

SUMMARY
Arterial stiffness is emerging as an important risk marker for poor brain aging and dementia through its associations with cerebral small vessel disease, stroke, β-amyloid deposition, brain atrophy and cognitive impairment. Arterial stiffness directly relates the detrimental effects of hypertension on peripheral organs with dire consequences for the extensive microvasculature structure of the kidneys and brain. In this review, we discuss the evidence linking arterial stiffness, hypertension and brain structural abnormalities in older adults. In particular, we discuss the potential mechanisms linking arterial stiffness to brain β-amyloid deposition and dementia and potential therapeutic strategies to prevent hypertension’s adverse effects on the brain.
Keywords: Alzheimer’s disease, arterial stiffness, β-amyloid deposition, brain aging, dementia, hypertension
Alzheimer’s disease (AD) and vascular dementia are the most common forms of cognitive impairment in the elderly [1]. Efforts to find treatments for AD have yielded only modest benefits, likely because longstanding AD pathological processes induce irreversible neurological compromise. While early-onset AD (age onset <65 years) is primary defined by inheritable genetic risk factors: PSEN1, PSEN2 and APP; late-onset AD shares more modifiable environmental risk factors, particularly those responsible for cardiovascular disease (see Table 1) [2]. Although traditionally considered clinically distinct from each other with mutually exclusive mechanisms [3], late-onset AD is increasingly regarded as a pathological process that is strongly influenced by cerebrovascular dysfunction. Even the major single gene associated with increased late-onset AD risk, APOE, was earlier identified as a risk gene for cardiovascular diseases [4]. The pathogenic mechanisms underlying these two conditions appear to be connected through cerebral hypoperfusion, blood–brain barrier compromise, inflammatory cell infiltration and hemorrhagic susceptibility [5–9]. Chief among these factors are those that are associated with hypertension.
Table 1.
Risk factors for Alzheimer’s disease and cardiovascular disease.
| Genetic and environmental factors | Early-onset Alzheimer’s disease | Late-onset Alzheimer’s disease | Cardiovascular disease |
|---|---|---|---|
| Adverse risk factors | |||
| Midlife hypertension | Yes, midlife | Yes | |
| Arterial stiffness | Yes | Yes | |
| Diabetes mellitus | Yes | Yes | |
| Hyperlipidemia | Yes, midlife | Yes | |
| Smoking, current | Yes | Yes | |
| Head injury, with loss of consciousness | Yes | No | |
| Obesity | Yes, midlife | Yes | |
| Protective factors | |||
| Moderate alcohol consumption | Yes | Yes | |
| High level of physical activity | Yes | Yes | |
| Education >15 years | Yes | No | |
| Statin drugs | Yes | Yes | |
| Nonsteroidal anti-inflammatory drugs | Yes | No, adverse | |
| Unmodifiable risk factors | |||
| Age | Yes | Yes | Yes |
| Family history of Alzheimer’s disease | Yes | Yes | Yes |
| APOE | Yes | Yes | |
| PSEN1 | Yes | ||
| PSEN2 | Yes | ||
| APP | Yes | ||
Several decades of research implicates hypertension and cardiovascular risk factors as potential modifiable risk factors in the development of stroke [10], AD and AD-associated pathology [11]. Yet, few studies have made convincing progress in identifying the underlying mechanisms connecting the seemingly disparate processes of hypertension and AD pathology. Recent studies show that high blood pressure is linked to brain β-amyloid (Aβ) deposition in older adults and suggest that arterial stiffness plays a central role in this relationship [12–15]. Arterial stiffness constitutes a reduced capacity of arteries to flex and accommodate the increase in blood ejected from the heart during systole. This lack of arterial compliance transmits more pulsatile pressure deeper into the periphery and to the microvasculature of distal-end organ systems. Arterial stiffness appears to be a novel and important risk factor underlying the effect of blood pressure on the brain and uniting several clinical phenotypes, including: stroke, cerebrovascular disease, AD and dementia. In this review, we summarize the potential role of arterial stiffening in the pathogenesis of cognitive impairment and dementia through its potential roles in hypertension’s effects on the brain, cerebrovascular disease and AD pathology. We reviewed the published literature using Medline search terms: ‘blood pressure’ and ‘amyloid’ and ‘brain’ limited to human studies, which returned the five observational studies reviewed herein. We expand the scope beyond human studies in order to discuss the potential mechanisms underlying the novel association between arterial stiffness and brain Aβ deposition. Arterial stiffness is an important modifiable risk factor for dementia and maladaptive brain aging. Therefore, we discuss potential interventions to slow, reverse or prevent the aspects of dementia associated with vascular disease.
Understanding the links between hypertension & AD
Nearly two decades of research has shown that hypertension increases the risk of AD and AD pathology. In general, data from observational epidemiology studies on hypertension and dementia are most positive, but inconsistent [16]. Observational studies of incident dementia tend to rely on hypertension defined by a single physiologic measurement and self-report of diagnosis, to the detriment of potential misclassification bias. These studies are summarized in an excellent systematic review and meta-analysis by Power et al. [16]. In short, prospective epidemiology studies do not provide clear evidence for a relationship between late-life blood pressure and AD. Although the evidence is relatively weak, the meta-data suggests that hypertension in midlife (midlife diastolic, but not midlife systolic) may have an adverse effect on risk of incident AD. A recent study from the Atherosclerosis Risk in Communities (ARIC) study showed that elevated midlife systolic BP (but not late-life BP) was associated with greater cognitive decline over 20 years of follow-up, a relationship that may be stronger in white participants than African–Americans [17]. Furthermore, the late-life blood pressure levels were unrelated to midlife levels. In contrast, elevated late-life blood pressure may actually be beneficial. This effect may be a result of reverse causation in observational studies and fits well with data showing late-life changes in systolic and diastolic blood pressure with age [18,19]; therefore, the relevance of blood pressure as a risk factor in late-life is limited.
A considerable amount of speculation remains regarding the relationship between hypertension and dementia, most of which is attributable to observational studies with clinically defined cognitive outcomes. In contrast, data from autopsy studies and neuroimaging studies provide a more convincing picture (reviewed in Power et al. [16]). A number of autopsy studies [20,21] have shown that the probability of manifesting clinical dementia for a given level of AD pathology increases with the presence of cerebrovascular pathology, both of which are strongly linked to hypertension [22–30] and arterial stiffness (discussed in detail in the ‘Arterial stiffness & the brain’ section). While there is little mechanistic evidence supporting the observed relationships between blood pressure with the formation of Tau proteins or neurofibrillary tangles in the brain [31,32], blood pressure may be related to AD pathogenesis or progression through mechanisms involving Aβ synthesis, clearance or its deposition as plaques [33]. Uncontrolled hypertension appears to predict the level of neurofibrillary tangles and neuritic Aβ plaques [34–36] in the brain, which implies hypertension could have direct effects on AD pathology.
Blood pressure is also related to atrophy in AD-prone brain regions. A recent meta-analysis of blood pressure and brain volume reduction showed that higher blood pressure levels are associated with greater total, cortical and hippocampal brain volume reduction, which was evident in both cross-sectional and longitudinal studies [37]. While the mechanisms underlying this association are unclear, some evidence suggests subcortical vascular pathology on cortical neuronal apoptosis underlie brain atrophy [38] and the reduced clearance of Aβ from the brain.
Factors underlying hypertension
The way that hypertension is assessed appears to affect the strength of its relationships with dementia and evidence of its pathology in patients. In research studies, the assessment of hypertension using a sphygmomanometer to obtain the brachial artery blood pressure is always preferable to self-report of hypertension; however, a seated assessment of blood pressure at a single time point to define hypertension is still subject to several physiologic antecedents and therefore substantial bias, especially the declines evident in the elderly adults mentioned above. There are also several distinct subtypes of hypertension, including: essential hypertension, isolated systolic, diastolic driven, etc. demonstrating that hypertension can represent several different etiologies. Furthermore, blood pressure is confounded by variation in its individual components that are discussed in detail below.
Arterial blood pressure is primarily a result of cardiac output and peripheral resistance as shown in Figure 1. Cardiac output is split into stroke volume and heart rate. During resting blood pressure assessment, cardiac output is intentionally minimized through the control heart rate by a predefined rest period. The exception comes in the form of ‘white coat hypertension’ where the individual’s sympathetic response increases the heart rate and vascular tension. This bias is overcome with more intensive continuous ambulatory blood pressure assessments outside the office.
Figure 1.

Diagram relating arterial pressure with brain abnormalities.
BP: Blood pressure.
To understand the contribution of peripheral resistance it is essential to appreciate the effects of aging on vascular biology. Arteries of the body vary not only in size but in composition depending upon their location along the arterial tree. We will briefly discuss the effects of arterial aging in three main groups of arteries: large elastic central elastic, medium muscular peripheral arteries and small arterioles.
In healthy young individuals, the central elastic arteries (e.g., aorta and carotid artery) expand and recoil effectively within each cardiac cycle. The elasticity produces a Windkessel Effect to dampen and transform the hemodynamic pulsatility into continuous blood flow through the capillaries [39]. Normally there is a steep gradient of increasing pressure along the artery with increasing distance from the heart. This pressure gradient which produces ‘reflected waves’ as the forward pulse waves is reflected back to the heart at each arterial branching and distal reductions in vessel caliber. Abnormal timing of the reflected wave can move shift its return to the heart into systole. The heart must pump harder to overcome added pressure, referred to as the augmentation index. This accelerates arterial aging. Arterial aging predominantly and almost exclusively affects the proximal portions of the aortic arch branches of the head and upper limbs [39] which progressively stiffen and dilate with age [40–42]. The progressive rupture and complete loss of elastin fibers in the medial coat of the aorta leads to a progressive loss of central elasticity. The usual steep gradient of arterial stiffness from the heart is lost when central pressure exceeds the peripheral resistance. This shift results in the propagation of the forward wave further into the periphery, usually occurring around the age of 60 years [43]. This increases the demands on distal vascular compensation.
As greater central stiffness transmits more pulsatile pressure to the peripheral arteries, they must adapt to handle the pressure. Peripheral arteries are muscular and stiffer than central arteries. They modulate flow based on tube size and smooth muscle constriction. The muscular arteries are also more susceptible to the metabolic demands of the surrounding muscle and tissue and may be more influenced by insulin signaling [44]. The small arterioles lack any muscularity and are more or less passive recipients of pulsatile flow. They normally carry blood with minimal pulsatile expansion and at steady constant flow velocity [45]. If the peripheral muscular arteries cannot compensate for the increased pulsatile energy, it reaches the fragile arterioles in the end microvasculature. Distal vascular stress results in the loss of elasticity with age in the arteries of the brain as well. Older adults have less elastin expression and function than arteries compared with middle-aged patients, in addition to a loss of distensibility in response to changes in pressure [46]. This often leads to damage in the form of microvascular disease in the kidneys and brain [47]. High pulsations in the microvessels of brain and kidneys cause damage to the endothelium with shedding of endothelial cells (the ultimate endothelial dysfunction), causing subsequent thrombosis and microinfarction [47]. High pulsatile pressure stretches and damages all classes of arteries, similar to the effects seen in malignant hypertension and intracerebral hemorrhage [31] resulting in lacunar stroke [24], microhemorrhage [23] and white matter hyperintensities (WMH) on MRI [22,25], which are commonly found at autopsy in persons with AD.
Measurement of arterial stiffness
Arterial stiffness can be measured noninvasively either locally or regionally by several means and across several arterial beds. Pulse wave velocity is the classic method of noninvasively studying age-related and disease-related changes in arterial stiffness. Pulse wave velocity (PWV) is the foot by foot measurement of the speed of travel of the arterial pulse along an arterial segment [48] and expressed in (meters/second × second; Figure 2). The current gold standard for measuring central or ‘aortic’ arterial stiffness is the carotid-femoral PWV (cfPWV) which determines the PWV at two sites along the carotid and femoral arteries [40]. An alternative form of central PWV, the heart-femoral PWV (hfPWV) is derived from the same measurement. Purely peripheral forms of PWV are measured at femoral and ankle arterial sites. The brachial ankle (baPWV) measurement is growing in popularity as a systemic measures of arterial stiffness due to the fact that it crosses several arterial beds and integrates arterial function across the central elastic arteries as well as the muscular peripheral arteries (see Laurent et al. [40] for an excellent discussion of PWV relative to other methods used to assess arterial stiffness).
Figure 2.
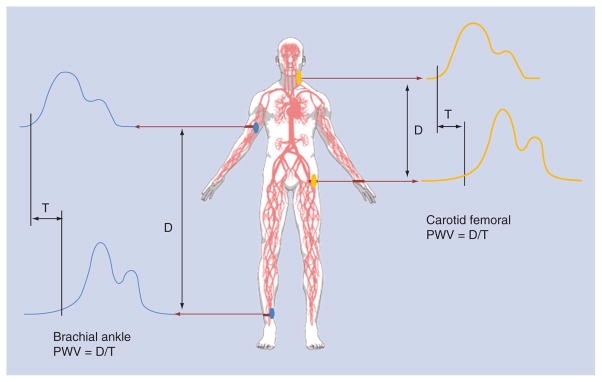
Diagram arterial pulse wave velocity measures.
D: Distance; PWV: Pulse wave velocity: T: Time.
Arterial stiffness & the brain
As a result of its extensive vasculature, the brain is a high-flow, low-resistance organ that is continuously exposed to the mechanical forces of cardiac pulsations [47]. In essence, arterial stiffness is the mechanism that relates the pulsatile force of the heart to the brain [43]. The relationships between arterial disease and brain structural abnormalities are summarized in a conceptual model presented in Figure 1. Greater arterial stiffness as measured by PWV is consistently associated cognitive impairment and cognitive decline (please see the excellent review by Rabkin [49]). Increased PWV is directly related to the degree of cognitive impairment independent of factors that affect cognitive function such as age and education. Importantly PWV is predictive of the development of cognitive impairment with higher PWV predicting the greatest decline in cognitive function [49–52].
Arterial stiffness & brain structure
Central stiffness as measured by high cfPWV has been associated with lower total brain volume, the presence and extent of WMH, lacunar infarcts and cerebral microbleeds visible on MRI in both hypertensive and normotensive participants [14,53–57]. Central arterial stiffening is also associated with microbleeds [54]. Microbleeds are often present with concomitant pathology such as: the presence of lacunar infarcts, hemorrhage and white matter changes in nearly all arterial territories. Hypertension, as evident by increased systolic blood pressure, increases the risk of microbleeds in the territory of the posterior cerebral artery and the deep and infratentorial locations [58]. It is speculated the cerebral Aβ angiopathy may be responsible for the microbleeds in the lobar area of brain [58]. Carotid plaques and increasing carotid lumen diameter as measured by intima-media thickness are less-specific markers of local carotid stiffness; yet, they are associated with increasing prevalence of lacunar infarcts and increasing WMH volume [59].
Changes in white matter microstructure are indicative of white matter integrity are thought to occur prior to WMH [60]. White matter integrity is quantified by diffusion tensor imaging and is associated with elevated blood pressure [61]. Arterial stiffness is proposed to underlie the strong relationship between elevated blood pressure and white matter integrity [61] eventually leading to visible WMH. The relationship between central arterial stiffness and white matter integrity may be focal, localized to individual tracts, but detectable in multiple ethnic groups [62].
Arterial stiffness is associated with the extent & progression of Aβ deposition in the brain
AD pathology is characterized by the protein aggregation of Tau and fibrillar Aβ in the brain that precedes atrophy and onset of cognitive impairment. In AD, Aβ proteins deposit as fibrillar Aβ plaques in the neocortex, while subcortical and vascular Aβ deposition is also present [63]. In contrast, Tau protein deposits start in the entorhinal region and follow the stages detailed by Braak and Braak [64]. Aβ pathology is necessary, but not sufficient for AD. Yet, AD pathology is rarely pure and does not occur in isolation. Estimates suggest that more than 50% of dementia cases are mixed pathology [20]. AD often manifests as co-occurring neuropathology (e.g., α-synuclein, lewy bodies, etc.) with other neurodegenerative disorders like Parkinson’s disease and other forms of dementia [65] and with evidence of cerebrovascular disease. In particular, the deposition of vascular Aβ described as cerebral amyloid angiopathy (CAA) is emerging as an important marker of risk for AD, microinfarction, microhemorrhage and macrohemorrhage of the brain and vascular cognitive impairment [66].
The use of Aβ-specific ligands in PET has opened new avenues for AD pathology research (reviewed by Kantarchi [67]). Arguably one of the most important contributions of Aβ-PET imaging in recent years was empirical evidence that brain Aβ-deposition does not reside solely in the domain of AD pathology, but is also a component of brain aging [68,69]. Recent studies show hypertension is associated with the extent of Aβ deposition in the brain measured by Aβ-PET [12–13,70]; however, these findings are inconsistent regarding the pressure variant. The extent of Aβ deposition was positively associated with higher systolic blood pressure [12], and higher diastolic blood pressure [13]. These two studies concluded that these initial relationships between blood pressure and brain Aβ were consistent across cognitive groups (normal, MCI and AD) [13] and could potentially be attributed to underlying arterial stiffness [12]. The effect of clinically diagnosed hypertension on brain Aβ burden may differ by APOE genotype, with the greatest Aβ burden among clinically diagnosed hypertensive APOE ε4 allele carriers [70].
Our group examined the relationship between Aβ-PET and several measures of arterial stiffness, blood pressure, atherosclerosis and brain structure in a group of elderly nondemented participants. Arterial stiffness was associated with the extent of Aβ-PET measured cross-sectionally and 2 years prior, but these associations differed by vascular bed. Greater systemic arterial stiffness, as measured by baPWV was strongly associated with the extent of Aβ-PET at both baseline [14] and 2 years later at follow-up [15]. Each standard deviation increase in baPWV increased the odds of being Aβ-positive by twofold at baseline and more than fourfold at follow- up. These associations were independent of differences in age, gender, resting systolic blood pressure, BMI and antihypertensive medication use. Interestingly, diastolic blood pressure was not associated with the extent of Aβ deposition at either timepoint in this study. Systolic blood pressure was marginally associated with Aβ-PET at baseline, but not at follow-up. Additional adjustment for current systolic blood pressure or pulse pressure did not appreciably attenuate the associations between systemic stiffness and Aβ-PET. On the other hand, central measures of arterial stiffness (cfPWV) were not associated with Aβ-PET at either baseline or follow-up, but seemed more closely related to evidence of white matter disease and the progression of Aβ deposition over time. Measures of arterial stiffness are not statistically different by cognitive status (mild cognitive impairment [MCI] or normal) in this group of nondemented older adults [14].
White matter burden and Aβ-PET show weak [71,72] or no correlation with each other [14,73–74]. However, Aβ and WHMs can carry independent but equal weight in an AD diagnosis [72]. It appears that measures of central stiffness cfPWV are strongly associated with WMH volume (cfPWV, hfPWV, systolic blood pressure and mean arterial pressure); while, measures of systemic (baPWV) and peripheral (faPWV) are not associated with WMH volume [14]. In contrast, PWV appears to be related to both WMH and Aβ-PET in independent, parallel processes [14] that may be most destructive when co-occuring [75,76]. Individuals with the combined neuropathology of WMH and Aβ-PET had the highest central and systemic arterial disease [14]. These initial data suggest that arterial stiffness may be a central factor relating independent processes of cerebrovascular disease and brain Aβ deposition in the brain.
Longitudinal Aβ-PET studies are beginning to emerge in the literature. Initial studies suggest that Aβ accumulates in all cognitive groups (normal, MCI and AD), but it is highest in the AD groups [77]. There is a monotonoic increase in Aβ accumulation across groups of cognitive function and with age. Extensive Aβ deposition precedes cognitive impairment, and is associated with APOE genotype, age and a higher risk of cognitive decline [78]. Population-based studies with Aβ-PET shows the prevalence of AD-like Aβ deposition is between 20 and 34% in cognitively normal older adults [67] and increases dramatically with increasing age [15,69]. Recently, we showed that 9th decade of life the proportion of nondemented older adults with AD-like Aβ deposition increased from 45 to 75% over 2 years of follow-up [13]. In this group, greater arterial stiffness was associated with the progressive accumulation of Aβ over 2 years of follow-up [15]. Measures of central stiffness were a strong indicator of the progression of brain Aβ, independent of the effects of age, gender, BMI, time between PET scan and current blood pressure measurements. It is important to note that in cognitively normal adults, the rate of Aβ accumulation does not predict the rate of neurodegeneration and the onset of cognitive symptoms [79]. The onset of clinical symptoms of dementia are influenced by genetic and vascular risk factors, not solely the appearance of AD pathology which is recognized to begin years and even decades before the onset of clinical symptoms [79].
Arterial stiffness versus atherosclerosis
Arterial stiffness and changes in vascular structure can occur from and be accelerated by atherosclerosis, which is a major risk factor for stroke [80]. Atherosclerotic plaques deposited in the media of the vessel wall changes both the regional blood flow and the local stiffness, and the calcification of atherosclerotic plaques can be imaged reliably with computed tomography (CT). Several studies show that CT-assessed coronary calcification is associated with cerebral atrophy on MRI, poorer cognitive performance [81,82] and worse microstructural integrity [83]. These relationships appear to be evident with both the extracranial and intracranial carotid arteries [84].
The presence of regional carotid atherosclerosis alone may be a risk factor for AD and dementia through its contributions to cerebrovascular disease. In fact, AD cases have a greater degree of cerebral artery (circle of Willis) occlusion than controls, and there was a positive correlation between the degree of arterial stenosis and neurofibrillary tangle score [7]. Regional peripheral carotid atherosclerosis is best imaged by carotid ultrasound when used to identify carotid plaques, identify vascular occlusions and measure carotid intima media thickness (IMT). Greater IMT of the carotid and brachiocephalic trunk are associated with deep subcortical white matter hyperintensity [85] and brain infarcts [80]. Individuals with evidence of coronary or aortic disease may not be at increased risk of AD; however, intracranial atherosclerosis is a confirmed, strong risk factor for dementia [86]. To date, no studies comparing atherosclerosis to brain Aβ deposition are available in the published literature. In the PPG study [69], we did not find a relationship between either carotid IMT and Aβ deposition in the brain, or its accumulation over time [Hughes TM et al., Unpublished Data] Future studies will have to confirm this apparent lack of association.
Perspectives for future research
Research surrounding the novel associations between arterial stiffness and brain Aβ deposition has considerable growth potential, and aspects relating these two complex multifactorial pathologies need to be evaluated through more detailed studies in the future. An important question raised by these initial studies is whether younger age groups with hypertension and advanced arterial stiffness show Aβ deposits earlier than individuals with healthy arteries. If so, there is considerable room for the treatment of hypertension and arterial stiffness in the prevention of AD and dementia. Future research will also need more comprehensive assessments of cerebrovascular disease beyond white matter lesions, such as cerebral microbleeds and microinfarcts, which are cerebrovascular markers more closely related to Aβ pathology in the brain. More detail regarding the shared mechanisms linking arterial stiffness, cerebrovascular disease and Alzheimer’s type pathology will further our knowledge of these seemingly disparate conditions.
Potential shared mechanisms linking arterial stiffness, cerebrovascular disease & Alzheimer’s-type pathology
It is possible that arterial stiffness is directly and indirectly related to Aβ deposition for several reasons which have yet to be fully addressed in the literature. For instance, there are a myriad of structural, cellular and genetic factors that contribute to arterial stiffness, including the roles of the scaffolding proteins, extracellular matrix, inflammatory molecules, endothelial cell function and reactive oxidant species, factors which are affected by: atherosclerosis, glucose regulation, chronic renal disease, salt and changes in neurohormonal regulation (as described in an excellent review by Zieman et al. [87]). All of these factors may provide pathways and potential mediators of the novel relationships between peripheral arterial stiffness, cerebrovascular disease and brain Aβ deposition.
Arterial stiffness may first produce cerebrovascular disease that in turn initiates abnormalities in Aβ production or clearance in the brain as suggested by the hypertensive rate model [88]. This suggests there should be a strong relationship between evidence of cerebrovascular disease and Aβ deposition in the brain among individuals with high vascular stiffness. Initial studies suggest white matter lesions and Aβ deposition are not related in the elderly [14,76]. It is likely that arterial stiffness contributes to cerebrovascular disease and Aβ deposition via independent mechanisms [75], therefore, contributing to brain abnormalities in multiple ways. Other markers of cerebrovascular disease may be more informative. Based on neuropathology, the leading candidate would be cerebral microbleeds, which are associated with both arterial stiffness and Aβ deposition in the microvasculature [89]. As of yet, no studies have evaluated the associations between microbleeds and brain Aβ in vivo. Additionally, cerebral microinfarcts are found in all brain regions, especially in the watershed areas of the cerebral cortex. Microinfarcts are common in age-related vascular diseases. They are present more in patients with vascular dementia (weighted average: 62%), AD (43%) and demented patients with mixed Alzheimer-type and cerebrovascular pathology (33%) compared with nondemented older individuals (24%). Microinfarcts are pathologically described as minute foci with neuronal loss, gliosis, pallor or more cystic lesions. Sizes vary from 50 μm to a few mm. Recent advances in high-field microstructural neuroimaging provide promising biomarkers [90,91] that may reveal greater detail of the potential relationships between brain Aβ deposition and cerebrovascular disease. Detection of these lesions in vivo with MRI would have a high potential impact for future pathophysiological studies of brain aging [6].
Arterial stiffness may alter cerebral blood flow leading to areas of abnormal blood flow and insufficiencies. It is uncertain how alterations in blood flow affects Aβ synthesis, aggregation or deposition in the human brain; however, animal studies suggest cerebral ischemia may be involved in the pathogenesis of AD through upregulation of Aβ precursor protein gene expression [92–94], promotion of Aβ precursor protein cleavage into Aβ peptides [95] or reduction in soluble Aβ clearance [96]. Arterial stiffness may also worsen as a consequence of Aβ deposition in the vasculature, particularly in the brain. In Aβ angiopathies (e.g. CAA), Aβ interacts with vascular matrix proteins and deposits within the walls of leptomeningeal and cerebral arteries, leading to alterations in the composition of the basement membrane and the eventual destruction of vascular smooth muscles within the tunica media [97]. CAA is common in the aging process and usually harmless. In some patients with severe CAA, however, the Aβ deposits cause the blood vessel walls to crack, causing microbleeds and severe hemorrhagic stroke. It is important to note that Aβ is produced throughout the body and brain and peripherally derived Aβ may contribute to CAA initiation and progression.
It is also feasible that arterial stiffness is directly related to Aβ deposition through its production or clearance from the brain. Vascular activation of endothelial cells relates systemic vascular dynamics to brain microvasculature. This process as it relates to arterial aging is already well reviewed by Wang and Lakatta [98] but it also implicated in AD as well. For example, the cerebral microcirculation is pathologically activated with brain endothelial cells overexpressing a wide variety of bioactive, neurotoxic and/or inflammatory proteins including thrombin, VEGF, ANGPT-2, TNF-α, TGF-β, IL-1β, IL-6, IL-8, MCP-1, HIF-1α, matrix metalloproteinases and integrins [99]. Vascular activation in the AD brain likely has deleterious consequences for neuronal health as many of these mediators are directly or indirectly neurotoxic. Interesting recent work suggest blood products (e.g., fibrinogen) may be involved in not just in vascular complications and stroke but also in AD pathogenesis. Fibrinogen is normally found circulating in blood, but in AD patients it also found deposited with Aβ in the brain parenchyma and cerebral blood vessels. Aβ and fibrinogen appear to interact, leading to increased fibrinogen aggregation, Aβ fibrillization and the formation of degradation-resistant fibrin clots [100].
Arterial stiffness may have direct effects on Aβ clearance from the brain. Murine models show that Aβ is cleared from the brain via the paravascular exchange of cerebrospinal fluid (CSF) with perivascular interstitial fluid (ISF), which has been dubbed the glymphatic system. In this system, the ISF fluid flows along the perivascular space of the cerebrovasculature [97,101–103]. This process is shown to be 60% more efficient at night [104], suggesting that sleep plays a major role in the removal of Aβ other waste products from the brain. It is unknown if the efficiency of the glymphatic system is related to nocturnal blood pressure patterns. The recent observation that cerebral arterial pulsatility contributes to glymphatic exchange suggests the possibility that reductions in arterial pulsatility due to arterial stiffening and cerebrovascular disease may also contribute to failure of the clearance of interstitial waste, including Aβ, from the aged brain [97]. This inability to clear soluble Aβ from the brain may further contribute to CAA. Further, cerebral arteries from older adults exhibit reduced mechanical compliance, a loss of elastin [46], and in some cases CAA. In these cases, such alterations in the arterial wall may slow Aβ clearance by inhibiting fluid exchange.
Modeling of glymphatic flow rates suggests that the pulsatile flow of ISF is driven by the reflected wave occurring as a result of the pulse wave. Initial work suggests that the role of the glymphatic system is to remove soluble waste products from the brain via the CSF. Recently Iliff et al. posited that the loss of arterial pulsatility reducing solute clearance may be compounded by the effect of paravascular solute deposition (such as Aβ) further reducing cerebral arterial pulsatility. Together the two mechanisms may constitute a feed forward pathogenic loop which in turn promotes synaptic dysfunction and neurodegeneration [102]. If borne true, treatments that target cerebrovascular compliance may protect against neurodegenerative processes by promoting solute clearance via the CSF and reducing Aβ deposition [102].
No treatment options tailored to arterial stiffness
For many years, therapy of hypertension was serendipitous, with the success of β-adrenoceptor antagonists (β-blockers) and diuretics not envisaged when they were first introduced. The most successful antihypertensive drugs now used – angiotensin-converting enzyme (ACE) inhibitors, angiotensin II type 1 receptor antagonists (angiotensin receptor blockers [ARBs]), renin inhibitors, calcium channel antagonists (calcium channel blockers [CCBs]), aldosterone receptor antagonists – have been synthesized to interact with known pathophysiological mechanisms. The most successful agents act predominantly as arterial vasodilators, reducing tone in conduit arteries with lesser effect on peripheral resistance, thus reducing pulsatile as well as mean blood pressure.
Renal microvascular disease is traditionally viewed in terms of local disturbance of the renin–angiotensin system, with need for treatment with ACE inhibitors or ARB drugs. Cerebral microvascular disease is regarded as a result of mechanical damage that should be targeted with antihypertensives and statins. Recent research suggests that microvascular disease at the two sites may share similar etiologies and may warrant similar treatment [105]. Targeting arterial stiffness may reduce the damage at these sites. However, clinical trials of antihypertensive therapy suggests that the long-term treatment with ARB and ACE inhibitors may provide some benefit for reducing pulse pressure and isolated systolic hypertension, but do not appear to improve central arterial stiffness [106]. Incidentally, meta-analyses of studies investigating the ability of antihypertensives to prevent age-related dementia show mixed results, suggesting either no benefit [107] or a beneficial effect [108]. The utility of antihypertensives on preventing cognitive decline likely varies by class and drug target, even when the effect of blood pressure lowering does not [108]. The rank-order of benefit on overall cognition is highest for ARBs followed by β-blockers, diuretics and angiotensin-converting enzyme inhibitors over placebo [108]. Antihypertensive treatment appears to be associated with an enhancement of all cognitive function, except language and possibly visuospacial abilites [108].
Antihypertensives act as a class target blood pressure and may have minimal effects on preventing continual accumulation of arterial stiffness. Current antihypertensive therapies are not designed to replace the lost aortic elastin or improve elasticity. As a result they have little effect on age-related arterial stiffening. Arterial stiffness may be a unique therapeutic approach for preventing Aβ deposition and dementia.
While the hemodynamic impact of arterial stiffness may not be easily drugable, arterial stiffness has several contributing metabolic factors that are all particularly amenable to lifestyle intervention. A number of lifestyle changes and therapies that reduce arterial stiffness are presented, including weight loss, exercise, salt reduction and reduced alcohol consumption and neuroendocrine therapies. Neuroendocrine-directed therapies to reduce arterial stiffness include those targeting the renin–angiotensin system, natriuretic peptides, insulin modulators, as well as novel therapies that target advanced glycation end products [87]. Any of which may be involved directly in the production, aggregation or clearance of Aβ form the brain.
Nonpharmacologic interventions for arterial stiffness
Increased aortic stiffness also plays a well-identified role in obesity and metabolic syndrome [109]. Its progression with age is proportional to the number of metabolic risk factors the individual posesses [109]. Lifestyle interventions including exercise and dietary interventions are attractive because they may reduce arterial stiffness by a variety of mechanisms (detailed in a recent review by Sacre) [110]. The potential additive effects, such as exercise and dietary interventions on arterial stiffness need to be studied in greater detail. Aerobic exercise training increases arterial compliance and reduces systolic blood pressure [111] in young and older adults [110]. Weight loss and corresponding improvements in insulin sensitivity are important factors targets for intervening on arterial stiffness. Significant weight loss results in improvements in central PWV [112]. Yet, weight loss alone may not have the impact on arterial stiffness that improving insulin sensitivity alone may have on vascular tone, specifically within the muscular arteries of the periphery. Weight loss and improvements in insulin appear to have synergistic effects on improving systemic stiffness [44]. Insulin has significant vasoactive properties on the resistive large arteries and precapillary arterioles which reduces the amplitude and timing of the carotid pulse under conditions of euglycemic clamp [113]. In patients with insulin resistance and metabolic syndrome, these disrupted physiologic mechanisms may contribute to the development of systolic hypertension and stiffening of the arteries [109]. Further, self-report data suggest that 68% of adults with diabetes also have hypertension [114]. The additional roles of insulin affecting the endothelium, renin–angiotensin system and oxidative stress cannot be ignored [109] and should be investigated in their roles in AD pathogenesis.
Conclusion & future perspective
Arterial stiffness provides an important link between hypertension and dementia because it serves as the driving force behind hypertension’s effects on the peripheral microvasculature of the brain and kidneys. Arterial stiffness is a well-established risk factor for white matter disease and is also associated with evidence of microvascular damage in the brain. Recent studies also show blood pressure and arterial stiffness are associated with brain Aβ deposition in elderly adults. While the relationships between blood pressure and Aβ deposition are inconsistent, the associations between arterial stiffness and Aβ deposition in the brain are robust that are independent of common potential confounders. The connections between arterial blood pressure, cerebrovascular disease and Aβ accumulation appear to be independent and centrally driven. It is likely that the effects of central stiffness on the brain are mediated by changes in the peripheral vasculature. Vascular contributions to brain abnormalities are likely to have direct effects on cerebral blood flow leading to areas of hyper- and hypo-perfusion and microvascular damage contributing to aggregation of blood products, endothelial dysfunction and impaired Aβ clearance. While brain Aβ and white matter disease appear to occur independently of each other, individuals with the presence of both have the highest arterial stiffness, suggesting arterial stiffness might contribute to the two processes independently causing a ‘double-hit’ to the brain. The findings relating arterial stiffness come from only one cohort and need to be replicated. Ideal cohorts for replication include younger cohorts of middle-age to early elderly adults designed to evaluate differences in Aβ aggregation between hypertensive and normotensive groups or clinical trials of blood pressure treatment. Longitudinal studies of Aβ deposition suggest a long-protracted development of Aβ deposition over decades (Figure 3) [78,115] and this relationship may be modified by ApoE genotype. Future studies need adequate power and recruitment to determine if the associations between arterial stiffness and brain Aβ accumulation differ by ApoE genotype.
Figure 3.

A conceptual model of vascular factors related to arterial stiffness and β-amyloid deposition in the brain across adulthood.
PWV: Pulse wave velocity.
Data taken from [15].
Understanding the relationship that arterial stiffness has to brain Aβ deposition is an opportunity to understand Aβ pathology in AD and aging. Currently there are no means to prevent or slow AD, with few known modifiable risk factors for AD and far fewer that have been related to AD pathology. If Aβ turns out to play a necessary and sufficient role in AD, then preventing arterial stiffness may be a unique therapeutic target for preventing Aβ deposition and AD. Current antihypertensive therapies have little effect of aortic stiffness despite robust improvements in pulse pressure. Yet, antihypertensive therapy may have important impact on preventing cognitive declines and may even prevent dementia. One potentially important treatment option is to slow the progression of arterial stiffening through the consistent and sustained treatment of hypertension throughout life, particularly at middle age. Trials like SPRINT will provide some insights into blood pressure treatment strategies aimed at intensive versus standard blood pressure goals on arterial stiffness, cognition and brain structure.
Practice points.
Alzheimer’s disease seldom results from a pure Alzheimer-type pathology and often contains one or more vascular pathologies.
Recent studies suggest that blood pressure and arterial stiffness plays a role in dementia risk and β-amyloid deposition in the brain.
Future studies are needed to determine if arterial stiffening promotes β-amyloid deposition through vascular damage, impaired β-amyloid clearance via paravascular drainage or other mechanisms.
Taken together these studies suggest that long-term treatment of hypertension at first sign will be important potential modifiers of disease course through β-amyloid and cerebrovascular mechanisms.
Longitudinal studies of antihypertensive therapy suggest that angiotensin-converting enzyme inhibitors and angiotensin receptor blockers have differential effects on dementia risk and promote improvements in arterial stiffness.
Footnotes
For reprint orders, please contact: reprints@futuremedicine.com
No writing assistance was utilized in the production of this manuscript.
Financial & competing interests disclosure
This work was supported by a NIA T32 postdoctoral training grant (T32 AG000181, TM Hughes) from the NIH. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
Papers of special note have been highlighted as:
• of interest; •• of considerable interest
- 1.Fotuhi M, Hachinski V, Whitehouse PJ. Changing perspectives regarding late-life dementia. Nat Rev Neurol. 2009;5(12):649–658. doi: 10.1038/nrneurol.2009.175. [DOI] [PubMed] [Google Scholar]
- 2.Kloppenborg RP, Van Den Berg E, Kappelle LJ, Biessels GJ. Diabetes and other vascular risk factors for dementia: which factor matters most? A systematic review. Eur J Pharmacol. 2008;585(1):97–108. doi: 10.1016/j.ejphar.2008.02.049. [DOI] [PubMed] [Google Scholar]
- 3.Erkinjuntti T, Gauthier S. The concept of vascular cognitive impairment. Front Neurol Neurosci. 2009;24:79–85. doi: 10.1159/000197886. [DOI] [PubMed] [Google Scholar]
- 4.Gungor Z, Anuurad E, Enkhmaa B, Zhang W, Kim K, Berglund L. Apo E4 and lipoprotein-associated phospholipase A2 synergistically increase cardiovascular risk. Atherosclerosis. 2012;223(1):230–234. doi: 10.1016/j.atherosclerosis.2012.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beach TG, Wilson JR, Sue LI, et al. Circle of Willis atherosclerosis: association with Alzheimer’s disease, neuritic plaques and neurofibrillary tangles. Acta Neuropathol. 2007;113(1):13–21. doi: 10.1007/s00401-006-0136-y. [DOI] [PubMed] [Google Scholar]
- 6.Brundel M, De Bresser J, Van Dillen JJ, Kappelle LJ, Biessels GJ. Cerebral microinfarcts: a systematic review of neuropathological studies. J Cereb Blood Flow Metab. 2012;32(3):425–436. doi: 10.1038/jcbfm.2011.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roher AE, Esh C, Kokjohn TA, et al. Circle of Willis atherosclerosis is a risk factor for sporadic Alzheimer’s disease. Arterioscler Thromb Vasc Biol. 2003;23(11):2055–2062. doi: 10.1161/01.ATV.0000095973.42032.44. [DOI] [PubMed] [Google Scholar]
- 8.Roher AE, Kuo YM, Esh C, et al. Cortical and leptomeningeal cerebrovascular amyloid and white matter pathology in Alzheimer’s disease. Mol Med. 2003;9(3–4):112–122. [PMC free article] [PubMed] [Google Scholar]
- 9.Yarchoan M, Xie SX, Kling MA, et al. Cerebrovascular atherosclerosis correlates with Alzheimer pathology in neurodegenerative dementias. Brain. 2012;135(Pt 12):3749–3756. doi: 10.1093/brain/aws271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Macmahon S, Peto R, Cutler J, et al. Blood pressure, stroke, and coronary heart disease. Part 1: prolonged differences in blood pressure: prospective observational studies corrected for the regression dilution bias. Lancet. 1990;335(8692):765–774. doi: 10.1016/0140-6736(90)90878-9. [DOI] [PubMed] [Google Scholar]
- 11.Wiesmann M, Kiliaan AJ, Claassen JA. Vascular aspects of cognitive impairment and dementia. J Cereb Blood Flow Metab. 2013;33(11):1696–1706. doi: 10.1038/jcbfm.2013.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Langbaum JB, Chen K, Launer LJ, et al. Blood pressure is associated with higher brain amyloid burden and lower glucose metabolism in healthy late middle-age persons. Neurobiol Aging. 2012;33(4):827, e811–e829. doi: 10.1016/j.neurobiolaging.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toledo JB, Toledo E, Weiner MW, et al. Cardiovascular risk factors, cortisol, and amyloid-beta deposition in Alzheimer’s Disease Neuroimaging Initiative. Alzheimers Dement. 2012;8(6):483–489. doi: 10.1016/j.jalz.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hughes TM, Kuller LH, Barinas-Mitchell EJM, et al. Pulse wave velocity is associated with β-amyloid deposition in the brains of very elderly adults. Neurology. 2013;81(19):1711–1718. doi: 10.1212/01.wnl.0000435301.64776.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hughes TM, Kuller LH, Barinas-Mitchell EJ, et al. Arterial stiffness and beta-amyloid progression in nondemented elderly adults. JAMA Neurol. 2014;71(5):562–568. doi: 10.1001/jamaneurol.2014.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16••.Power MC, Weuve J, Gagne JJ, Mcqueen MB, Viswanathan A, Blacker D. The association between blood pressure and incident Alzheimer disease: a systematic review and meta-analysis. Epidemiology. 2011;22(5):646–659. doi: 10.1097/EDE.0b013e31822708b5. Comprehensive systematic review and meta-analysis of 18 populations exmining the association bewteen of blood pressure and Alzheimer’s disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gottesman RF, Schneider AL, Albert M, et al. Midlife hypertension and 20 year cognitive change: the atherosclerosis risk in communities neurocognitive study. JAMA Neurol. 2014;71(10):1218–1218. doi: 10.1001/jamaneurol.2014.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stewart R, Xue QL, Masaki K, et al. Change in blood pressure and incident dementia: a 32 year prospective study. Hypertension. 2009;54(2):233–240. doi: 10.1161/HYPERTENSIONAHA.109.128744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joas E, Backman K, Gustafson D, et al. Blood pressure trajectories from midlife to late life in relation to dementia in women followed for 37 years. Hypertension. 2012;59(4):796–801. doi: 10.1161/HYPERTENSIONAHA.111.182204. [DOI] [PubMed] [Google Scholar]
- 20.Schneider JA, Wilson RS, Bienias JL, Evans DA, Bennett DA. Cerebral infarctions and the likelihood of dementia from Alzheimer disease pathology. Neurology. 2004;62(7):1148–1155. doi: 10.1212/01.wnl.0000118211.78503.f5. [DOI] [PubMed] [Google Scholar]
- 21.Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease: the Nun Study. JAMA. 1997;277(10):813–817. [PubMed] [Google Scholar]
- 22.De Leeuw FE, De Groot JC, Oudkerk M, et al. Hypertension and cerebral white matter lesions in a prospective cohort study. Brain. 2002;125(Pt 4):765–772. doi: 10.1093/brain/awf077. [DOI] [PubMed] [Google Scholar]
- 23.Viswanathan A, Chabriat H. Cerebral microhemorrhage. Stroke. 2006;37(2):550–555. doi: 10.1161/01.STR.0000199847.96188.12. [DOI] [PubMed] [Google Scholar]
- 24.Kazui S, Levi CR, Jones EF, Quang L, Calafiore P, Donnan GA. Risk factors for lacunar stroke: a case-control transesophageal echocardiographic study. Neurology. 2000;54(6):1385–1387. doi: 10.1212/wnl.54.6.1385. [DOI] [PubMed] [Google Scholar]
- 25.Veldink JH, Scheltens P, Jonker C, Launer LJ. Progression of cerebral white matter hyperintensities on MRI is related to diastolic blood pressure. Neurology. 1998;51(1):319–320. doi: 10.1212/wnl.51.1.319. [DOI] [PubMed] [Google Scholar]
- 26.Longstreth WT, Jr, Bernick C, Manolio TA, Bryan N, Jungreis CA, Price TR. Lacunar infarcts defined by magnetic resonance imaging of 3660 elderly people: the Cardiovascular Health Study. Arch Neurol. 1998;55(9):1217–1225. doi: 10.1001/archneur.55.9.1217. [DOI] [PubMed] [Google Scholar]
- 27.Prabhakaran S, Wright CB, Yoshita M, et al. Prevalence and determinants of subclinical brain infarction: the Northern Manhattan Study. Neurology. 2008;70(6):425–430. doi: 10.1212/01.wnl.0000277521.66947.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vermeer SE, Koudstaal PJ, Oudkerk M, Hofman A, Breteler MM. Prevalence and risk factors of silent brain infarcts in the population-based Rotterdam Scan Study. Stroke. 2002;33(1):21–25. doi: 10.1161/hs0102.101629. [DOI] [PubMed] [Google Scholar]
- 29.Vernooij MW, Van Der Lugt A, Ikram MA, et al. Prevalence and risk factors of cerebral microbleeds: the Rotterdam Scan Study. Neurology. 2008;70(14):1208–1214. doi: 10.1212/01.wnl.0000307750.41970.d9. [DOI] [PubMed] [Google Scholar]
- 30.Poels MM, Vernooij MW, Ikram MA, et al. Prevalence and risk factors of cerebral microbleeds: an update of the Rotterdam scan study. Stroke. 2010;41(Suppl 10):S103–S106. doi: 10.1161/STROKEAHA.110.595181. [DOI] [PubMed] [Google Scholar]
- 31.Diaz-Ruiz C, Wang J, Ksiezak-Reding H, et al. Role of hypertension in aggravating abeta neuropathology of AD type and tau-mediated motor impairment. Cardiovasc Psychiatry Neurol. 2009;2009:107286. doi: 10.1155/2009/107286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Glodzik L, Rusinek H, Pirraglia E, et al. Blood pressure decrease correlates with tau pathology and memory decline in hypertensive elderly. Neurobiol Aging. 2014;35(1):64–71. doi: 10.1016/j.neurobiolaging.2013.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iadecola C, Gorelick PB. Converging pathogenic mechanisms in vascular and neurodegenerative dementia. Stroke. 2003;34(2):335–337. doi: 10.1161/01.str.0000054050.51530.76. [DOI] [PubMed] [Google Scholar]
- 34.Petrovitch H, Ross GW, Steinhorn SC, et al. AD lesions and infarcts in demented and non-demented Japanese–American men. Ann Neurol. 2005;57(1):98–103. doi: 10.1002/ana.20318. [DOI] [PubMed] [Google Scholar]
- 35.Hoffman LB, Schmeidler J, Lesser GT, et al. Less Alzheimer disease neuropathology in medicated hypertensive than nonhypertensive persons. Neurology. 2009;72(20):1720–1726. doi: 10.1212/01.wnl.0000345881.82856.d5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sparks DL, Scheff SW, Liu H, Landers TM, Coyne CM, Hunsaker JC., 3rd Increased incidence of neurofibrillary tangles (NFT) in non-demented individuals with hypertension. J Neurol Sci. 1995;131(2):162–169. doi: 10.1016/0022-510x(95)00105-b. [DOI] [PubMed] [Google Scholar]
- 37••.Beauchet O, Celle S, Roche F, et al. Blood pressure levels and brain volume reduction: a systematic review and meta-analysis. J Hypertens. 2013;31(8):1502–1516. doi: 10.1097/HJH.0b013e32836184b5. A systematic review and meta-analysis of 28 studies examining associations between blood pressure and brain volume. [DOI] [PubMed] [Google Scholar]
- 38.Viswanathan A, Gray F, Bousser MG, Baudrimont M, Chabriat H. Cortical neuronal apoptosis in CADASIL. Stroke. 2006;37(11):2690–2695. doi: 10.1161/01.STR.0000245091.28429.6a. [DOI] [PubMed] [Google Scholar]
- 39.Nichols W, O’Rourke M, Vlachopoulos C. McDonald’s Blood Flow in Arteries. 6. Hodder Arnold; London, UK: 2011. [Google Scholar]
- 40••.Laurent S, Cockcroft J, Van Bortel L, et al. Expert consensus document on arterial stiffness: methodological issues and clinical applications. Eur Heart J. 2006;27(21):2588–2605. doi: 10.1093/eurheartj/ehl254. European expert concensus on approaches to measure arterial stiffness. [DOI] [PubMed] [Google Scholar]
- 41.Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises. Part I: aging arteries: a ‘set up’ for vascular disease. Circulation. 2003;107(1):139–146. doi: 10.1161/01.cir.0000048892.83521.58. [DOI] [PubMed] [Google Scholar]
- 42.O’Rourke MF, Hashimoto J. Mechanical factors in arterial aging: a clinical perspective. J Am Coll Cardiol. 2007;50(1):1–13. doi: 10.1016/j.jacc.2006.12.050. [DOI] [PubMed] [Google Scholar]
- 43.Mitchell GF. Effects of central arterial aging on the structure and function of the peripheral vasculature: implications for end-organ damage. J Appl Physiol. 2008;105(5):1652–1660. doi: 10.1152/japplphysiol.90549.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hughes TM, Althouse AD, Niemczyk NA, Hawkins MS, Kuipers AL, Sutton-Tyrrell K. Effects of weight loss and insulin reduction on arterial stiffness in the SAVE trial. Cardiovasc Diabetol. 2012;11:114. doi: 10.1186/1475-2840-11-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O’rourke MF, Safar ME, Dzau V. The cardiovascular continuum extended: aging effects on the aorta and microvasculature. Vasc Med. 2010;15(6):461–468. doi: 10.1177/1358863X10382946. [DOI] [PubMed] [Google Scholar]
- 46.Fonck E, Feigl GG, Fasel J, et al. Effect of aging on elastin functionality in human cerebral arteries. Stroke. 2009;40(7):2552–2556. doi: 10.1161/STROKEAHA.108.528091. [DOI] [PubMed] [Google Scholar]
- 47.O’Rourke MF, Safar ME. Relationship between aortic stiffening and microvascular disease in brain and kidney: cause and logic of therapy. Hypertension. 2005;46(1):200–204. doi: 10.1161/01.HYP.0000168052.00426.65. [DOI] [PubMed] [Google Scholar]
- 48.Safar M, O’Rourke M. Arterial stiffness in hypertension. In: Birkenhager W, Reid J, editors. Handbook of Hypertension. Elsevier; London, UK: 2006. [Google Scholar]
- 49••.Rabkin SW. Arterial stiffness: detection and consequences in cognitive impairment and dementia of the elderly. J Alzheimers Dis. 2012;32(3):541–549. doi: 10.3233/JAD-2012-120757. Systemic review of twelve studies examining the associations between arterial stiffness and cognitive impairment. [DOI] [PubMed] [Google Scholar]
- 50.Elias MF, Robbins MA, Budge MM, Abhayaratna WP, Dore GA, Elias PK. Arterial pulse wave velocity and cognition with advancing age. Hypertension. 2009;53(4):668–673. doi: 10.1161/HYPERTENSIONAHA.108.126342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kearney-Schwartz A, Rossignol P, Bracard S, et al. Vascular structure and function is correlated to cognitive performance and white matter hyperintensities in older hypertensive patients with subjective memory complaints. Stroke. 2009;40(4):1229–1236. doi: 10.1161/STROKEAHA.108.532853. [DOI] [PubMed] [Google Scholar]
- 52.Waldstein SR, Rice SC, Thayer JF, Najjar SS, Scuteri A, Zonderman AB. Pulse pressure and pulse wave velocity are related to cognitive decline in the Baltimore Longitudinal Study of Aging. Hypertension. 2008;51(1):99–104. doi: 10.1161/HYPERTENSIONAHA.107.093674. [DOI] [PubMed] [Google Scholar]
- 53.Henskens LH, Van Oostenbrugge RJ, Kroon AA, De Leeuw PW, Lodder J. Brain microbleeds are associated with ambulatory blood pressure levels in a hypertensive population. Hypertension. 2008;51(1):62–68. doi: 10.1161/HYPERTENSIONAHA.107.100610. [DOI] [PubMed] [Google Scholar]
- 54.Henskens LH, Kroon AA, Van Oostenbrugge RJ, et al. Increased aortic pulse wave velocity is associated with silent cerebral small-vessel disease in hypertensive patients. Hypertension. 2008;52(6):1120–1126. doi: 10.1161/HYPERTENSIONAHA.108.119024. [DOI] [PubMed] [Google Scholar]
- 55.Coutinho T, Turner ST, Kullo IJ. Aortic pulse wave velocity is associated with measures of subclinical target organ damage. JACC Cardiovasc Imaging. 2011;4(7):754–761. doi: 10.1016/j.jcmg.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Poels MM, Zaccai K, Verwoert GC, et al. Arterial stiffness and cerebral small vessel disease: the Rotterdam Scan Study. Stroke. 2012;43(10):2637–2642. doi: 10.1161/STROKEAHA.111.642264. [DOI] [PubMed] [Google Scholar]
- 57.Tsao CW, Seshadri S, Beiser AS, et al. Relations of arterial stiffness and endothelial function to brain aging in the community. Neurology. 2013;81(11):984–991. doi: 10.1212/WNL.0b013e3182a43e1c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jia Z, Mohammed W, Qiu Y, Hong X, Shi H. Hypertension increases the risk of cerebral microbleed in the territory of posterior cerebral artery: a study of the association of microbleeds categorized on a basis of vascular territories and cardiovascular risk factors. J Stroke Cerebrovasc Dis. 2014;23(1):e5–e11. doi: 10.1016/j.jstrokecerebrovasdis.2012.12.016. [DOI] [PubMed] [Google Scholar]
- 59.Brisset M, Boutouyrie P, Pico F, et al. Large-vessel correlates of cerebral small-vessel disease. Neurology. 2013;80(7):662–669. doi: 10.1212/WNL.0b013e318281ccc2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maillard P, Carmichael O, Fletcher E, Reed B, Mungas D, Decarli C. Coevolution of white matter hyperintensities and cognition in the elderly. Neurology. 2012;79(5):442–448. doi: 10.1212/WNL.0b013e3182617136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gons RA, De Laat KF, Van Norden AG, et al. Hypertension and cerebral diffusion tensor imaging in small vessel disease. Stroke. 2010;41(12):2801–2806. doi: 10.1161/STROKEAHA.110.597237. [DOI] [PubMed] [Google Scholar]
- 62.Rosano C, Watson N, Chang Y, et al. Aortic pulse wave velocity predicts focal white matter hyperintensities in a biracial cohort of older adults. Hypertension. 2013;61(1):160–165. doi: 10.1161/HYPERTENSIONAHA.112.198069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tiraboschi P, Hansen LA, Thal LJ, Corey-Bloom J. The importance of neuritic plaques and tangles to the development and evolution of AD. Neurology. 2004;62(11):1984–1989. doi: 10.1212/01.wnl.0000129697.01779.0a. [DOI] [PubMed] [Google Scholar]
- 64.Braak H, Braak E, Bohl J. Staging of Alzheimer-related cortical destruction. Eur Neurol. 1993;33(6):403–408. doi: 10.1159/000116984. [DOI] [PubMed] [Google Scholar]
- 65.Jellinger KA. Neuropathological aspects of Alzheimer disease, Parkinson disease and frontotemporal dementia. Neurodegener Dis. 2008;5(3–4):118–121. doi: 10.1159/000113679. [DOI] [PubMed] [Google Scholar]
- 66.Gorelick PB, Scuteri A, Black SE, et al. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the american heart association/american stroke association. Stroke. 2011;42(9):2672–2713. doi: 10.1161/STR.0b013e3182299496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67••.Kantarci K. Molecular Imaging of Alzheimer Disease Pathology. AJNR Am J Neuroradiol. 2014;35(6 Suppl):S12–S7. doi: 10.3174/ajnr.A3847. Review of amyloid imaging in Alzheimer’s disease research studies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lim YY, Maruff P, Pietrzak RH, et al. Abeta and cognitive change: examining the preclinical and prodromal stages of Alzheimer’s disease. Alzheimers Dement. 2014;10(6):743.e1–751.e1. doi: 10.1016/j.jalz.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 69.Mathis CA, Kuller LH, Klunk WE, et al. In vivo assessment of amyloid-beta deposition in nondemented very elderly subjects. Ann Neurol. 2013;73(6):751–761. doi: 10.1002/ana.23797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rodrigue KM, Rieck JR, Kennedy KM, Devous MD, Sr, Diaz-Arrastia R, Park DC. Risk factors for beta-amyloid deposition in healthy aging: vascular and genetic effects. JAMA Neurol. 2013;70(5):600–606. doi: 10.1001/jamaneurol.2013.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wirth M, Villeneuve S, Haase CM, et al. Associations between Alzheimer disease biomarkers, neurodegeneration, and cognition in cognitively normal older people. JAMA Neurol. 2013;70(12):1512–1519. doi: 10.1001/jamaneurol.2013.4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Provenzano FA, Muraskin J, Tosto G, et al. White matter hyperintensities and cerebral amyloidosis: necessary and sufficient for clinical expression of Alzheimer disease? JAMA Neurol. 2013;70(4):455–461. doi: 10.1001/jamaneurol.2013.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chao LL, Decarli C, Kriger S, et al. Associations between white matter hyperintensities and beta amyloid on integrity of projection, association, and limbic fiber tracts measured with diffusion tensor MRI. PLoS ONE. 2013;8(6):e65175. doi: 10.1371/journal.pone.0065175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee MJ, Seo SW, Na DL, et al. Synergistic Effects of Ischemia and beta-amyloid burden on cognitive decline in patients with subcortical vascular mild cognitive impairment. JAMA Psychiatry. 2014;71(4):412–422. doi: 10.1001/jamapsychiatry.2013.4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Provenzano FA, Muraskin J, Tosto G, et al. White matter hyperintensities and cerebral amyloidosis: necessary and sufficient for clinical expression of alzheimer disease? JAMA Neurology. 2013;70(4):455–461. doi: 10.1001/jamaneurol.2013.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Marchant NL, Reed BR, Sanossian N, et al. The aging brain and cognition: contribution of vascular injury and aβ to mild cognitive dysfunction. JAMA Neurology. 2013;70(4):488–495. doi: 10.1001/2013.jamaneurol.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Villain N, Chetelat G, Grassiot B, et al. Regional dynamics of amyloid-beta deposition in healthy elderly, mild cognitive impairment and Alzheimer’s disease: a voxelwise PiB-PET longitudinal study. Brain. 2012;135(Pt 7):2126–2139. doi: 10.1093/brain/aws125. [DOI] [PubMed] [Google Scholar]
- 78.Villemagne VL, Pike KE, Chetelat G, et al. Longitudinal assessment of Aβ and cognition in aging and Alzheimer disease. Ann Neurol. 2011;69(1):181–192. doi: 10.1002/ana.22248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jack CR, Jr, Holtzman DM. Biomarker modeling of Alzheimer’s disease. Neuron. 2013;80(6):1347–1358. doi: 10.1016/j.neuron.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Touboul PJ, Labreuche J, Vicaut E, Amarenco P, Investigators G. Carotid intima-media thickness, plaques, and Framingham risk score as independent determinants of stroke risk. Stroke. 2005;36(8):1741–1745. doi: 10.1161/01.STR.0000174490.23495.57. [DOI] [PubMed] [Google Scholar]
- 81.Rosano C, Naydeck B, Kuller LH, Longstreth WT, Jr, Newman AB. Coronary artery calcium: associations with brain magnetic resonance imaging abnormalities and cognitive status. J Am Geriatr Soc. 2005;53(4):609–615. doi: 10.1111/j.1532-5415.2005.53208.x. [DOI] [PubMed] [Google Scholar]
- 82.Vidal JS, Sigurdsson S, Jonsdottir MK, et al. Coronary artery calcium, brain function and structure: the AGES-Reykjavik study. Stroke. 2010;41(5):891–897. doi: 10.1161/STROKEAHA.110.579581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bos D, Vernooij MW, Elias-Smale SE, et al. Atherosclerotic calcification relates to cognitive function and to brain changes on magnetic resonance imaging. Alzheimers Dement. 2012;8(5 Suppl):S104–S111. doi: 10.1016/j.jalz.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 84.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81(2):741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 85.Isa K, Sakima A, Sakima H, Nakachi K, Kinjyo K, Ohya Y. Association between the intima-media thickness of the brachiocephalic trunk and white matter hyperintensity in brain MRI. Hypertens Res. 2013;36(11):980–984. doi: 10.1038/hr.2013.72. [DOI] [PubMed] [Google Scholar]
- 86.Dolan H, Crain B, Troncoso J, Resnick SM, Zonderman AB, Obrien RJ. Atherosclerosis, dementia, and Alzheimer disease in the Baltimore Longitudinal Study of Aging cohort. Ann Neurol. 2010;68(2):231–240. doi: 10.1002/ana.22055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zieman SJ, Melenovsky V, Kass DA. Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler Thromb Vasc Biol. 2005;25(5):932–943. doi: 10.1161/01.ATV.0000160548.78317.29. [DOI] [PubMed] [Google Scholar]
- 88.Schreiber S, Drukarch B, Garz C, et al. Interplay between age, cerebral small vessel disease, parenchymal amyloid-beta, and tau pathology: longitudinal studies in hypertensive stroke-prone rats. J Alzheimers Dis. 2014;42(Suppl 3):S205–S215. doi: 10.3233/JAD-132618. [DOI] [PubMed] [Google Scholar]
- 89.Cullen KM, Kocsi Z, Stone J. Microvascular pathology in the aging human brain: evidence that senile plaques are sites of microhaemorrhages. Neurobiol Aging. 2006;27(12):1786–1796. doi: 10.1016/j.neurobiolaging.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 90.Van Veluw SJ, Zwanenburg JJ, Engelen-Lee J, et al. In vivo detection of cerebral cortical microinfarcts with high-resolution 7T MRI. J Cereb Blood Flow Metab. 2013;33(3):322–329. doi: 10.1038/jcbfm.2012.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Van Veluw SJ, Zwanenburg JJ, Hendrikse J, Van Der Kolk AG, Luijten PR, Biessels GJ. High resolution imaging of cerebral small vessel disease with 7 T MRI. Acta Neurochir Suppl. 2014;119:125–130. doi: 10.1007/978-3-319-02411-0_21. [DOI] [PubMed] [Google Scholar]
- 92.Nihashi T, Inao S, Kajita Y, et al. Expression and distribution of beta amyloid precursor protein and beta amyloid peptide in reactive astrocytes after transient middle cerebral artery occlusion. Acta Neurochir (Wien) 2001;143(3):287–295. doi: 10.1007/s007010170109. [DOI] [PubMed] [Google Scholar]
- 93.Jin K, Mao XO, Eshoo MW, et al. Microarray analysis of hippocampal gene expression in global cerebral ischemia. Ann Neurol. 2001;50(1):93–103. doi: 10.1002/ana.1073. [DOI] [PubMed] [Google Scholar]
- 94.Shi J, Yang SH, Stubley L, Day AL, Simpkins JW. Hypoperfusion induces overexpression of beta-amyloid precursor protein mRNA in a focal ischemic rodent model. Brain Res. 2000;853(1):1–4. doi: 10.1016/s0006-8993(99)02113-7. [DOI] [PubMed] [Google Scholar]
- 95.Saido TC, Yokota M, Maruyama K, et al. Spatial resolution of the primary beta-amyloidogenic process induced in postischemic hippocampus. J Biol Chem. 1994;269(21):15253–15257. [PubMed] [Google Scholar]
- 96.Weller RO, Yow HY, Preston SD, Mazanti I, Nicoll JA. Cerebrovascular disease is a major factor in the failure of elimination of Abeta from the aging human brain: implications for therapy of Alzheimer’s disease. Ann NY Acad Sci. 2002;977:162–168. doi: 10.1111/j.1749-6632.2002.tb04812.x. [DOI] [PubMed] [Google Scholar]
- 97.Weller RO, Djuanda E, Yow HY, Carare RO. Lymphatic drainage of the brain and the pathophysiology of neurological disease. Acta Neuropathol. 2009;117(1):1–14. doi: 10.1007/s00401-008-0457-0. [DOI] [PubMed] [Google Scholar]
- 98.Wang M, Jiang L, Monticone RE, Lakatta EG. Proinflammation: the key to arterial aging. Trends Endocrinol Metab. 2014;25(2):72–79. doi: 10.1016/j.tem.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99•.Grammas P, Martinez J, Sanchez A, et al. A new paradigm for the treatment of alzheimer’s disease: targeting vascular activation. J Alzheimers Dis. 2014;40(3):619–630. doi: 10.3233/JAD-2014-132057. Influential position paper on the potential roles of vascular activation in Alzheimer’s disease. [DOI] [PubMed] [Google Scholar]
- 100.Cortes-Canteli M, Zamolodchikov D, Ahn HJ, Strickland S, Norris EH. Fibrinogen and altered hemostasis in Alzheimer’s disease. J Alzheimers Dis. 2012;32(3):599–608. doi: 10.3233/JAD-2012-120820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Carare RO, Bernardes-Silva M, Newman TA, et al. Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol Appl Neurobiol. 2008;34(2):131–144. doi: 10.1111/j.1365-2990.2007.00926.x. [DOI] [PubMed] [Google Scholar]
- 102•.Iliff JJ, Wang M, Zeppenfeld DM, et al. Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J Neurosci. 2013;33(46):18190–18199. doi: 10.1523/JNEUROSCI.1592-13.2013. Excellent description of the role of arterial pulsation in solute clearance from the murine brain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Iliff JJ, Wang M, Liao Y, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med. 2012;4(147):147ra111. doi: 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Xie L, Kang H, Xu Q, et al. Sleep drives metabolite clearance from the adult brain. Science. 2013;342(6156):373–377. doi: 10.1126/science.1241224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Safar ME, O’Rourke MF. Pulse pressure and antihypertensive agents. Hypertension. 2005;46(2):e6. doi: 10.1161/01.HYP.0000179215.13289.55. author reply e6–e7. [DOI] [PubMed] [Google Scholar]
- 106.Mackenzie IS, Mceniery CM, Dhakam Z, Brown MJ, Cockcroft JR, Wilkinson IB. Comparison of the effects of antihypertensive agents on central blood pressure and arterial stiffness in isolated systolic hypertension. Hypertension. 2009;54(2):409–413. doi: 10.1161/HYPERTENSIONAHA.109.133801. [DOI] [PubMed] [Google Scholar]
- 107.Mcguinness B, Todd S, Passmore P, Bullock R. Blood pressure lowering in patients without prior cerebrovascular disease for prevention of cognitive impairment and dementia. Cochrane Database Syst Rev. 2009;(4):CD004034. doi: 10.1002/14651858.CD004034.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108••.Levi Marpillat N, Macquin-Mavier I, Tropeano AI, Bachoud-Levi AC, Maison P. Antihypertensive classes, cognitive decline and incidence of dementia: a network meta-analysis. J Hypertens. 2013;31(6):1073–1082. doi: 10.1097/HJH.0b013e3283603f53. Excellent review and meta-analysis of antihypertensive class effects on cognitive decline and dementia. [DOI] [PubMed] [Google Scholar]
- 109.Safar ME, Balkau B, Lange C, et al. Hypertension and vascular dynamics in men and women with metabolic syndrome. J Am Coll Cardiol. 2013;61(1):12–19. doi: 10.1016/j.jacc.2012.01.088. [DOI] [PubMed] [Google Scholar]
- 110.Sacre JW, Jennings GL, Kingwell BA. Exercise and dietary influences on arterial stiffness in cardiometabolic disease. Hypertension. 2014;63(5):888–893. doi: 10.1161/HYPERTENSIONAHA.113.02277. [DOI] [PubMed] [Google Scholar]
- 111.Graham MR, Evans P, Davies B, Baker JS. Arterial pulse wave velocity, inflammatory markers, pathological GH and IGF states, cardiovascular and cerebrovascular disease. Vasc Health Risk Manag. 2008;4(6):1361–1371. doi: 10.2147/vhrm.s3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rider OJ, Tayal U, Francis JM, et al. The effect of obesity and weight loss on aortic pulse wave velocity as assessed by magnetic resonance imaging. Obesity (Silver Spring) 2010;18(12):2311–2316. doi: 10.1038/oby.2010.64. [DOI] [PubMed] [Google Scholar]
- 113.Yki-Jarvinen H, Westerbacka J. Insulin resistance, arterial stiffness and wave reflection. Adv Cardiol. 2007;44:252–260. doi: 10.1159/000096746. [DOI] [PubMed] [Google Scholar]
- 114.Fast Facts: National Diabetes Fact Sheet. 2011 http://www.cdc.gov/diabetes/pubs/pdf.
- 115.Villemagne VL, Burnham S, Bourgeat P, et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 2013;12(4):357–367. doi: 10.1016/S1474-4422(13)70044-9. [DOI] [PubMed] [Google Scholar]