

Abstract
Pendred syndrome (PS) is characterized by autosomal recessive inheritance of goiter associated with a defect of iodide organification, hearing loss, enlargement of the vestibular aqueduct (EVA), and mutations of the SLC26A4 gene. However, not all EVA patients have PS or SLC26A4 mutations. Two mutant alleles of SLC26A4 are detected in ¼ of North American or European EVA populations, one mutant allele is detected in another ¼ of patient populations, and no mutations are detected in the other ½. The presence of two mutant alleles of SLC26A4 is associated with abnormal iodide organification, increased thyroid gland volume, increased severity of hearing loss, and bilateral EVA. The presence of a single mutant allele of SLC26A4 is associated with normal iodide organification, normal thyroid gland volume, less severe hearing loss and either bilateral or unilateral EVA. When other underlying correlations are accounted for, the presence of a cochlear malformation or the size of EVA does not have an effect on hearing thresholds. This is consistent with observations of an Slc26a4 mutant mouse model of EVA in which hearing loss is independent of endolymphatic hydrops or inner ear malformations. Segregation analyses of EVA in families suggest that the patients carrying one mutant allele of SLC26A4 have a second, undetected mutant allele of SLC26A4, and the probability of a sibling having EVA is consistent with its segregation as an autosomal recessive trait. Patients without any mutations are an etiologically heterogeneous group in which siblings have a lower probability of having EVA. SLC26A4 mutation testing can provide prognostic information to guide clinical surveillance and management, as well as the probability of EVA affecting a sibling.
Keywords: SLC26A4, Pendred syndrome, Genetic testing, Goiter, Hearing loss, Vestibular aqueduct, Genotype-phenotype correlation
Pendred Syndrome and Nonsyndromic Hearing Loss With Enlargement of the Vestibular Aqueduct
Pendred syndrome (PS) is characterized by autosomal recessive inheritance of goiter and hearing loss, first reported in two sisters by Pendred1 in 1896. Fraser2 estimated this syndrome accounted for 5.6% of congenital hearing loss in his series of 2355 children. The causative gene for PS was mapped to chromosome 7q in 19963 and identified as SLC26A4 in 19974. Molecular testing for SLC26A4 mutations and temporal bone imaging have established that PS is always accompanied by inner ear deformities, with enlargement of the vestibular aqueduct (EVA) as the most penetrant feature5 (Figure 1). The identification of SLC26A4 mutations associated with PS suggested a possible association of nonsyndromic hearing loss with EVA (NSEVA) with mutations of this gene. Usami et al6 identified SLC26A4 mutations in sporadic and familial cases of NSEVA, showing that SLC26A4 mutations are commonly associated with NSEVA. These observations were confirmed in numerous studies of large cohorts of PS and NSEVA patients from different ethnic populations7-11.
Figure 1. Schematic illustration of the relationship of the vestibular aqueduct with the endolymphatic sac and duct.
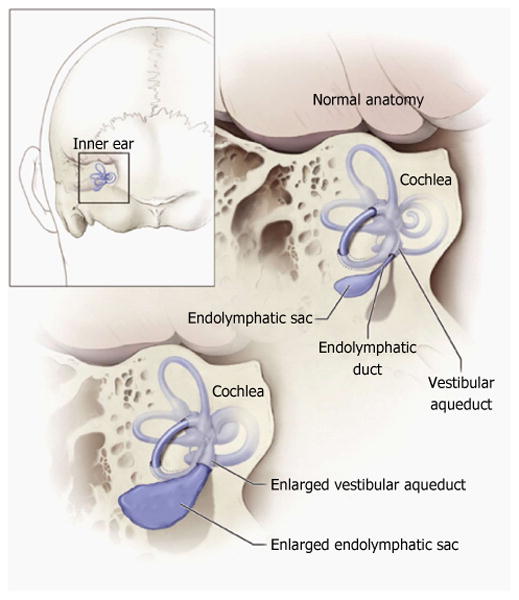
Normal anatomy of the inner ear structures is shown above. Pathologic enlargement of the endolymphatic sac and abnormal enlargement of the vestibular aqueduct are shown below. Some ears with enlargement of the vestibular aqueduct also have a reduced number of cochlear turns. Reproduced from http://www.nidcd.nih.gov/health/hearing/vestAque.htm.
Most clinicians now rely upon molecular testing of SLC26A4 for the etiologic diagnosis of PS and NSEVA. There are over 200 reported mutations in SLC26A4 associated with sporadic and familial forms of PS and NSEVA. Furthermore, a large-scale study demonstrated mutations of SLC26A4 in approximately 5%-10% of individuals with childhood deafness among several large global populations12. This percentage is coincident with Fraser's phenotypic estimate of the prevalence of PS2. However, in North American and European populations, SLC26A4 mutations cannot be detected in up to one half of patients with hearing loss and EVA, while only one mutant SLC26A4 allele is identified in one fourth of patients9-11,13. EVA has also been detected in a subset of patients with branchio-oto-renal or branchio-oto syndrome14, Waardenburg syndrome15, and deafness associated with the recessive form of distal renal tubular acidosis16. However, there is no published evidence that mutations of the genes underlying these syndromes cause PS or NSEVA.
SLC26A4 encodes a transmembrane protein, called pendrin, comprised of 780 amino acids and 12 or more predicted membrane-spanning domains4,17-21. Mouse Slc26a4 is expressed in a restricted tissue distribution that includes the inner ear, thyroid, kidney, lung, and several other organs4. Pendrin has been shown to exchange anions across the plasma membrane in several heterologous expression systems. Physiologically predominant functional modes are thought to include Cl-/I- exchange in the thyroid22 and Cl-/HCO3- exchange in the inner ear23. This anion exchange activity is critical during late embryonic and early postnatal development of the inner ear24. A variety of cellular details of the pathogenic events have been described25-28. Here we summarize the clinical phenotypes, genetics, and a novel mouse model of EVA.
Correlation of SLC26A4 Genotype with Thyroid Phenotype
The pathogenesis of goiter in PS is thought to be a thyroidal iodine organification defect29. The goiter tends to be diffuse at first, but later becomes nodular2. The organification defect can be detected by measuring the discharge of inorganic radioiodide from the thyroid after administration of potassium perchlorate. Potassium perchlorate is a competitive inhibitor of the sodium-iodide symporter, which transports iodide into thyroid folliculocytes across their basolateral membrane. An abnormally high discharge of iodide from the thyroid gland in response to perchlorate administration is a relatively specific finding for the clinical diagnosis of PS. For decades, it was the gold standard for the diagnosis of PS. Goiter, an abnormal perchlorate discharge, or both is identified in one third to one fourth of patients with hearing loss and EVA30,31. Goiter is an incompletely penetrant feature of PS. Furthermore, an onset during adolescence is typical2,32. The distinction between PS and NSEVA can therefore be difficult to make during childhood. This problem is exacerbated by the insensitivity of the physical examination for detection of goiter. While ultrasound examination with volume determinations may be helpful, normal gland size varies with age, and volume determinations have typically not been reported in a normalized fashion. In addition, goiter of other etiologies is common in some regions and populations, leading to phenocopies that increase the potential for misdiagnosis33.
SLC26A4 mutations are responsible for both PS and some cases of NSEVA, which suggested a possible correlation between particular types of mutations and the presence of the goiter8,34. Scott et al7 concluded that normal thyroid function in NSEVA patients is the consequence of residual pendrin activity encoded by hypofunctional SLC26A4 variants as compared to functional null alleles in PS patients. However, subsequent studies of cohorts with EVA and hearing loss failed to support this hypothesis8,35. Alternatively, a correlation between clinical phenotype and the number of mutant alleles of SLC26A4 has been suggested. With a definition of PS as > 15% discharge of iodide 2 to 3 h after administration of perchlorate, there was strong correlation between PS and the presence of two (M2) mutant SLC26A4 alleles, while NSEVA was associated with either one (M1) or zero (M0) mutant alleles9,10. Moreover, a multivariate analysis concluded that thyroid gland volume is primarily dependent on the presence of two mutant alleles of SLC26A4, at least in pediatric (< 10 years old) EVA patients30.
Correlation of SLC26A4 Genotype with Auditory Phenotype
Radiologically detectable inner ear deformities are often considered to be pathologic changes that contribute directly to congenital deafness. Inner ear deformities were first reported by Mondini36 in a temporal bone histopathological study in 1791. For centuries afterwards, the term “Mondini dysplasia” was often used for any inner ear malformation. Over many years, the classification and interpretation of inner ear anomalies, especially cochlear deformities, were based on a linear developmental model in which a developmental arrest occurred during embryogenesis37,38.
However, certain observations do not support the developmental arrest model for all inner ear malformations. The vestibular aqueduct (VA) is a narrow bony canal that opens onto the medial surface of the temporal bone and contains the endolymphatic sac and duct (Figure 1). The VA continues to grow throughout fetal life, but does not reach its full mature size before birth39. Some temporal bone studies indicate that the VA continues to grow postnatally in size until 3 years of age40,41. These observations were inconsistent with the hypothesis of arrested development38. Kim et al42 reported that EVA and scala media expansion occurred at embryonic day 14 in the Slc26a4-null mouse model. Their model postulated that enlargement depends on disruption of the normal balance between endolymph secretion and absorption in the labyrinth and endolymphatic sac. They speculated that lumen enlargement might be a form of hydrops caused by increased endolymphatic osmotic pressure due to impaired resorptive ion transport. This observation suggested that a developmental distortion, as well as arrest, occurs during fetal embryogenesis, thus explaining the concomitance of EVA and Mondini dysplasia.
EVA in humans is conventionally defined as a VA diameter exceeding 1.5 mm, measured at the midpoint between the common crus and external aperture (Figure 2). This original radiologic criterion was proposed by Valvassori et al43 in 1978. A recent study demonstrated that 1.0 mm is a more sensitive criterion for EVA44. EVA can occur as an isolated anomaly, as well as in combination with other inner ear deformities45,46. Inner ear deformities have been detected in 20%-30% of patients with congenital deafness46-49. EVA is the most common inner ear deformity, recognized in approximately 5%-15% of ears of deaf children49-52.
Figure 2. Right temporal bone of a patient with enlargement of the vestibular aqueduct.
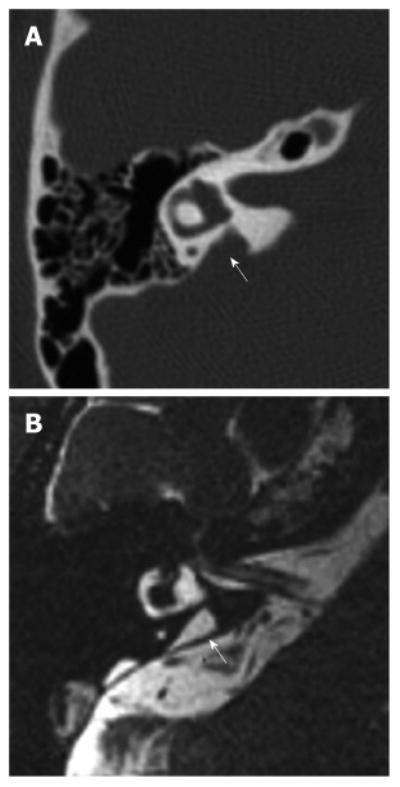
A: Axial computer tomography image of a right temporal bone with an enlarged vestibular aqueduct (arrow); B: Equivalent magnetic resonance image of the same temporal bone showing an enlarged endolymphatic duct (arrow). Reproduced from http://www.nidcd.nih.gov/health/hearing/Pages/eva.aspx.
No significant association has been reported between the type or number of mutant alleles of SLC26A4 and the presence of cochlear anomalies10,45,53. In contrast, two mutant alleles of SLC26A4 (M2) are tightly correlated with bilateral EVA, while unilateral EVA is correlated with only one (M1) or zero (M0) mutant allele of SLC26A49. Unilateral EVA is found with and without other inner ear deformities and is two to six times less frequent than bilateral EVA in North American and European populations34,54-56.
The hearing loss associated with SLC26A4 mutations is predominantly sensorineural or mixed, asymmetric, with an onset in the first few years of life. The degree of hearing loss can vary from mild to profound31,38,45,55. The hearing loss often shows fluctuation and overall downward progression that can be precipitated by minor head trauma or barotrauma. Hearing loss progression has been observed in 36%-88% of ears and fluctuation has been observed in 30%-92% of ears associated with SLC26A4 mutations10,31,45. Almost one half of the ears with fluctuating hearing loss eventually showed overall progressive loss of hearing. Even in the ears with normal to moderate hearing loss, hearing loss could progress at the rate of about 1 dB/year, with no apparent effect of environment factors45.
No significant relationship has been reported between the degree of hearing loss and the type of mutation or the presence of cochlear deformities, whereas the degree of hearing loss associates significantly with the number of mutant alleles of SLC26A49,10,45,55. The presence of two mutant alleles (M2) is associated with more severe hearing loss than only one (M1) or zero (M0) mutant alleles. Most reports have failed to reveal significant effects of number of mutant alleles of SLC26A4 or the presence or absence of cochlear anomalies on longitudinal hearing10,45. The degree of hearing loss does not correlate with the degree of enlargement of the VA or its contents, the endolymphatic duct45,57. This strongly suggests that endolymphatic hydrops is not a direct cause of hearing loss. Although others have reported potential correlations of radiologic findings with hearing loss phenotypes58, these conclusions were based upon univariate analyses that did not account for underlying factors and correlations such as SLC26A4 genotype, age, and other genetic diagnoses.
Pathogenesis of Hearing Loss Associated with Eva
Although hearing loss is often sensorineural, bone conduction threshold testing can reveal a mixed (conductive plus sensorineural) hearing loss at low frequencies associated with normal tympanometry and middle ear findings59-62, and an abnormal vestibular evoked myogenic potential result63. These findings are thought to be due to a “third window” effect upon sound transmission within the labyrinth64.
The pathogenesis of sensorineural component in hearing loss ears with EVA has been enigmatic. It was initially believed that trauma or barotrauma increases intracranial pressure with reflux of the contents of the endolymphatic sac and duct into the scala media through the enlarged endolymphatic duct. However, there is little evidence to support this theory, as obliteration of the endolymphatic sac and duct does not reverse or even prevent further hearing loss in patients with EVA38. It has also been suggested that sudden drops of hearing might be caused by rupture of Reissner's membrane38, hemorrhage in the endolymphatic sac65 or a fistulous round window membrane66. There may be occasional examples of these pathogenic mechanisms, but recent research indicates that the underlying mechanism is more often attributable to an intrinsic disruption of endolymphatic homeostasis.
Studies of an Slc26a4-null mouse model suggested scala media expansion and endolymphatic acidosis are early consequences of a lack of pendrin expression67,68. Subsequently, oxidative stress, abnormal cell stretching, impaired cell-to-cell communication, and loss of KCNJ10 expression occur in the stria vascularis, associated with a reduced endocochlear potential (EP) and hearing loss23,69-71.
Slc26a4 is expressed in multiple non-sensory cell populations of the cochlea, vestibular labyrinth, and endolymphatic sac and duct70,72,73. The Foxi1 gene encodes a fork-head transcription factor74, which regulates transcription of Slc26a4 in the endolymphatic sac and duct75-77, but not in the cochlea or vestibular labyrinth. The observation of EVA and deafness in a Foxi1-null mouse, in which pendrin is expressed in the cochlea and vestibular labyrinth but not in the endolymphatic sac, suggested that pendrin expression in the endolymphatic sac is essential for the acquisition of normal hearing75.
Slc26a4- and Foxi1-null mice are profoundly deaf with severe inner ear malformations and degenerative changes that do not model the less severe human phenotype. Choi et al24 reported a binary transgenic mouse line with doxycycline-inducible pendrin expression, in which pendrin expression during embryonic day 16.5 to postnatal day 2 was necessary and sufficient to acquire normal hearing at 1 mo of age. Lack of pendrin during this period could lead to endolymphatic acidification, loss of the EP and mild to severe hearing loss, even without significant scala media expansion or EVA. The timing of pendrin expression could be manipulated to generate mice with unilateral or asymmetric hearing loss associated with minimal, if any, EVA and no other morphogenetic anomalies (Figure 3). Since this latter model more closely approximated the human phenotype, endolymphatic acidification appears to be more important than scala media expansion for the pathogenesis of hearing loss. Although there are no histopathological specimens from patients with isolated EVA to corroborate these observations in mouse models, it seems doubtful that endolymphatic hydrops plays a direct causative role in the hearing loss associated with EVA78.
Figure 3. Morphology of the endolymphatic sac and duct and vestibular aqueduct in Slc26a4 mutant mouse models of enlargement of the vestibular aqueduct.
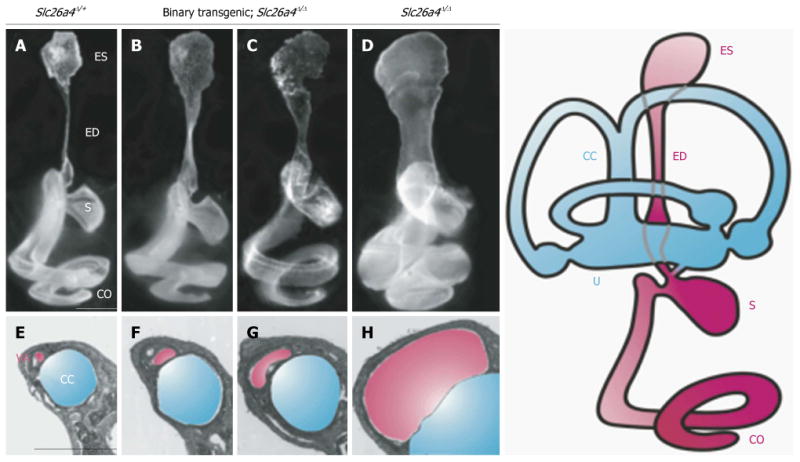
Slc26a4Δ/+ normal control (A and E), binary transgenic; Slc26a4Δ/Δ (B, C, F and G), or Slc26a4Δ/Δ mutant control mice (D and H) were sacrificed at P3 for paint-fill analysis (A-D) or between P28 and P109 for cross-sectional histopathology of the vestibular aqueduct (VA, shaded pink) adjacent to the common crus (CC, shaded blue; E–H). Scale bars: 500 μm (A, applies to A–D; E, applies to E–H). Manipulating pendrin expression in binary transgenic; Slc26a4Δ/Δ mice results in less enlargement of the endolymphatic duct and sac and vestibular aqueduct (B, C, F and G). ES: Endolymphatic sac; ED: Endolymphatic duct; S: Saccule; U: Utricle; CO: Cochlea. Reproduced with modification from Choi et al24.
Etiology of Eva in Patients with Non-Diagnostic SLC26A4 Genotypes
A single mutant allele of SLC26A4 is unlikely to be sufficient to cause hearing loss and EVA. There are no published reports of vertical co-segregation of EVA with a single mutant allele of SLC26A4 or of sporadic cases associated with a single de novo mutant allele of SLC26A49. To elucidate the genetic causes and recurrence probability of EVA in families of probands with non-diagnostic SLC26A4 genotypes (M1 or M0), Choi et al79 compared segregation ratios of EVA in M1 and M0 families with M2 families. A segregation ratio is a measure of the frequency of the phenotype among a proband's siblings and, thus, provides an estimate of recurrence probability in siblings. The segregation ratio of EVA in M1 families was not significantly different from that in M2 families, consistent with the predicted ratio (25%) for an autosomal recessive trait with full penetrance and viability. The results suggested the existence of a second, undetected SLC26A4 mutation in the M1 families79. It is also possible that a single pathogenic mutation of SLC26A4 might cause EVA in combination with a mutation in another gene9. Yang et al80 described digenic heterozygosity for mutations of SLC26A4 and FOXI177 or KCNJ10 in EVA patients. However, these results have not been reproduced in other studies of EVA cohorts54,81-83 and the pathogenic potential of FOXI1 and KCNJ10 variants thus remains undetermined84,85. Furthermore, SLC26A4-linked polymorphic DNA markers co-segregated with EVA in M1 families. This result is consistent with the hypothesis that current mutation analyses are failing to detect mutations that affect SLC26A4 or its expression on the apparently wild type allele of SLC26A4 in M1 families. Taken together, the data suggest that there is a second, undetected mutation of SLC26A4 that alters a promoter or enhancer or creates a cryptic splice site within an intron. Alternatively, epigenetic modifications of SLC26A4 such as DNA methylation might repress transcription86 and account for the observed co-segregation of EVA and SLC26A4 in M1 families. The correlation of the absence of goiter, and less severe inner ear deformities and hearing loss with M1 genotypes may reflect undetected mutant or epigenetically-modified alleles of SLC26A4 that act as hypomorphic alleles with residual function79, in a tissue - or time-specific manner24, or a combination of these mechanisms.
In M0 families, the segregation ratio was significantly lower than in M2 families and there was discordant inheritance of SLC26A4-linked DNA markers with EVA. These results suggested etiologic heterogeneity that includes environmental causes, mutations in other genes, or a combination of these factors79. Congenital cytomegalovirus (CMV) infection can produce a very similar auditory phenotype to that associated with EVA87. However, congenital CMV infection was ruled out as a common or significant cause of EVA88.
Genetic Testing for Eva
Most patients want to know the cause of their hearing loss and have a positive attitude toward genetic testing89-91. Genetic testing for SLC26A4 mutations can provide useful information for EVA patients. In some families, it may alleviate parental anxiety or guilt about the cause of hearing loss in their children. Second, it can guide the decision to longitudinally monitor the thyroid gland for enlargement or dysfunction. Third, it can be used to estimate the severity of hearing loss10,45,55. Fourth, it provides data for genetic counseling about recurrence probability, and the relative likelihood that EVA would be unilateral or bilateral if it does affect a sibling.
Assuming full viability and full penetrance of EVA in persons with two mutant alleles of SLC26A4, the probability of EVA in the sibling of an M2 EVA proband is 25%. Similarly, the probability of EVA in a sibling of a heterozygous (M1) proband with hearing loss and EVA is statistically indistinguishable from that for a sibling of an M2 proband79. The probability of EVA in a sibling of an M0 proband is significantly less than that for a sibling of an M1 or M2 proband, although the probability (about 11%) is not zero79. In the NIH cohort of EVA subjects, when EVA was observed in M0 sibling pairs, the siblings were often monozygotic or dizygotic twins. It is not clear if this reflects ascertainment bias or a relationship of twinning with the development of EVA.
We conclude that genetic testing for SLC26A4 mutations can be beneficial for some patients with EVA. However, it should always follow pre-test counseling so that patients and parents understand what testing can and cannot reveal. Pre-test counseling should also include a discussion of potential risks, including the possibility that testing may reveal unexpected biological relationships, implied carrier status in relatives, or potential insurance or employment discrimination. It is rare for otolaryngologists to have the time and expertise to conduct pre- and post-test counseling for genetic testing. A genetic counselor can provide pre- and post-test counseling, as well as educate the patient and family about genetics and inheritance. Genetic counselors can also collect pedigree and medical information90,91.
Future Directions
The advent of massively parallel DNA sequencing (also known as “next-generation” DNA sequencing) provides clinicians and researchers with the ability to sequence entire genomes or entire coding regions of genomes (also known as “exomes”). This opportunity also presents a challenge: the interpretation of DNA sequence variants of unknown pathogenicity. In the absence of conclusive genetic evidence linking mutations of genes other than SLC26A4 to EVA, direct Sanger di-deoxy sequencing of SLC26A4 currently remains the most efficient and reliable routine diagnostic test for the etiology of EVA. In the future, research should be directed toward identifying or confirming other genetic causes of EVA. Another avenue of research is to identify the etiologic, probably genetic, co-factors that cause EVA in patients with one detectable mutant allele of SLC26A4.
Conclusion
Genetic testing for SLC26A4 mutations in patients with hearing loss associated with EVA can provide useful information for establishing the etiology of the hearing loss, prognosis, clinical surveillance and management of the thyroid gland, and counseling families about the probability of EVA in one or both ears and severity of hearing loss in siblings of patients with EVA. The most informative aspect of an SLC26A4 genotype is the number of mutant alleles, since this shows the strongest correlation with the severity of hearing loss, laterality (unilateral vs bilateral) of EVA, thyroid gland volume, and recurrence probability. Patients with two mutant alleles of SLC26A4 typically have bilateral EVA, more severe hearing loss, a thyroid iodide organification defect associated with increased thyroid gland volume, and a 25% recurrence probability of EVA for each sibling. Patients with one mutant allele have unilateral or bilateral EVA, less severe hearing loss, on average, in the ear(s) with EVA, a normal thyroid gland, and a recurrence probability that is similar to that of patients with two mutant alleles. Patients with no mutations of SLC26A4 have thyroid and auditory phenotypes that are indistinguishable from those in patients with one mutant allele, but the probability of EVA in their siblings is much lower. Therefore even a “negative” SLC26A4 mutation test result can provide useful diagnostic, prognostic, and familial recurrence information.
Acknowledgments
We thank Thomas B Friedman and Inna A Belyantseva for critical review of the manuscript.
Supported by NIH intramural research funds Z01-DC-000039, Z01-DC-000060 and Z01-DC-000064, NIH grants R01-DK43495 and P30-DK34854, Kansas State University CVM-SMILE and the Kansas City Area Life Science Institute
Footnotes
Author contributions: Ito T and Griffith AJ reviewed the literature and wrote the initial draft of the manuscript; Muskett J, Chattaraj P, Choi BY, Lee KY, Zalewski CK, King KA, Li X, Wangemann P, Shawker T, Brewer CC and Alper SL critically reviewed and contributed to content and revision of the article.
Core tip: Enlargement of the vestibular aqueduct (EVA) is a common inner ear anomaly. We review the correlation of phenotype with genotype of SLC26A4. SLC26A4 mutations are the most prevalent known cause of hearing loss associated with EVA. The number of mutated alleles is correlated with the presence or absence of a thyroid iodination defect, thyroid gland volume, severity of hearing loss, laterality (bilateral vs unilateral) of the inner ear anomaly, and probability of recurrence of EVA in a sibling. We discuss the risks and benefits of genetic testing and counseling for affected patients. These concepts may be of broad interest to otolaryngologists, audiologists and other clinicians.
Contributor Information
Taku Ito, Otolaryngology Branch, National Institute on Deafness and Other Communication Disorders, National Institutes of Health, Rockville, MD 20850, United States.
Julie Muskett, Otolaryngology Branch, National Institute on Deafness and Other Communication Disorders, National Institutes of Health, Rockville, MD 20850, United States.
Parna Chattaraj, Otolaryngology Branch, National Institute on Deafness and Other Communication Disorders, National Institutes of Health, Rockville, MD 20850, United States.
Byung Yoon Choi, Laboratory of Molecular Genetics, National Institute on Deafness and Other Communication Disorders, National Institutes of Health, Rockville, MD 20850, United States.
Kyu Yup Lee, Otolaryngology Branch, National Institute on Deafness and Other Communication Disorders, National Institutes of Health, Rockville, MD 20850, United States.
Christopher K Zalewski, Otolaryngology Branch, National Institute on Deafness and Other Communication Disorders, National Institutes of Health, Rockville, MD 20850, United States.
Kelly A King, Otolaryngology Branch, National Institute on Deafness and Other Communication Disorders, National Institutes of Health, Rockville, MD 20850, United States.
Xiangming Li, Anatomy and Physiology Department, Kansas State University, Manhattan, KS 66506, United States.
Philine Wangemann, Anatomy and Physiology Department, Kansas State University, Manhattan, KS 66506, United States.
Thomas Shawker, Diagnostic Radiology Department, Warren G. Magnuson Clinical Center, National Institutes of Health, Bethesda, MD 20892, United States.
Carmen C Brewer, Otolaryngology Branch, National Institute on Deafness and Other Communication Disorders, National Institutes of Health, Rockville, MD 20850, United States.
Seth L Alper, Renal Division, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA 02215, United States.
Andrew J Griffith, Otolaryngology Branch, National Institute on Deafness and Other Communication Disorders, National Institutes of Health, Rockville, MD 20850, United States.
References
- 1.Pendred V. Deaf-mutism and goiter. Lancet. 1896;2:532. [Google Scholar]
- 2.Fraser GR. Association of congenital deafness with goitre (Pendred's syndrome) a study of 207 families. Ann Hum Genet. 1965;28:201–249. doi: 10.1111/j.1469-1809.1964.tb00479.x. [DOI] [PubMed] [Google Scholar]
- 3.Coyle B, Coffey R, Armour JA, Gausden E, Hochberg Z, Grossman A, Britton K, Pembrey M, Reardon W, Trembath R. Pendred syndrome (goitre and sensorineural hearing loss) maps to chromosome 7 in the region containing the nonsyndromic deafness gene DFNB4. Nat Genet. 1996;12:421–423. doi: 10.1038/ng0496-421. [DOI] [PubMed] [Google Scholar]
- 4.Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, Adawi F, Hazani E, Nassir E, Baxevanis AD, Sheffield VC, Green ED. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS) Nat Genet. 1997;17:411–422. doi: 10.1038/ng1297-411. [DOI] [PubMed] [Google Scholar]
- 5.Phelps PD, Coffey RA, Trembath RC, Luxon LM, Grossman AB, Britton KE, Kendall-Taylor P, Graham JM, Cadge BC, Stephens SG, Pembrey ME, Reardon W. Radiological malformations of the ear in Pendred syndrome. Clin Radiol. 1998;53:268–273. doi: 10.1016/S0009-9260(98)80125-6. [DOI] [PubMed] [Google Scholar]
- 6.Usami S, Abe S, Weston MD, Shinkawa H, Van Camp G, Kimberling WJ. Non-syndromic hearing loss associated with enlarged vestibular aqueduct is caused by PDS mutations. Hum Genet. 1999;104:188–192. doi: 10.1007/s004390050933. [DOI] [PubMed] [Google Scholar]
- 7.Scott DA, Wang R, Kreman TM, Andrews M, McDonald JM, Bishop JR, Smith RJ, Karniski LP, Sheffield VC. Functional differences of the PDS gene product are associated with phenotypic variation in patients with Pendred syndrome and non-syndromic hearing loss (DFNB4) Hum Mol Genet. 2000;9:1709–1715. doi: 10.1093/hmg/9.11.1709. [DOI] [PubMed] [Google Scholar]
- 8.Tsukamoto K, Suzuki H, Harada D, Namba A, Abe S, Usami S. Distribution and frequencies of PDS (SLC26A4) mutations in Pendred syndrome and nonsyndromic hearing loss associated with enlarged vestibular aqueduct: a unique spectrum of mutations in Japanese. Eur J Hum Genet. 2003;11:916–922. doi: 10.1038/sj.ejhg.5201073. [DOI] [PubMed] [Google Scholar]
- 9.Pryor SP, Madeo AC, Reynolds JC, Sarlis NJ, Arnos KS, Nance WE, Yang Y, Zalewski CK, Brewer CC, Butman JA, Griffith AJ. SLC26A4/PDS genotype-phenotype correlation in hearing loss with enlargement of the vestibular aqueduct (EVA): evidence that Pendred syndrome and non-syndromic EVA are distinct clinical and genetic entities. J Med Genet. 2005;42:159–165. doi: 10.1136/jmg.2004.024208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Albert S, Blons H, Jonard L, Feldmann D, Chauvin P, Loundon N, Sergent-Allaoui A, Houang M, Joannard A, Schmerber S, Delobel B, Leman J, Journel H, Catros H, Dollfus H, Eliot MM, David A, Calais C, Drouin-Garraud V, Obstoy MF, Tran Ba Huy P, Lacombe D, Duriez F, Francannet C, Bitoun P, Petit C, Garabédian EN, Couderc R, Marlin S, Denoyelle F. SLC26A4 gene is frequently involved in nonsyndromic hearing impairment with enlarged vestibular aqueduct in Caucasian populations. Eur J Hum Genet. 2006;14:773–779. doi: 10.1038/sj.ejhg.5201611. [DOI] [PubMed] [Google Scholar]
- 11.Campbell C, Cucci RA, Prasad S, Green GE, Edeal JB, Galer CE, Karniski LP, Sheffield VC, Smith RJ. Pendred syndrome, DFNB4, and PDS/SLC26A4 identification of eight novel mutations and possible genotype-phenotype correlations. Hum Mutat. 2001;17:403–411. doi: 10.1002/humu.1116. [DOI] [PubMed] [Google Scholar]
- 12.Park HJ, Shaukat S, Liu XZ, Hahn SH, Naz S, Ghosh M, Kim HN, Moon SK, Abe S, Tukamoto K, Riazuddin S, Kabra M, Erdenetungalag R, Radnaabazar J, Khan S, Pandya A, Usami SI, Nance WE, Wilcox ER, Riazuddin S, Griffith AJ. Origins and frequencies of SLC26A4 (PDS) mutations in east and south Asians: global implications for the epidemiology of deafness. J Med Genet. 2003;40:242–248. doi: 10.1136/jmg.40.4.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coyle B, Reardon W, Herbrick JA, Tsui LC, Gausden E, Lee J, Coffey R, Grueters A, Grossman4 A, Phelps PD, Luxon L, Kendall-Taylor P, Scherer SW, Trembath RC. Molecular analysis of the PDS gene in Pendred syndrome. Hum Mol Genet. 1998;7:1105–1112. doi: 10.1093/hmg/7.7.1105. [DOI] [PubMed] [Google Scholar]
- 14.Chen A, Francis M, Ni L, Cremers CW, Kimberling WJ, Sato Y, Phelps PD, Bellman SC, Wagner MJ, Pembrey M. Phenotypic manifestations of branchio-oto-renal syndrome. Am J Med Genet. 1995;58:365–370. doi: 10.1002/ajmg.1320580413. [DOI] [PubMed] [Google Scholar]
- 15.Madden C, Halsted MJ, Hopkin RJ, Choo DI, Benton C, Greinwald JH. Temporal bone abnormalities associated with hearing loss in Waardenburg syndrome. Laryngoscope. 2003;113:2035–2041. doi: 10.1097/00005537-200311000-00034. [DOI] [PubMed] [Google Scholar]
- 16.Berrettini S, Neri E, Forli F, Panconi M, Massimetti M, Ravecca F, Sellari-Franceschini S, Bartolozzi C. Large vestibular aqueduct in distal renal tubular acidosis. High-resolution MR in three cases. Acta Radiol. 2001;42:320–322. doi: 10.1080/028418501127346710. [DOI] [PubMed] [Google Scholar]
- 17.Dossena S, Rodighiero S, Vezzoli V, Nofziger C, Salvioni E, Boccazzi M, Grabmayer E, Bottà G, Meyer G, Fugazzola L, Beck-Peccoz P, Paulmichl M. Functional characterization of wild-type and mutated pendrin (SLC26A4), the anion transporter involved in Pendred syndrome. J Mol Endocrinol. 2009;43:93–103. doi: 10.1677/JME-08-0175. [DOI] [PubMed] [Google Scholar]
- 18.Ohana E, Shcheynikov N, Yang D, So I, Muallem S. Determinants of coupled transport and uncoupled current by the electrogenic SLC26 transporters. J Gen Physiol. 2011;137:239–251. doi: 10.1085/jgp.201010531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gillam MP, Sidhaye AR, Lee EJ, Rutishauser J, Stephan CW, Kopp P. Functional characterization of pendrin in a polarized cell system. Evidence for pendrin-mediated apical iodide efflux. J Biol Chem. 2004;279:13004–13010. doi: 10.1074/jbc.M313648200. [DOI] [PubMed] [Google Scholar]
- 20.Royaux IE, Suzuki K, Mori A, Katoh R, Everett LA, Kohn LD, Green ED. Pendrin, the protein encoded by the Pendred syndrome gene (PDS), is an apical porter of iodide in the thyroid and is regulated by thyroglobulin in FRTL-5 cells. Endocrinology. 2000;141:839–845. doi: 10.1210/endo.141.2.7303. [DOI] [PubMed] [Google Scholar]
- 21.Alper SL, Sharma AK. The SLC26 gene family of anion transporters and channels. Mol Aspects Med. 2013;34:494–515. doi: 10.1016/j.mam.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scott DA, Wang R, Kreman TM, Sheffield VC, Karniski LP. The Pendred syndrome gene encodes a chloride-iodide transport protein. Nat Genet. 1999;21:440–443. doi: 10.1038/7783. [DOI] [PubMed] [Google Scholar]
- 23.Wangemann P, Nakaya K, Wu T, Maganti RJ, Itza EM, Sanneman JD, Harbidge DG, Billings S, Marcus DC. Loss of cochlear HCO3- secretion causes deafness via endolymphatic acidification and inhibition of Ca2+ reabsorption in a Pendred syndrome mouse model. Am J Physiol Renal Physiol. 2007;292:F1345–F1353. doi: 10.1152/ajprenal.00487.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi BY, Kim HM, Ito T, Lee KY, Li X, Monahan K, Wen Y, Wilson E, Kurima K, Saunders TL, Petralia RS, Wangemann P, Friedman TB, Griffith AJ. Mouse model of enlarged vestibular aqueducts defines temporal requirement of Slc26a4 expression for hearing acquisition. J Clin Invest. 2011;121:4516–4525. doi: 10.1172/JCI59353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Griffith AJ, Wangemann P. Hearing loss associated with enlargement of the vestibular aqueduct: mechanistic insights from clinical phenotypes, genotypes, and mouse models. Hear Res. 2011;281:11–17. doi: 10.1016/j.heares.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wangemann P. The role of pendrin in the development of the murine inner ear. Cell Physiol Biochem. 2011;28:527–534. doi: 10.1159/000335113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dossena S, Nofziger C, Tamma G, Bernardinelli E, Vanoni S, Nowak C, Grabmayer E, Kössler S, Stephan S, Patsch W, Paulmichl M. Molecular and functional characterization of human pendrin and its allelic variants. Cell Physiol Biochem. 2011;28:451–466. doi: 10.1159/000335107. [DOI] [PubMed] [Google Scholar]
- 28.Dror AA, Brownstein Z, Avraham KB. Integration of human and mouse genetics reveals pendrin function in hearing and deafness. Cell Physiol Biochem. 2011;28:535–544. doi: 10.1159/000335163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morgans ME, Trotter WR. Association of congenital deafness with goitre; the nature of the thyroid defect. Lancet. 1958;1:607–609. doi: 10.1016/s0140-6736(58)90866-3. [DOI] [PubMed] [Google Scholar]
- 30.Madeo AC, Manichaikul A, Reynolds JC, Sarlis NJ, Pryor SP, Shawker TH, Griffith AJ. Evaluation of the thyroid in patients with hearing loss and enlarged vestibular aqueducts. Arch Otolaryngol Head Neck Surg. 2009;135:670–676. doi: 10.1001/archoto.2009.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suzuki H, Oshima A, Tsukamoto K, Abe S, Kumakawa K, Nagai K, Satoh H, Kanda Y, Iwasaki S, Usami S. Clinical characteristics and genotype-phenotype correlation of hearing loss patients with SLC26A4 mutations. Acta Otolaryngol. 2007;127:1292–1297. doi: 10.1080/00016480701258739. [DOI] [PubMed] [Google Scholar]
- 32.Reardon W, Coffey R, Chowdhury T, Grossman A, Jan H, Britton K, Kendall-Taylor P, Trembath R. Prevalence, age of onset, and natural history of thyroid disease in Pendred syndrome. J Med Genet. 1999;36:595–598. [PMC free article] [PubMed] [Google Scholar]
- 33.Kopp P, Arseven OK, Sabacan L, Kotlar T, Dupuis J, Cavaliere H, Santos CL, Jameson JL, Medeiros-Neto G. Phenocopies for deafness and goiter development in a large inbred Brazilian kindred with Pendred's syndrome associated with a novel mutation in the PDS gene. J Clin Endocrinol Metab. 1999;84:336–341. doi: 10.1210/jcem.84.1.5398. [DOI] [PubMed] [Google Scholar]
- 34.Azaiez H, Yang T, Prasad S, Sorensen JL, Nishimura CJ, Kimberling WJ, Smith RJ. Genotype-phenotype correlations for SLC26A4-related deafness. Hum Genet. 2007;122:451–457. doi: 10.1007/s00439-007-0415-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.López-Bigas N, Melchionda S, de Cid R, Grifa A, Zelante L, Govea N, Arbonés ML, Gasparini P, Estivill X. Identification of five new mutations of PDS/SLC26A4 in Mediterranean families with hearing impairment. Hum Mutat. 2001;18:548. doi: 10.1002/humu.1238. [DOI] [PubMed] [Google Scholar]
- 36.Mondini C, Hartley GJ, Phelps PD. Minor Works of Carlo Mondini: The Anatomical Section of a Boy Born Deafa: Opuscula Caroli Mundini: Anatomica Surdi Nati Sectio: Carolus Mundinus. Am J Otol. 1997;18:288–293. [PubMed] [Google Scholar]
- 37.Jackler RK, Luxford WM, House WF. Congenital malformations of the inner ear: a classification based on embryogenesis. Laryngoscope. 1987;97:2–14. doi: 10.1002/lary.5540971301. [DOI] [PubMed] [Google Scholar]
- 38.Jackler RK, De La Cruz A. The large vestibular aqueduct syndrome. Laryngoscope. 1989;99:1238–1242. doi: 10.1288/00005537-198912000-00006. discussion 1242-1243. [DOI] [PubMed] [Google Scholar]
- 39.Pyle GM. Embryological development and large vestibular aqueduct syndrome. Laryngoscope. 2000;110:1837–1842. doi: 10.1097/00005537-200011000-00014. [DOI] [PubMed] [Google Scholar]
- 40.Kodama A, Sando I. Postnatal development of the vestibular aqueduct and endolymphatic sac. Ann Otol Rhinol Laryngol Suppl. 1982;96:3–12. [PubMed] [Google Scholar]
- 41.Fujita S, Sando I. Postnatal development of the vestibular aqueduct in relation to the internal auditory canal. Computer-aided three-dimensional reconstruction and measurement study. Ann Otol Rhinol Laryngol. 1994;103:719–722. doi: 10.1177/000348949410300910. [DOI] [PubMed] [Google Scholar]
- 42.Kim HM, Wangemann P. Failure of fluid absorption in the endolymphatic sac initiates cochlear enlargement that leads to deafness in mice lacking pendrin expression. PLoS One. 2010;5:e14041. doi: 10.1371/journal.pone.0014041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valvassori GE, Clemis JD. The large vestibular aqueduct syndrome. Laryngoscope. 1978;88:723–728. doi: 10.1002/lary.1978.88.5.723. [DOI] [PubMed] [Google Scholar]
- 44.Boston M, Halsted M, Meinzen-Derr J, Bean J, Vijayasekaran S, Arjmand E, Choo D, Benton C, Greinwald J. The large vestibular aqueduct: a new definition based on audiologic and computed tomography correlation. Otolaryngol Head Neck Surg. 2007;136:972–977. doi: 10.1016/j.otohns.2006.12.011. [DOI] [PubMed] [Google Scholar]
- 45.King KA, Choi BY, Zalewski C, Madeo AC, Manichaikul A, Pryor SP, Ferruggiaro A, Eisenman D, Kim HJ, Niparko J, Thomsen J, Butman JA, Griffith AJ, Brewer CC. SLC26A4 genotype, but not cochlear radiologic structure, is correlated with hearing loss in ears with an enlarged vestibular aqueduct. Laryngoscope. 2010;120:384–389. doi: 10.1002/lary.20722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu CC, Chen YS, Chen PJ, Hsu CJ. Common clinical features of children with enlarged vestibular aqueduct and Mondini dysplasia. Laryngoscope. 2005;115:132–137. doi: 10.1097/01.mlg.0000150691.85387.3f. [DOI] [PubMed] [Google Scholar]
- 47.Jensen J. Malformations of the inner ear in deaf children. A tomographic and clinical study. Acta Radiol Diagn (Stockh) 1968;(Suppl 286):3+. [PubMed] [Google Scholar]
- 48.Mafong DD, Shin EJ, Lalwani AK. Use of laboratory evaluation and radiologic imaging in the diagnostic evaluation of children with sensorineural hearing loss. Laryngoscope. 2002;112:1–7. doi: 10.1097/00005537-200201000-00001. [DOI] [PubMed] [Google Scholar]
- 49.Antonelli PJ, Varela AE, Mancuso AA. Diagnostic yield of high-resolution computed tomography for pediatric sensorineural hearing loss. Laryngoscope. 1999;109:1642–1647. doi: 10.1097/00005537-199910000-00018. [DOI] [PubMed] [Google Scholar]
- 50.Okumura T, Takahashi H, Honjo I, Takagi A, Mitamura K. Sensorineural hearing loss in patients with large vestibular aqueduct. Laryngoscope. 1995;105:289–293. doi: 10.1288/00005537-199503000-00012. [DOI] [PubMed] [Google Scholar]
- 51.Arcand P, Desrosiers M, Dubé J, Abela A. The large vestibular aqueduct syndrome and sensorineural hearing loss in the pediatric population. J Otolaryngol. 1991;20:247–250. [PubMed] [Google Scholar]
- 52.Madden C, Halsted M, Benton C, Greinwald J, Choo D. Enlarged vestibular aqueduct syndrome in the pediatric population. Otol Neurotol. 2003;24:625–632. doi: 10.1097/00129492-200307000-00016. [DOI] [PubMed] [Google Scholar]
- 53.Wu CC, Yeh TH, Chen PJ, Hsu CJ. Prevalent SLC26A4 mutations in patients with enlarged vestibular aqueduct and/or Mondini dysplasia: a unique spectrum of mutations in Taiwan, including a frequent founder mutation. Laryngoscope. 2005;115:1060–1064. doi: 10.1097/01.MLG.0000163339.61909.D0. [DOI] [PubMed] [Google Scholar]
- 54.Jonard L, Niasme-Grare M, Bonnet C, Feldmann D, Rouillon I, Loundon N, Calais C, Catros H, David A, Dollfus H, Drouin-Garraud V, Duriez F, Eliot MM, Fellmann F, Francannet C, Gilbert-Dussardier B, Gohler C, Goizet C, Journel H, Mom T, Thuillier-Obstoy MF, Couderc R, Garabédian EN, Denoyelle F, Marlin S. Screening of SLC26A4, FOXI1 and KCNJ10 genes in unilateral hearing impairment with ipsilateral enlarged vestibular aqueduct. Int J Pediatr Otorhinolaryngol. 2010;74:1049–1053. doi: 10.1016/j.ijporl.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 55.Madden C, Halsted M, Meinzen-Derr J, Bardo D, Boston M, Arjmand E, Nishimura C, Yang T, Benton C, Das V, Smith R, Choo D, Greinwald J. The influence of mutations in the SLC26A4 gene on the temporal bone in a population with enlarged vestibular aqueduct. Arch Otolaryngol Head Neck Surg. 2007;133:162–168. doi: 10.1001/archotol.133.2.162. [DOI] [PubMed] [Google Scholar]
- 56.Mori T, Westerberg BD, Atashband S, Kozak FK. Natural history of hearing loss in children with enlarged vestibular aqueduct syndrome. J Otolaryngol Head Neck Surg. 2008;37:112–118. [PubMed] [Google Scholar]
- 57.Griffith AJ, Arts A, Downs C, Innis JW, Shepard NT, Sheldon S, Gebarski SS. Familial large vestibular aqueduct syndrome. Laryngoscope. 1996;106:960–965. doi: 10.1097/00005537-199608000-00009. [DOI] [PubMed] [Google Scholar]
- 58.Campbell AP, Adunka OF, Zhou B, Qaqish BF, Buchman CA. Large vestibular aqueduct syndrome: anatomic and functional parameters. Laryngoscope. 2011;121:352–357. doi: 10.1002/lary.21278. [DOI] [PubMed] [Google Scholar]
- 59.Arjmand EM, Webber A. Audiometric findings in children with a large vestibular aqueduct. Arch Otolaryngol Head Neck Surg. 2004;130:1169–1174. doi: 10.1001/archotol.130.10.1169. [DOI] [PubMed] [Google Scholar]
- 60.Govaerts PJ, Casselman J, Daemers K, De Ceulaer G, Somers T, Offeciers FE. Audiological findings in large vestibular aqueduct syndrome. Int J Pediatr Otorhinolaryngol. 1999;51:157–164. doi: 10.1016/s0165-5876(99)00268-2. [DOI] [PubMed] [Google Scholar]
- 61.Nakashima T, Ueda H, Furuhashi A, Sato E, Asahi K, Naganawa S, Beppu R. Air-bone gap and resonant frequency in large vestibular aqueduct syndrome. Am J Otol. 2000;21:671–674. [PubMed] [Google Scholar]
- 62.Sato E, Nakashima T, Lilly DJ, Fausti SA, Ueda H, Misawa H, Uchida Y, Furuhashi A, Asahi K, Naganawa S. Tympanometric findings in patients with enlarged vestibular aqueducts. Laryngoscope. 2002;112:1642–1646. doi: 10.1097/00005537-200209000-00021. [DOI] [PubMed] [Google Scholar]
- 63.Zhou G, Gopen Q, Kenna MA. Delineating the hearing loss in children with enlarged vestibular aqueduct. Laryngoscope. 2008;118:2062–2066. doi: 10.1097/MLG.0b013e31818208ad. [DOI] [PubMed] [Google Scholar]
- 64.Merchant SN, Nakajima HH, Halpin C, Nadol JB, Lee DJ, Innis WP, Curtin H, Rosowski JJ. Clinical investigation and mechanism of air-bone gaps in large vestibular aqueduct syndrome. Ann Otol Rhinol Laryngol. 2007;116:532–541. doi: 10.1177/000348940711600709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim M, Kim J, Kim SH, Kim SC, Jeon JH, Lee WS, Kim UK, Kim HN, Choi JY. Hemorrhage in the endolymphatic sac: a cause of hearing fluctuation in enlarged vestibular aqueduct. Int J Pediatr Otorhinolaryngol. 2011;75:1538–1544. doi: 10.1016/j.ijporl.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 66.Belenky WM, Madgy DN, Leider JS, Becker CJ, Hotaling AJ. The enlarged vestibular aqueduct syndrome (EVA syndrome) Ear Nose Throat J. 1993;72:746–751. [PubMed] [Google Scholar]
- 67.Kim HM, Wangemann P. Epithelial cell stretching and luminal acidification lead to a retarded development of stria vascularis and deafness in mice lacking pendrin. PLoS One. 2011;6:e17949. doi: 10.1371/journal.pone.0017949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Everett LA, Belyantseva IA, Noben-Trauth K, Cantos R, Chen A, Thakkar SI, Hoogstraten-Miller SL, Kachar B, Wu DK, Green ED. Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Hum Mol Genet. 2001;10:153–161. doi: 10.1093/hmg/10.2.153. [DOI] [PubMed] [Google Scholar]
- 69.Singh R, Wangemann P. Free radical stress-mediated loss of Kcnj10 protein expression in stria vascularis contributes to deafness in Pendred syndrome mouse model. Am J Physiol Renal Physiol. 2008;294:F139–F148. doi: 10.1152/ajprenal.00433.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Royaux IE, Belyantseva IA, Wu T, Kachar B, Everett LA, Marcus DC, Green ED. Localization and functional studies of pendrin in the mouse inner ear provide insight about the etiology of deafness in pendred syndrome. J Assoc Res Otolaryngol. 2003;4:394–404. doi: 10.1007/s10162-002-3052-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wangemann P, Kim HM, Billings S, Nakaya K, Li X, Singh R, Sharlin DS, Forrest D, Marcus DC, Fong P. Developmental delays consistent with cochlear hypothyroidism contribute to failure to develop hearing in mice lacking Slc26a4/pendrin expression. Am J Physiol Renal Physiol. 2009;297:F1435–F1447. doi: 10.1152/ajprenal.00011.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Everett LA, Morsli H, Wu DK, Green ED. Expression pattern of the mouse ortholog of the Pendred's syndrome gene (Pds) suggests a key role for pendrin in the inner ear. Proc Natl Acad Sci USA. 1999;96:9727–9732. doi: 10.1073/pnas.96.17.9727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wangemann P, Itza EM, Albrecht B, Wu T, Jabba SV, Maganti RJ, Lee JH, Everett LA, Wall SM, Royaux IE, Green ED, Marcus DC. Loss of KCNJ10 protein expression abolishes endocochlear potential and causes deafness in Pendred syndrome mouse model. BMC Med. 2004;2:30. doi: 10.1186/1741-7015-2-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pierrou S, Hellqvist M, Samuelsson L, Enerbäck S, Carlsson P. Cloning and characterization of seven human forkhead proteins: binding site specificity and DNA bending. EMBO J. 1994;13:5002–5012. doi: 10.1002/j.1460-2075.1994.tb06827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hulander M, Kiernan AE, Blomqvist SR, Carlsson P, Samuelsson EJ, Johansson BR, Steel KP, Enerbäck S. Lack of pendrin expression leads to deafness and expansion of the endolymphatic compartment in inner ears of Foxi1 null mutant mice. Development. 2003;130:2013–2025. doi: 10.1242/dev.00376. [DOI] [PubMed] [Google Scholar]
- 76.Hulander M, Wurst W, Carlsson P, Enerbäck S. The winged helix transcription factor Fkh10 is required for normal development of the inner ear. Nat Genet. 1998;20:374–376. doi: 10.1038/3850. [DOI] [PubMed] [Google Scholar]
- 77.Yang T, Vidarsson H, Rodrigo-Blomqvist S, Rosengren SS, Enerback S, Smith RJ. Transcriptional control of SLC26A4 is involved in Pendred syndrome and nonsyndromic enlargement of vestibular aqueduct (DFNB4) Am J Hum Genet. 2007;80:1055–1063. doi: 10.1086/518314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Merchant SN, Adams JC, Nadol JB. Pathophysiology of Meniere's syndrome: are symptoms caused by endolymphatic hydrops? Otol Neurotol. 2005;26:74–81. doi: 10.1097/00129492-200501000-00013. [DOI] [PubMed] [Google Scholar]
- 79.Choi BY, Madeo AC, King KA, Zalewski CK, Pryor SP, Muskett JA, Nance WE, Butman JA, Brewer CC, Griffith AJ. Segregation of enlarged vestibular aqueducts in families with non-diagnostic SLC26A4 genotypes. J Med Genet. 2009;46:856–861. doi: 10.1136/jmg.2009.067892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang T, Gurrola JG, Wu H, Chiu SM, Wangemann P, Snyder PM, Smith RJ. Mutations of KCNJ10 together with mutations of SLC26A4 cause digenic nonsyndromic hearing loss associated with enlarged vestibular aqueduct syndrome. Am J Hum Genet. 2009;84:651–657. doi: 10.1016/j.ajhg.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mercer S, Mutton P, Dahl HH. Identification of SLC26A4 mutations in patients with hearing loss and enlarged vestibular aqueduct using high-resolution melting curve analysis. Genet Test Mol Biomarkers. 2011;15:365–368. doi: 10.1089/gtmb.2010.0177. [DOI] [PubMed] [Google Scholar]
- 82.Wu CC, Lu YC, Chen PJ, Yeh PL, Su YN, Hwu WL, Hsu CJ. Phenotypic analyses and mutation screening of the SLC26A4 and FOXI1 genes in 101 Taiwanese families with bilateral nonsyndromic enlarged vestibular aqueduct (DFNB4) or Pendred syndrome. Audiol Neurootol. 2010;15:57–66. doi: 10.1159/000231567. [DOI] [PubMed] [Google Scholar]
- 83.Pera A, Villamar M, Viñuela A, Gandía M, Medà C, Moreno F, Hernández-Chico C. A mutational analysis of the SLC26A4 gene in Spanish hearing-impaired families provides new insights into the genetic causes of Pendred syndrome and DFNB4 hearing loss. Eur J Hum Genet. 2008;16:888–896. doi: 10.1038/ejhg.2008.30. [DOI] [PubMed] [Google Scholar]
- 84.Yang T, Smith RJ. The c.-103T>C variant in the 5′-UTR of SLC26A4 gene: a pathogenic mutation or coincidental polymorphism? Hum Mutat. 2009;30:1469–1470. doi: 10.1002/humu.21097. [DOI] [PubMed] [Google Scholar]
- 85.Choi BY, Alper SL, Griffith AJ. Response to: The c.-103T>C variant in the 5′-UTR of SLC26A4 gene: a pathogenic mutation or coincidental polymorphism? Hum Mutat. 2009;30:1471. doi: 10.1002/humu.21098. [DOI] [PubMed] [Google Scholar]
- 86.Xing M, Tokumaru Y, Wu G, Westra WB, Ladenson PW, Sidransky D. Hypermethylation of the Pendred syndrome gene SLC26A4 is an early event in thyroid tumorigenesis. Cancer Res. 2003;63:2312–2315. [PubMed] [Google Scholar]
- 87.Morton CC, Nance WE. Newborn hearing screening--a silent revolution. N Engl J Med. 2006;354:2151–2164. doi: 10.1056/NEJMra050700. [DOI] [PubMed] [Google Scholar]
- 88.Pryor SP, Demmler GJ, Madeo AC, Yang Y, Zalewski CK, Brewer CC, Butman JA, Fowler KB, Griffith AJ. Investigation of the role of congenital cytomegalovirus infection in the etiology of enlarged vestibular aqueducts. Arch Otolaryngol Head Neck Surg. 2005;131:388–392. doi: 10.1001/archotol.131.5.388. [DOI] [PubMed] [Google Scholar]
- 89.Abe S, Noguchi Y, Kitamura K. What do patients with hereditary deafness think of genetic studies? Auris Nasus Larynx. 2010;37:422–426. doi: 10.1016/j.anl.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 90.Brunger JW, Murray GS, O'Riordan M, Matthews AL, Smith RJ, Robin NH. Parental attitudes toward genetic testing for pediatric deafness. Am J Hum Genet. 2000;67:1621–1625. doi: 10.1086/316901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Withrow KA, Tracy KA, Burton SK, Norris VW, Maes HH, Arnos KS, Pandya A. Impact of genetic advances and testing for hearing loss: results from a national consumer survey. Am J Med Genet A. 2009;149A:1159–1168. doi: 10.1002/ajmg.a.32800. [DOI] [PubMed] [Google Scholar]