

Abstract
Acute myeloid leukemia (AML) is the most common form of adult leukemia. The transcription factor fusion CBFβ-SMMHC (core binding factor β and the smooth-muscle myosin heavy chain), expressed in AML with the chromosome inversion inv(16)(p13q22), outcompetes wild-type CBFβ for binding to the transcription factor RUNX1, deregulates RUNX1 activity in hematopoiesis, and induces AML. Current inv(16) AML treatment with nonselective cytotoxic chemotherapy results in a good initial response but limited long-term survival. Here, we report the development of a protein-protein interaction inhibitor, AI-10-49, that selectively binds to CBFβ-SMMHC and disrupts its binding to RUNX1. AI-10-49 restores RUNX1 transcriptional activity, displays favorable pharmacokinetics, and delays leukemia progression in mice. Treatment of primary inv(16) AML patient blasts with AI-10-49 triggers selective cell death. These data suggest that direct inhibition of the oncogenic CBFβ-SMMHC fusion protein may be an effective therapeutic approach for inv(16) AML, and they provide support for transcription factor targeted therapy in other cancers.
Acute myeloid leukemia (AML) is the most common form of adult leukemia (1). Long-term survival for AML remains poor and varies with the mutational composition of the leukemic cells. The transcription factor fusion CBFb-SMMHC (fusion of core binding factor β and smooth-muscle myosin heavy chain), expressed in AML with the chromosome inversion inv(16)(p13q22), cooperates with activating mutations in components of cytokine signaling pathways in leukemia transformation (2–5). CBFb is a component of the heterodimeric transcription factor core binding factor, where it binds to RUNX proteins and enhances their affinity for DNA (6), and the resulting complex plays a key role in regulating hematopoiesis (7). CBFβ-SMMHC outcompetes CBFb for binding to RUNX1 (8), deregulates RUNX1 transcription factor activity in hematopoiesis, and induces AML. Current inv(16) AML treatment with nonselective cytotoxic chemotherapy results in a good initial response but limited long-term survival. Studies in mice and patient samples support the concept that inv(16) is a driver mutation that generates preleukemic progenitor cells that, upon acquisition of additional cooperating mutations, progress to leukemia (3, 4, 9-12).
To develop a targeted inhibitor of CBFβ-SMMHC function, we used a previously described fluorescence resonance energy transfer (FRET) assay (13) with Venus-CBFβ-SMMHC replacing Venus-CBFb (fig. S1) to screen the National Cancer Institute, NIH, Diversity Set for compounds that inhibit the binding of CBFβ-SMMHCto theRUNX1 Runt domain. This screen identified the active compound AI-4-57 with a 50% inhibitory concentration (IC50) of 22 μM, whereas AI-4-88, a derivative lacking the methoxy functionality, is inactive (Table 1). Changes in the chemical shifts in a nuclear magnetic resonance (NMR) spectrum of a protein upon binding of a small molecule are a powerful method to confirm binding to a protein. We recorded two-dimensional 2D 15N-1H heteronuclear single quantum coherence (HSQC) spectra and 1D saturation transfer difference (STD) NMR experiments of AI-4-57 with CBFβ and the Runt domain. No interaction was observed for the Runt domain, but we can demonstrate chemical shift perturbations in the HSQC spectrumof CBFβ upon addition ofAI-4-57 (Fig. 1A) and no changes upon addition of the inactive derivative AI-4-88 (fig. S2), which establishes that the compound binds to CBFβ. Chemical shift perturbations in the backbone and in two aromatic side chains [tryptophan at position 113 (W113) and tyrosine at position 96 (Y96)] indicate that the compound binds in a site spatially close to CBFβ but not on the protein-protein interaction surface on CBFβ, that is, it acts in an allosteric manner to inhibit binding (fig. S3).
Table 1.
Chemical structures and IC50 values of AI-10-49 and related compounds, determined by using the FRET assay.
| Compound Name | Compound Structure | FRET IC50 (μM) |
|---|---|---|
| AI-4-88 |
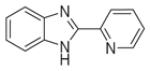
|
>240 |
| AI-4-57 |
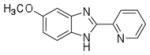
|
22 ± 8 |
| AI-10-11 |
|
14 ± 4 |
| AI-10-19 |
|
>2.5 |
| AI-4-83 |
|
0.35 ± 0.05 |
| AI-4-82 |
|
0.44 ± 0.07 |
| AI-4-71 |
|
0.37 ± 0.07 |
| AI-10-47 |
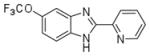
|
2.0 ± 0.03 |
| AI-10-49 |
|
0.26 ± 0.01 |
Fig. 1. Development of potent selective inhibitor of CBFβ-SMMHC-RUNX binding.

(A) 15N-1H HSQC spectrum (peaks correspond to all NH moieties of protein) of CBFβ alone (blue) and CBFβ + AI-4-57 (red). (B) Schematic diagram for the application of polyvalency to develop a specific and potent inhibitor of CBFβ-SMMHC-RUNX binding. Equations refer to predicted KD values for a bivalent inhibitor binding to CBFβ and CBFβ-SMMHC. Ceff is the effective local concentration, which depends on the distance between CBFβ domains in the oligomeric CBFβ-SMMHC. (C) FRET assay measurements for bivalent inhibitors with varying linker lengths with 10 nM Cerulean-Runt domain and 10 nM Venus-CBFβ-SMMHC.The y axis is the ratio of emission intensities at 525 and 474 nm. Three independent measurements were performed, and their average and standard deviation were used for IC50 data fitting. (D) FRETassay measurements of inhibition of CBFβ-SMMHC- RUNX binding for AI-4-57 and AI-4-83 with 10 nM Cerulean-Runt domain and 10 nM Venus-CBFβ-SMMHC. Data for AI-4-83 are the same as presented in (C). Data for these two compounds are presented separately for clarity of comparison to one another. Left y axis is the ratio of emission intensities at 525 and 474 nm. Right y axis indicates the FRET ratios observed with addition of 100 nM and 1000 nM untagged CBFβ, corresponding to roughly 1-fold and 10-fold dissociation of CBFβ-SMMHC and Runt domain [CBFβ-SMMHC binds with 7-fold the affinity of CBFβ (8)]. Three independent measurements were performed, and their average and standard deviation were used for IC50 data fitting. (E) Dose-dependent effect of a 24-hour treatment of ME-1 cells with bivalent inhibitors with varying linker lengths measured by MTT assay and normalized to the DMSO-treated group. Each symbol represents the mean of triplicate experiments; error bars represent the SD. (F and G) Dose-dependent effect of AI-10-47 (red) and AI-10-49 (black) treatment for 48 hours; (F) ME-1 cells were assessed by annexin V and 7-amino-actinomycin (7AAD) viability staining, and (G) human bone marrow cells were assessed by MTT assay. The data were normalized to the DMSOtreated group. Each data point represents the mean of triplicate experiments; error bars represent the SD.
We have shown that a reduced dosage of CBFβ in the presence of a CBFβ-SMMHC knockin allele enhances leukemogenesis in mice (14) and argue that selectivity for CBFβ-SMMHC versus CBFβ is critical for in vivo utility. To achieve such specificity, we have taken advantage of the state of the two in solution: CBFβ-SMMHC is oligomeric, whereas CBFβ is monomeric (8, 15) and have applied the principles of polyvalency (16, 17) to develop derivatives of AI-4-57 with enhanced potency and selectivity (Fig. 1B). Substitutions at the five position of the pyridine ring do not affect activity (Table 1), sowe have utilized polyethylene glycol-based linkers at this position to create bivalent derivatives with 5-, 7-, 10-, and 16-atom linker lengths (Table 1). Measurement of the IC50 values with the FRET assay (Fig. 1C) shows that the five-atom linker compound has less activity, but the longer linker compounds show potent inhibition with the seven-atom linker inhibitor (AI-4-83) yielding a 350 nM IC50, which corresponds to a 63-fold enhancement over themonovalent compound (Fig. 1D and Table 1). In addition, AI-4-83 achieves >10-fold dissociation of CBFβ-SMMHC andRUNX1 Runt domain at saturating concentrations (Fig. 1D).
The activity of the bivalent inhibitors on cell growth was tested in three human leukemia cell lines [ME-1, inv(16) cell line; Kasumi-1, t(8;21) cell line; and U937, lymphoma cell line] by using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) conversion assay. Mimicking the results obtained with the FRET assay, even in terms of relative efficacy, growth of inv(16) cell line ME-1 was sensitive to compounds AI-4-71, AI-4-83, and AI-4-82 but not to AI-10-19 (Fig. 1E). In contrast, growth of non-inv(16) cell lines U937 and Kasumi-1 was unaffected over the same concentration range (fig. S4, A and B), which demonstrated a high degree of specificity and suggested that the activity of these bivalent compounds was on target.
Analysis of the pharmacokinetic properties of AI-4-57 showed that the compound has a short half-life (t½ = 37 min) in mouse plasma (fig. S5) and that loss of the methyl group from the methoxy functionality is the primary metabolite. Trifluoromethoxy (CF3O) substitutions have been shown to be less reactive (18, 19), so we synthesized AI-10-47 with this substitution. FRET measurements show that this substitution actually enhances the activity of the monovalent compound (Table 1). Measurements of stability in liver microsomes showed that AI-10-47 reduced the metabolic liability and so justified the synthesis of the bivalent derivative AI-10-49 (Table 1).AI-10-49 is potent (FRET IC50=260nM) (Table 1) [isothermal titration calorimetry (ITC) measurements yielded a dissociation constant (KD) = 168 nM] (fig. S6), has improved in vivo pharmacokinetic properties (t½ = 380min) (fig. S5), and has enhanced inhibitory activity on ME-1 cell growth (IC50 = 0.6 mM) (Fig. 1F) compared with the parent protonated bivalent compound AI-4-83 (IC50 of ~3 μM) (Fig. 1E). Note that AI-10-49 showed negligible activity (IC50 > 25 μM) in normal human bone marrow cells (Fig. 1G), which indicated a robust potential therapeutic window. In a panel of 11 human leukemia cell lines, ME-1 cells were the only cell line highly sensitive to AI-10-49 (fig. S7).
The specificity of AI-10-49 in disrupting endogenous RUNX1 binding to CBFβ-SMMHC versus CBFβ was assessed in ME-1 cells. AI-10-49 effectively dissociatedRUNX1 from CBFβ-SMMHC, with 90% dissociation after 6 hours of treatment, whereas it had only a modest effect on CBFβ-RUNX1 association (Fig. 2A). The stability of RUNX1, CBFβ, andCBFβ-SMMHCwas not affected by AI-10-49 (fig. S8A). Expression of the RUNX1-regulated genes RUNX3, CSF1R, and CEBPA is repressed by CBFβ-SMMHC in inv(16)AML (20-22). Previous studies have shown decreased RUNX1 binding to target genes in the presence of CBFβ-SMMHC (23, 24), which suggests that CBFβ-SMMHC represses RUNX1 target genes by blocking binding ofRUNX1 to targetDNAsites (Fig. 2B). Consistent with this model, chromatin-immunoprecipitation (ChIP) assays showed that treatment of ME-1 cells for 6 hours with AI-10-49 increased RUNX1 occupancy 8-, 2.2-, and 8-fold at the RUNX3, CSF1R, and CEBPA promoters, respectively, whereas no enrichment was observed at control loci (Fig. 2C and fig. S8, B and C). In accordance with this, treatment of ME-1 cells for 6 or 12 hours with AI-10-49 increased expression of RUNX3, CSF1R, and CEBPA but had no effect on control gene PIN1 (Fig. 2D).Neither of these effectswas observed in inv(16)-negative U937 cells. These data establish AI-10-49 selectivity in inhibiting CBFβ-SMMHC binding to RUNX1 and validate our approach of using bivalent inhibitors to achieve this specificity.
Fig. 2. Specificity of AI-10-49 activity on CBFβ-SMMHC-RUNX1 binding.

(A) Effect of 1 μM AI-10-49 on CBFβ-RUNX1 and CBFβ-SMMHC-RUNX1 binding at 3 and 6 hours in ME-1 cells, measured by coimmunoprecipitation (quantification of three experiments is shown on the right). (B) Schematic of the effect of CBFβ-SMMHC on RUNX1 occupancy and target gene expression and the effect of AI-10-49 on occupancy and expression. (C) Chromatin immunoprecipitation assay showing RUNX1 occupancy on RUNX3, CSF1R, and CEBPA in ME-1 and U937 cells treated with 1 μM AI-10-49 for 6 hours and represented as fold enrichment relative to DMSOtreated cells. Each symbol represents the mean of triplicate experiments; error bars represent the SD. (D) Relative expression (qRT-PCR) of RUNX3, CSF1R, and CEBPA in ME-1 and U937 cells treated with 1 μM AI-10-49 for 6 and 12 hours, and normalized to the DMSO control group. Each symbol represents the mean of triplicate experiments; error bars represent the SD. For all panels, significance was calculated as unpaired t-test, *P < 0.05, or ***P < 0.001.
Up to 90% of inv(16) AML patients have cooperating mutations in components of the receptor tyrosine kinase pathway, including N-RAS and c-Kit (25). We have recently developed an efficient mouse model of inv(16) AML, by combining the conditional NrasLSL-G12D and CbfbMYH11 alleles (26). To test the effects of AI-10-49 administration in vivo, we transplantedmice with Cbfb+/MYH11:Ras+/G12D leukemic cells, waited 5 days for engraftment, and then treated mice with vehicle [dimethyl sulfoxide (DMSO)] or 200 mg/kg of body weight AI-10-49 for 10 days, and assessed the effect on disease latency. As shown in Fig. 3A, vehicle-treatedmice succumbed to leukemia with a median latency of 33.5 days, whereas AI-10-49-treated mice survived significantly longer (median latency = 61 days; P = 2.7 × 10−6). Thus, transient treatment with AI-10-49 reduces leukemia expansion in vivo. Although we have not assessed toxicity after longterm exposure, after 7 days of administration of AI-10-49, we observe no evidence of toxicity (figs. S9 to S11).
Fig. 3. Activity of AI-10-49 in inv(16)mousemodel and inv(16) AML patient samples.

(A) Kaplan-Meier survival curve of mice (n = 11 per group) transplanted with 2 × 103 cbfb+/MYH11;Ras+/G12D leukemic cells and treated between days 5 and 15 posttransplantation (blue arrows)with DMSO(black line) or 200 mg/kg of body weight per day AI-10-49 (red line); Statistics described in the statistical methods section. (B) Percent viability (annexin V and 7AAD assay) relative to vehicle control (DMSO) for CD34+ purified primary human inv(16) AML samples treated for 48 hours with either AI-10-49 or AI-10-47 at the indicated concentrations. Each symbol represents the average for an individual sample from duplicate treatments. The line represents the mean; error bars represent the SD. (C) Percent viability (annexin V and 7AAD assay) relative to vehicle control (DMSO) for primary human AML samples with normal karyotype treated for 48 hours with either AI-10-49 or AI-10-47 at the indicated concentrations. Each symbol represents the average for an individual sample from duplicate treatments.The line represents the mean of all biological replicates; error bars represent the SD. (D) Percentage of colony-forming units (CFUs) after treatment with AI-10-49 relative to vehicle control (DMSO) for primary human AML cells. Each symbol represents the average for an individual sample from duplicate treatments; error bars represent the SD. (E) Percent CFUs to vehicle control (DMSO) for CD34+ purified primary human inv(16) AML samples treated with either AI-10-49 or AI-10-47 at the indicated concentrations. CFU assays were performed in triplicate. Error bars represent the SD. (F) Percent CFUs to vehicle control (DMSO) for CD34+ purified primary human AML samples with normal karyotype treated with either AI-10-49 or AI-10-47 at the indicated concentrations. CFU assays were performed in triplicate. Error bars represent the SD. (G) Percent CFUs to vehicle control (DMSO) for CD34+ purified primary CD34+ cord blood cells treated with either AI-10-49 or AI-10-47 at the indicated concentrations. Significance calculated as unpaired t test, *P < 0.05 or ***P < 0.001.
To test the potential utility of AI-10-49 for use in human inv(16) leukemia treatment, we evaluated the survival of four primary inv(16) AML cell samples treated for 48 hours with a dose range ofmonovalent AI-10-47 and bivalent AI-10-49. As shown in Fig. 3B, the viability of inv(16) patient cells was reduced by treatment with AI-10-49 at 5 and 10 μM concentrations (individual dose-response experiments are shown in fig. S12). Note that the bivalent AI-10-49 was more potent than the monovalent compound AI-10-47 and so recapitulated the effects observed in the human inv(16) cell line ME-1. In contrast, the viability of normal karyotype AML sampleswas not affected by AI-10-49 treatment (Fig. 3C). Analysis of an additional set of five AML samples revealed that AI-10-49 treatment specifically reduces the viability of inv(16) leukemic cells without having an apparent effect on their differentiation (fig. S13). AI-10-49 specificity was also evident when we assessed the ability of AML cells to form colonies by evaluating colony-forming units (CFUs) after compound exposure. The ability of inv(16) AML cells to form CFUs was selectively reduced by AI-10-49 when compared with normal karyotype and t(8;21) AML patient samples (Fig. 3D). This inhibitory effect was dose-dependent (40 and 60% at 5 and 10 μM, respectively) (Fig. 3E), whereas there was no change in CFUs of AML cells treated with AI-10-47, AML cells with normal karyotype (Fig. 3F), or CD34+ cord blood cells (Fig. 3G). These studies show that AI-10-49 selectively inhibits viability and CFU capacity in inv(16) AML blasts, whereas it has negligible effects on AML blasts with normal karyotype or, importantly, on normal human hematopoietic progenitors.
Dysregulated gene expression is a hallmark of cancer and is particularly important for the maintenance of cancer stem-cell properties, such as self-renewal, which lead to relapse. As such, the targeting of proteins that drive transcriptional dysregulation, so-called “transcription therapy,” represents an avenue for drug development with immense potential. A number of fusion proteins involving transcription factors have been identified as drivers of disease in leukemia (27); sarcoma (28); and, recently, in prostate cancer (29), which provide excellent targets for therapeutic intervention. Our results provide a proof-ofprinciple for this approach, as AI-10-49 specifically inhibits CBFβ-SMMHC-RUNXbinding and shows efficacy against CBFβ-SMMHC-driven leukemia in mice with no obvious side effects. Specificity of action is a key component in the development of a targeted drug. Imatinib, for example, shows excellent specificity, and its efficacy in chronic myelogenous leukemia is clearly a result of effective inhibition of the BCR-ABL fusion protein that drives chronic myelogenous leukemia. However, even this highly selective agent inhibits both the BCR-ABL fusion protein, aswell aswild-typeABL. The “Holy Grail,” as it were, of targeted therapy with fusion or mutated protein drivers of cancer is to achieve inhibition of the fusion or mutated protein with little to no effect on the wild-type protein. This study demonstrates that AI-10-49 represents an example of such selectivity for inv(16) leukemia, as it inhibits CBFβ-SMMHC activity while having a minimal effect on CBFβ function. There are relatively few examples of drugs targeting transcription factors. ATRA (all-transretinoic acid) for RAR (retinoic acid receptor) fusions in leukemia and MDM2-p53 inhibitors are successful examples; however, neither of these has the selectivity of AI-10-49. In addition, AI-10-49 has the key properties of a high-quality chemical probe recently outlined by Frye (30)–namely, a clear molecular profile of activity, mechanism of action, identity of active species, and proven utility. Development of agents, like AI-10-49, which can inhibit the driver mutation(s) in specific types of cancer, is essential for better therapeutic outcomes for patients.
In summary, AI-10-49 is a potent and specific first-generation CBFβ-SMMHC lead compound that induces cell death in inv(16) leukemic cells. The work described here provides additional evidence that transcription factor drivers of cancer can be directly targeted.
Supplementary Material
ACKNOWLEDGMENTS
We thank P. Bradley for assisting in the linker-length analyses in cell lines, R. Delwel for providing patient AML cells, and L. Zhu for performing the statistical analysis of mouse transplantation studies. This work was supported by grants from the National Cancer Institute, NIH (R01 CA140398), to J.H.B., L.H.C., and R.A.R.; (R01 CA096983) to L.H.C.; and a Specialized Center of Research grant from the Leukemia and Lymphoma Society to J.H.B. (SCOR 7006). L.H.C. is the recipient of a Scholar Award from the Leukemia & Lymphoma Society (grant 1334-08) and J.A.P. of a Scholar Award from the American Society of Hematology. M.L.G. is funded by the NIH through the NIH Director's New Innovator Award Program, 1 DP2 OD007399-01. J.H.B., A.I., and J.G. are coinventors on a U.S. patent (US8748618 B2) for AI-10-49 and related analogs. A patent application (10775546.4) has also been filed in Europe.
REFERENCES
- 1.Kumar CC. Genes Cancer. 2011;2:95–107. doi: 10.1177/1947601911408076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu P, et al. Science. 1993;261:1041–1044. doi: 10.1126/science.8351518. [DOI] [PubMed] [Google Scholar]
- 3.Castilla LH, et al. Nat. Genet. 1999;23:144–146. doi: 10.1038/13776. [DOI] [PubMed] [Google Scholar]
- 4.Castilla LH, et al. Proc. Natl. Acad. Sci. U.S.A. 2004;101:4924–4929. doi: 10.1073/pnas.0400930101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ravandi F, Burnett AK, Agura ED, Kantarjian HM. Cancer. 2007;110:1900–1910. doi: 10.1002/cncr.23000. [DOI] [PubMed] [Google Scholar]
- 6.Adya N, Castilla LH, Liu PP. Semin. Cell Dev. Biol. 2000;11:361–368. doi: 10.1006/scdb.2000.0189. [DOI] [PubMed] [Google Scholar]
- 7.de Bruijn MF, Speck NA. Oncogene. 2004;23:4238–4248. doi: 10.1038/sj.onc.1207763. [DOI] [PubMed] [Google Scholar]
- 8.Lukasik SM, et al. Nat. Struct. Biol. 2002;9:674–679. doi: 10.1038/nsb831. [DOI] [PubMed] [Google Scholar]
- 9.Kuo YH, et al. Cancer Cell. 2006;9:57–68. doi: 10.1016/j.ccr.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 10.Kottaridis PD, et al. Blood. 2002;100:2393–2398. doi: 10.1182/blood-2002-02-0420. [DOI] [PubMed] [Google Scholar]
- 11.Shih LY, et al. Leukemia. 2008;22:303–307. doi: 10.1038/sj.leu.2404995. [DOI] [PubMed] [Google Scholar]
- 12.Nakano Y, et al. Br. J. Haematol. 1999;104:659–664. doi: 10.1046/j.1365-2141.1999.01256.x. [DOI] [PubMed] [Google Scholar]
- 13.Gorczynski MJ, et al. Chem. Biol. 2007;14:1186–1197. doi: 10.1016/j.chembiol.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 14.Heilman SA, Kuo YH, Goudswaard CS, Valk PJ, Castilla LH. Cancer Res. 2006;66:11214–11218. doi: 10.1158/0008-5472.CAN-06-0959. [DOI] [PubMed] [Google Scholar]
- 15.Huang X, Peng JW, Speck NA, Bushweller JH. Nat. Struct. Biol. 1999;6:624–627. doi: 10.1038/10670. [DOI] [PubMed] [Google Scholar]
- 16.Mammen M, Choi SK, Whitesides GM. Angew. Chem. Int. Ed. 1998;37:2754–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 17.Kiessling LL, Gestwicki JE, Strong LE. Angew. Chem. Int. Ed. Engl. 2006;45:2348–2368. doi: 10.1002/anie.200502794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leroux FR, Manteau B, Vors JP, Pazenok S. Beilstein J. Org. Chem. 2008;4:13. doi: 10.3762/bjoc.4.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manteau B, Pazenok S, Vors JP, Leroux FR. J. Fluor. Chem. 2010;131:140–158. [Google Scholar]
- 20.Cheng CK, et al. Blood. 2008;112:3391–3402. doi: 10.1182/blood-2008-02-137083. [DOI] [PubMed] [Google Scholar]
- 21.Guo H, Ma O, Speck NA, Friedman AD. Blood. 2012;119:4408–4418. doi: 10.1182/blood-2011-12-397091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang DE, et al. Mol. Cell. Biol. 1994;14:8085–8095. doi: 10.1128/mcb.14.12.8085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cao W, et al. Oncogene. 1997;15:1315–1327. doi: 10.1038/sj.onc.1201305. [DOI] [PubMed] [Google Scholar]
- 24.Markus J, et al. Cancer Res. 2007;67:992–1000. doi: 10.1158/0008-5472.CAN-06-2964. [DOI] [PubMed] [Google Scholar]
- 25.Haferlach C, et al. Leukemia. 2010;24:1065–1069. doi: 10.1038/leu.2010.22. [DOI] [PubMed] [Google Scholar]
- 26.Xue L, Pulikkan JA, Valk PJ, Castilla LH. Blood. 2014;124:426–436. doi: 10.1182/blood-2013-12-541730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Look AT. Science. 1997;278:1059–1064. doi: 10.1126/science.278.5340.1059. [DOI] [PubMed] [Google Scholar]
- 28.Ladanyi M. Diagn. Mol. Pathol. 1995;4:162–173. doi: 10.1097/00019606-199509000-00003. [DOI] [PubMed] [Google Scholar]
- 29.Hessels D, Schalken JA. Curr. Urol. Rep. 2013;14:214–222. doi: 10.1007/s11934-013-0321-1. [DOI] [PubMed] [Google Scholar]
- 30.Frye SV. Nat. Chem. Biol. 2010;6:159–161. doi: 10.1038/nchembio.296. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.