

Abstract
Chromosomal microarray analysis is now commonly used in clinical practice to identify copy number variants (CNVs) in the human genome. We report our experience with the use of the 105K and 180K oligonucleotide microarrays in 215 consecutive patients referred with either autism or autism spectrum disorders (ASD) or developmental delay/learning disability for genetic services at the University of Kansas Medical Center during the past 4 years (2009–2012). Of the 215 patients [140 males and 75 females (male/female ratio = 1.87); 65 with ASD and 150 with learning disability], abnormal microarray results were seen in 45 individuals (21%) with a total of 49 CNVs. Of these findings, 32 represented a known diagnostic CNV contributing to the clinical presentation and 17 represented non-diagnostic CNVs (variants of unknown significance). Thirteen patients with ASD had a total of 14 CNVs, 6 CNVs recognized as diagnostic and 8 as non-diagnostic. The most common chromosome involved in the ASD group was chromosome 15. For those with a learning disability, 32 patients had a total of 35 CNVs. Twenty-six of the 35 CNVs were classified as a known diagnostic CNV, usually a deletion (n = 20). Nine CNVs were classified as an unknown non-diagnostic CNV, usually a duplication (n = 8). For the learning disability subgroup, chromosomes 2 and 22 were most involved. Thirteen out of 65 patients (20%) with ASD had a CNV compared with 32 out of 150 patients (21%) with a learning disability. The frequency of chromosomal microarray abnormalities compared by subject group or gender was not statistically different. A higher percentage of individuals with a learning disability had clinical findings of seizures, dysmorphic features and microcephaly, but not statistically significant. While both groups contained more males than females, a significantly higher percentage of males were present in the ASD group.
Keywords: Autism spectrum disorders (ASD), Developmental delay, Learning disability, Chromosomal microarray analysis, Copy number variant (CNV)
Introduction
Classical autism, first described in 1943 by Kanner, belongs to a group of heterogeneous disorders known as autism spectrum disorders (ASD) [1, 2]. Autism spectrum disorders are characterized by impairment in three domains: social interaction, communication skills, and restricted repetitive and stereotyped patterns of behavior, interests, and activities. The onset of these impairments begins before the age of 3 years [2]. About 40% of individuals with ASD also have a learning disability [3].
The etiology of ASD is complex and involves genetic factors, epigenetics, and the environment. Single gene disorders are recognized as causative in less than 20 percent of subjects with ASD while the remaining have other causative genetic or polygenic factors which may be impacted by epigenetic changes influenced by the environment [4,5]. Thus, the role of genetic testing and clinical genetic evaluation for individuals with ASD is emphasized when identifying a cause. A diagnostic yield reported in the literature ranges from 6 to 40% [4–7] with the most common single gene disorders being fragile X syndrome and tuberous sclerosis [8]. Chromosomal microarray analysis in the clinical setting is now recommended as a first tier test for children and adults presenting with ASD [9] to improve the diagnostic yield. Microarray testing can detect copy number variation and type (i.e., deletions or duplications), size (e.g., < 1 Mb) and presence of known genes within the chromosome region.
Historically, the recurrence risk for ASD in families in which one child has ASD has varied from 4% if the first affected child is female to 7% if the first affected child is male [10] but more recent evidence indicates that the gender and functioning of the older sibling does not predict ASD outcome [11]. The recurrence rate of ASD for families in which two children are diagnosed with ASD is significantly higher, estimated at 25 to 30% with recent evidence indicating that first degree relatives of those with ASD are also at an increased risk for ASD-related characteristics [12]. Studies of identical twins in which one twin is diagnosed with ASD have shown at least 60% concordance [10].
Developmental delay involves any significant lag in physical, cognitive, communication, social, emotional, and/or adaptive skills [13]. Global developmental delay is defined as performing at more than two standard deviations below same-aged peers in two or more developmental domains [14] and affects between 1 to 3 percent of children [15,16]. Many children with global developmental delay will also develop intellectual disability which is classified as having an IQ below 70. For our study, we will group infants and children with developmental delay and older children and adults with intellectual disability into a single category referred to as learning disability. Various studies have examined the etiology of learning disabilities with chromosomal microarray analysis which is now considered as a first tier test for children and adults in the clinic setting [9]. Herein, we report our experience over the past 4 years using chromosomal microarray analysis to identify copy number variation (deletions/duplications) in consecutive patients referred with ASD or learning disability and presenting for genetic services at a rural-based Midwestern academic medical center in the United States.
Patient Data
We studied 215 consecutive patients (140 males and 75 females; mean age ± SD = 10y ± 9.7y; age range = 5 months to 52 years) referred for genetic services to the Clinical Genetics setting at the University of Kansas Medical Center (KUMC), Kansas City, Kansas during the past 4 years (2009–2012) with autism spectrum disorders (ASD, N = 65) or learning disability (N = 150). The patients were unrelated based on family history obtained by a genetic counselor at the time of clinical genetics evaluation. Patients were referred from private practice settings (family medicine, pediatricians, internists, psychiatrists/psychologists), hospitals, and medical centers in the Kansas City and surrounding Midwestern region of the nation. KUMC is a primary and tertiary care academic center and includes the University of Kansas Hospital and Clinics. In 2012, KUMC reported 530,918 outpatient encounters and 28,331 inpatient discharges representing all 105 counties in Kansas, a rural population-based state. The majority of counties in the adjoining state of Missouri were also represented.
Methods
We obtained peripheral blood samples in EDTA tubes and sent by overnight delivery to the clinically approved and certified commercially available CombiMatrix Diagnostics Laboratory (Irvine, CA) for DNA isolation for 105K or 180K oligonucleotide microarray analysis. The 105K array contained more than 99,000 probes and the 180K array contained more than 170,000 probes covering coding and non-coding human genome sequences. The average spatial resolution between probes for the 105K array was approximately 21 Kb, while that of the 180K array was approximately 16 Kb. A copy number change was identified when more than 6 consecutive probes were involved in a segment with a maximum of contiguous probe spacing of 1 Mb. The patient DNA copy number was compared to a reference diploid sex-matched DNA sample. Targeted evaluation of copy number changes involving more than 6 probes was performed in all regions of the genome with confirmation of abnormal results by BAC aCGH or FISH probes targeted to the identified region. Most parental testing was performed by using FISH probes. Analysis was performed using Nexus Copy Number software (BioDiscovery, Hawthorne, CA).
We conducted a clinical genetics evaluation, including a physical and dysmorphologic examination, and obtained pregnancy, medical, family, and social histories from each patient. We then summarized the chromosomal microarray data for comparison with findings from the clinical genetics evaluation. The reason for referral was ASD or learning disability. Those found with a recognized syndrome such as Down syndrome, fragile X syndrome, or single gene disorders (e.g., neurofibromatosis, tuberous sclerosis) were not included in the analysis and not a focus of this study.
Among families in which more than one affected family member was evaluated, microarray results from only one affected family member were included. For patients with abnormal microarray results, parental testing (e.g., FISH analysis) for the deletion and/or duplication was undertaken when possible to determine the origin of the diagnostic finding or variant of unknown significance.
Statistical Analysis
Analysis of clinical and microarray data was performed using SAS Statistical Analysis Software Version 9.2 (SAS Institute Inc., Cary, North Carolina, USA). Statistical significance was tested for differences in categorical distributions using the Fisher’s Exact test and mean differences using ANOVA.
Results
Our study included 215 consecutive male and female patients presenting with autism spectrum disorders (ASD) or learning disability over the past four years seeking genetic services at the University of Kansas Medical Center. Both study groups contained more male than female subjects but statistically significantly more males were observed in the ASD group (52 males, 13 females) compared with the learning disability group (88 males, 62 females; Fisher’s Exact test, p < 0.003). We found that approximately one out of every five patients had an abnormal microarray finding (deletion/duplication) identified using either the 105K or 180K oligonucleotide microarray. By searching genome variant databases, including the UCSC Genome Browser (http://genome.ucsc.edu/cgi-bin/hgGateway), the Database of Genomic Variants (http://projects.tcag.ca/cgi-bin/variation/gbrowse/hg18/), Online Mendelian Inheritance in Man (http://www.ncbi.nlm.nih.gov/omim), DECIPHER (http://decipher.sanger.ac.uk/), CombiTrak (CombiMatrix’s internal database of over 20,000 samples) and dbVar (http://www.ncbi.nlm.nih.gov/dbvar/) along with cited published literature, the genetic change was characterized as diagnostic copy number variant (CNV) when reported previously to be associated with disease such as ASD or learning disability or due to a non-diagnostic CNV or variant of unknown significance at the time of the report. Chromosome 15 was the most common chromosome involved in the ASD group, as previously reported [18], while chromosomes 2 and 22 were most involved in those with learning disabilities.
Although a higher frequency of chromosomal microarray abnormalities were observed in females (N = 20 or 27%) compared with males (N = 25 or 18%), no statistically significant differences were observed in the frequency when compared by subject group (Fisher’s Exact test, p = 1.0) or gender (Fisher’s Exact test, p = 0.16). For patients presenting with ASD, 13 of 65 (20%) were found to have an abnormality on microarray analysis (Table 1). The 13 patients with ASD and abnormal microarray results had a total of 14 findings, including 6 diagnostic abnormalities (36%) and 8 variants of unknown significance (64%) (Tables 1 and 2). Of the 13 patients with ASD and abnormal results, 3 (23%) had a family history of ASD, 3 (23%) had macrocephaly, and 4 (31%) had dysmorphic features (Table 3). None of the patients with ASD and abnormal microarray results had a history of seizures. The mean ± SD size of the abnormality (deletion/duplication) on the microarray results for those with ASD was 966 ± 1464 Kb.
Table 1.
Summary of Abnormal Microarray Data for Autism Spectrum Disorders (N=13 of 65 subjects)
| Age | Gender | Clinical features | Del/Dup | Parent of origin | Chromosome location | Chromosome coordinates | Size | Number of genes in deletion or duplication | Selected gene(s) and/or chromosome regions of interest | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| 5y | M | Dysmorphism | del + | unknown | 2p13.3-p12 | 72,554,557–78,190,171 | 5.6 Mb | 76 | 2p12 b MRPL19 |
Scerri et al. [19] |
| 11y | F | Obesity, macrocephaly | del + | paternal | 2p16.3 | 51,027,443–51,123,408 | 96 Kb | 1 | NRXN1 a,b | Bena et al. [20] |
| 12y* | M | ADHD, OCD, easy bruising, irregular sleep disturbances | del − | unknown | 4q35.2 | 187,471,061- 188,661,633 | 1.2 Mb | 2 | FAT1 a | Abou et al. [21] Chien et al. [22] Youngs et al. [23] |
| 9y | M | Dysmorphism | dup − dup − |
unknown, non-paternal unknown, non-paternal |
7p12.1 15q26.1 |
51,000,278–51,236,004 87,456,775–87,706,924 |
236 Kb 250 Kb |
1 4 |
COBL
a POLG |
Griswold et al. [24] Uusimaa et al. [25] |
| 11y | M | Dysmorphism | del + | unknown | 7q11.23 | 72,360,373–73,795,527 | 1.4 Mb | 26 | 7q11.23 b (Williams syndrome) | Meral at al. [26] |
| 9y | M | None | dup − | de novo | 12p13.33 | 2,350,189–2,626,105 | 276 Kb | 1 | CACNA1C b | Cross-Disorder Group of the Pychiatric Genetics Consortium [27] Lu et al. [28] |
| 9y** | M | None | dup + | maternal | 15q11.2 | 19,623,685–20,325,942 | 702 Kb | 5 | 15q11.2 a,b | Burnside et al. [29] |
| 20y | M | None | dup − | unknown | 15q11.2 | 20,306,985–20,860,610 | 554 Kb | 8 | 15q11.2 a,b | Burnside et al. [29] |
| 27y | M | None | dup − | maternal | 15q13.3 | 29,792,719–30,340,762 | 548 Kb | 1 | 15q13.3 b CHRNA7 | Szafranski et al. [30] Williams et al. [31] |
| 26y | M | Dysmorphism | dup + | paternal | 16p11.2 | 29,514,266–30,239,774 | 726 Kb | 42 | 16p11.2 a,b | Weiss et al. [32] |
| 41y*** | M | Macrocephaly | dup + | unknown | 16p13.2 | 6,936,805–6,990,017 | 53 Kb | 1 | A2BP1 a | Martin et al. [33] Butler et al. [34] |
| 4y | F | None | dup − | maternal | 19p13.2 | 10,867,787–11,685,678 | 818 Kb | 26 | SMARCA4 b | Tsurusaki et al. [35] |
| 11y | M | Dysmorphism | del − | unknown | Xp21.3 | 28,719,810–28,757,691 | 38 Kb | 1 | IL1RAPL1 b | Franek et al. [36] Behnecke et al. [37] |
patient previously reported [23],
patient previously reported [29],
patient previously reported [34]
+ = CNVs (deletions or duplications) that were reported previously to be associated with disease or disorder
− = CNVs (deletions or duplications) not previously reported at the time of genetic testing
autism spectrum disorders (ASD);
learning disability
Chromosome coordinates from UCSC hg18 Human Genome (Mar. 2006). Available from: http://genome.ucsc.edu.
Table 2.
Summary of Abnormal Microarray Data for our Subjects
| Autism Spectrum Disorder N=65 | Developmental Delay with Learning Deficits N=150 | Total N=215 | |
|---|---|---|---|
| Microarray Abnormality | 14 | 36 | 50** |
|
| |||
| Diagnostic CNV | 6 | 27 | 33 |
|
| |||
| Deletion | 3 | 20 | 23 |
| Duplication | 3 | 6 | 9 |
| Other | 1 (XY female) | 1 | |
|
| |||
| Non-diagnostic CNV* | 8 | 9 | 17 |
|
| |||
| Deletion | 2 | 1 | 3 |
| Duplication | 6 | 8 | 14 |
Non-diagnostic CNV now referred to as variant of unknown significance
50 CNVs identified in 45 subjects [13 with ASD including a 9-year-old male with two variants of unknown significance and 32 with developmental delay including a 9-month-old female with three diagnostic CNVs, a 9-year-old male with two diagnostic CNVs and a 2-year-old female with two variants of unknown significance].
Fisher Exact Test (p = 0.09) was performed and indicated a possible trend for more diagnostic CNVs in the developmental delay group but not significant.
Table 3.
Summary of the Clinical Findings Seen in our Subjects with Abnormal Microarray Results
| Clinical Finding | Autism Spectrum Disorders & Abnormal Microarray N=13/65 (20%) | Developmental Delay/Learning Deficits & Abnormal Microarray N=32/150 (21%) | All Subjects with Abnormal Microarray N=45/215 (21%) | Fisher’s Exact Test P value |
|---|---|---|---|---|
| Seizures | 0 | 4 (13%) | 4 (9%) | 0.31 |
| Dysmorphic Features | 4 (31%) | 18 (56%) | 22 (49%) | 0.19 |
| Microcephaly | 0 | 8 (25%) | 8 (18%) | 0.08 |
| Macrocephaly | 3 (23%) | 1 (3%) | 4 (9%) | 0.07 |
| Positive Family History | 3 (23%) | 9 (28%) | 12 (27%) | 0.49 |
For patients presenting with learning disability, 32 of 150 (21%) were found to have an abnormality on microarray analysis (Table 4). The 32 patients with abnormal results had a total of 35 findings, including 26 diagnostic abnormalities (74%) and 9 variants of unknown significance (26%) (Tables 2 and 4). Of the 32 patients with learning disability and abnormal results, 9 (28%) had a family history of developmental delay, 8 (25%) had microcephaly, 1 (3%) had macrocephaly, 18 (56%) had dysmorphic features, and 4 (13%) had a history of seizures (Table 3). No obvious gender differences were seen in the frequency of seizures, dysmorphic features, microcephaly, macrocephaly or positive family history between the two subject groups. The mean ± SD size of the abnormality (deletion/duplication) on the microarray results of infants and younger children with developmental delay or older children and adults with an intellectual disability was 2.90 ± 2.87 Mb. The mean age of the microarray abnormality in those with learning disability was significantly larger than that seen in ASD (F = 2.6; p < 0.03).
Table 4.
Summary of Abnormal Microarray Data for Learning Disability (N=32 of 150 subjects)
| Age | Gender | Clinical features |
Del/ Dup |
Parent of origin |
Chromosome location |
Chromosome coordinates |
Size | Number of genes in deletion or duplication |
Selected gene(s) and/or chromosome regions of interest |
Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| 1.5y | M | Dysmorphism | dup + | unknown | 1q21.3-q23.1 | 150,406,974–156,451,142 | 6.0 Mb | 169 |
GATAD2B
b HAX1 b LMNA |
De Ligt et al. [38] Matsubara et al. [39] Hattori et al. [40] |
|
| ||||||||||
| 18y | M | Microcephaly, dysmorphism | del + | de novo | 2p21-p16.3 | 44,726,451–48,990,449 | 4.3 Mb | 27 | 2p16.2p21 b | Sanders et al. [41] |
|
| ||||||||||
| 2y | M | Microcephaly, dysmorphism | del + | unknown | 2q13 | 110,850,204–112,829,316 | 2.0 Mb | 12 | 2q13 b ACOXL, BCL2L11 |
Yu et al. [42] |
|
| ||||||||||
| 3y | F | Macrocephaly, obesity | dup − | paternal | 2q13 | 111,104,335–112,879,277 | 1.8 Mb | 11 | 2q13 b ACOXL, BCL2L11 |
Yu et al. [42] |
|
| ||||||||||
| 42y* | M | Seizures | del + | unknown | 2q33.1-q34 | 203,191,088–210,989,186 | 7.8 Mb | 46 | 2q33.3q34 b MAP2 b |
Brandau et al. [43] Pescuccia et al. [44] |
|
| ||||||||||
| 1y | F | Seizures | del + | maternal | 2q37.3 | 237,763,137–242,951,149 | 5.2 Mb | 58 | 2q37.3 b HDAC b |
Kitsiou-Tzeli et al. [45] Williams et al. [46] |
|
| ||||||||||
| 1y | F | None | del + | maternal | 3p26.2 | 3,982,658–4,937,707 | 955 Kb | 4 |
ITPR1
b SUMF1 a,b |
DiGregorio et al. [47] Vardarajan et al. [48] |
|
| ||||||||||
| 19y | F | Dysmorphism | dup − | unknown | 3q27.3 | 187,712,288–189,031,898 | 1.3 Mb | 22 |
ADIPOQ KNG1 |
Breitfeld et al. [49] |
|
| ||||||||||
| 8y** | F | Dysmorphism | del + | de novo | 3q29 | 197,174,369–198,842,531 | 1.7 Mb | 24 | 3q29 a,b PAK2, DLG1, FBXO45 |
Dasouki et al. [50] Sagar et al. [51] |
|
| ||||||||||
| 25y | F | Dysmorphism | dup − | unknown | 6q26 | 162,247,153–162,594,464 | 347 Kb | 1 | PARK2 a,b | Scheuerle & Wilson [52] Mariani et al. [53] |
|
| ||||||||||
| 3y | M | None | del + | de novo | 9p13.3-p13.1 | 33,394,668–38,459,158 | 5.1 Mb | 89 | 9p13 b | Niemi et al. [54] |
|
| ||||||||||
| 6y | M | Dysmorphism | del + | de novo | 9p13.3-p13.1 | 33,521,700–38,781,172 | 5.3 Mb | 86 | 9p13 b | Niemi et al. [54] |
|
| ||||||||||
| 5y | M | Dysmorphism | del + | unknown | 11q24.1-q25 | 122,813,651–134,452,384 | 11.6 Mb | 93 | 11q24.1 b (Jacobsen syndrome) | Manolakos et al. [55] |
|
| ||||||||||
| 9y | M | Microcephaly, cataract, dysmorphism | del + del + |
paternal maternal |
13q12.12 15q11.2 |
22,385,973–23,818,065 20,290,385–20,640,325 |
1.4 Mb 350 Kb |
10 5 |
SACS
b 15q11.2 a,b |
Breckpot et al. [56] Burnside et al. [29] |
|
| ||||||||||
| 5y | F | Dysmorphism | del − | unknown, non- maternal | 15q26.3 | 98,800,411–99,434,987 | 634 Kb | 5 | LINS b | Akawi et al. [57] |
|
| ||||||||||
| 9m | M | Dysmorphism | del + | maternal | 16p11.2 | 28,730,299–29,009,896 | 280 Kb | 10 | 16p11.2 a,b SH2B1 b |
Bachmann-Gagescu [58] |
|
| ||||||||||
| 17y | M | Dysmorphism | dup + | maternal | 16p13.12-p13.11 | 14,651,493–16,504,719 | 1.9 Mb | 28 | 16p13.11 a,b | Ramalingam et al. [59] |
|
| ||||||||||
| 8y | M | Microcephaly | del + | unknown | 16q24.3 | 87,920,905–88,102,506 | 182 Kb | 1 | 16q24.3 b ANKRD11 |
Sacharow et al. [60] |
|
| ||||||||||
| 9m | F | Microcephaly, dysmorphism | dup + | unknown | 17pter-p13.1 | 0–8,261,267 | 8.3 Mb | 216 |
DLG4
a FXR2b |
Feyder et al. [61] Schluth-Bolard et al. [62] |
|
NLGN2
a USP6 a YWHAE a,b |
Pettem et al. [63] Tentler et al. [64] Capra et al. [65] Bruno et al. [66] |
|||||||||
| del + | unknown | 20p11.21 | 23,050,819–23,557,142 | 506 Kb | 10 | 20p11.21 b GZF1 |
D’Angelo et al. [67] | |||
|
| ||||||||||
| 3y | F | Microcephaly, dysmorphism | dup + | unknown | 17p12-p11.2 | 11,826,994–16,316,855 | 4.5 Mb | 24 | 17p11.2 a,b PMP22 |
Doco-Fenzy et al. [68] |
|
| ||||||||||
| 46y | F | Seizures | dup − | unknown | 17p13.3 | 1,383,290–1,537,874 | 155 Kb | 4 | PRPF8 | Liu & Zack [69] |
|
| ||||||||||
| 17y | F | Microcephaly, dysmorphism | del + | unknown | 18q22.3-qter | 70,596,888–76,117,153 | 5.5 Mb | 20 |
18q23
b GALR1 MBP ZNF407 a,b |
Margarit et al. [70] Ren et al. [71] |
|
| ||||||||||
| 6y | F | Seizures | dup + | paternal | 22q11.21 | 17,002,559–20,140,494 | 3.1 Mb | 62 | 22q11.21 b | Bittel et al. [72] Alberti et al. [73] |
|
| ||||||||||
| 32y | F | Dysmorphism | del + | unknown, non- maternal | 22q11.21 | 17,036,698–18,739,379 & 18,927,136–19,967,262 | 2.7 Mb | 67 | 22q11.2 b (DiGeorge/velocardiofacial syndrome) | Bittel et al. [72] Shprintzen et al. [74] |
|
| ||||||||||
| 8y | F | None | del + | unknown | 22q11.21 | 17,145,346–19,958,163 | 2.8 Mb | 51 | 22q11.2 b (DiGeorge/velocardiofacial syndrome) | Bittel et al. [72] Shprintzen et al. [74] |
|
| ||||||||||
| 4y | F | Dysmorphism | del + | de novo | 22q13.32-q13.33 | 47,953,197–49,691,432 | 1.7 Mb | 40 | 22q13 b (Phelan-McDermid syndrome) SHANK3 | Dhar et al. [75] |
|
| ||||||||||
| 8y | F | None | del + | de novo | 22q13.33 | 49,379,810–49,691,432 | 312 Kb | 7 | 22q13 b (Phelan-McDermid syndrome) SHANK3 | Dhar et al. [75] |
|
| ||||||||||
| 15y*** | M | Dysmorphism | del + | maternal | Xp21.3-p21.2 | 29,134,047–30,084,234 | 950 Kb | 1 | IL1RAPL1 b | Nawara et al. [76] Youngs et al. [77] |
|
| ||||||||||
| 7y | M | None | dup − | maternal | Xp22.2 | 13,337,055–13,800,520 | 463 Kb | 6 | TRAPPC2 | Gedeon et al. [78] |
|
| ||||||||||
| 2y | M | Immune deficiency | dup − | paternal | Xp22.33/Yp11.32 | 525,304–1,228,569 | 703 Kb | 1 | SHOX | Thomas et al. [79] |
|
| ||||||||||
| 2y | F | Microcephaly | dup − | unknown, non-maternal | Xq25 | 122,801,654–123,444,057 | 642 Kb | 4 |
Xq25
b STAG2 XIAP |
Phillippe et al. [80] |
| dup − | maternal | 2q13 | 110,046,870–111,145,135 | 1.1 Mb | 14 | NPHP1 b | Baris et al. [81] | |||
|
| ||||||||||
| 6y | F | None | NA + | NA | XY female | NA | NA | |||
patient reported within (see Clinical Case Report),
patient previously reported [50],
patient previously reported [77]
+ = CNVs (deletions or duplications) that were reported previously to be associated with disease or disorder
− = CNVs (deletions or duplications) not previously reported at the time of genetic testing
autism spectrum disorders (ASD);
learning disability
Chromosome coordinates from UCSC hg18 Human Genome (Mar. 2006). Available from: http://genome.ucsc.edu.
Clinical Case Report
We describe a 42-year old white male with a learning disability, as a representative example of a patient identified with an abnormal chromosomal microarray result in our study. This patient had moderate intellectual disability, autistic features, and intermittent explosive disorder. The parents were unavailable and therefore the prenatal and early childhood histories were unrecorded. Medical history included congenital absence of the right kidney, hypothyroidism, type II diabetes, pernicious anemia, and history of seizures (petit mal and grand mal). He did not graduate from high school. He received special education training and has limited verbal communication. He lives in a group home setting for the intellectually disabled. EEGs performed at age 16 years and at age 22 years noted no epileptiform discharges. A head CT at age 24 years was normal. At age 25 years, he sustained a closed head injury from a bicycle accident caused by a seizure. A head CT performed shortly after the injury showed small right parietal contusions with a minimal amount of surrounding edema. A complete blood count with differential, coagulation studies, and lipids were normal at that time. A comprehensive metabolic panel showed low potassium and high CO2 levels. An EEG obtained at age 29 years showed a rare focal epileptiform discharge in the left central area and mild diffuse slow wave abnormalities indicating diffuse cerebral dysfunction. He was previously diagnosed with an intestinal volvulus and had an ischemic area in the sigmoid colon which required removal with placement of a colostomy. At age 41 years of age, he experienced a nose bleed and aspiration pneumonia following a dental procedure in which general anesthesia was used. He was hospitalized for 4 weeks due to pneumonia, and a lung CT scan showed bilateral pneumonia, pleural effusion, and right lung abnormality.
Physical examination revealed a normal head circumference (40th centile), normal height (50th centile), and normal weight (75th centile). He did have two posterior hair whorls and premature graying. Malar hypoplasia, downslanting palpebral fissures, bilateral ptosis, a high arched palate and missing teeth were seen along with a deviated nasal septum (injury-related) with an elongated nose, attached ear lobes, and hallux valgus deformity of both feet (Figure 1). He had brown coloration of both legs below the knees including the feet with sparse hair on the legs. The 180K microarray showed a 2q33.1-q34 deletion (7.8 Mb in size) containing 46 genes (see Figure 2). Parents were not available for testing to determine if this deletion was inherited or de novo in origin.
Figure 1.
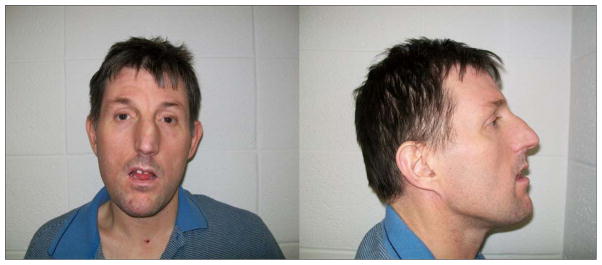
Frontal and profile facial views of the proband with a 7.8 Mb deletion at chromosome 2q33.1-q34 region at 42 years of age showing malar hypoplasia, ptosis, downslanting palpebral fissures, an elongated abnormal nose, Cupid’s bow appearance to upper lip, dental anomalies, and attached ear lobes.
Figure 2.

An array comparative genomic hybridization (aCGH) was carried out using DNA Array - Oligo 180K oligonucleotide array (CombiMatrix Diagnostics, Irvine, CA) and showed a 7.8 Mb deletion at 2q33.1-q34 (203,191,088-210,989,186 bp from the p terminus). The 46 genes found to be deleted in this chromosome region are listed alphabetically. The first (FAM117B) and last (MYL1) genes located in the deleted region are highlighted at the top of figure.
We previously reported a similar but smaller deletion (2q33.3-q34, 3.7 Mb in size) in a young male with autistic and dysmorphic features, including downslanting palpebral fissures, mild right ptosis, a prominent nasal tip, abnormal ears, Cupid’s bow of upper lip, dental anomalies, malar hypoplasia, and a high forehead [43]. Several of the cranio-facial and developmental features seen in that report were in common with our 42-year-old male with an overlapping but larger 2q33.1-q34 deletion. This chromosome region contains genes deleted in both subjects and involved the WNT pathway for organ development (FZD5), calcium regulation (ALS2CR8, PTH2R), transcription (KLF7, EEF1B2), muscle function and energy production (IDH1, MYL1, RPE, ACADL, NDUFS1) thought to contribute to their clinical presentations.
Discussion
Our study describes our experience of 215 consecutive patients with ASD or learning disability and presenting for genetic services at an academic medical center in the rural-based Midwestern region of the United States. We also describe a representative clinical case report identified using chromosomal microarray analysis in this patient population. The overall diagnostic yield in our study was 21% for patients with ASD or learning disability. This yield is similar to the 18.2% yield reported by Shen et al. [83] in microarray studies with 932 patients with ASD. Schaefer et al. [7] further reported their experience and found significant copy number abnormalities in 22% (14 of 68) of ASD subjects. In our study the size of the CNVs seen in each subject group varied. However, the size of the CNVs in the ASD group (966 ± 1464 Kb) was significantly smaller than seen in the learning disability group (2.90 ± 2.87 Mb). The significance or meaning of this observation in microarray analysis is unclear but may relate to differences in genetic causation (i.e., single gene in ASD versus larger genomic deletions or duplications involving more than one gene in those with learning disability).
Prior to chromosomal microarray studies in ASD reported in the literature, Miles and Hillman [6] tested 94 children for genetic causes and found that 6% (6 of 94) had identifiable genetic disorders while Herman et al. [84] reported genetic causes in 10% (7 of 71) of ASD subjects. Schaefer and Lutz [4] reported positive genetic findings in 40% (13 of 32) of subjects with ASD including 5% with a high resolution chromosome abnormality, 5% with fragile X syndrome, 5% with Rett syndrome, 3% with PTEN gene mutations, 10% with other genetic syndromes such as tuberous sclerosis, and about 10% with small deletions or duplications not detectable with high resolution chromosome analysis. Diagnostic yields are now being reported with microarray analysis showing a wide range of deletions and duplications while the most common chromosomal abnormality associated with non-syndromal autism prior to chromosomal microarray analysis was a maternal duplication of the 15q11-q13 region which accounted for 5% of cases with autism [18]. Large microdeletions in the chromosome 16p11.2 and 22q regions also accounted for another 1% of cases.
Unexplained learning disability/autism spectrum disorders associated with dysmorphic features in pediatric patients were studied by Battaglia et al. [9] using chromosomal microarray analysis and found 91 CNVs ranging in size from 1 Mb to 60 Kb in 77 (or 22%) of 349 patients. Additionally, Aggarwal et al. [85] reported that 58% (196 of 338) of their patients with developmental delay or intellectual disability had an identifiable genetic cause. These causes included Down and microdeletion syndromes and unbalanced and balanced chromosomal rearrangements in 33% (112 of 338) of their subjects. Non-chromosomal syndromes, such as fragile X syndrome, tuberous sclerosis, Noonan syndrome, and Cornelia de Lange syndrome were recognized in an additional 10% (32 of 338). Various neurometabolic disorders were identified in 10% (34 of 338) with the remaining subjects classified as having structural central nervous system defects, cerebral palsy, environmental insults, or idiopathic intellectual disability. A separate report by Michelson et al. [16] on individuals with learning disability found the diagnostic yield for karyotype studies to be at least 4%, and the diagnostic yield for fragile X testing was approximately 2% for a full mutation. However, with chromosomal microarray analysis, they found diagnostic abnormalities in 8% of subjects with learning disabilities [16] and 11% in those with learning disability and dysmorphic features, congenital anomalies, or neurologic symptoms.
Selected genes in the deletion/duplication detected by microarray analysis and/or chromosome regions of interest were identified by searching the medical literature for biological functions of involved genes and OMIM [Online Mendelian Inheritance in Man (www.ncbi.nlm.nih.gov/omim)], as a comprehensive, authoritative compendium of human genes and genetic phenotype including chromosome regions and genomic coordinates that is freely available online and updated daily. The goal was to search for information about CNVs, chromosome regions, genes involved and their function (if known) along with recognized syndromes with ASD or learning disability. Those genes present in the deletion or duplication regions were studied to determine if they could play a role in neurological development or function (i.e., ASD or learning disability) when disturbed by searching published medical literature reports, websites, OMIM or previously reported as a feature of a recognized genetic syndrome (e.g, Williams syndrome). For example, for 13 of the 65 subjects within the ASD group, selected genes and/or chromosome regions of interest were found, see Table 1, with literature citations listed for each subject. Recognized genetic syndromes (e.g., Williams syndrome) were noted with disturbed genes known to contribute to ASD and also genes found in deletions/duplications reported on more than one occasion to cause ASD (e.g., NRXN1, CACNA1C). Similarly, 32 subjects with learning disability were found to have CNV deletions/duplications by microarray analysis with selected genes or chromosome regions of interest known to play a role in their clinical presentation (see Table 4). Known genetic syndromes included DiGeorge/velocardiofacial syndrome. Selected genes included NLGN2, IL1RAP1, ANKRD11 and PARK2 known to play a role in neurological development or function and when disturbed can account for learning disability are listed in Table 4.
The diagnostic yield of chromosomal microarray analysis for individuals with ASD or learning disability in our study, and results reported by others, is consistently greater than the diagnostic yield of chromosomal karyotype studies alone or for fragile X DNA testing [16, 83]. As genetic technology continues to improve with advances in testing platforms and access to next generation exome sequencing, further genetic lesions will be identified, reported, and characterized as a cause of ASD or learning disability in patients presenting for genetic services. This information will be important for medical management and therapy with more accurate and specific genetic counseling for affected individuals and at-risk family members. The authors encourage the reporting of other microarray experiences at academic medical centers with similar patients presenting in the clinical setting for genetic services to increase our knowledge base, thereby impacting on the quality of life and outcome for affected individuals and their families.
References
- 1.Kanner L. Autistic psychopathy in childhood. Nerv Child. 1943;2:217–250. [Google Scholar]
- 2.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4. Washington D.C: American Psychiatric Association Press; 2000. [Google Scholar]
- 3.Autism and Developmental Disabilities Monitoring Network Surveillance Year 2008 Principal Investigators; Centers for Disease Control and Prevention. Prevalence of Autism Spectrum Disorders-Autism and Developmental Disabilities Monitoring Network, 14 sites, United States, 2008. MMWR Surveill Summ. 2012;61:1–19. [PubMed] [Google Scholar]
- 4.Schaefer GB, Lutz RE. Diagnostic yield in the clinical genetic evaluation of autism spectrum disorders. Genet Med. 2006;8:549–556. doi: 10.1097/01.gim.0000237789.98842.f1. [DOI] [PubMed] [Google Scholar]
- 5.El-Fishawy P, State MW. The genetics of autism: key issues, recent findings, and clinical implications. Psychiatr Clin North Am. 2010;33:83–105. doi: 10.1016/j.psc.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miles JH, Hillman RE. Value of a clinical morphology examination in autism. Am J Med Genet. 2000;91:245–253. [PubMed] [Google Scholar]
- 7.Schaefer GB, Starr L, Pickering D, Skar G, Dehaai K, Sanger WG. Array comparative genomic hybridization findings in a cohort referred for an autism evaluation. J Child Neurol. 2010;25:1498–1503. doi: 10.1177/0883073810370479. [DOI] [PubMed] [Google Scholar]
- 8.Devlin B, Scherer SW. Genetic architecture in autism spectrum disorder. Curr Opin in Genet Dev. 2012;22:229–237. doi: 10.1016/j.gde.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 9.Battaglia A, Doccini V, Bernardini L, Novelli A, Loddo S, Capalbo A, et al. Confirmation of chromosomal microarray as a first-tier clinical diagnostic test for individuals with developmental delay, intellectual disability, autism spectrum disorders and dysmorphic features. Eur J Paediatr Neurol. 2013 May 24; doi: 10.1016/j.ejpn.2013.04.010. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 10.Folstein SE, Piven J. Etiology of autism: genetic influences. Pediatrics. 1991;87:767–73. [PubMed] [Google Scholar]
- 11.Ozonoff S, Young GS, Carter A, Messinger D, Yirmiya N, Zwaigenbaum L, et al. Recurrence risk for autism spectrum disorders: a Baby Siblings Research Consortium study. Pediatrics. 2011;128:e488–95. doi: 10.1542/peds.2010-2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Messinger D, Young GS, Ozonoff S, Dobkins K, Carter A, Zwaigenbaum L, et al. Beyond autism: a baby siblings research consortium study of high-risk children at three years of age. J Am Acad Child Adoles Psych. 2013;52:300–308. doi: 10.1016/j.jaac.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. [Accessed 2013 April 11];Individuals with Disabilities Education Improvement Act, 20 U.S.C. § 1400 et seq [Internet] Available from: http://idea.ed.gov/download/statute.html.
- 14.Petersen MC, Kube DA, Palmer FB. Classification of developmental delays. Sem Ped Neurol. 1998;5:2–14. doi: 10.1016/s1071-9091(98)80012-0. [DOI] [PubMed] [Google Scholar]
- 15.Flore LA, Milunsky JM. Updates in the genetic evaluation of the child with global developmental delay or intellectual disability. Sem Ped Neurol. 2012;19:173–80. doi: 10.1016/j.spen.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 16.Michelson DJ, Shevell MI, Sherr EH, Moeschler JB, Gropman AL, Ashwal S. Evidence report: genetic and metabolic testing on children with global developmental delay: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2011;77:1629–1635. doi: 10.1212/WNL.0b013e3182345896. [DOI] [PubMed] [Google Scholar]
- 17.Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749–764. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schroer RJ, Phelan MC, Michaelis RC, Crawford EC, Skinner SA, Cuccaro M, et al. Autism and maternally derived aberrations of chromosome 15q. Am J Med Genet. 1998;76:327–336. doi: 10.1002/(sici)1096-8628(19980401)76:4<327::aid-ajmg8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 19.Scerri TS, Darki F, Newbury DF, Whitehouse AJO, Peyrard-Janvid M, Mattson H, et al. The dyslexia candidate locus on 2p12 is associated with general cognitive ability and white matter structure. PLoS ONE. 2012;7:e50321. doi: 10.1371/journal.pone.0050321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bena F, Bruno DL, Eriksson M, van Ravenswaaij-Arts C, Stark Z, Dijkhuizen T, et al. Molecular and clinical characterization of 25 individuals with exonic deletions of NRXN1 and comprehensive review of the literature. Am J Med Genet Part B. 2013;162:388–403. doi: 10.1002/ajmg.b.32148. [DOI] [PubMed] [Google Scholar]
- 21.Abou JR, Becker T, Georgi A, Feulner T, Schumacher J, Stromaier J, et al. Genetic variation of the FAT gene at 4q35 is associated with bipolar affective disorder. Mol Psychiatry. 2008;13:277–284. doi: 10.1038/sj.mp.4002111. [DOI] [PubMed] [Google Scholar]
- 22.Chien WH, Gau SSF, Wu YY, Huang US, Fang JS, Chen YJ, et al. Identification and molecular characterization of two novel chromosomal deletions associated with autism. Clin Genet. 2012;78:449–456. doi: 10.1111/j.1399-0004.2010.01395.x. [DOI] [PubMed] [Google Scholar]
- 23.Youngs EL, Henkhaus RS, Hellings JA, Butler MG. 12-year-old boy with a 4q35.2 microdeletion and involvement of MTNR1A, FAT1, and F11 genes. Clin Dysmorphol. 2012;21:93–96. doi: 10.1097/MCD.0b013e32834e9216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Griswold AJ, Ma D, Cukier HN, Nations LD, Schmidt MA, Chung RH, et al. Evaluation of copy number variations reveals novel candidate genes in autism spectrum disorder-associated pathways. Hum Mole Genet. 2012;21:3513–3523. doi: 10.1093/hmg/dds164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Uusimaa J, Gowda V, McShane A, Smith C, Evans J, Shrier A, et al. Prospective study of POLG mutations presenting in children with intractable epilepsy: prevalence and clinical features. Epilepsia. 2013;54:1002–1011. doi: 10.1111/epi.12115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Merla G, Brunetti-Pierri N, Micale L, Fusco C. Copy number variants at Williams-Beuren syndrome 7q11.23 region. Hum Genet. 2010;128:3–26. doi: 10.1007/s00439-010-0827-2. [DOI] [PubMed] [Google Scholar]
- 27.Cross-Disorder Group of the Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381:1371–1379. doi: 10.1016/S0140-6736(12)62129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu AT, Dai X, Martinez-Agosto JA, Cantor RM. Support for calcium channel gene defects in autism spectrum disorders. Mol Autism. 2012;3:18. doi: 10.1186/2040-2392-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burnside RD, Pasion R, Mikhail FM, Carroll AJ, Robin NH, Youngs EL, et al. Microdeletion/microduplication of proximal 15q11.2 between BP1 and BP2: a susceptibility region for neurological dysfunction including developmental and language delay. Hum Genet. 2011;130:517–528. doi: 10.1007/s00439-011-0970-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szafranski P, Schaaf CP, Person RE, Gibson IB, Xia Z, Mahadevan S, et al. Structures and molecular mechanisms for common 15q13. 3 microduplications involving CHRNA7: benign or pathological? Hum Muta. 2010;31:840–850. doi: 10.1002/humu.21284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Williams NM, Franke B, Mick E, Anney RJL, Freitag CM, Gill M, et al. Genome -wide analysis of copy number variants in attention deficit hyperactivity disorder: The role of rare variants and duplications at 15q13.3. Am J Psych. 2012;169:195–204. doi: 10.1176/appi.ajp.2011.11060822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, et al. Association between microdeletion and microduplication at 16p11.2 and autism. New Engl J Med. 2008;358:667–775. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 33.Martin CL, Duvall JA, Ilkin Y, Simon JS, Arreaza MG, Wilkes K, et al. Cytogenetic and molecular characterization of A2BP1/FOX1 as a candidate gene for autism. Am J Med Genet Part B. 2007;144B:869–876. doi: 10.1002/ajmg.b.30530. [DOI] [PubMed] [Google Scholar]
- 34.Butler MG, Youngs EL, Roberts JL, Hellings JA. Assessment and treatment in autism spectrum disorders: a focus on genetics and psychiatry. Autism Res Treat. doi: 10.1155/2012/242537. Epub 2012 May 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsurusakia Y, Okamotob N, Ohashic H, Mizunod S, Matsumotoe N, Makitaf Y, et al. Coffin–Siris syndrome is a SWI/SNF complex disorder. Clin Genet. 2013 Jul 1; doi: 10.1111/cge.12225. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 36.Franek KJ, Butler J, Johnson J, Simensen R, Friez MJ, Bartel F, et al. Deletion of the immunoglobulin domain of IL1RAPL1 results in nonsyndromic X-linked intellectual disability associated with behavioral problems and mild dysmorphism. Am J Med Genet Part A. 2011;155A:1109–1114. doi: 10.1002/ajmg.a.33833. [DOI] [PubMed] [Google Scholar]
- 37.Behnecke A, Hinderhofer K, Bartsch O, Numann A, Ipach ML, Damatova N, et al. Intragenic deletions of IL1RAPL1: Report of two cases and review of the literature. Am J Med Genet Part A. 2011;155A:372–379. doi: 10.1002/ajmg.a.33656. [DOI] [PubMed] [Google Scholar]
- 38.De Ligt J, Willemsen MH, van Bon BWM, Kleefstra T, Yntema HG, Kroes T, et al. Diagnostic exome sequencing in persons with severe intellectual disability. New Engl J Med. 2012;367:1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- 39.Matsubara K, Imai K, Okada S, Miki M, Ishikawa N, Tsumura M, et al. Severe developmental delay and epilepsy in a Japanese patient with severe congenital neutropenia due to HAX1 deficiency. Haematologica. 2007;92:e123–e1235. doi: 10.3324/haematol.11973. [DOI] [PubMed] [Google Scholar]
- 40.Hattori A, Komaki H, Kawatani M, Sakuma H, Saito Y, Nakagawa E, et al. A novel mutation in the LMNA gene causes congenital muscular dystrophy with dropped head and brain involvement. Neuromuscul Disord. 2012;22:149–151. doi: 10.1016/j.nmd.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 41.Sanders SR, Dawson AJ, Vust A, Hryshko M, Tomiuk M, Riordan D, et al. Interstitial deletion of chromosome 2p16.2p21. Clin Dysmorphol. 2003;12:183–185. doi: 10.1097/01.mcd.0000065051.36236.e9. [DOI] [PubMed] [Google Scholar]
- 42.Yu HE, Hawash K, Picker J, Stoler J, Urion D, Wu BL, et al. A recurrent 1.71 Mb genomic imbalance at 2q13 increases the risk of developmental delay and dysmorphism. Clin Genet. 2012;81:257–264. doi: 10.1111/j.1399-0004.2011.01637.x. [DOI] [PubMed] [Google Scholar]
- 43.Brandau DT, Lund M, Cooley LD, Sanger WG, Butler MG. Autistic and dysmorphic features associated with a submicroscopic 2q33.3-q34 interstitial deletion detected by array comparative genomic hybridization. Am J Med Genet A. 2008;146A:521–524. doi: 10.1002/ajmg.a.32153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pescuccia C, Melonia I, Bruttinia M, Ariania F, Longoa I, Maria F, et al. Chromosome 2 deletion encompassing the MAP2 gene in a patient with autism and Rett-like features. Clin Genet. 2003;64:497–501. doi: 10.1046/j.1399-0004.2003.00176.x. [DOI] [PubMed] [Google Scholar]
- 45.Kitsiou-Tzeli S, Sismani C, Ioannides M, Bashiardes S, Ketoni A, Touliatou V, et al. Array-CGH analysis and clinical description of 2q37.3 de novo subtelomeric deletion. Eur J Med Genet. 2007;50:73–78. doi: 10.1016/j.ejmg.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 46.Williams SR, Aldred MA, Der Kaloustian VM, Halal F, Gowans G, McLeod DR, et al. Haploinsufficiency of HDAC4 causes Brachydactyly Mental Retardation Syndrome, with Brachydactyly Type E, developmental delays, and behavioral problems. Am J Hum Genet. 2010;87:219–228. doi: 10.1016/j.ajhg.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Di Gregorio E, Orsi L, Godani M, Vaula G, Jensen S, Salmon E, et al. Two Italian families with ITPR1 gene deletion presenting a broader phenotype of SCA15. Cerebellum. 2010;9:115–123. doi: 10.1007/s12311-009-0154-0. [DOI] [PubMed] [Google Scholar]
- 48.Vardarajan BN, Eran A, Jung JY, Kunkel LM, Wall DP. Haplotype structure enables prioritization of common markers and candidate genes in autism spectrum disorder. Transl Psychiatry. 2013;3:e262. doi: 10.1038/tp.2013.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Breitfeld J, Stumvoll M, Kovacs P. Genetics of adiponectin. Biochimie. 2012;94:2157–2163. doi: 10.1016/j.biochi.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 50.Dasouki MJ, Lushington GH, Hovanes K, Casey J, Gorre M. The 3q29 microdeletion syndrome: report of three new unrelated patients and in silico “RNA binding” analysis of the 3q29 region. Am J Med Genet Part A. 2011;155A:1654–1660. doi: 10.1002/ajmg.a.34080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sagar A, Bishop JR, Tessman DC, Guter S, Martin CL, Cook EH. Co-occurrence of autism, childhood psychosis, and intellectual disability associated with a de novo 3q29 microdeletion. Am J Med Genet Part A. 2013;161A:845–849. doi: 10.1002/ajmg.a.35754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scheuerle A, Wilson K. PARK2 copy number aberrations in two children presenting with autism spectrum disorder: further support of an association and possible evidence for a new microdeletion/microduplication syndrome. Am J Med Genet Part B. 2011;156B:413–420. doi: 10.1002/ajmg.b.31176. [DOI] [PubMed] [Google Scholar]
- 53.Mariani M, Crosti F, Redaelli S, Fossati C, Piras R, Biondi A, et al. Partial duplication of the PARK2 gene in a child with developmental delay and her normal mother: a second report. Am J Med Genet Part B. 2013;162B:485–486. doi: 10.1002/ajmg.b.32173. [DOI] [PubMed] [Google Scholar]
- 54.Niemi AK, Kwan A, Hudgins L, Cherry AM, Manning MA. Report of two patients and further characterization of interstitial 9p13 deletion--a rare but recurrent microdeletion syndrome? Am J Med Genet Part A. 2012;158A:2328–2335. doi: 10.1002/ajmg.a.35536. [DOI] [PubMed] [Google Scholar]
- 55.Manolakos E, Orru S, Neroutsou R, Kefalas K, Louizou E, Papoulidis I, et al. Detailed molecular and clinical investigation of a child with a partial deletion of chromosome 11 (Jacobsen syndrome) Mol Cytogenet. 2009;2:26. doi: 10.1186/1755-8166-2-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Breckpot J, Takiyama Y, Thienpont B, Van Vooren S, Vermeesch JR, Ortibus E, et al. A novel genomic disorder: a deletion of the SACS gene leading to spastic ataxia of Charlevoix-Saguenay. Eur J Hum Genet. 2008;16:1050–1054. doi: 10.1038/ejhg.2008.58. [DOI] [PubMed] [Google Scholar]
- 57.Akawi NA, Al-Jasmi F, Al-Shamsi AM, Ali BR, Al-Gazali L. LINS, a modulator of the WNT signaling pathway, is involved in human cognition. Orphanet J Rare Dis. 2013;8:87. doi: 10.1186/1750-1172-8-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bachmann-Gagescu R, Mefford HC, Cowan C, Glew GM, Hing AV, Wallace S, et al. Recurrent 200-kb deletions of 16p11.2 that include the SH2B1 gene are associated with developmental delay and obesity. Genet Med. 2010;12:641–647. doi: 10.1097/GIM.0b013e3181ef4286. [DOI] [PubMed] [Google Scholar]
- 59.Ramalingam A, Zhou XG, Fiedler SD, Brawner SJ, Joyce JM, Liu HY, et al. 16p13.11 duplication is a risk factor for a wide spectrum of neuropsychiatric disorders. J Hum Genet. 2011;56:541–544. doi: 10.1038/jhg.2011.42. [DOI] [PubMed] [Google Scholar]
- 60.Sacharow S, Li D, Fan YS, Tekin M. Familial 16q24.3 microdeletion involving ANKRD11 causes a KBG-like syndrome. Am J Med Genet Part A. 2012;158A:547–552. doi: 10.1002/ajmg.a.34436. [DOI] [PubMed] [Google Scholar]
- 61.Feyder M, Karlsson RM, Mathur P, Lyman M, Bock R, Momenan R, et al. Association of mouse Dlg4 (PSD-95) gene deletion and human DLG4 gene variation with phenotypes relevant to autism spectrum disorders and Williams’ syndrome. Am J Psych. 2010;167:1508–1517. doi: 10.1176/appi.ajp.2010.10040484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schluth-Bolard C, Sanlaville D, Labalme A, Till M, Morin I, Touraine R, et al. 17p13.1 microdeletion involving the TP53 gene in a boy presenting with mental retardation but no tumor. Am J Med Genet Part A. 2010;152A:1278–1282. doi: 10.1002/ajmg.a.33316. [DOI] [PubMed] [Google Scholar]
- 63.Pettem KL, Yokomaku D, Takahashi H, Ge Y, Craig AM. Interaction between autism-linked MDGAs and neuroligins suppresses inhibitory synapse development. J Cell Biol. 2013;200:321–336. doi: 10.1083/jcb.201206028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tentler D, Johannesson T, Johansson M, Råstam M, Gillberg C, Orsmark C, et al. A candidate region for Asperger syndrome defined by two 17p breakpoints. Eur J Hum Genet. 2003;11:189–195. doi: 10.1038/sj.ejhg.5200939. [DOI] [PubMed] [Google Scholar]
- 65.Capra V, Mirabelli-Badenier M, Stagnaro M, Rossi A, Tassano E, Gimelli S, et al. Identification of a rare 17p13.3 duplication including the BHLHA9 and YWHAE genes in a family with developmental delay and behavioural problems. BMC Med Genet. 2012;13:93. doi: 10.1186/1471-2350-13-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bruno DL, Anderlid BM, Lindstrand A, van Ravenswaaij-Arts C, Ganesamoorthy D, Lundin J, et al. Further molecular and clinical delineation of co-locating 17p13.3 microdeletions and microduplications that show distinctive phenotypes. J Med Genet. 2010;47:299–311. doi: 10.1136/jmg.2009.069906. [DOI] [PubMed] [Google Scholar]
- 67.D’Angelo CS, de Oliveira MA, de Castro CI, Koiffmann CP. Molecular cytogenetic characterization of an inherited maternal duplication 20p11.21p13 associated with a small 20p11.21 deletion. Am J Med Genet Part A. 2010;152A:3197–3202. doi: 10.1002/ajmg.a.33741. [DOI] [PubMed] [Google Scholar]
- 68.Doco-Fenzy M, Holder-Espinasse M, Bieth E, Magdelaine C, Vincent MC, Khoury M, et al. The clinical spectrum associated with a chromosome 17 short arm proximal duplication (dup 17p11.2) in three patients. Am J Med Genet Part A. 2008;146:917–924. doi: 10.1002/ajmg.a.32195. [DOI] [PubMed] [Google Scholar]
- 69.Liu MM, Zack DJ. Alternative splicing and retinal degeneration. Clin Genet. 2013;84:142–149. doi: 10.1111/cge.12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Margarit E, Morales C, Rodríguez-Revenga L, Monné R, Badenas C, Soler A, et al. Familial 4.8 MB deletion on 18q23 associated with growth hormone insufficiency and phenotypic variability. Am J Med Genet A. 2012;158A:611–616. doi: 10.1002/ajmg.a.34221. [DOI] [PubMed] [Google Scholar]
- 71.Ren CM, Liang Y, Wei F, Zhang YN, Zhong SQ, Gu H, et al. Balanced translocation t(3;18)(p13;q22.3) and points mutation in the ZNF407 gene detected in patients with both moderate non-syndromic intellectual disability and autism. Biochim Biophys Acta. 2013;1832:431–438. doi: 10.1016/j.bbadis.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 72.Bittel DC, Yu S, Newkirk H, Kibiryeva N, Holt A, 3rd, Butler MG, et al. Refining the 22q11.2 deletion breakpoints in DiGeorge syndrome by aCGH. Cytogenet Genome Res. 2009;124:113–120. doi: 10.1159/000207515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Alberti CR, Falco M, Cali F, Schinoccac P, Galesi O, Spalletta A, et al. 1.5 Mb de novo 22q11. 21 microduplication in a patient with cognitive deficits and dysmorphic facial features. Clin Genet. 2007;71:177–182. doi: 10.1111/j.1399-0004.2007.00750.x. [DOI] [PubMed] [Google Scholar]
- 74.Shprintzen RJ. Velo-cardio-facial syndorme: 30 years of study. Dev Disabil Res Rev. 14:3–10. doi: 10.1002/ddrr.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dhar SU, del Gaudio D, German JR, Peters SU, Ou Z, Bader PI, et al. 22q13.3 deletion syndrome: clinical and molecular analysis using array CGH. Am J Med Genet Part A. 2010;152A:573–581. doi: 10.1002/ajmg.a.33253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nawara M, Klapecki J, Borg K, Jurek M, Moreno S, Tryfon J, et al. Novel mutation of IL1RAPL1 gene in a nonspecific X-linked mental retardation (MRX) family. Am J Med Genet Part A. 2008;146A:3167–3172. doi: 10.1002/ajmg.a.32613. [DOI] [PubMed] [Google Scholar]
- 77.Youngs EL, Henkhaus R, Hellings JA, Butler MG. IL1RAPL1 gene deletion as a cause of X-linked intellectual disability and dysmorphic features. Eur J Med Genet. 2012;55:32–36. doi: 10.1016/j.ejmg.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gedeon AK, Tiller GE, Le Merrer M, Heuertz S, Tranebjaerg L, Chitayat D, et al. The molecular basis of X-linked spondyloepiphyseal dysplasia tarda. Am J Hum Genet. 2001;68:1386–1397. doi: 10.1086/320592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Thomas NS, Harvey JF, Bunyan DJ, Rankin J, Grigelioniene G, Bruno DL, et al. Clinical and molecular characterization of duplications encompassing the human SHOX gene reveal a variable effect on stature. Am J Med Genet Part A. 2009;149A:1407–1414. doi: 10.1002/ajmg.a.32914. [DOI] [PubMed] [Google Scholar]
- 80.Philippe A, Malan V, Jacquemont ML, Boddaert N, Bonnefont JP, Odent S, et al. Xq25 duplications encompassing GRIA3 and STAG2 genes in two families convey recognizable X-linked intellectual disability with distinctive facial appearance. Am J Med Genet Part A. 2013;161A:1370–1375. doi: 10.1002/ajmg.a.35307. [DOI] [PubMed] [Google Scholar]
- 81.Baris H, Bejjani BA, Tan WH, Coulter DL, Martin JA, Storm AL, et al. Identification of a novel polymorphism--the duplication of the NPHP1 (nephronophthisis 1) gene. Am J Med Genet Part A. 2006;140A:1876–1879. doi: 10.1002/ajmg.a.31390. [DOI] [PubMed] [Google Scholar]
- 82.Shen Y, Dies KA, Holm IA, Bridgemohan C, Sobeih MM, Caronna EB, Miller KJ, et al. Clinical genetic testing for patients with autism spectrum disorders. Pediatrics. 2010;125:e727–735. doi: 10.1542/peds.2009-1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Herman GE, Henninger N, Ratliff-Schaub K, Pastore M, Fitzgerald S, McBride KL. Genetic testing in autism: how much is enough? Genet Med. 2007;9:268–274. doi: 10.1097/gim.0b013e31804d683b. [DOI] [PubMed] [Google Scholar]
- 84.Aggarwal S, Bogula VR, Mandal K, Kumar R, Phadke SR. Aetiologic spectrum of mental retardation & developmental delay in India. Ind J Med Res. 2012;136:436–444. [PMC free article] [PubMed] [Google Scholar]