY-153 and CWY-155 to generate CWY-157 and CWY-159 cells, respectively. The CWY-156 was mated with CWY-154 to generate CWY-158 cells. Appropriate selection media (SD−trp, ClonNAT (100 mg/liter); SD−trp, G418 (200 mg/liter); and SD−trp, G418 (200 mg/liter), ClonNAT 100 mg/liter) were used to select for OM45-GFP::TRP1-containing haploid colonies with taz1Δ yme1Δ or single gene mutations.
Plasmid Construction
To express TAZ1 exogenously from its own promoter, its coding sequence, along with its upstream (340 bp from translation start site) and downstream (200 bp from stop codon) flanking regions, was PCR-amplified and subcloned to the pRS416 plasmid. The primers TazF_BglII_8140454 (GGAAGATCTATTTTGTCACCTCGTAGGAGC) and TazR_XhoI_815730 (CCGCTCGAGCAAGGCCTTTTCTGGTCTTT), which included the BglII and XhoI restriction enzyme sequence overhangs, respectively, were used to PCR-amplify the specified DNA insert from genomic DNA extracted from CWY-150 for ligation into plasmid pRS416. The ligation mixture was electroporated into DH10B Escherichia coli (Invitrogen) and plated onto LB plates with 100 mg/liter ampicillin. Plasmids were isolated from colonies and checked by restriction digest for the successful generation of a pRS416 TAZ1 plasmid. The TAZ1 gene was sequenced to ensure polymerase fidelity.
Synthetic Genetic Array (SGA) Analysis
In S. cerevisiae, there are ∼5,000 non-essential genes (of ∼6,400 total genes), where deletion of any one these non-essential genes in a haploid yeast cell does not affect its viability. At present, ∼4,800 mutants (deletion collection), each carrying a single non-essential gene deletion, are available as an ordered array from EUROSCARF (European Saccharomyces cerevisiae Archive for Functional Analysis). In each deletion collection mutant, the deleted gene is replaced by a dominant kanamycin-resistance selectable marker (KANMX4). A query strain carrying a nourseothricin resistance marker (NATMX4) that replaces the ORF of a gene of interest, in our case taz1Δ::NATMX4, is also generated. SGA analysis, with the aid of robotics, facilitates large scale mating between the query strain and each of the deletion collection mutants to generate an array of 4,800 diploid cells, each a haploid for the gene of interest and every non-essential yeast gene. Because the disrupted genes are replaced with antibiotic resistance markers, double mutants can be easily selected on medium with both G418 (200 mg/liter; Invitrogen) and ClonNAT (100 mg/liter; Werner BioAgents). Among the 4,700 double mutants, some mutants may not be viable, or they may grow much more slowly than normal, indicating a synthetic lethal or synthetic sick interaction between the genes. This type of result generally indicates that the two genes either act in a single large complex or function in parallel compensatory pathways. Identification of such genetic interactions has been successfully used in the past to define gene function (23).
SGA analysis was conducted as previously described (23); the query strain CBY-303 was generated by replacing the TAZ1 coding region with the NATMX4 cassette in MATα Y2454 yeast cells by PCR-based gene disruption as described above. This strain was then robotically crossed with each of the MATa deletion collection mutants, and the array of generated diploids was selected on plates with YPD medium containing G418 and ClonNAT. Meiosis was induced in these diploids by robotically pinning them to sporulation medium (2% agar, 1% potassium acetate, 0.1% yeast extracts, 0.05% glucose, supplemented with uracil, histidine, and leucine). After 5 days of sporulation at 22 °C, MATa haploid spores that derived from mating between the query strain and the deletion collection strains were specifically selected by incubating the spores in SD medium lacking histidine and arginine but containing canavanine (SD−His/Arg, canavanine (50 mg/liter)) for 2 days at 30 °C. The MATα query and the MATa deletion collection mutants are histidine auxotrophs and do not grow in the absence of histidine. The query strain carries the HIS3 under the control of the MATa-activated promoter (MFApr), such that HIS3 is only expressed in MATa cells. Therefore, growth in SD medium without histidine will only support the growth of MATa haploid spores that resulted from mating between the query and one of the deletion collection mutants. In addition, unlike the deletion mutants, the query strain has a defective arginine permease (can1Δ) and cannot take up arginine or its analogue canavanine from the growth medium. When canavanine instead of arginine is present in the growth medium, CAN1 cells incorporate it into cells, causing lethality. Therefore, inclusion of canavanine (50 mg/liter) in the haploid selection medium provides further insurance that only haploid spores and not CAN1/can1Δ diploids are selected for further analysis. After another round of selection in haploid selection medium, the cells were pinned to SD medium with G418 (SD−His/Arg, G418 (200 mg/liter), canavanine (50 mg/liter)) for a day before they were finally transferred to agar plates containing both G418 and ClonNAT (SD−His/Arg, G418 (200 mg/liter), ClonNAT (100 mg/liter), canavanine (50 mg/liter)) for 2 days at 30 °C. The double mutant haploid arrays were manually screened for lethality and growth defects, and validity of the observed genetic interactions was checked by random spore analysis and/or tetrad analysis.
Random Sporulation Assay to Confirm TAZ1-aggravating Gene Deletions
The 35 genes that showed genetic interaction with taz1Δ and had a role in the mitochondria were selected for random sporulation assays. The CBY-303 strain was manually mated with each of 35 single gene mutants from the deletion collection, and the resulting diploids were selected on agar plates with YPD (G418 (200 mg/liter), ClonNAT (100 mg/liter)). The diploids were suspended at ∼2 × 107 cells/ml in sporulation broth (1% potassium acetate, 0.1% yeast extracts, 0.05% glucose, supplemented with uracil, histidine, and leucine) and then allowed to sporulate for 5–6 days. The spores were harvested and suspended at ∼5 × 108 cells/ml in distilled water containing 100 μg/ml zymolase 100T and incubated for 15 min at 30 °C to completely digest the ascus wall. After that, 0.5-ml aliquots were transferred to 1.5-ml Eppendorf tubes and then pelleted at 14,000 × g for 30 s; the supernatant was disposed of; and the pellets were resuspended in 1 ml of distilled water, centrifuged as before, and then resuspended in 0.1 ml of fresh distilled water. This mixture was vigorously vortexed for 2 min; because the spores are far more hydrophobic than vegetative cells, they clump together and also adhere to polypropylene walls of the 1.5-ml Eppendorf tubes. The vegetative cells were discarded by several washings in distilled water before the adhered spores were resuspended by the addition of 1 ml of water containing 0.01% Nonidet P-40 and sonicated in ice for 1–3 min. The purified spores were diluted, and about 200 spores were evenly plated on plates containing SD−His/Arg; SD−His/Arg, G418 (200 mg/liter); and SD−His/Arg, G418 (200 mg/liter, ClonNAT (100 mg/liter) and allowed to grow at 30 °C. The colonies in each of the plates were counted after 3–5 days manually and with the help of Quantity One software (Bio-Rad VersaDoc). Spores that carried double mutants with true genetic interaction with TAZ1 typically did not grow in SD−His/Arg, G418 (200 mg/liter), ClonNAT (100 mg/liter) plates.
Tetrad Analysis to Confirm the Genetic Interaction between TAZ1 and YME1
Tetrad analysis was performed to determine the validity and strength of the negative genetic interaction between TAZ1 and YME1. Yeast strains CWY-153 (W3031-A taz1Δ::NATMX4) and CWY-154 (W3031-α yme1Δ::KANMX4) were mated on plates with YPD medium at 25 °C. The resulting diploids were selected on YPD plates containing G418 (200 mg/liter) and ClonNAT (100 mg/liter) for 2 days. After that, cells were sporulated for 5–6 days at 22 °C in sporulation medium (2% agar, 1% potassium acetate, 0.1% yeast extracts, 0.05% glucose, supplemented with leucine, tryptophan, uracil, adenine, and histidine at 50% of normal requirement). With an inoculation needle, spores were suspended in 50 μl of zymolase 100T solution (0.5 mg/ml zymolase in 1 m sorbitol). This mixture was incubated at 30 °C to enable the partial digestion of the ascus walls. After 10 min, the digestion was stopped by placing the tube on ice. The spore mixture was then diluted with distilled water to 0.5–1 ml before they were spread onto YPD plates. Under a microdissection microscope, intact ascii, each containing four spores, were teased with a microdissection needle and placed at distinct positions within the YPD plate. The dissected spores were then allowed to germinate into haploid colonies for 2–3 days at 25 °C. The resulting colonies were then replica-plated onto YPD plates containing G418 (200 mg/liter) and ClonNAT (100 mg/liter) for 2 days. There, growth was determined, and their sensitivity to G418 and ClonNAT was used to infer the genetic makeup of each strain.
Steady State Analysis of Phospholipid Levels
Yeast phospholipids were obtained from isolated mitochondria and analyzed by one-dimensional TLC as described previously (24, 25). Briefly, 3-ml cultures of the indicated yeast strains were initiated at 0.025 A600 in either YPD or YPL broth in the presence of 10 μCi of 32Pi/ml (orthophosphoric acid). The cultures were allowed to grow for 5–6 generations at 30 °C to achieve steady state 32P incorporation into each phospholipid class of the yeast cell to reflect its relative mass compared with total phospholipids. The cells were harvested and suspended in 0.3 ml of MTE buffer (0.65 m mannitol, 20 mm Tris, pH 8.0, and 1 mm EDTA) containing 1 mm PMSF and an appropriate concentration of complete protease inhibitors (Roche Applied Science). In the presence of 0.2 ml of 0.5-mm acid-washed glass beads, the cells were then mechanically fractured by high speed vortexing for 1 h. Unbroken cells and glass beads were first sedimented by centrifugation at 300 × g for 5 min at 4 °C; the resulting supernatant was then centrifuged at 13,000 × g for 5 min at 4 °C to a obtain the crude mitochondrial fraction. Lipids were extracted from equal amounts of crude mitochondrial preparation (as determined by liquid scintillation) by the Folch method. Briefly, 1.5 ml of 2:1 chloroform/methanol was added to the extract and agitated for 1 h at room temperature; after that, 0.3 ml of distilled water was added to this mixture, vortexed, and centrifuged at 2,200 rpm for 20 min to separate the aqueous and organic phases. To the organic phase, 0.25 ml of 1:1 methanol/distilled water was added and vortexed to mix the phases. The mixture was centrifuged as before to collect the organic phase, which was then dried under a stream of liquid nitrogen. The dried samples were resuspended in chloroform and loaded onto prewashed (with 1:1 chloroform/methanol) and pretreated (with 1.8% boric acid in absolute ethanol) LK5 silica gel 150 A TLC plates (Whatman). Lipids were separated for ∼2 h by placing the TLC plates in a chromatography tank containing 30:35:7:35 (v/v/v/v) chloroform/ethanol/water/triethylamine. After the first run, the plates were removed from the tank, dried completely in a fume hood, and then rerun for the second time in the same direction to achieve adequate resolution. Dried TLC plates were wrapped in plastic wrap and exposed to X-Omat Blue film (Eastman Kodak Co.) to visualize phospholipids by autoradiography. To quantitate the steady state levels of phospholipids, the same TLC plates were exposed to a phosphor imager screen (Bio-Rad) for 30 min before it was scanned and quantified by the Storm scan system (Molecular Dynamics) and ImageQuant software, respectively.
Mitophagy-monitoring Assay
Yeast OM45 encodes a MOM protein (Om45) that is expressed in stationary phase cells grown in medium with a non-fermentable carbon source. C terminus GFP tagging of chromosomal OM45 (Om45-GFP) does not affect its normal mitochondrial localization; however, mitophagy results in Om45-GFP accumulation in the vacuole. Hence, tracking the Om45-GFP signal from the mitochondria to the vacuole upon nitrogen starvation (mitophagy induction) is used to assess mitophagy by microscopy. Also, GFP, unlike other most other proteins, is relatively resistant to vacuolar degradation, and it persists as free GFP after the degradation of tagged protein (Om45). Therefore, after nitrogen starvation, estimation of free GFP versus Om45-GFP levels by Western blotting with anti-GFP serum is used to determine mitophagy efficiency, as was shown previously (22).
Yeast strains CWY-156, CWY-157, CWY-158, and CWY-159 were subjected to mitophagy induction by nitrogen starvation or growth to post-log/stationary phase. To induce mitophagy by nitrogen starvation or growth to post-log phase, the indicated strains were first grown in 2 ml of YPD medium, and once they reached 0.5 A600, they were harvested by centrifugation, washed twice in distilled water, and then resuspended in 6 ml of YPL at 0.2 A600. They were then incubated at 25 °C under constant shaking until they reached stationary phase (0.8–1.5 A600). After that, an aliquot of cells (3 ml) from each strain was collected, and cells were immediately pelleted and washed once with GTE (20% glucose, 50 mm Tris-HCl, pH 7.4, and 1 mm EDTA) buffer before they were stored at −70 °C. The remaining culture was washed thoroughly in distilled water three times to remove traces of YPL medium and resuspended in 3 ml of SD−N medium lacking nitrogen or resuspended in 3 ml of fresh YPL medium and grown to post-log phase. After 24 h or 3–5 days, the cells were harvested by centrifugation and marked as post-nitrogen starvation or post-log phase, respectively.
For Western blots, 1 ml of GTE buffer containing 1 mm PMSF and an appropriate concentration of complete protease inhibitors (Roche Applied Science) was added to the cell pellets saved above. The cells were then lysed using a bead beater (Bio-Spec) in the presence of 0.2 ml of 0.5-mm acid-washed glass beads. The glass beads and unbroken cells were pelleted by centrifugation at 300 × g for 5 min at 4 °C; the resulting supernatant was then denatured in sample buffer (150 mm Tris-HCl, pH 8.8, 6% SDS, 25% glycerol, 6 mm EDTA, 0.5% 2-mercaptoethanol, and 0.05% bromphenol blue). The denatured proteins were then separated by 10% SDS-PAGE and transferred to PVDF membranes as described before. The blots were then subjected to Western blotting with (1:10,000) GFP anti-sera (Clontech) to estimate the levels of Om45-bound and free GFP in samples from before and after mitophagy induction.
To assess mitophagy by fluorescence microscopy, the indicated strains were grown as described for Om45-GFP Western blotting, albeit in 2-ml culture volumes. Cells before and after nitrogen starvation were harvested by slow centrifugation at 1,000 × g for 1 min and then resuspended in 50 μl of PBS. To visualize Om45-GFP signal, 3-μl aliquots were spotted on glass slides, covered with coverslips, and viewed under a Zeiss Axiovert 200M microscope through the GFP filter. The images were captured with a Zeiss Axio Cam HR coupled to Zeiss Axiovision version 4.5 software. The accumulation of GFP signal in the vacuole after starvation was confirmed by superimposing the images of the vacuoles captured under the differential interference contrast filter over the GFP images. Further, the extent of mitophagy induction was quantified by manually counting at least 250 cells for each strain studied before and after mitophagy induction from three separate experiments.
Isolation of Yeast Mitochondria
One-liter cultures of yeast strains were grown in YPL medium at 30 °C until they reached an A600 of 1–2. The cells were harvested by centrifugation at 2,500 × g for 5 min at room temperature, and mitochondria were isolated by differential centrifugation as detailed previously (26). Briefly, harvested cells were washed once in distilled water, resuspended in 30 ml of freshly prepared TD buffer (100 mm Tris-sulfate, pH 9.4, 10 mm DTT), and incubated for 5 min at 30 °C with gentle shaking. These cells were then collected by centrifugation (2,500 × g for 5 min) and resuspended in SP buffer (1.2 m sorbitol, 20 mm potassium phosphate, pH 7.4). After that, spheroplasts were generated by treatment with zymolase 100T (1.5 mg/g cells) for 30–60 min at 30 °C with gentle shaking. Spheroplasts were harvested by centrifugation at 2,500 × g for 5 min at 4 °C and then washed twice with 40 ml of ice-cold SP buffer. After the washes, the spheroplasts were resuspended in 60 ml of ice-cold SH buffer (0.6 m sorbitol, 20 mm HEPES-KOH, pH 7.4) with 1 mm PMSF and appropriate concentrations of complete protease inhibitors (Roche Applied Science). The suspension was transferred to a large piston glass homogenizer (Potter-Elvehjem type tissue grinders) and homogenized with 25 strokes; the homogenized mixture was then subjected to centrifugation twice at 2,500 × g for 5 min at 4 °C. The supernatants from the previous steps were pooled together in a fresh 50-ml glass tube and pelleted by centrifugation at 12,000 × g for 10 min at 4 °C. Using a small-scale glass homogenizer, the pellet was first carefully resuspended in 1 ml of SH buffer and then resuspended in 25 ml of ice-cold SH buffer. This suspension was further centrifuged at 2,500 × g for 5 min at 4 °C, and the supernatant was saved and then pelleted at 12,000 × g for 10 min at 4 °C. The resulting pellet (mitochondria) was resuspended in 0.5 ml of SH buffer and divided into aliquots containing 0.1–0.3 mg of protein (estimated by a Coomassie (Bradford) protein assay kit (Pierce)). The aliquots were flash frozen in liquid nitrogen and stored at −70 °C until further analysis.
Blue Native PAGE
The status of yeast respiratory chain supercomplexes was assessed by BN-PAGE as described previously with minor modifications (27). Frozen mitochondria (0.2 mg of protein) were thawed and suspended in 0.04 ml of 50 mm NaC1, 5 mm 6-aminohexanoic acid, 50 mm imidazole/HC1, pH 7.0. To solubilize membranes, 10% solution of dodecylmaltoside at a detergent/protein ratio of 2 g/g was added to this mixture, and it was incubated for 20 min in ice. The suspension was centrifuged at 20,000 × g for 20 min at 4 °C, the supernatant with solubilized protein was saved, and 1 μl of 5% Coomassie Blue G-250 (Pierce) was added to the supernatant, which was then separated on a 3–12% precast BisTris native polyacrylamide gel (Invitrogen, BN1001BOX) using an XCell sure lock mini-cell gel running tank (Invitrogen) by the application of constant current at 150 V for ∼2 h in a cold room. To improve detection of faint protein bands and to maximize protein transfer to PVDF membranes, the dark cathode buffer with 0.02% Coomassie Blue G-250 was replaced with light cathode buffer (0.0002% Coomassie Blue G-250) once the proteins reached one-third of the running distance in BN-PAGE. A diluted aliquot of ferritin, which runs as two visible (brown) bands with molecular masses of 440 and 880 kDa (which matches the mass of ETC complex IV monomer and ETC III2IV2 supercomplex), was separated along with the samples by BN-PAGE to serve as a molecular marker (28). The proteins were then transferred to PVDF membranes using a semi-dry electroblotting apparatus in the presence of electrode buffer containing 5 mm Tricine, 7.5 mm imidazole, pH 7.0, at 4 °C for 90 min. The blot was then probed by Western blotting with 1:5,000 dilutions of anti-Cox2 antibody (MitoSciences) to detect the status of yeast respiratory chain supercomplexes.
Electron Microscopy
Yeast strains CWY-150 (W3031-a), CWY-153 (W3031-a taz1Δ:NATMX4), CWY-154 (W3031-α yme1Δ:KANMX4), and CWY-155 (W3031-a taz1Δ:NATMX4 yme1Δ:KANMX4) were grown in 5 ml of SC medium with 2% lactate to early stationary phase (0.9–1.0 A600). The cells were then harvested and washed with 1 ml of distilled water before they were suspended in 1 ml of 1.5% KMnO4. This mixture was kept under constant shaking on an orbital shaker. After 20 min, the cells were pelleted at 14,000 × g for 1 min and then washed twice in 1 ml of distilled water. The resulting pellet was incubated in 1 ml of 1% sodium periodate for 15 min under constant shaking. At the end of the incubation, the cells were pelleted as before and washed once with distilled water. One ml of 1% NH4Cl was added to the washed cells, and they were kept under constant shaking for another 10 min before they were washed and suspended in 0.5 ml of PBS. The KMnO4-fixed cells were gradually dehydrated after washes in increasing concentrations of ethanol. Dried samples were then infiltrated with LR White Resin, which was then embedded into capsules and left to dry at 60 °C for 2 days. Once the resin hardened, thin sections were cut using an LKB Huxley ultramicrotome. To assess mitochondrial morphology, the sections were viewed and captured using a JEOL JEM 1230 transmission electron microscope at 80 kV and a Hamamatsu ORCA-HR digital camera, respectively. The observed mitochondrial morphological alterations were quantitated by manually counting at least 150 mitochondria for each strain studied.
RESULTS
A Genome-wide Screen Identifies Genes Required for Fitness in Cells Lacking Tafazzin Function
The biochemical function of the tafazzin protein as an MLCL transacylase is conserved from yeast to humans. We inactivated the TAZ1 gene in a yeast strain designed to facilitate high throughput genome-wide screens. The taz1Δ strain constructed possessed the expected biochemical phenotype typical of a taz1Δ strain, an increase in MLCL level with a concomitant decrease in CL, and expression of TAZ1 from the pRS416 plasmid (low copy) under the control of its own promoter was able to restore the CL and MLCL levels in the taz1Δ mutant to wild type (data not shown). To determine processes that are required for cellular fitness in the absence of tafazzin function, we performed an SGA screen using this S. cerevisiae query strain with the TAZ1 gene inactivated. SGA analysis allows for the construction of cells where the TAZ1 gene is inactivated along with each non-essential yeast gene, creating a panel of ∼5,000 double mutants. These double mutants were analyzed for those that resulted in a decrease in cell growth in fermentable medium, a condition where cells lacking only the TAZ1 gene grow at a normal rate.
The SGA screen identified 35 genes whose inactivation decreased growth of taz1Δ cells and had a known role in mitochondrial function. To rule out false positive results due to the ability of yeast cells to accumulate second site suppressors during the screen, the double mutants were recreated using standard yeast genetics mating techniques and analyzed by random sporulation to determine whether their inactivation decreased growth of cells lacking tafazzin function. This analysis identified seven genes, ORT1, ISA2, GGC1, FRE4, SIT1, TOM5, and YME1, that, when inactivated, decreased growth in cells lacking tafazzin function. This list was expanded to 11 genes by our testing of predicted negative genetic interactions based on ORT1 participating in in the argininosuccinate shunt pathway (Table 2).
TABLE 2.
Genes whose inactivation results in decreased fitness in cells lacking tafazzin function
| Standard name | Systematic name | SGD description |
|---|---|---|
| Mitochondrial quality control | ||
| YME1 | YPR024W | Catalytic subunit of the i-AAA protease complex; responsible for degradation of unfolded or misfolded mitochondrial gene products; also has a role in intermembrane space protein folding; mutation causes an elevated rate of mitochondrial turnover |
| Arginine metabolism | ||
| ORT1 | YOR130C | Ornithine transporter of the mitochondrial inner membrane; exports ornithine from mitochondria as part of arginine biosynthesis; human ortholog is associated with hyperammonemia-hyperornithinemia-homocitrullinuria (HHH) syndrome |
| ARG3 | YJL088W | Ornithine carbamoyltransferase; also known as carbamoylphosphate:l-ornithine carbamoyltransferase; catalyzes the biosynthesis of the arginine precursor citrulline. |
| CPA1 | YOR303W | Small subunit of carbamoyl phosphate synthetase; carbamoyl phosphate synthetase catalyzes a step in the synthesis of citrulline, an arginine precursor; translationally regulated by an attenuator peptide encoded by YOR302W within the CPA1 mRNA 5′-leader. |
| CPA2 | YJR109C | Large subunit of carbamoyl phosphate synthetase; carbamoyl phosphate synthetase catalyzes a step in the synthesis of citrulline, an arginine precursor |
| ARG1 | YOL058W | Arginosuccinate synthetase; catalyzes the formation of l-argininosuccinate from citrulline and l-aspartate in the arginine biosynthesis pathway; potential Cdc28p substrate. |
| Mitochondrial iron metabolism | ||
| ISA2 | YPR067W | Protein required for maturation of mitochondrial [4Fe-4S] proteins; functions in a complex with Isa1p and possibly Iba57p; localizes to the mitochondrial intermembrane space; overexpression of ISA2 suppresses grx5 mutations. |
| GGC1 | YDL198C | Mitochondrial GTP/GDP transporter; essential for mitochondrial genome maintenance; has a role in mitochondrial iron transport. |
| Mitochondrial iron metabolism | ||
| FRE4 | YNR060W | Ferric reductase; reduces a specific subset of siderophore-bound iron prior to uptake by transporters; expression induced by low iron levels. |
| SIT1 | YEL065W | Ferrioxamine B transporter; member of the ARN family of transporters that specifically recognize siderophore-iron chelates; transcription is induced during iron deprivation and diauxic shift; potentially phosphorylated by Cdc28p |
| Mitochondrial protein import | ||
| TOM5 | YPR133W-A | Component of the TOM (translocase of outer membrane) complex; responsible for recognition and initial import of all mitochondrially directed proteins; involved in transfer of precursors from the Tom70p and Tom20p receptors to the Tom40p pore |
Specific Mitochondrial Processes Are Required for Fitness of Cells Lacking Tafazzin Function
The SGA screen provided insight into mitochondrial processes that need to be functional to ensure robust cell growth in the absence of tafazzin function. Deletion of genes involved in iron metabolism, namely ISA2 and GGC1, was identified by our SGA screen. ISA2 encodes a mitochondrial protein required for the maturation of Fe-S proteins. GGC1 encodes a mitochondrial GTP/GDP transporter that is also known to import iron from the IMS to the matrix, where the maturation of Fe-S-containing proteins occurs. Iron uptake in yeast is dependent on a family of iron siderophore transporters that import ferrous ion-containing siderophores into the cell. These siderophores are then acted upon by a family of ferric reductases that reduce them to free ferric ions for use by the cell. Our SGA analysis also determined that deletion of either SIT1 (siderophore transporter) or FRE4 (ferric reductase) in a taz1Δ mutant decreased growth. In addition, a recent report determined that CL is required for Fe-S biogenesis, and in yeast cells unable to synthesis CL at all (crd1Δ mutants), there is a decrease in the activities of enzymes that require Fe-S cofactors, including enzymes within the TCA cycle and the ETC (29). In humans, Fe-S biosynthetic disorders lead to hypertrophic cardiomyopathy and heart failure (30), phenotypes similar to those observed for Barth syndrome patients. The mechanism linking Fe-S biogenesis to CL is not clear. Our results point to a role for Fe-S biogenesis as an important contributor to cell fitness in the absence of the inability to remodel CL due to TAZ1 deficiency.
Our SGA screen also determined that in the absence of tafazzin function, there was a requirement for ORT1, a MIM ornithine transporter that exports ornithine from the mitochondria to the cytosol that is part of the argininosuccinate shunt pathway. We went on to analyze other structural genes within the argininosuccinate shunt and determined that ARG1, ARG3, CPA1, CPA2, and CPA1 attenuator peptide were all required for cell fitness in the absence of TAZ1. Through the Krebs cycle, the argininosuccinate shunt produces TCA intermediates fumarate and malate. It is known that a decrease in TCA cycle function occurs in Barth syndrome (29). We speculate that further decreasing TCA efficiency or an accumulation of argininosuccinate intermediates themselves results in the observed synthetic sickness when this pathway is inactivated in cells lacking tafazzin function. A second option is that defects in nitrogen metabolism or sensing, such as the metabolism of major sensing molecules used by yeast, such as glutamine, could result in alterations in downstream processes, such as autophagy/mitophagy.
The TOM (translocase of outer membrane) complex is located in the MOM and is responsible for the initiation of the import of mitochondrial targeted proteins. Tom5 is part of this, and we determined that inactivation of the TOM5 gene decreased growth of cells lacking the TAZ1 gene. This genetic interaction was confirmed and analyzed in detail by others (6), who determined that inactivation of Taz1 decreases mitochondrial protein import through the MOM.
The SGA screen performed by us also determined that deletion of YME1 in taz1Δ cells caused a growth defect. YME1 is a mitochondrial quality control protease that degrades misfolded mitochondrial IMS and MIM proteins, including misfolded tafazzin mutant proteins that cause Barth syndrome (31). Yme1 is a member of the ATP- and Zn2+-dependent family of proteases that contain a C-terminal proteolytic domain and an ATPase (AAA) domain (32). The protease domain of Yme1 faces the IMS of the mitochondria, and it is thus often referred to as i-AAA protease. The N terminus of Yme1 is in the mitochondrial matrix with a single transmembrane domain traversing the MIM (32). Similar to phenotypes observed for yeast cells lacking the TAZ1 gene, loss of YME1 function in yeast causes growth defects in non-fermentable medium at 37 °C (33) and alterations to mitochondrial morphology (33). Based on these phenotypic similarities, we surmised that TAZ1 and YME1 may function to regulate a crucial process that is exacerbated upon inactivation of both genes simultaneously. We went on to study the nature of this genetic interaction in detail.
Mitochondrial Quality Control Is Necessary for Cell Fitness in Cells Lacking Tafazzin
The genetic interaction between YME1 and TAZ1 was further confirmed by tetrad analysis (Fig. 2A). The YME1 open reading frame was replaced with a kanamycin resistance marker (KanMX) by homologous recombination. The yme1Δ mutant with the KanMX cassette was then mated with taz1Δ yeast, where the TAZ1 gene had been inactivated with the NatMX cassette, conferring resistance to nourseothricin. The resulting diploid cells were forced to undergo meiosis, and the haploid spores from each meiotic division were separated by tetrad dissection and allowed to grow as individual colonies on YPD medium for 3 days. Tetrad analysis showed that the double mutants grew very slowly compared with wild type and each single mutant (Fig. 2A). The double mutant colonies from tetrads were further tested for their growth phenotype by a second growth assay, where serial dilutions of wild type, taz1Δ, yme1Δ, and taz1Δ yme1Δ yeast strains were spotted onto YPD plates, and cells were allowed to grow for 3 days. The taz1Δ yme1Δ cells grew much more slowly, providing further confirmation that YME1 inactivation decreases the growth of yeast cells lacking tafazzin function (Fig. 2B).
FIGURE 2.

TAZ1 and YME1 double mutant cells are synthetically sick. A, tetrad separation of meiotic spores from a cross between taz1Δ and yme1Δ yeast shows slow growing taz1Δ yme1Δ mutants (indicated by white circles) in fermentable YPD medium. Each column has four colonies arising from a single tetrad (ascus) that resulted from the cross between taz1Δ and yme1Δ cells. The single mutants taz1Δ and yme1Δ carry nourseothricin and kanamycin resistance markers, respectively. Growth resistance to the addition of both the antibiotics G418 and nourseothricin to YPD medium confirmed the genetic make up of the double mutant colonies. B, spot assay shows that taz1Δ yme1Δ double mutants are synthetically sick in YPD medium. The indicated strains were grown to early stationary phase, 0.9–1.0 A600, and then resuspended in SC medium at 0.45 A600. This dilution was then serially diluted (1:10) and spotted on YPD plates and grown for 3–5 days at 25 °C. C, the indicated strains were grown to early stationary phase, 0.9–1.0 A600, and then suspended in minimal medium at 0.45 A600. This dilution was then serially diluted (1:10) and spotted on SC medium with 2% lactate and grown for 3–5 days at 25 and 37 °C.
Similar to taz1Δ cells, yeast cells lacking Yme1 function have been shown to grow poorly at 37 °C in medium containing a non-fermentable carbon source (34). To determine whether this growth defect was exacerbated in cells lacking both Taz1 and Yme1 function, the growth of wild type, taz1Δ, yme1Δ, and taz1Δ yme1Δ yeast strains in SC medium with 2% lactate as the sole carbon source was determined at 25 and 37 °C. As reported above, the yme1Δ cells grew normally at 25 °C, and similar to the taz1Δ mutant, their growth was severely decreased compared with that of wild type cells at 37 °C (Fig. 2C). The taz1Δ yme1Δ yeast displayed a growth defect at 25 °C far more severe than either single mutant alone, and, similar to taz1Δ cells, failed to grow at 37 °C. Yme1 is required for the fitness of cells lacking tafazzin function. Growth defects upon inactivation of the YME1 gene are clearly apparent in taz1Δ cells on fermentable medium and are exacerbated by growth on a non-fermentable carbon source.
Inactivation of Yme1 in taz1Δ Cells Does Not Alter ETC Supercomplex Formation
In yeast lacking tafazzin function, there is a defect in ETC supercomplex stability with an increase in free complex IV monomer and a concomitant decrease in III2IV2 level (15). In addition, it has been reported that the loss of tafazzin results in a slight decrease in respiration and a slight increase in ROS, consistent with a less efficient ETC (17, 18). This defect may be due to the requirement for CL to facilitate ADP-ATP carrier activity and oxidative phosphorylation as CL directly binds to the ADP-ATP carrier and is required for its activity (35, 36). CL has also been shown to bind and regulate proper folding of ETC complex I, complex III, complex IV, and complex V (37, 38). We sought to determine whether inactivation of the mitochondrial quality control protein Yme1 increased the defect in ETC supercomplex formation due to loss of function of tafazzin.
BN-PAGE was performed on mitochondrial membrane extracts, and ETC supercomplex assembly was determined by Western blot (Fig. 3). Cox2 is a subunit of complex IV and was used previously to determine that there is more free complex IV monomer and less supercomplex III2IV2 in taz1Δ yeast. As expected, inactivation of TAZ1 caused an accumulation of free complex IV monomer and a decrease in III2IV2 levels (Fig. 3), but surprisingly, although yme1Δ deletion by itself did not affect the formation of supercomplexes, inactivation of YME1 in taz1Δ yeast cells prevented the accumulation free complex IV and restored the formation of III2IV2 supercomplexes to wild type levels (Fig. 3). This implies that the growth defect observed upon loss of Yme1 function in taz1Δ cells is not due to exacerbation of a defect in supercomplex III2IV2 stability observed in taz1Δ cells.
FIGURE 3.
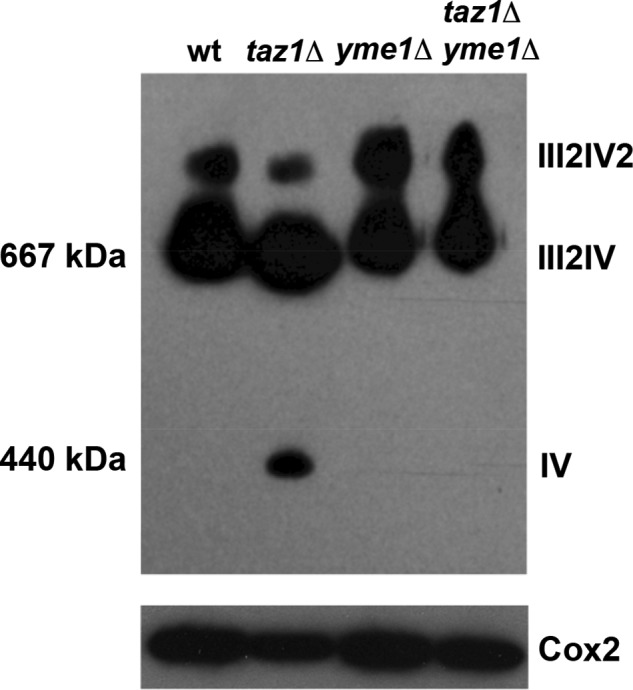
Supramolecular association of ETC III and IV into III2IV2 supercomplex is hampered in taz1Δ, which could be reversed by YME1 deletion. A, representative BN-PAGE gel that was transferred to nitrocellulose membrane and probed with an antibody against Cox2. Briefly, mitochondria were isolated and solubilized with dodecylmaltoside from the indicated strains and separated by BN-PAGE in the presence of Coomassie Blue dye in the gel as described under “Materials and Methods.” Proteins were then transferred to a PVDF membrane that was probed with antisera against Cox2 to ascertain the formation of III2IV2 and III2IV ETC supercomplexes. Cox2 is one of the subunits of ETC IV; hence, antiserum against it is effective in assessing the efficiency of complex IV to associate with III to form higher order supercomplexes. TAZ1 (lane 2) deletion increases the accumulation of free IV monomer with a concomitant reduction in the formation of III2IV2 supercomplex. Interestingly, deletion of YME1 in taz1Δ yeast was able to restore III2IV2 supercomplex formation, preventing the accumulation of IV monomer. B, to assess the level of Cox2, equal amounts of mitochondrial extracts were separated under denaturing conditions (SDS-PAGE), and an antibody against Cox2 was used to determine the levels of Cox2 by Western blot. PI, phosphatidylinositol; PS, phosphatidylserine; PC, phosphatidylcholine.
Inactivation of Yme1 in taz1Δ Cells Does Not Further Alter Mitochondrial Phospholipid Levels
Tafazzin is a MLCL transacylase, and a conserved hallmark of loss of tafazzin function is the accumulation of MLCL and a decrease in CL. Further, Yme1 has been shown to regulate the level of the mitochondrial enzyme Psd1 (39), which synthesizes the major mitochondrial phospholipid phosphatidylethanolamine (PE). To determine whether phospholipid content was aberrant in taz1Δ yme1Δ yeast, we analyzed the mitochondrial phospholipid content in wild type, taz1Δ, yme1Δ, and taz1Δ yme1Δ yeast by labeling cellular phospholipids with 32P to steady state, such that the incorporation of 32P reflected relative phospholipid mass. CL and MLCL levels were not different between the taz1Δ yme1Δ yeast and the single taz1Δ mutants (Fig. 4), other than a small increase in the level of PE in the taz1Δ yme1Δ strain. The decreased growth of taz1Δ yme1Δ cells does not appear to be due to overt changes in mitochondrial phospholipid levels.
FIGURE 4.
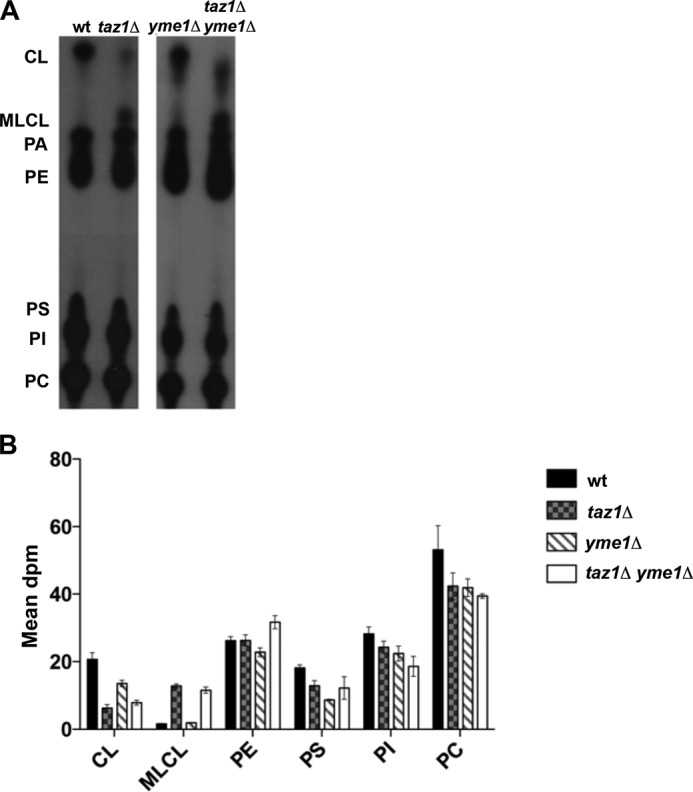
Double mutant taz1Δ yme1Δ yeast did not show any significant changes to steady state CL and MLCL levels when compared with taz1Δ mutants. A, representative TLC autoradiogram of the steady state phospholipid levels of various yeast strains. The indicated yeast strains were suspended in YPL medium at 0.025 A600 in the presence of 10 μCi of 32Pi/ml and were grown for 5–6 generations at 30 °C to achieve steady state labeling of phospholipids. At the end of the growth period, phospholipids were extracted from mitochondria, separated by TLC, and visualized by autoradiography. B, quantitation data of steady state phospholipid levels from two independent TLC analyses of the indicated yeast strains using a STORM PhosphorImager.
Inactivation of Yme1 in taz1Δ Cells Results in Mitochondrial Structural Abnormalities
The loss of tafazzin function has been shown to cause abnormal mitochondrial morphology with MIM adhesions, cristae that are vacuolated, and defective cristae arrangement in yeast and mammalian cells (40). Loss of function of Yme1 has been shown to result in punctate mitochondria that lacked reticulated cristae structures (41). To determine whether taz1Δ yme1Δ cells had mitochondrial morphology defects that were more extreme than either single mutant alone, electron microscopy (EM) was performed. Wild type yeast mitochondria were normal with a defined reticulated network (Fig. 5A). In our yeast strain, there were limited defects in the mitochondrial morphology of taz1Δ cells, but they did contain a significant proportion of elongated mitochondria. The loss of Yme1 in yeast led to abnormally shaped mitochondria with vacuolated cristae, and their reticulated arrangement was also lost in most of these aberrantly shaped mitochondria (Fig. 5, A and B). Alterations to mitochondrial shape and cristae arrangement were much more pronounced in taz1Δ yme1Δ cells (Fig. 5, A and B). Also, giant and swollen mitochondria in close proximity to the vacuole were also noted in some of the taz1Δ yme1Δ cells (Fig. 5B), and many of the taz1Δ yme1Δ cells had large multivacuolar structures. These observations indicate that deletion of YME1 in taz1Δ yeast causes severe defects to mitochondrial morphology, resulting in a larger proportion of cells with giant mitochondria that lack cristae, with a subset of these lying adjacent to the vacuole.
FIGURE 5.

Electron microscopy reveals severe mitochondrial morphological abnormalities in taz1Δ yme1Δ double mutant yeast. Wild type (W) and mutant strains were grown to early stationary phase (0.9–1.0 A600) in SC medium with 2% lactate and then processed for electron microscopy as described under “Materials and Methods.” Briefly, the harvested cells were treated with 1.5% KMnO4 and 1% sodium periodate before they were ethanol-dried and processed for electron microscopy using an LKB Huxley Ultramicrotome. The cells were then viewed using a JEOL JEM 1230 transmission electron microscope, and the images were captured with a Hamamatsu ORCA-HR digital camera. A, panels show a representative EMs. Wild type and taz1Δ yeast harbored predominantly healthy mitochondria with an increase in the presence of healthy but elongated mitochondria in taz1Δ yeast. Both yme1Δ and taz1Δ yme1Δ yeast harbored abnormally shaped mitochondria with vacuolated cristae. These defects were pronounced in taz1Δ yme1Δ cells, which also harbored cells with giant mitochondria, and further, many of the cells were not intact or were dying with no visible mitochondria. Arrows, vacuole (V) and mitochondria (M). B, tabulation of mitochondrial morphologies from at least 150 mitochondria for each strain.
Mitophagy Is Inefficient upon Inactivation of Yme1 in Cells Lacking Tafazzin Function
Aberrant mitochondria are selectively taken up by the vacuole to be degraded by a process termed mitophagy (42). Our EM results revealed that the taz1Δ yme1Δ yeast cells accumulate a significant number of aberrant mitochondria, and some were in close proximity to the vacuolar membrane, suggesting that these large mitochondria might be undergoing either an increased rate of mitophagy or might be unable to complete mitophagy. We determined whether mitophagy could proceed effectively in taz1Δ yme1Δ cells.
To monitor mitophagy, we used a well characterized mitophagy assay that tracks vacuolar uptake and degradation of a GFP-tagged MOM protein, Om45-GFP (22). Under normal growth conditions, there is very little mitophagy occurring in yeast cells; however, upon nitrogen starvation, mitophagy is induced. When the wild type and mutant yeast cells were grown under normal conditions, Om45-GFP resided predominantly at the mitochondria, indicating that mitophagy was not occurring (Fig. 6, A and B). Cells were then washed and resuspended in nitrogen-free medium for 24 h to induce mitophagy. Fluorescence microscopy revealed that in more than 90% of wild type and taz1Δ the GFP signal was predominantly at the vacuole, indicative of mitophagy occurring in these cells; this was reduced to 50% in yme1Δ cells, implying a slightly impaired ability to carry out mitophagy (Fig. 6, A and B). Notably, in the taz1Δ yme1Δ yeast cells, less than 10% of cells possessed GFP in their vacuole, signifying a substantial impairment in mitophagy due to nitrogen starvation upon inactivation of Yme1 function in the absence of tafazzin function.
FIGURE 6.

Mitophagy is inefficient in taz1Δ yme1Δ yeast. The Om45 allele was tagged with a GFP epitope in all of the indicated strains, and these cells were grown in YPL medium to 0.8–1.5 A600. The cells were then washed twice in distilled water, and an aliquot of cells was saved and processed for microscopy and Western blotting prior to resuspension and growth in SD−N medium to induce N2 starvation. After 24 h of growth in SD−N medium, the cells were harvested and processed for fluorescence microscopy. A, representative fluorescence and differential interference contrast microscopy, indicating the cellular localization of Om45-GFP before and after 24 h of nitrogen starvation. After nitrogen starvation, Om45-GFP was predominantly localized to the vacuole (an indication of mitophagy) in the majority of the cells of all of the yeast strains except for taz1Δyme1Δ yeast (arrows indicate cells with GFP signal outside the vacuole). B, the number of GFP-positive mitochondria and vacuoles from 250 cells were determined from three experiments for each strain under each growth condition. The S.E. was less than 5% for each experiment.
Once mitochondria are taken up by the vacuole, Om45-GFP is cleaved to Om45 and free GFP. Unlike most proteins, free GFP is relatively stable in the vacuole. The difference in the amount of free GFP and Om45-GFP is used to estimate the efficiency of mitophagy by Western blot versus GFP (Fig. 7A). Before nitrogen starvation, only full-length Om45-GFP was detected in any of the yeast strains (Fig. 7A). After nitrogen starvation, both free and Om45-bound GFP signal could be detected; however, the amount of free GFP in taz1Δ yme1Δ yeast cells was significantly lower than that in wild type, taz1Δ, and yme1Δ cells (Fig. 7A). Loss of function of either Taz1 or Yme1 allowed mitophagy to occur; however, loss of both resulted in inefficient mitophagy upon nitrogen starvation.
FIGURE 7.
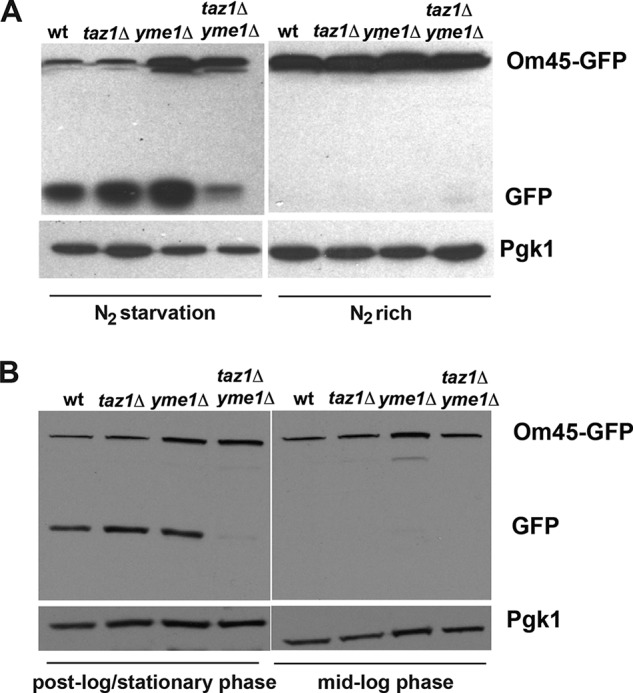
Mitophagy induced by nitrogen starvation or cells grown to stationary phase is defective in taz1Δ yme1Δ yeast. A, an anti-GFP antibody was used to perform Western blotting to ascertain the levels of Om45-GFP and free GFP in the indicated yeast strains before and after nitrogen starvation. Western blotting indicated lower levels of free GFP in taz1Δ yme1Δ cells when compared with wild type and single mutants, indicating inefficient mitophagy in these cells. An antibody against Pgk1 was used to assess equal protein loading among the different lanes. B, yeast strains with a chromosomally tagged Om45-GFP allele were grown to post-log phase for 3–5 days and processed for Western blotting with an anti-GFP antibody to determine the levels of Om45-GFP and free GFP at mid-log and stationary phase. Western blotting indicated a marked reduction in the levels of free GFP in taz1Δ yme1Δ cells when compared with wild type and single mutants, indicating a severe mitophagy defect in these cells. An antibody against Pgk1 was used to assess equal protein loading among the different lanes.
Mitophagy in yeast can also be induced by growth to post-log/stationary phase. Similar to the results observed upon nitrogen starvation, there was a substantial decrease in the appearance of free GFP in the taz1Δ yme1Δ cells compared with wild type, taz1Δ, and yme1Δ cells (Fig. 7B), indicating impaired mitophagy upon inactivation of YME1 in the absence of tafazzin function.
The taz1Δ yme1Δ Yeast Strain Is Sensitive to H2O2
Loss of tafazzin protein has been observed to result in a small decrease in ETC efficiency and a slight increase in ROS. ROS-damaged mitochondria are removed from the cell by mitophagy. We sought to determine whether inactivation of Yme1 in taz1Δ cells results in increased sensitivity to ROS and/or increased ROS generation, resulting in damage to cellular constituents.
To determine whether there was an increase in de novo ROS generation, the amount of protein carbonyl content was determined in taz1Δ yme1Δ cells and compared with wild type, taz1Δ, or yme1Δ cells (Fig. 8A). Increased ROS generation introduces carbonyl groups to the side chains of amino acid residues in proteins; therefore, measuring the cellular protein carbonyl content serves as a direct measure of ROS-induced damage. The taz1Δ cells have previously been shown to contain increased protein carbonyl content when grown in medium with a non-fermentable carbon source (17), and in our study, the protein carbonyl content was also observed to increase in taz1Δ yeast cells (Fig. 8A). The amount of protein carbonyl content in taz1Δ yme1Δ cells was similar to that observed in taz1Δ cells (Fig. 8A), although this was higher than that found in wild type and yme1Δ cells. Thus, inactivation of Yme1 does not further increase the generation of ROS in cells lacking tafazzin function.
FIGURE 8.
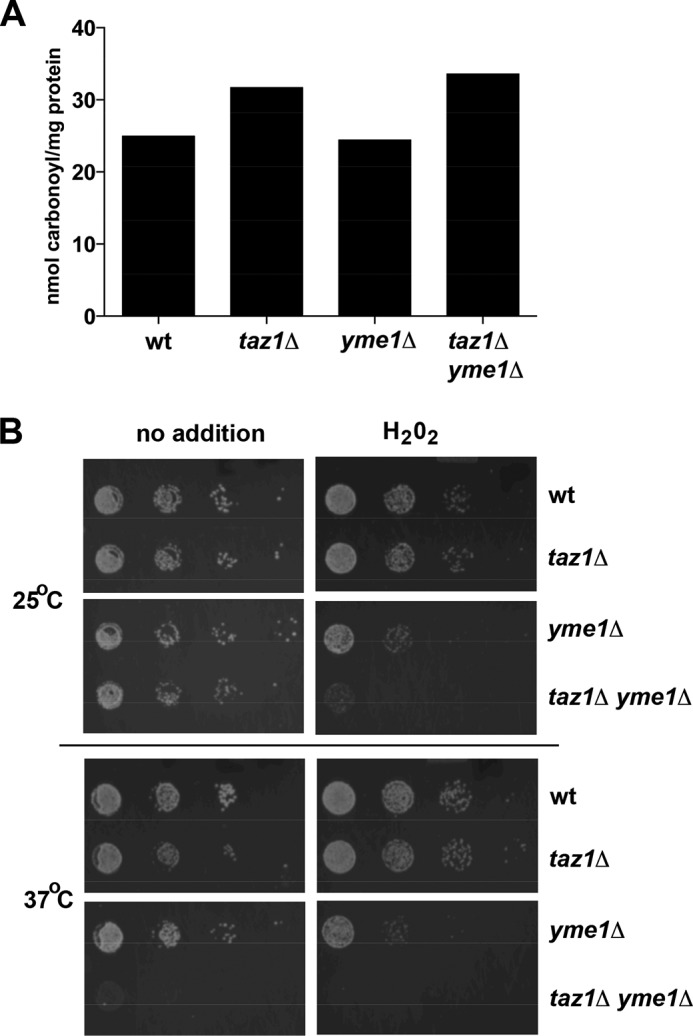
The taz1Δ yme1Δ yeast strain is sensitive to H2O2. A, oxidative stress causes the introduction of carbonyl groups to side chains of amino acid residues in proteins. Protein carbonylation was measured by the spectrophotometric estimation of protein-bound 2,4-dinitrophenylhydrazone (DNP) formed by the conversion of 2,4-dinitrophenylhydrazine to 2,4-dinitrophenylhydrazone by protein carbonyl groups. Yeast cells were grown to 0.9–1.0 A600 in YPL medium, and after that, proteins were extracted and estimated for carbonyl content using the protein carbonyl content assay kit (BioVision). The carbonyl content was then normalized to the amount of protein in each sample and expressed as nmol of carbonyl/mg of protein. The protein content in all of the samples was measured by the Bradford protein assay using the BCA assay kit (BioVision). Results are from three separate experiments, each with a S.E. of less than 5% of the mean. B, yeast strains were grown to early stationary phase (0.9–1.0 A600) in SC medium and then resuspended in the same medium to 0.45 A600, serially diluted (1:10), and spotted on YPL medium with or without 2.5 mm H2O2 and grown for 3–5 days at 25 or 37 °C. The presence of 2.5 mm H2O2 in the growth medium affects the growth of yme1Δ and taz1Δ yme1Δ cells; however, the growth defect is more pronounced in yeast lacking both TAZ1 and YME1.
To address the question of whether taz1Δ yme1Δ cells were more sensitive to ROS, cells were grown in the presence or absence of 2.5 mm H2O2. In the presence of H2O2, the taz1Δ yme1Δ cells barely grew (Fig. 8B), and although H2O2 also decreased growth of yme1Δ cells, it was not to the extent observed in taz1Δ yme1Δ cells. The decrease in growth of the taz1Δ yeast strain was similar to the decrease in growth observed in wild type cells (Fig. 8B). Inactivation of the TAZ1 gene does not alter sensitivity to H2O2, whereas inactivation of YME1 results in a slight increase in H2O2 sensitivity; however, inactivation of both TAZ1 and YME1 together results in a dramatic increase in H2O2 sensitivity.
DISCUSSION
A genome-wide screen identified processes that participate with Taz1 to regulate cell growth. Focusing on the role of the i-AAA protease Yme1, we determined that (i) tafazzin and Yme1 together are required for efficient growth on a fermentable carbon source, and this growth defect is exacerbated when mitochondrial respiration is the main source of energy; (ii) together, these genes are necessary for the maintenance of normal mitochondrial morphology; (iii) cells lacking both genes are more sensitive to H2O2; and (iv) together, both genes are required for efficient mitophagy induced by nitrogen starvation or entry into post-log/stationary phase.
Loss of both CL and PE has been shown to affect mitochondrial biogenesis and cause synthetic lethality in yeast (43). Yme1 has been shown to regulate the turnover of proteins such as Psd1, Ups1, and Ups2, which are known regulators of both PE and CL levels in yeast (39, 44). Psd1 catalyzes the conversion of phosphatidylserine to PE in the mitochondria. Both Ups1 and Ups2 reside in the IMS and are proteolytic targets of Yme1 (45), and Ups1 has been shown to transport phosphatidic acid (PA) from MOM to MIM to supply PA for CL synthesis (46), and Ups2, by an unidentified mechanism, has been shown to regulate PE levels in yeast (45). Surprisingly, we did not observe gross changes to mitochondrial phospholipid levels in the taz1Δ yme1Δ cells compared with the taz1Δ single mutant other than a slight increase in PE content. Thus, major alterations in mitochondrial phospholipid levels do not appear to be a major driver of why loss of YME1 function in taz1Δ cells exacerbates defects in growth and mitochondrial morphology.
The ETC complexes III and IV associate to form supercomplexes. In yeast lacking TAZ1, complex IV has been shown to dissociate from complex III, and this is surmised to cause a leak of electrons from the latter complex to act as source of ROS (15). In this study, we also showed that the supercomplex formation is destabilized in the taz1Δ mutant. Surprisingly, loss of Yme1 function stabilized supercomplex formation in taz1Δ cells. Because both CL and PE are non-hexagonal forming lipids, the slight increase in PE in the taz1Δ yme1Δ cells could allow for the observed return to normal supercomplex III2IV2 stability because CL has been demonstrated to be able to be substituted by other non-hexagonal forming lipids to maintain ETC stability.
The majority of yme1Δ cells contained normal mitochondria, but 25% of the total mitochondria accounted for were abnormally shaped with loss of reticulated cristae structures. These observations are in agreement with previous studies (33, 41) in yeast lacking YME1. In taz1Δ cells, the majority of the mitochondria were normal; however, ∼20% of the total mitochondria were elongated but did not show any alterations in their MIM cristae structures. This observation is contradictory to one study (40) that reported abnormal mitochondria with cristae reticulations and MIM adhesion but in agreement with a recent study (47) that assessed mitochondrial morphology of tafazzin deficiency in various yeast strain backgrounds where loss of tafazzin produced only mild alterations to mitochondrial morphology. In our study, mitochondrial morphological defects in the taz1Δ yme1Δ cells were more severe than in either yme1Δ or taz1Δ cells alone. Approximately 60% of the total mitochondria were abnormal, and moreover, a few cells possessed giant mitochondria that were in close proximity to the vacuole.
Abnormal mitochondria are an increased source of ROS production, and failure to clear these mitochondria has been shown to increase the oxidative stress on the yeast cell (42). Protein carbonylation was increased in both the taz1Δ and taz1Δ yme1Δ double mutant; however, there was no increase in taz1Δ yme1Δ cells compared with taz1Δ cells. This implies that there is not an increase in the production of ROS by the abnormal mitochondria that accumulate in taz1Δ yme1Δ cells. However, the taz1Δ yme1Δ were far more sensitive to the ROS generator H2O2 compared with either single mutant alone, implying that there is a defect in H2O2 scavenging. At present, how loss of YME1 causes H2O2 scavenging defects and how this phenotype is aggravated by the loss of Taz1 function are not clear. However, because mitophagy is inefficient in taz1Δ yme1Δ cells, their sensitivity to H2O2 could be due to the effect of accumulation of damaged mitochondria.
In addition to YME1, SGA analysis also identified that TAZ1 has negative genetic interactions with genes regulating iron metabolism and with structural genes for the argininosuccinate shunt. The argininosuccinate shunt is a major means by which cells regulate nitrogen metabolism and sensing, and nitrogen sensing is a major driver of mitophagy. In light of our determination that cells lacking tafazzin function are more prone to altered mitophagy upon inactivation of YME1, this may point to a more general theme regarding an increase in susceptibility to mitophagy to cells with inactive tafazzin. Our observed genetic interaction with genes regulating iron metabolism is interesting because a previous study using crd1Δ cells that cannot synthesize any CL showed genetic interactions between CL and iron metabolism, including Fe-S-requiring enzymes of the TCA cycle and ETC (29). Because both the TCA cycle and ETC are partially compromised in cells lacking tafazzin function, further compromise of both is a likely scenario by which defective Fe-S assembly results in decreased cell growth.
In summary, this study has pointed to several new processes that are required to be efficient to allow for normal growth of cells lacking tafazzin function that will drive further research. One of these new processes, mitochondrial quality control through the i-AAA protease Yme1, was investigated in detail and found to be necessary to maintain mitochondrial morphology and to drive mitophagy in cells lacking tafazzin function.
Acknowledgments
We thank Dr. J. Pedro Fernandez-Murray for helpful comments during the course of this work and Dr. Kanki (Kyushu University School of Medical Sciences) for kindly providing the Om45-GFP strains.
This work was supported by Genome Canada, Genome Atlantic, and the Barth Syndrome Foundation of Canada. This study was also supported by a Canadian Institutes of Health Research grant (to C. R. M.).
- MLCL
- monolysocardiolipin
- CL
- cardiolipin
- ETC
- electron transport chain
- ROS
- reactive oxygen species
- MIM
- mitochondrial inner membrane
- MOM
- mitochondrial outer membrane
- IMS
- intermembrane space
- TCA
- tricarboxylic acid
- NAT
- neurseothricin
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- BN-PAGE
- blue native PAGE
- SGA
- synthetic genetic array
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- PE
- phosphatidylethanolamine
- PA
- phosphatidic acid.
REFERENCES
- 1. Clarke S. L. N., Bowron A., Gonzalez I. L., Groves S. J., Newbury-Ecob R., Clayton N., Martin R. P., Tsai-Goodman B., Garratt V., Ashworth M., Bowen V. M., McCurdy K. R., Damin M. K., Spencer C. T., Toth M. J., Kelley R. I., Steward C. G. (2013) Barth syndrome. Orphanet. J. Rare Dis. 8, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jefferies J. L. (2013) Barth syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 10.1002/ajmg.c.31372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Neuwald A. F. (1997) Barth syndrome may be due to an acyltransferase deficiency. Curr. Biol. 7, R465–R466 [DOI] [PubMed] [Google Scholar]
- 4. Xu Y., Malhotra A., Ren M., Schlame M. (2006) The enzymatic function of tafazzin. J. Biol. Chem. 281, 39217–39224 [DOI] [PubMed] [Google Scholar]
- 5. Schneiter R. (1999) Electrospray ionization tandem mass spectrometry (ESI-MS/MS) analysis of the lipid molecular species composition of yeast subcellular membranes reveals acyl chain-based sorting/remodeling of distinct molecular species en route to the plasma membrane. J. Cell Biol. 146, 741–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gebert N., Joshi A. S., Kutik S., Becker T., McKenzie M., Guan X. L., Mooga V. P., Stroud D. A., Kulkarni G., Wenk M. R., Rehling P., Meisinger C., Ryan M. T., Wiedemann N., Greenberg M. L., Pfanner N. (2009) Mitochondrial cardiolipin involved in outer-membrane protein biogenesis: implications for Barth syndrome. Curr. Biol. 19, 2133–2139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Horvath S. E., Daum G. (2013) Lipids of mitochondria. Prog. Lipid Res. 52, 590–614 [DOI] [PubMed] [Google Scholar]
- 8. Schlame M., Towbin J. A., Heerdt P. M., Jehle R., DiMauro S., Blanck T. J. J. (2002) Deficiency of tetralinoleoyl-cardiolipin in Barth syndrome. Ann. Neurol. 51, 634–637 [DOI] [PubMed] [Google Scholar]
- 9. Phoon C. K. L., Acehan D., Schlame M., Stokes D. L., Edelman-Novemsky I., Yu D., Xu Y., Viswanathan N., Ren M. (2012) Tafazzin knockdown in mice leads to a developmental cardiomyopathy with early diastolic dysfunction preceding myocardial noncompaction. J. Am. Heart Assoc. 10.1161/JAHA.111.000455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xu Y., Condell M., Plesken H., Edelman-Novemsky I., Ma J., Ren M., Schlame M. (2006) A Drosophila model of Barth syndrome. Proc. Natl. Acad. Sci. U.S.A. 103, 11584–11588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gu Z., Valianpour F., Chen S., Vaz F. M., Hakkaart G. A., Wanders R. J. A., Greenberg M. L. (2004) Aberrant cardiolipin metabolism in the yeast taz1 mutant: a model for Barth syndrome. Mol. Microbiol. 51, 149–158 [DOI] [PubMed] [Google Scholar]
- 12. Khuchua Z., Yue Z., Batts L., Strauss A. W. (2006) A zebrafish model of human Barth syndrome reveals the essential role of tafazzin in cardiac development and function. Circ. Res. 99, 201–208 [DOI] [PubMed] [Google Scholar]
- 13. Acehan D., Xu Y., Stokes D. L., Schlame M. (2007) Comparison of lymphoblast mitochondria from normal subjects and patients with Barth syndrome using electron microscopic tomography. Lab. Invest. 87, 40–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McKenzie M., Lazarou M., Thorburn D. R., Ryan M. T. (2006) Mitochondrial respiratory chain supercomplexes are destabilized in Barth syndrome patients. J. Mol. Biol. 361, 462–469 [DOI] [PubMed] [Google Scholar]
- 15. Brandner K., Mick D. U., Frazier A. E., Taylor R. D., Meisinger C., Rehling P. (2005) Taz1, an outer mitochondrial membrane protein, affects stability and assembly of inner membrane protein complexes: implications for Barth syndrome. Mol. Biol. Cell 16, 5202–5214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gonzalvez F., D'Aurelio M., Boutant M., Moustapha A., Puech J.-P., Landes T., Arnauné-Pelloquin L., Vial G., Taleux N., Slomianny C., Wanders R. J., Houtkooper R. H., Bellenguer P., Møller I. M., Gottlieb E., Vaz F. M., Manfredi G., Petit P. X. (2013) Barth syndrome: cellular compensation of mitochondrial dysfunction and apoptosis inhibition due to changes in cardiolipin remodeling linked to tafazzin (TAZ) gene mutation. Biochim. Biophys. Acta. 1832, 1194–1206 [DOI] [PubMed] [Google Scholar]
- 17. Chen S., He Q., Greenberg M. L. (2008) Loss of tafazzin in yeast leads to increased oxidative stress during respiratory growth. Mol. Microbiol. 68, 1061–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Claypool S. M., Oktay Y., Boontheung P., Loo J. A., Koehler C. M. (2008) Cardiolipin defines the interactome of the major ADP/ATP carrier protein of the mitochondrial inner membrane. J. Cell Biol. 182, 937–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Johnston J., Kelley R. I., Feigenbaum A., Cox G. F., Iyer G. S., Funanage V. L., Proujansky R. (1997) Mutation characterization and genotype-phenotype correlation in Barth syndrome. Am. J. Hum. Genet. 61, 1053–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ronvelia D., Greenwood J., Platt J., Hakim S., Zaragoza M. V. (2012) Intrafamilial variability for novel TAZ gene mutation: Barth syndrome with dilated cardiomyopathy and heart failure in an infant and left ventricular noncompaction in his great-uncle. Mol. Genet. Metab. 107, 428–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lorenz M. C., Muir R. S., Lim E., McElver J., Weber S. C., Heitman J. (1995) Gene disruption with PCR products in Saccharomyces cerevisiae. Gene 158, 113–117 [DOI] [PubMed] [Google Scholar]
- 22. Kanki T., Kang D., Klionsky D. J. (2009) Monitoring mitophagy in yeast: the Om45-GFP processing assay. Autophagy 5, 1186–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tong A. H., Evangelista M., Parsons A. B., Xu H., Bader G. D., Pagé N., Robinson M., Raghibizadeh S., Hogue C. W., Bussey H., Andrews B., Tyers M., Boone C. (2001) Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294, 2364–2368 [DOI] [PubMed] [Google Scholar]
- 24. Claypool S. M., McCaffery J. M., Koehler C. M. (2006) Mitochondrial mislocalization and altered assembly of a cluster of Barth syndrome mutant tafazzins. J. Cell Biol. 174, 379–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vaden D. L., Gohil V. M., Gu Z., Greenberg M. L. (2005) Separation of yeast phospholipids using one-dimensional thin-layer chromatography. Anal. Biochem. 338, 162–164 [DOI] [PubMed] [Google Scholar]
- 26. Diekert K., de Kroon A. I., Kispal G., Lill R. (2001) Isolation and subfractionation of mitochondria from the yeast Saccharomyces cerevisiae. Methods Cell Biol. 65, 37–51 [DOI] [PubMed] [Google Scholar]
- 27. Schägger H. (2001) Blue-native gels to isolate protein complexes from mitochondria. Methods Cell Biol. 65, 231–244 [DOI] [PubMed] [Google Scholar]
- 28. Swamy M., Siegers G. M., Minguet S., Wollscheid B., Schamel W. W. A. (2006) Blue native polyacrylamide gel electrophoresis (BN-PAGE) for the identification and analysis of multiprotein complexes. Sci. STKE 2006, pl4. [DOI] [PubMed] [Google Scholar]
- 29. Patil V. A., Fox J. L., Gohil V. M., Winge D. R., Greenberg M. L. (2013) Loss of cardiolipin leads to perturbation of mitochondrial and cellular iron homeostasis. J. Biol. Chem. 288, 1696–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tsai C.-L., Barondeau D. P. (2010) Human frataxin is an allosteric switch that activates the Fe-S cluster biosynthetic complex. Biochemistry 49, 9132–9139 [DOI] [PubMed] [Google Scholar]
- 31. Whited K., Baile M. G., Currier P., Claypool S. M. (2013) Seven functional classes of Barth syndrome mutation. Hum. Mol. Genet. 22, 483–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Langer T. (2000) AAA proteases: cellular machines for degrading membrane proteins. Trends Biochem. Sci. 25, 247–251 [DOI] [PubMed] [Google Scholar]
- 33. Campbell C. L., Tanaka N., White K. H., Thorsness P. E. (1994) Mitochondrial morphological and functional defects in yeast caused by yme1 are suppressed by mutation of a 26S protease subunit homologue. Mol. Biol. Cell 5, 899–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Thorsness P. E., White K. H., Fox T. D. (1993) Inactivation of YME1, a member of the ftsH-SEC18-PAS1-CDC48 family of putative ATPase-encoding genes, causes increased escape of DNA from mitochondria in Saccharomyces cerevisiae. Mol. Cell Biol. 13, 5418–5426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Beyer K., Nuscher B. (1996) Specific cardiolipin binding interferes with labeling of sulfhydryl residues in the adenosine diphosphate/adenosine triphosphate carrier protein from beef heart mitochondria. Biochemistry 35, 15784–15790 [DOI] [PubMed] [Google Scholar]
- 36. Beyer K., Klingenberg M. (1985) ADP/ATP carrier protein from beef heart mitochondria has high amounts of tightly bound cardiolipin, as revealed by phosphorus-31 nuclear magnetic resonance. Biochemistry 24, 3821–3826 [DOI] [PubMed] [Google Scholar]
- 37. Houtkooper R. H., Vaz F. M. (2008) Cardiolipin, the heart of mitochondrial metabolism. Cell Mol. Life Sci. 65, 2493–2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schlame M., Rua D., Greenberg M. L. (2000) The biosynthesis and functional role of cardiolipin. Prog. Lipid Res. 39, 257–288 [DOI] [PubMed] [Google Scholar]
- 39. Nebauer R., Schuiki I., Kulterer B., Trajanoski Z., Daum G. (2007) The phosphatidylethanolamine level of yeast mitochondria is affected by the mitochondrial components Oxa1p and Yme1p. FEBS J. 274, 6180–6190 [DOI] [PubMed] [Google Scholar]
- 40. Claypool S. M., Boontheung P., McCaffery J. M., Loo J. A., Koehler C. M. (2008) The cardiolipin transacylase, tafazzin, associates with two distinct respiratory components providing insight into Barth syndrome. Mol. Biol. Cell 19, 5143–5155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Campbell C. L., Thorsness P. E. (1998) Escape of mitochondrial DNA to the nucleus in yme1 yeast is mediated by vacuolar-dependent turnover of abnormal mitochondrial compartments. J. Cell Sci. 111, 2455–2464 [DOI] [PubMed] [Google Scholar]
- 42. Kurihara Y., Kanki T., Aoki Y., Hirota Y., Saigusa T., Uchiumi T., Kang D. (2012) Mitophagy plays an essential role in reducing mitochondrial production of reactive oxygen species and mutation of mitochondrial DNA by maintaining mitochondrial quantity and quality in yeast. J. Biol. Chem. 287, 3265–3272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Joshi A. S., Thompson M. N., Fei N., Hüttemann M., Greenberg M. L. (2012) Cardiolipin and mitochondrial phosphatidylethanolamine have overlapping functions in mitochondrial fusion in Saccharomyces cerevisiae. J. Biol. Chem. 287, 17589–17597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tamura Y., Endo T., Iijima M., Sesaki H. (2009) Ups1p and Ups2p antagonistically regulate cardiolipin metabolism in mitochondria. J. Cell Biol. 185, 1029–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Potting C., Wilmes C., Engmann T., Osman C., Langer T. (2010) Regulation of mitochondrial phospholipids by Ups1/PRELI-like proteins depends on proteolysis and Mdm35. EMBO J. 29, 2888–2898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Connerth M., Tatsuta T., Haag M., Klecker T., Westermann B., Langer T. (2012) Intramitochondrial transport of phosphatidic acid in yeast by a lipid transfer protein. Science 338, 815–818 [DOI] [PubMed] [Google Scholar]
- 47. Baile M. G., Sathappa M., Lu Y.-W., Pryce E., Whited K., McCaffery J. M., Han X., Alder N. N., Claypool S. M. (2014) Unremodeled and remodeled cardiolipin are functionally indistinguishable in yeast. J. Biol. Chem. 289, 1768–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]