

Abstract
High molecular weight multiblock copolymers are synthesized as robust polymer fibers via interfacial bioorthogonal polymerization employing the rapid cycloaddition of s-tetrazines with strained trans-cyclooctenes. When cell-adhesive peptide was incorporated in the tetrazine monomer, the resulting protein-mimetic polymer fibers provide guidance cues for cell attachment and elongation.
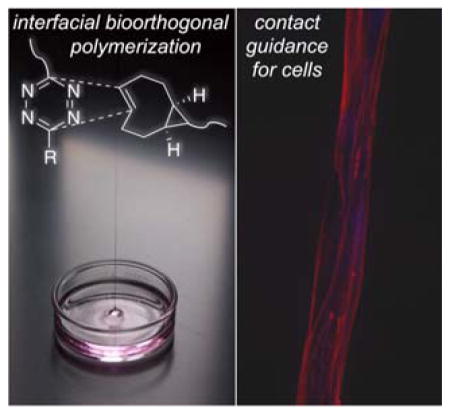
Keywords: multiblock copolymer, interfacial polymerization, bioorthogonal, fibers, guidance cues
Block copolymers—covalently connected polymer chains of distinct chemical compositions—exhibit advanced physical, mechanical and biological properties that often exceed the properties of individual building blocks.[1, 2] While di- and triblock copolymers have been extensively investigated, less attention has been devoted to multiblock copolymers, most likely due to their more involved syntheses.[3] Recent advances in living radical polymerization[2, 4] have enabled the synthesis of multiblock copolymers of up to 20 repeats, via an elegant one-pot approach by sequential addition of different monomers.[5] Because these multiblock copolymers were synthesized via the chain-growth mechanism, they are limited to vinyl monomers and require protecting groups for reactive side chains. Moreover, individual blocks must be relatively short to facilitate complete monomer consumption and an efficient addition of the next block.
An alternative approach to the synthesis of multiblock copolymers is to link polymeric precursors carrying complementary functional groups via a step-growth polymerization strategy. Bioorthogonal reactions —unnatural reactions that proceed efficiently in biological context[6–8] —provide an attractive route for the construction of complex polymers with backbone diversity and complexity through step-growth polymerization. Matyjaszewski and coworkers demonstrated the first synthesis of step-growth polymers by CuI-catalyzed alkyneazide cycloaddition (CuAAC)[9] using homobifunctional (α,ω-diazido-terminated, plus propargyl ether) or heterotelechelic (α-azide, ω-alkyne) polystyrene prepared by atom transfer radical polymerization (ATRP).[10] Most recently, multiblock alternating copolymers of poly(ethylene oxide) (PEO) and green fluorescent protein were synthesized by CuAAC, and under appropriate processing conditions, the hybrid copolymer self-assembled into micrometer-long fibrous aggregates.[11] Our group has explored the utility of CuAAC for the construction of multiblock copolymers using diverse sets of synthetic and peptidic building blocks, such as poly(tert-butyl acrylate), polystyrene, poly(ethylene glycol) (PEG) and elastin-derived peptides.[12–15] Separately, thiol/norbornene photochemistry has been employed for the synthesis of mixed polystyrene, poly(ethylene oxide) and poly(dimethyl siloxane) multiblock copolymers.[16] In all these cases, oligomers with average repeats of 4–5 were obtained, and a high percentage of cyclic oligomers was found in the products.[17] Moreover, additional post-polymerization modification or processing steps are necessary to produce matrices or fibers for tissue engineering applications.
Bioorthogonal reactions involving strained alkenes with tetrazines, first described by us and others in 2008,[18, 19] have emerged as an important tool for biomedical research.[20] Tetrazine ligations of conformationally strained trans-cyclooctene (sTCO) derivatives developed in the Fox group are the fastest bioorthogonal reactions reported to date, with rate constants (k2) that can exceed 106 M−1s−1.[21–23] Tetrazine-norbornene chemistry has been utilized in polymer synthesis and hydrogel fabrication,[24–26] and trans-cyclooctene has been used to modify polymers and nanoparticles for imaging applications.[27, 28] Recently, our group described the first interfacial cross-linking reactions based on a bioorthogonal reaction with a rapid rate constant (k2) of 2.86 x 105 M−1s−1 (Figure 1A). Without triggers or templates, this provided a method for creating and patterning biomaterials through diffusion-controlled gelation at liquid-gel interfaces.[29] However, trans-cyclooctene-tetrazine ligation has not been used for polymerization purposes.
Figure 1.
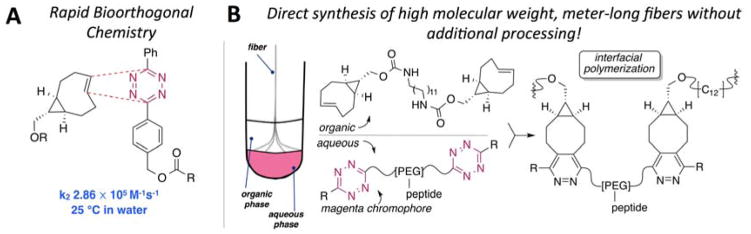
(A) The cycloaddition involving sTCO and a diphenyl-s-tetrazine proceeds with rapid kinetics. (B) Schematic description of interfacial bioorthogonal polymerization between phase-separated monomer solutions to produce multiblock copolymer fibers.
Classically, interfacial polymerization is based on reactions that are fast but poorly selective,[30] thereby precluding the incorporation of biological molecules during the polymerization. Herein, we describe the first example of interfacial bioorthogonal polymerization— the use of bioorthogonal chemistry to create multiblock copolymer fibers through rapid reaction between sTCO and a diphenyl-s-tetrazine derivative at an immiscible liquid-liquid interface (Figure 1). Interfacial polymerization of carefully chosen monomeric building blocks with differential solubility produces high molecular weight, multiblock copolymer fibers with a semicrystalline microstructure. By contrast, direct solution polymerization gives copolymers with a large fraction of low molecular weight cyclic products. Atomic force microscopy (AFM) was used to characterize the microstructure and stiffness of the polymer fibers. The bioorthogonal nature of the reaction permits facile incorporation of biomolecules during the polymerization to fine-tune the properties of the resulting materials to induce desired cellular responses under in vitro cell culture conditions. Specifically, polymerization of monomers containing fibronectin-derived integrin-binding peptide side chains produced protein-mimetic fibers that direct the attachment and alignment of cells of both mesenchymal and epithelial origins.
The monomers for interfacial polymerization were prepared from precursors that are readily available on a multigram scale and react with a rapid rate (Figure 1A). A bis-sTCO monomer (1) with a hydrophobic spacer was designed to be soluble in organic media, and bis-tetrazine monomers (2a and 2b) with PEG spacers were designed to be water soluble and suited for the incorporation of peptidic side chains. PEG spacers with molecular weights of 7.5 kDa and 3.5 kDa were used to prepare monomers 2a and 2b, respectively, providing comparable lengths for these monomers. We postulated that the fast reactivity between Tz and sTCO would enable interfacial polymerization, with the ability to draw functionalized multiblock polymer fibers from the liquid-liquid interface. The concept and experimental design are illustrated in Figure 1B, and the structures of the monomers (1, 2a and 2b), along with the polymer products, 3a and 3b, are displayed in Figure 2A. In our design, a fibronectin-derived cell-adhesive peptide with a basic sequence of GRGDSP[31, 32] was strategically placed dangling from the backbone so that after interfacial polymerization, the peptide would serve as the side chain for greater accessibility to integrins on the cell surface.[12] The overall molecular weights of 2a and 2b are comparable to ensure a similar diffusion rate.
Figure 2.

(A) Chemical structures of bis-sTCO (1) and bis-tetrazine monomers (2a, 2b) and the repeating unit of the interfacial products (3a, 3b). (B) Photograph showing an ethyl acetate solution (colorless) of bis-sTCO 1 (2.0 mg/mL, 3.6 mM) overlaid on an aqueous solution (pink) of bis-tetrazine 2a (2.0 mg/mL, 0.25 mM). (C) Photograph showing multiblock copolymer fibers (arrow, colorless) being pulled out of the immiscible interface.(D–G): Photographs showing the time course of solution (THF) polymerization of 1 and 2a. (H) GPC traces of 3a produced by interfacial (black) and solution (blue) polymerization.
When an ethyl acetate solution of 1 (3 mL, 2.0 mg/mL (3.6 mM)) was overlaid on an aqueous solution of 2a (3 mL, 2.0 mg/mL (0.25 mM)), a colorless multiblock copolymer was formed instantly at the interface between the two immiscible solutions. An excess of the bis-sTCO 1 was employed because this monomer is wicked from the top phase as the fiber is drawn, causing the bis-sTCO concentration to drop during the fiber pulling experiment. As shown in Figure 2C, polymer fibers were pulled from the interface and collected on a rotating frame at 20 RPM (see video in the supporting information). Meter-long microfibers have been continuously drawn from the interface without breaking until approximately 70% of 2a was consumed, determined by monitoring the UV absorbance of tetrazine chromophore in solutions before and after polymerization. A number of water-immiscible solvents, including ethyl ether, ethyl acetate, hexanes and toluene, were evaluated for the interfacial polymerization process. All of these solvents successfully gave polymer films at the interface, with ethyl acetate proving most conducive to fiber production due to its moderate volatility and high solvation power toward 1. In addition, ethyl acetate keeps the polymer film at the interface in a swollen state (as evidenced by a smaller fiber diameter for the dry fibers as compared to the freshly drawn fibers), facilitating rapid monomer diffusion towards the polymer chain ends at the interface.
Polymers produced by interfacial polymerization were characterized by gel permeation chromatography (GPC) using THF as the mobile phase and narrow disperse PEO as the standards. It was necessary to reflux the fibers in THF in order to completely dissolve them. As shown in Figure 2H, the interfacially polymerized polymer has a broad molecular weight distribution, as is characteristic for step-growth polymerization. The GPC trace was deconvoluted into two peaks, both with a Gaussian distribution. The respective number average molecular weights (Mn) calculated are 69.0 and 262.5 kDa, corresponding to an average of 7.5 and 28.7 repeating units (Table 1), respectively. These values are significantly higher than those calculated from previously reported step-growth polymers prepared from CuAAC or thiol/ene chemistry.[10, 13, 16] The interfacial process resulted in multiblock copolymers with molecular weights on a par with those synthesized by controlled radical polymerization of vinyl monomers,[33, 34] but with the added advantage that bioorthogonal chemistry used for the polymerization can tolerate a broad range of biological functionalities without need for protection/deprotection schemes. The incorporation of biological peptide did not compromise the polymerization process, as high molecular weight polymer 3b (Mn = 235 kDa) was also obtained using interfacial polymerization (Figure S7). However, the broad nature and the tailing of the GPC trace for 3b suggest that the analyses may be complicated by the non-covalent interactions of the polymer with the GPC stationary phase and an artificially broad representation of the molecular weight distribution.
Table 1.
GPC analyses of multiblock copolymer, 3a, synthesized by interfacial and solution phase polymerization.
| Polymerization Strategies | Interfacial1 | Solution2 | ||||||
|---|---|---|---|---|---|---|---|---|
| Overall | Deconvoluted Peaks | Overall | Deconvoluted Peaks | |||||
| Mn([kDa])3 | 180.1 | 69.0 | 262.5 | 22.0 | 3.4 | 9.7 | 11.9 | 76.1 |
| Mw([kDa]) | 543.0 | 105.6 | ||||||
| Number of Repeats4 | 19.7 | 7.5 | 28.7 | 2.4 | - | 1.1 | 1.3 | 8.3 |
| PDI | 3.0 | 4.8 | ||||||
| Relative Percentage5 | 100% | 45% | 55% | 100% | 59% | 6% | 11% | 24% |
For interfacial polymerization, monomer 1 was dissolved in ethyl acetate at a concentration of 2.0 mg/mL (3.6 mM) and monomer 2a was dissolved in water at a concentration of 2.0 mg/mL (0.25 mM);
For solution polymerization, monomer 1 and 2a, dissolved in THF separately at a concentration of 2.0 mg/mL, were mixed at a molar ratio of 1/1 to give a final monomer concentration of 0.23 mM;
The molecular weights (Mn and Mw) were calculated from GPC based on PEO standards;
The molecular weight of the repeating unit in the multiblock copolymer is calculated as 9,142 Da.
Relative percentages were calculated based on the areas under the GPC curves.
A solution polymerization was carried using the same monomer pair (1 and 2a) at concentrations comparable to the interfacial process using equimolar amounts of 1 and 2a in THF (Figure 2D–G). At the point of contact where 1 was first added to 2a, the initially pink solution instantaneously became colorless, indicating an immediate local consumption of the pink tetrazine chromophore. Within three minutes in the absence of any agitation, the entire polymerization solution was nearly colorless. This observation clearly indicates that the reaction of Tz with sTCO is significantly faster than the diffusion of the monomeric species. The GPC trace for the solution phase product (Figure 2H) was deconvoluted into four peaks, with the highest molecular weight fraction having 8–10 repeating units, but representing only 24% of the polymeric product by integration (Table 1). A major fraction (59% by integration) of the solution polymerization product eluted at 14.5 min — a longer retention time than monomer 2a (Figure S6). Plausibly, this may indicate the formation of cyclic byproducts with smaller hydrodynamic volumes than the oligomeric starting material.[10] Doubling the monomer concentration while maintaining the overall stoichiometry in solution polymerization gave rise to a polymer product with a similar multimodal GPC curve, although the same curve fitting procedure revealed a slightly higher portion (35%) of the high molecular weight fraction (Figure S6, Table S1).
The high molecular weight of the interfacial products is a direct consequence of the rapid, sTCO-based tetrazine ligation, which enables diffusion controlled polymerization at the liquid-liquid interface. In solution phase step growth polymerization, high molecular weight products can only be obtained when the reaction stoichiometry is strictly maintained and the polymerization is driven to close to 100% conversion.[30] During interfacial polymerization, the stoichiometry is naturally maintained at the interface. As polymers are removed from the interface, additional monomers continue to diffuse into the interface and attach to the growing chain ends to produce high molecular weight products. Overall, the interfacial bioorthogonal polymerization serves as a powerful strategy to yield high molecular weight multiblock copolymer products. The modular approach also permits straightforward incorporation of peptidic cues for contact guidance of cells. Thus, when peptide-containing tetrazine monomer (2b) is used in place of 2a, peptide conjugated multiblock hybrid copolymer (3b, Figure 2A) fibers are produced.
Fibers were collected in a consistent and reproducible fashion throughout the course of polymerization over the entire collecting frame (2 cm wide, Figure 3A–B). Crosshatched meshes (Figure 3C) were generated by simply changing the axis of rotation of the collecting frame. The collected fibers can be readily transferred to a glass-supported silicone well (Figure 3D) for cell culture purposes (see supporting information for details). The polymeric fibers are optically birefringent (Figure 3E–F) as evidenced by the change of fiber color when the fiber orientation is rotated relative to polarized incident light.[35] This observation implies the presence of locally ordered crystalline structures in the fibers, which were induced during the fiber pulling process. Scanning electron microscopy (SEM) imaging (Figure 3G–H) showed a similar morphology for 3a and 3b fibers, with the majority of the fibers having a diameter in the range of 6–11 μm (Figure 3I) although a small population of thicker fibers is also present, possibly due to fiber merging during the pulling process. Collectively, these findings illustrate how interfacial bioorthogonal polymerization can be used to fabricate aligned or woven fibers of uniform diameter through a simple pulling process.
Figure 3.

(A) Multiblock copolymer fibers collected on a copper frame. (B–C) Dry polymer fibers visualized under light microscope. (D) Fibers secured in a silicone well. (E–F) Polymer fibers imaged under polarized light. Fibers were aligned at a 45° angle relative to the polarizer. Fibers appear yellow (E) and blue (F) as a result of subtractive and additive interference, respectively. (G–H) SEM micrographs of 3a (G) and 3b (H) fibers. (I) SEM histogram depicting the size distribution of multiblock copolymer fiber 3a. The histograms for 3a and 3b largely overlap.
The polymer fibers were further subjected to thermal, morphological and mechanical analyses. Differential scanning calorimetry (DSC) experiments revealed broad melting transitions centered around 53 °C and 34 °C (Figure 4A) for 3a and 3b, respectively, indicating the semicrystalline nature of the polymers. The DSC thermograms of PEG-based bis-tetrazine monomers 2a and 2b show sharp endotherms at 55 °C and 43 °C (Figure S8), corresponding to the respective melting transition of the PEG chains. Compared to monomers 2a and 2b, polymers 3a and 3b had a broader melting peak and a lower melting enthalpy. On the other hand, the second heating cycle of the bis-TCO monomer 1 did not reveal any melting endotherms (Figure S8). Collectively, our DSC results imply that the PEG and the aliphatic blocks contribute to the crystalline and the amorphous domains of the multiblock copolymers. The crystalline structure may also be reinforced by the propensity for hydrogen bonding and aromatic-aromatic interactions in 2a and 2b due to the presence of amide and aromatic residues, respectively. Analogously, non-covalent interactions have been invoked to explain polymer chain association and crystallinity in nylon interfacial polymerization.[30] Compared to 3a, the dangling peptide in 3b compromises the crystalline packing efficiency, as evidenced by a lower Tm and reduced enthalpy relative to that determined for 3a. A broad endortherm around −40 °C in 3b is postulated to be a glass transition. Such a transition was not seen in 3a, possibly due to its higher crystallinity.
Figure 4.

(A) DSC thermograms of 3a and 3b. (B–C) AFM height images of 3a (B) and 3b (C) fibers. (D) DMT modulus histograms for 3a and 3b fibers. Experimental data were curve-fitted with a Gaussian distribution.
Atomic force microscopy (AFM), operated in PeakForce Tapping mode, was employed for quantitative nanomechanical property mapping (QNM) of the polymer fibers.[36–38] The AFM height image for 3a fibers (Figure 4B) shows a semicrystalline polymer morphology with lamellar patterns, composed by densely packed crystalline domains appearing brighter than the surrounding amorphous interstitials. The uneven fiber surface introduced addition height difference across the scanned area. The surface of 3b fibers (Figure 4C) displays a more diffuse feature, with less discernible crystalline domains. AFM modulus mapping at a nanometer scale provides a measure of the local mechanical environment that is relevant to cells. The nanomechanical properties of the multiblock copolymer fibers were extrapolated from the AFM force-separation curves using a Hertzian model and taking into consideration adhesive forces.[39, 40] From the representative modulus histograms shown in Figure 4D, Young’s modulus was calculated as 120 ± 21 MPa for 3a fibers. The histogram for 3b fibers can be curve fitted into two populations with the estimated modulus of 106 ± 12 and 74 ± 5 MPa, respectively. The AFM results, in terms of the surface morphology and nanoscale stiffness, are in good agreement with our DSC observations with regard to the crystallinity of 3a and 3b. The modulus values measured for the multiblock copolymer fibers are comparable to semicrystalline polymers, such as poly(ε-caprolactone) and its derivatives,[40] widely used to fabricate fibrous scaffolds via electrospinning. [41]
The peptide-containing multiblock copolymer fiber (3b) was intended to simulate fibrous proteins found in the native extracellular matrices to provide biophysical and biochemical cues to cells. Here, we evaluated the ability of the synthetic fibers to promote the attachment and alignment of NIH 3T3 fibroblasts and myoepithelial-like cells. To this end, fibers were immobilized on poly(2-hydroxyethyl methacrylate)-coated substrate for cell culture purposes (see details in the supporting information). Three hours post seeding, fibroblasts attached to peptide-containing fibers, adopted a spindle shape, and oriented along the fiber (Figure 5A–C). Cells in close contact with the fiber developed exceptionally long and narrow processes (Figure 5B, white arrow) with actin stress fibers traversing the entire cell body. Those in close proximity to fiber-anchored cells had a much smaller cell body and formed cell clusters bridging neighboring fibers. In areas of high cell density, cells aggregated to form branched and interconnected multicellular networks, and individual cells within the cluster exhibited a stellate morphology with no preferential cell orientation. Cells at the edge of the cluster extended short processes (Figure 5C, white arrow) to interrogate their surroundings for potential contacts even though the substrate in between the fibers is cell-repellent. A similar observation has been reported in our previous work when mesenchymal stem cells were cultured on a hydrogel substrate containing cell-adhesive islands.[42]
Figure 5.
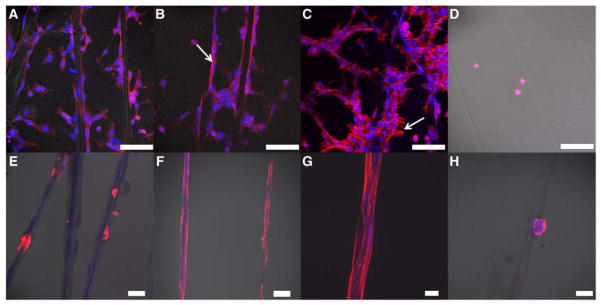
Confocal images of fibroblasts (A–D) and myoepithelial-like cells (E–H) cultured in the presence of multiblock copolymer fibers with (3b, A–C and E–G) and without (3a, D and H) the cell-adhesive peptide. F-actin and nuclei were stained with Phalloidin (red) and DRAQ 5 (blue), respectively. Fibroblasts were cultured for 3 h (A–D) and myoepithelial-like cells were cultured for 24 (E) and 60 h (F–H) before being imaged with a confocal microscope. Scale bar: A–F and H: 100 μm, G: 20 μm.
The ability of peptide-containing multiblock copolymer fibers to support the attachment of myoepithelial-like cells, isolated from healthy human salivary glands, was also evaluated. These cells did not attach to the synthetic fibers as rapidly as did fibroblasts; twenty-four hours post seeding, cells were loosely anchored on the fibers (Figure 5E), but had not undergone significant spreading. Sixty hours post seeding, cells became more spread out, developed long and narrow lamellipodia and oriented parallel to the long axis of the fiber (Figure 5F–G). In some cases, multiple cells formed a cohesive blanket enclosing the fiber. Myoepithelial cells did not bridge neighboring fibers through cell-cell contacts, and cells that did not attach directly to the fibers were washed off during the fixation process. When incubated with peptide free fibers (3a), both fibroblasts and myoepithelial-like cells remained round (Figure 5D, H) in suspension and on the substrate and the majority of unbound cells were washed off during the fixation process. Overall, the peptide-containing fibers present appropriate biochemical signals and topographical features for the anchorage and alignment of both fibroblasts and myoepithelial-like cells. The differences observed between the two types of cells can be attributed to the differences in cell origins, proliferative potentials, integrin expression levels and/or sensitivity to substrate stiffness. Our results highlight the importance of contact guidance for establishing appropriate cell-matrix and cell-cell interactions.
In summary, we have developed a novel strategy for the synthesis of hybrid multiblock copolymers employing sTCO-based tetrazine ligation. Judicious selection of the monomeric building blocks allowed for the polymerization to be carried out at the immiscible solvent interface. The interfacial bioorthogonal approach effectively overcomes limitations associated with solution phase step-growth polymerization to produce high molecular weight multiblock copolymers with an alternating molecular architecture. The interfacial strategy also enabled the simultaneous production of microfibers with aligned semicrystalline structures. The bioorthogonal nature of the tetrazine ligation permits facile incorporation of functional peptides into the multiblock copolymer to fine-tune its biological properties. While fibronectin-derived cell adhesive peptide was used here as a proof-of-concept illustration, our modular approach allows straightforward incorporation of any bioactive peptide sequences. These features, combined with the anisotropic characteristics of the microfibers and the straightforward fiber collection methods, render the synthetic fibers particularly attractive for cell culture and tissue engineering applications, potentially encouraging and controlling directional biological responses through contact guidance.
Supplementary Material
Acknowledgments
The authors acknowledge Dr. Chandran R. Sabanayagam for his expert assistance in AFM characterization, Ms. Jinglin Liu in Prof. David Martin’s lab for helpful discussions on the birefringence results and Mr. Daniel R. Zakheim in Drs. Robert L. Witt and Swati Pradhan-Bhatt’s group for providing the myoepithelial-like cells. This work was supported in part by NSF (DMR 1206310) and NIH (NIDCR R01 DE022969). Data were obtained with instrumentation supported by grants from NIH (P30GM110758, S10OD016267, S10RR026962) and NSF (CRIF:MU CHE 0840401, CHE-1229234).
Footnotes
Supporting Information is available online from the Wiley Online Library or from the author.
Contributor Information
Dr. Shuang Liu, Department of Materials Science and Engineering, University of Delaware, Newark, DE, 19716, USA.
Han Zhang, Department of Chemistry and Biochemistry, University of Delaware, Newark, DE, 19716, USA.
Roddel A. Remy, Department of Materials Science and Engineering, University of Delaware, Newark, DE, 19716, USA
Dr. Fei Deng, Department of Materials Science and Engineering, University of Delaware, Newark, DE, 19716, USA
Prof Michael E. Mackay, Department of Materials Science and Engineering, University of Delaware, Newark, DE, 19716, USA.
Prof Joseph M. Fox, Department of Materials Science and Engineering, University of Delaware, Newark, DE, 19716, USA, Department of Chemistry and Biochemistry, University of Delaware, Newark, DE, 19716, USA.
Prof Xinqiao Jia, Department of Materials Science and Engineering, University of Delaware, Newark, DE, 19716, USA.
References
- 1.Ruzette AV, Leibler L. Nat Mater. 2005;4:19. doi: 10.1038/nmat1295. [DOI] [PubMed] [Google Scholar]
- 2.Diaz RH, Ferguson AP. Block Copolymers: Phase Morphology, Material Applications and Future Challenges. Nova Science Publishers; New York: 2014. [Google Scholar]
- 3.Bates FS, Hillmyer MA, Lodge TP, Bates CM, Delaney KT, Fredrickson GH. Science. 2012;336:434. doi: 10.1126/science.1215368. [DOI] [PubMed] [Google Scholar]
- 4.Keddie DJ. Chem Soc Rev. 2014;43:496. doi: 10.1039/c3cs60290g. [DOI] [PubMed] [Google Scholar]
- 5.Gody G, Maschmeyer T, Zetterlund PB, Perrier S. Nat Commun. 2013;4:9. doi: 10.1038/ncomms3505. [DOI] [PubMed] [Google Scholar]
- 6.Shih HW, Kamber DN, Prescher JA. Curr Opin Chem Biol. 2014;21:103. doi: 10.1016/j.cbpa.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 7.King M, Wagner A. Bioconj Chem. 2014;25:825. doi: 10.1021/bc500028d. [DOI] [PubMed] [Google Scholar]
- 8.McKay CS, Finn MG. Chem Biol. 2014;21:1075. doi: 10.1016/j.chembiol.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kolb HC, Finn MG, Sharpless KB. Angew Chem-Int Edit. 2001;40:2004. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 10.Tsarevsky NV, Sumerlin BS, Matyjaszewski K. Macromolecules. 2005;38:3558. [Google Scholar]
- 11.Averick S, Karacsony O, Mohin J, Yong X, Moellers NM, Woodman BF, Zhu WP, Mehl RA, Balazs AC, Kowalewski T, Matyjaszewski K. Angew Chem-Int Edit. 2014;53:8050. doi: 10.1002/anie.201402827. [DOI] [PubMed] [Google Scholar]
- 12.Grieshaber SE, Farran AJ, Bai S, Kiick KL, Jia X. Biomacromolecules. 2012;13:1774. doi: 10.1021/bm3002705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grieshaber SE, Farran AJ, Lin-Gibson S, Kiick KL, Jia X. Macromolecules. 2009;42:2532. doi: 10.1021/ma802791z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greene AC, Zhu JH, Pochan DJ, Jia XQ, Kiick KL. Macromolecules. 2011;44:1942. doi: 10.1021/ma102869y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grieshaber SE, Paik BA, Bai S, Kiick KL, Jia X. Soft Matter. 2013;9:1589. doi: 10.1039/C2SM27496E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walker CN, Sarapas JM, Kung V, Hall AL, Tew GN. ACS Macro Letters. 2014;3:453. doi: 10.1021/mz5001288. [DOI] [PubMed] [Google Scholar]
- 17.Sumerlin BS, Tsarevsky NV, Louche G, Lee RY, Matyjaszewski K. Macromolecules. 2005;38:7540. [Google Scholar]
- 18.Blackman ML, Royzen M, Fox JM. J Am Chem Soc. 2008;130:13518. doi: 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Devaraj NK, Weissleder R, Hilderbrand SA. Bioconjugate Chem. 2008;19:2297. doi: 10.1021/bc8004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Devaraj NK, Weissleder R. Acc Chem Res. 2011;44:816. doi: 10.1021/ar200037t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lang K, Davis L, Wallace S, Mahesh M, Cox DJ, Blackman ML, Fox JM, Chin JW. J Am Chem Soc. 2012;134:10317. doi: 10.1021/ja302832g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Selvaraj R, Fox JM. Curr Opin Chem Biol. 2013;17:753. doi: 10.1016/j.cbpa.2013.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Darko A, Wallace S, Dmitrenko O, Machovina MM, Mehl RA, Chin JW, Fox JM. Chem Sci. 2014;5:3770. doi: 10.1039/C4SC01348D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hansell CF, Espeel P, Stamenovic MM, Barker IA, Dove AP, Du Prez FE, O'Reilly RK. J Am Chem Soc. 2011;133:13828. doi: 10.1021/ja203957h. [DOI] [PubMed] [Google Scholar]
- 25.Cok AM, Zhou H, Johnson JA. Macromol Symp. 2013;329:108. [Google Scholar]
- 26.Alge DL, Azagarsamy MA, Donohue DF, Anseth KS. Biomacromolecules. 2013;14:949. doi: 10.1021/bm4000508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haun JB, Devaraj NK, Hilderbrand SA, Lee H, Weissleder R. Nat Nano. 2010;5:660. doi: 10.1038/nnano.2010.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keliher EJ, Reiner T, Turetsky A, Hilderbrand SA, Weissleder R. ChemMedChem. 2011;6:424. doi: 10.1002/cmdc.201000426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang H, Dicker KT, Xu X, Jia X, Fox JM. ACS Macro Letters. 2014;3:727. doi: 10.1021/mz5002993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Odian G. Principles of Polymerization. John Wiley & Sons, Inc; Hoboken, NJ: 2004. [Google Scholar]
- 31.Akiyama SK, Aota S, Yamada KM. Cell Adhes Commun. 1995;3:13. doi: 10.3109/15419069509081275. [DOI] [PubMed] [Google Scholar]
- 32.Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular Biology of the Cell. Garland Science; New York: 2002. [Google Scholar]
- 33.Pietrasik J, Dong HC, Matyjaszewski K. Macromolecules. 2006;39:6384. [Google Scholar]
- 34.Soeriyadi AH, Boyer C, Nystrom F, Zetterlund PB, Whittaker MR. J Am Chem Soc. 2011;133:11128. doi: 10.1021/ja205080u. [DOI] [PubMed] [Google Scholar]
- 35.Gindl W, Martinschitz KJ, Boesecke P, Keckes J. Biomacromolecules. 2006;7:3146. doi: 10.1021/bm060698u. [DOI] [PubMed] [Google Scholar]
- 36.Hernandez JJ, Rueda DR, Garcia-Gutierrez MC, Nogales A, Ezquerra TA, Soccio M, Lotti N, Munari A. Langmuir. 2010;26:10731. doi: 10.1021/la100959j. [DOI] [PubMed] [Google Scholar]
- 37.Schon P, Bagdi K, Molnar K, Markus P, Pukanszky B, Vancso GJ. Eur Polym J. 2011;47:692. [Google Scholar]
- 38.Sweers KKM, van der Werf KO, Bennink ML, Subramaniam V. Nanoscale. 2012;4:2072. doi: 10.1039/c2nr12066f. [DOI] [PubMed] [Google Scholar]
- 39.Derjaguin BV, Muller VM, Toporov YP. J Colloid Interface Sci. 1975;53:314. [Google Scholar]
- 40.Yang X, Cui C, Tong Z, Sabanayagam CR, Jia X. Acta Biomater. 2013;9:8232. doi: 10.1016/j.actbio.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tong ZX, Sant S, Khademhosseini A, Jia XQ. Tissue Eng Part A. 2011;17:2773. doi: 10.1089/ten.tea.2011.0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jha AK, Xu X, Duncan RL, Jia X. Biomaterials. 2011;32:2466. doi: 10.1016/j.biomaterials.2010.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.