

Abstract
CRISPR/Cas systems mediate bacterial adaptive immune responses that evolved to protect bacteria from bacteriophage and other horizontally transmitted genetic elements. Several CRISPR/Cas systems exist but the simplest variant, referred to as Type II, has a single effector DNA endonuclease, called Cas9, which is guided to its viral DNA target by two small RNAs, the crRNA and the tracrRNA. Initial efforts to adapt the CRISPR/Cas system for DNA editing in mammalian cells, which focused on the Cas9 protein from Streptococcus pyogenes (Spy), demonstrated that Spy Cas9 can be directed to DNA targets in mammalian cells by tracrRNA:crRNA fusion transcripts called single guide RNAs (sgRNA). Upon binding, Cas9 induces DNA cleavage leading to mutagenesis as a result of error prone non-homologous end joining (NHEJ). Recently, the Spy Cas9 system has been adapted for high throughput screening of genes in human cells for their relevance to a particular phenotype and, more generally, for the targeted inactivation of specific genes, in cell lines and in vivo in a number of model organisms. The latter aim seems likely to be greatly enhanced by the recent development of Cas9 proteins from bacterial species such as Neisseria meningitidis and Staphyloccus aureus that are small enough to be expressed using adeno-associated (AAV)-based vectors that can be readily prepared at very high titers. The evolving Cas9-based DNA editing systems therefore appear likely to not only impact virology by allowing researchers to screen for human genes that affect the replication of pathogenic human viruses of all types but also to derive clonal human cell lines that lack individual gene products that either facilitate or restrict viral replication. Moreover, high titer AAV-based vectors offer the possibility of directly targeting DNA viruses that infect discrete sites in the human body, such as herpes simplex virus and hepatitis B virus, with the hope that the entire population of viral DNA genomes might be destroyed. In conclusion, we believe that the continued rapid evolution of CRISPR/Cas technology will soon have a major, possibly revolutionary, impact on the field of virology.
Keywords: CRISPR/Cas, Gene editing, Antiviral gene therapy, Sequence specific DNA cleavage
Introduction
Sequence specific DNA endonucleases were first identified in the 1960s as enzymes that restrict the ability of DNA bacteriophage to grow in particular bacterial isolates or species. Now called restriction endonucleases, these proteins recognize short sequences of DNA, often palindromic in nature, and cleave either within the recognition sequence or nearby to destroy the incoming bacteriophage DNA (Pingoud et al., 2014). The host DNA is protected by a modification enzyme that introduces a covalent modification of a base, for example methylation of an A residue, within the restriction enzyme recognition sequence that prevents restriction enzyme function. Incoming phage genomes that derive from bacteria lacking the relevant modification enzyme are in contrast highly susceptible to cleavage. While restriction endonucleases played a key role in the development of recombinant DNA research (Pingoud et al., 2014), even the longest restriction endonuclease recognition sequences are far too short to allow the specific, single site mutagenesis of large viral genomes or, particularly, the mammalian host cell genome.
The first technology to be developed with the potential to be useful in the selective modification of large viral or cellular DNA genomes was the zinc finger endonuclease (ZFN) (Gaj et al., 2013; Klug, 2010). Zinc fingers are ~30 amino acid long domains that have the potential to specifically recognize many, but not all, 3 base pair (bp) DNA sequences. Zinc fingers function in a modular manner and can therefore be arrayed to form a larger protein that can selectively bind DNA sequences up to 18 bp in length, thus opening up the possibility of binding unique sequences in the human genome. While ZFNs do not themselves cleave the bound DNA, they can be fused to a non- specific DNA endonuclease to allow DNA cleavage to occur (Gaj et al., 2013; Klug, 2010). The most common approach is to fuse the ZFN to the FokI endonuclease, which is only active upon formation of a FokI homodimer. By designing two ZFNs that target adjacent DNA sequences on opposite strands of the target genome, the binding of the two ZFNs can induce the dimerization of the fused FokI enzymes to generate an active dimer that cleaves precisely between the two binding sites. This dsDNA break is most commonly repaired by the error-prone non-homologous DNA end joining (NHEJ) pathway, which can result in small indels being introduced at the cleaved target site. Conversely, if NHEJ is precise, the target site will be reconstituted to allow a second round of cleavage, and this futile cycle will continue until a mutation is introduced that prevents ZFN recognition and further cleavage events.
While ZFNs have proven to be very useful for both mammalian cell DNA editing and inhibition of DNA virus replication, each zinc finger is not necessarily highly specific for the 3 bp target that it is designed to bind and zinc fingers specific for all possible 3 bp target sequences have not yet been derived (Gaj et al., 2013). To address this perceived deficiency, a second technology, transcription activator like endonucleases (TALENs) was developed, based on a distinct modular DNA binding motif. TALENs are made up of four ~34 aa domains, derived from a DNA binding protein found in the pathogenic plant bacterium Xanthomonas, that each recognize a single DNA base pair with high specificity (Christian et al., 2010; Gaj et al., 2013; Miller et al., 2011). As in the case of ZFNs, these modular domains can be arrayed to recognize essentially any DNA sequence. Again, the strategy is to construct two distinct DNA binding domains by sequential addition of single base pair-specific modules until a protein specific for the two chosen target sequences is derived. These target sequences are again chosen to be adjacent, on opposite DNA strands, and the two expressed TALENs again consist of fusions of these artificial DNA binding domains to the FokI endonuclease, to permit the highly specific cleavage of the DNA at a site located between the two TALEN recognition sequences (Christian et al., 2010; Miller et al., 2011). Each TALEN is generally designed to recognize an ~16 bp DNA sequence that is close to unique in the cell of interest and the TALEN heterodimer therefore recognizes an ~32 bp sequence, which is certainly large enough to ensure that no off-target cleavage events will occur. While perhaps more specific than ZFNs, TALENs require a rather laborious exercise in genetic engineering and are quite large, thus limiting the ability to express TALENs using viral vectors.
While ZFNs and TALENs remain useful reagents for targeted gene editing, it seems likely that they will in future be largely superseded by the newly developed CRISPR/Cas based systems, which are both highly specific and allow facile retargeting to new genomic loci (Barrangou and Marraffini, 2014; Cong et al., 2013; Hsu et al., 2014; Mali et al., 2013). Clustered regularly interspaced short palindromic repeats (CRISPR) loci are found in a wide range of bacteria and have now been shown to be transcribed to generate a family of targeting RNAs specific for a range of different DNA bacteriophage that can infect that bacterium (Barrangou and Marraffini, 2014; Hsu et al., 2014). In bacteria that express a type II CRISPR/Cas system, these phage-derived sequences are transcribed along with sequences from the adjacent constant region to give a CRISPR RNA (crRNA) which forms a complex with the invariant trans-activating RNA (tracrRNA), using sequence complementarity between the tracrRNA and the invariant part of the crRNA (Fig. 1). This heterodimer is then bound by the effector protein of the type II CRISPR/Cas systems, called Cas9. Cas9 has the ability to directly recognize a short DNA sequence, 5′-NGG-3′ for the commonly used Streptococcus pyogenes (Spy) Cas9 protein, called the protospacer adjacent motif (PAM) (Barrangou and Marraffini, 2014; Cong et al., 2013; Hsu et al., 2014; Mali et al., 2013). The Cas9 protein scans a target genome for the PAM sequence and then binds and queries the DNA for full 5′ sequence complementarity to the variable part of the crRNA. If detected, the Cas9 protein directly cleaves both strands of the target bacteriophage DNA ~3 bp 50 to the PAM, using two distinct protein domains: the Cas9 RuvC-like domain cleaves the non-complementary strand, while the Cas9 HNH nuclease domain cleaves the complementary strand (Gasiunas et al., 2012; Jinek et al., 2012). This dsDNA break then induces the degradation of the phage DNA genome and blocks infection (Fig. 1) (Garneau et al., 2010; Gasiunas et al., 2012; Sapranauskas et al., 2011).
Fig. 1.

The cleavage cycle of Cas9 RNA-guided DNA endonucleases. Starting at the upper left, Cas9 first binds either a native tracrRNA/crRNA complex or a synthetic sgRNA. The protospacer, or targeting portion of the crRNA/sgRNA, is depicted in red, the repeat region in black, and the tracrRNA portion in blue. Note the added joining loop shown in gray in the case of the sgRNA. Next, the sgRNA-loaded Cas9 protein scans a dsDNA target for a PAM (lower left), and upon encountering one it initiates sgRNA-complementarity interrogation of the 5′ adjacent DNA sequence. If both full seed sequence complementarity and the PAM are present, dsDNA cleavage occurs. Subsequent to cleavage, non-homologous end joining (NHEJ) will at times faithfully repair the lesion resulting in restoration of a fully functional DNA target that can re-enter the cleavage cycle. Alternatively, the error-prone NHEJ process can lead to mutagenesis of the seed target region, resulting in the formation of a cleavage refractory DNA target bearing an indel.
A key step forward in making the Spy Cas9 system more user-friendly for genetic engineering in human cells was the demonstration that the crRNA and tracrRNA could be linked by an artificial loop sequence to generate a fully functional small guide RNA (sgRNA) ~100 nt in length (Fig. 1) (Cong et al., 2013; Mali et al., 2013). Further work, including mutational analysis of DNA targets, has revealed that sequence specificity for Spy Cas9 relies both on the PAM and on full complementarity to the 3′ ~13 nt of the ~20 nt variable region of the sgRNA, with more 5′ sequences making only a minor contribution (Fig. 1). Spy Cas9 therefore has an ~15 bp (13 bp in the guide and 2bp in the PAM) sequence specificity which, while high, is generally not sufficient to entirely avoid a small number of potential off-target cleavage sites in the large genome present in human cells. Nevertheless, this is a high level of specificity and a small number of off-targets in non-transcribed regions of the human genome appear unlikely to be highly problematic, especially if due diligence is devoted to bioinformatic analysis of potential off-target cleavage sites. Moreover, this concern can be dealt with by mutating the Cas9 protein to inactivate one of the two independent HNH and RuvC nuclease sites, to generate a so-called “nickase” (Cong et al., 2013; Ran et al., 2013). It is then possible to target two nickase Cas9s to two closely proximal (<20 bp) sites on the two strands of the DNA target. Once nicked on both strands, the DNA will fall apart to give a staggered dsDNA break, analogous to what is obtained upon cleavage at a single recognition sequence using wild-type Spy Cas9, except that the DNA target specificity is now ~30 bp, amply sufficient to ensure complete specificity even in a large genome, such that present in human cells. As in the case of cleavage by ZFNs or TALENs, this dsRNA break is repaired by NHEJ resulting in an indel, which blocks further cleavage, or resulting in perfect repair, which results in an additional round of cleavage by Cas9 (Fig. 1).
While the Spy CRISPR/Cas system allows the specific cleavage and mutagenesis of host cell genes, it can also be used for other purposes. If a single-stranded DNA template complementary to the cleavage site is provided in trans, homologous DNA repair is favored over NHEJ, allowing the introduction of site-specific mutations or, perhaps even more usefully, the repair of defective human genes linked to particular disease states (Cong et al., 2013; Schwank et al., 2013; Wu et al., 2013). Moreover, it is possible to mutate Cas9 so it is no longer capable of DNA cleavage, yet retains full DNA binding capacity (Qi et al., 2013). These cleavage incompetent Cas9 proteins can then be fused to various effector domains, for example transcriptional activation or repression domains, and targeted to the promoter regions of cellular genes that you wish to turn on or off (Barrangou and Marraffini, 2014; Hsu et al., 2014; Qi et al., 2013). While CRISPR/Cas systems will therefore no doubt have a dramatic effect on numerous aspects of biology and medicine, our focus here is on the specific DNA cleavage activity of Cas9/sgRNA combinations and our thoughts on how they will affect the future of virology research and also potentially the treatment of specific viral diseases.
Using CRISPR/Cas to inactivate or mutate DNA virus genomes
The idea of using sequence-specific DNA endonucleases to target and destroy DNA viruses has been around for some time (Schiffer et al., 2012), with earlier work describing the use of ZFNs, TALENs or a third type of endonuclease, called a homing endonuclease, to target hepatitis B virus (HBV), human papillomavirus (HPV) and herpes simplex virus type 1 (HSV-1) with varying degrees of success (Aubert et al., 2014; Bloom et al., 2013; Chen et al., 2014; Cradick et al., 2010; Grosse et al., 2011; Mino et al., 2006; Weber et al., 2014). This work has now been extended, in our laboratory and elsewhere, to the use of Spy-derived Cas9/sgRNA combinations, delivered by lentiviral vector transduction or transfection, to induce the mutational inactivation of the DNA genomes of HBV, HPV and HIV-1 (Ebina et al., 2013; Hu et al., 2014a; Hu et al., 2014b; Kennedy et al., In press; Kennedy et al., 2014; Lin et al., 2014; Zhen et al., 2014). In the case of HPV, the efficient mutational inactivation of the viral E6 gene, which normally functions to block the activity of the cellular tumor suppressor p53, results in activation of the p53 transcription factor and its downstream effectors, resulting in cell cycle arrest and the apoptotic death of HPV-transformed cells (Kennedy et al., 2014; Zhen et al., 2014). Similarly, in the case of HBV, Cas9/sgRNA combinations targeted to the HBV reverse transcriptase (RT), core or surface antigen genes result in a marked inhibition of viral protein expression and loss of viral DNA molecules, including the covalently closed circular DNA (cccDNA) molecules that play a critical role in HBV persistence in patients even in the face of treatment with nucleoside-based inhibitors of RT function (Kennedy et al., In press; Lin et al., 2014). Finally, expression of HIV-1 specific Cas9/sgRNA combinations has been shown to both block de novo infection, by cleaving the newly synthesized HIV-1 proviral DNA intermediate prior to integration into the host cell genome, and to eradicate a pre-existing latent infection by targeting sequences in the HIV-1 long terminal repeat (LTR), thus inducing the excision of the provirus from the genome of the infected cell, leaving behind a single LTR copy (Ebina et al., 2013; Hu et al., 2014a).
While impressive, these studies were all performed in cultured cells and can at best be considered proof-of-principle. In order to be feasible as an in vivo treatment approach, two problems need to be addressed. Firstly, although current data suggest that off-target cleavage by Spy Cas9 is minimal, it is not zero and the safety of this approach in vivo needs to be further demonstrated (Hsu et al., 2013; Kuscu et al., 2014; Wu et al., 2014). Additional animal studies, above and beyond the encouraging results already reported, therefore need to be performed. Even more importantly, it is clear that methods for the efficient delivery of Cas9/sgRNA combinations to infected cells in vivo need to be developed. Previous work has largely focused on transfection, which is not feasible in vivo, or lentiviral vectors, which are difficult to produce at titers much above 108 infectious units (IU) per ml. We therefore believe that CRISPR/Cas-based approaches to the in vivo treatment of DNA virus infections will require the development of vectors based on adeno-associated virus (AAV), which can generally be concentrated to titers of ≥ 1014 viral particles per ml, a level of vector that has the potential to transduce all virus-infected cells in a patient, especially if these are all found in a single location, e.g., in the liver or specific neurons (Kotterman and Schaffer, 2014). Moreover, AAV-based vectors have a well-established record of safety and do not integrate at significant levels into the target cell genome, thus avoiding the potential for insertional activation of deleterious genes (Kotterman and Schaffer, 2014).
The problem with this approach is that AAV vectors have a maximum packaging capacity of ~4.7kb, and this includes the AAV inverted terminal repeats, which together occupy ~290 bp, leaving only ~4.4 kb for heterologous DNA. As the Spy Cas9 gene, including an essential added nuclear localization signal (NLS), is ~4.2 kb in size, this does not leave enough room for a pol II promoter and poly (A) addition site for Cas9 expression as well as a pol III promoter and sgRNA sequence. One possible approach to resolve this problem is to express the Cas9 protein from one AAV vector and the sgRNA(s) from a second AAV, but the required two hit kinetics for function are not ideal, especially in vivo (Swiech et al., 2014). Rather, the way forward is clearly to use one of the many smaller Cas9 proteins encoded by other bacterial species. In particular, Neisseria meningitidis (Nme) encodes a Cas9 protein with the PAM sequence 5′-NNNNGATT-3′ while Staphylococcus aureus (Sau) encodes a Cas9 with the PAM sequence 5′-NNGRRT-3′, where R is purine (Esvelt et al., 2013; Hou et al., 2013; Zhang and Ran, 2014). Both proteins are encoded by genes ~3.2 kb in length, leaving ample room for two sgRNA cassettes, in addition to all required regulatory elements, in an AAV vector context (Fig. 2). In our hands, Sau Cas9 is at least as active, or possibly more active, than Spy Cas9 on the same DNA target sequence and the sequence specificity of Sau Cas9 appears to be comparable to Spy Cas9 (data not shown). Therefore, it now seems entirely feasible to move to animal experiments that are designed to test the efficacy and safety of AAV-based Cas9/sgRNA vector systems.
Fig. 2.
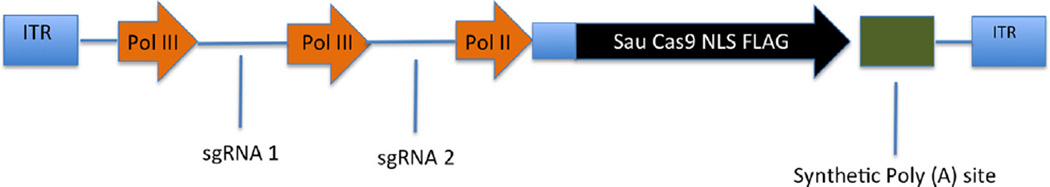
A potential AAV multiplex Cas9 delivery vector. The minimal Cas9 protein from S. aureus has a total coding size of ~3.2 kb, including an epitope tag and an NLS, and this shorter Cas9 enzyme therefore opens up substantial packaging space in AAV. Minimal Pol II and Pol III promoters and a synthetic Poly A sequence can be employed to minimize the nucleotide footprints of these cis-elements. In total, we have now generated small AAV-based CRISPR/Cas9 expression vectors that encode both the Sau Cas9 protein and two independent sgRNAs yet occupy <4.5 kb of packaging space, well below the AAV size limit of ~4.7 kb.
While a number of DNA viruses are potential targets for elimination using CRISPR/Cas, we will here focus on four viruses, i.e., HBV, HSV, HPV and HIV-1.
Hepatitis B virus
HBV remains a major public health problem, with over 300 million people chronically infected worldwide (Komatsu, 2014). These individuals have an ~25% risk of dying from the consequences of HBV infection, including hepatocellular carcinoma (HCC) and cirrhosis, and ~800,000 individuals are thought to die each year due to HBV. An excellent vaccine for HBV is available, but this is not helpful in individuals with pre-existing infection, is not fully effective at preventing vertical transmission and is not given to all children in resource limited contexts. As noted above, the HBV RT can be effectively inhibited by nucleoside-based drugs but this does not cure this infection due to the extraordinary stability of the viral episomal cccDNA intermediate (Werle-Lapostolle et al., 2004). Recent in vitro data demonstrate that HBV genomic DNA molecules, including the cccDNA, can be effectively cleaved and mutationally inactivated by Cas9/sgRNA combinations in cells undergoing either acute or chronic infections (Kennedy et al., In press) and preliminary evidence, using hydrodynamic injection to deliver both the HBV DNA and an HBV-specific Cas9/sgRNA combination to the mouse liver, is also consistent with the hypothesis that HBV-specific Cas9/sgRNA combinations can block HBV replication and eliminate the cccDNA pool if they can be effectively delivered to hepatocytes (Lin et al., 2014). AAV may be ideal for this task, as several AAV serotypes are naturally hepatotropic and even more highly hepatotropic AAV vectors have recently been isolated by “shuffling” AAV sequences in vivo (Lisowski et al., 2014). The next step is clearly to examine whether AAV-delivered Cas9/sgRNA combinations can effectively cure HBV in the humanized, immunodeficient mouse liver model system, the best available preclinical model for HBV replication in vivo (Dandri and Lutgehetmann, 2014).
Herpes simplex virus
HSV-1 infects ~70% of the U.S. population and about one third of affected individuals suffer from recurrent, primarily oral, cold sores. HSV-1 most commonly initially infects the oral mucosal epithelium, leading to a local productive infection, and then undergoes retrograde transport to the trigeminal ganglia, where it establishes a latent infection in a small number of sensory neurons that persists after the initial, productive infection is cleared by the host immune response (Roizman et al., 2007). During latency, the HSV-1 DNA genome is maintained as a nuclear episome, with from 1 to ~50 copies per latently infected neuron. At this point, the only region of the genome that is actively transcribed encodes the latency associated transcript LAT, which is processed to give rise to a single long non-coding RNA of ~2.1 kb, as well as 8 virally encoded miRNAs, that together are thought to regulate exit from latency (Umbach et al., 2008). Importantly, no viral proteins are made, thus preventing immune recognition of latently infected cells. Occasionally, generally after some form of stress, one or more latently infected neuron is activated to produce infectious virions that migrate down the axons of the reactivating neuron to the original site of infection, where they re-establish a transient productive infection that can lead to the formation of cold sores. While often no more than an irritation, HSV-1 infections can also lead to serious morbidity and HSV-1 keratitis represents the most common form of infectious blindness in the USA (Liesegang, 2001). A closely related virus, HSV-2, that is found in ~20% of the US population, has a similar replication cycle but generally is sexually transmitted and often infects the genital mucosa. In the case of HSV-2, latency is established in sensory neurons of the sacral ganglia and reactivation can lead to genital ulcers. Again, serious morbidity is rare but does occur in some individuals and neonatal HSV-2 infections acquired during vaginal delivery can be fatal (Roizman et al., 2007).
While there are several drugs that can treat productive HSV-1 or HSV-2 infections, generally by targeting viral DNA synthesis, latent HSV genomes are entirely refractory to current treatment regimens and it remains impossible to cure these infections. What is clearly needed is an approach that directly targets HSV-1 or HSV-2 episomal DNA for cleavage and elimination from latently infected neurons. AAV-delivered HSV-specific Cas9/sgRNA combinations appear ideal for this purpose (Chamberlin et al., 1998). In particular, several AAV serotypes, including AAV8, are able to infect almost all (> 95%) of the sensory neurons in the dorsal root ganglion after application of particles of a green fluorescent protein (gfp) expressing AAV8 vector to the rear footpad of the mouse (D. Bloom, personal communication). Latent HSV-1 infections of neurons in the mouse trigeminal ganglia (TGs) can be readily established and it is therefore possible to test whether transduction of these same TGs with AAV8-based vectors encoding HSV-1-specific Cas9/sgRNA combinations will result in a detectable reduction in viral DNA load and an inhibition in the ability of latent HSV-1 to reactivate after explant and culture of the infected TGs. Given the tight localization of HSV-1 and HSV-2 to the trigeminal and sacral ganglia respectively, given the low level of viral DNA genomes present in these cells, and given the ability to efficiently transduce sensory neurons with AAV8-based vectors, this seems like an ideal viral candidate for cure using CRISPR/Cas.
Human papilloma virus
Humans are infected by a wide variety of HPVs that, while normally innocuous, can also give rise to warts on the skin or genitalia (Howley and Lowry, 2007). Most HPV variants replicate as episomes in the basal epithelial layer of the skin, where the virus expresses exclusively non-structural proteins. When the infected precursor epithelial cell migrates towards the surface of the epidermis and undergoes differentiation into a keratinocyte, the productive HPV replication cycle is activated leading to the release of infectious HPV virions (Howley and Lowry, 2007).
As noted above, most HPVs are non-pathogenic yet there are also a small number of so-called high-risk HPV serotypes, of which the most prominent are HPV-16 and HPV-18, which together cause ~70% of all cervical cancers. In most HPV induced cancers, the HPV episome is found clonally integrated into the cell genome in a manner that destroys or deletes the viral E2 gene (Howley and Lowry, 2007). The role of the E2 protein is to bind to the HPV origin of replication, where it functions to ensure the distribution of HPV episomes to both daughter cells after cell division, and E2 also acts to regulate HPV early gene transcription. One key activity of E2 is to limit the expression of the HPV oncogenes E6 and E7, and disruption of E2 during integration into the host cell genome can lead to high, constitutive levels of E6 and E7 expression (Thierry and Howley, 1991). E6 functions to bind and destabilize the p53 tumor suppressor (Scheffner et al., 1990) while E7 similarly binds and destabilizes the Rb tumor suppressor (Mighty and Laimins, 2014) and these two functions play a critical role in the maintenance of HPV-transformed cells. Cancers associated with HPV infection include cervical carcinomas, which are almost always HPV-positive, as well as a substantial fraction of head and neck (H&N) cancers as well as anal cancers, all of which arise due to sexual transmission of HPV. While cervical cancers can be detected early using diagnostic tests and are generally readily cured by surgical intervention, this is not the case for H&N and anal cancers, which are usually treated by chemotherapy and radiation. While this is often successful, a substantial percentage of anal and H&N cancers recur locally, necessitating salvage surgery that can lead to significant morbidity. Therefore, a novel treatment regimen for chemoresistant HPV+ tumors would be highly desirable. Of note, almost all HPV+ H&N and anal cancers are HPV-16 positive, thus restricting the required sequence range for a possible HPV-specific AAV-based Cas9/sgRNA expression vector.
As noted above, both the HPV E6 and E7 proteins play a crucial role in HPV tumorigenesis by blocking the action of p53 and Rb, respectively (Mighty and Laimins, 2014). Consistent with this idea, inactivation of the E6 gene in the HPV-18+ cervical carcinoma cell line HeLa or the HPV-16+ cell line SiHa using Spy CRISPR/Cas has been found to result in induction of p53 expression followed by the expression of downstream targets of this cellular transcription factor, including the CDK inhibitor p21 and several activators of apoptosis, leading to cell cycle arrest and cell death (Kennedy et al., 2014; Zhen et al., 2014). Similarly, disruption of the E7 gene using CRISPR/Cas results in the increased expression of Rb, formation of Rb/E2F heterodimers and then the induction of cellular genes that induce senescence and cell death (Hu et al., 2014b; Kennedy et al., 2014). Therefore, the delivery of CRISPR/Cas combinations specific for HPV E6 and/or E7 by direct injection of high titer AAV vector preparations into the HPV+ tumors has the potential to serve as a novel, highly specific and effective therapy for chemoresistant HPV-16 induced anal and H&N tumors.
Human immunodeficiency virus
While highly active antiretroviral therapy (HAART) can reduce HIV-1 replication to levels below the detection limit, HIV-1 persists as a latent infection in a small number of resting CD4+ memory T cells (Chun et al., 1997). In these long lived cells, intact integrated HIV-1 proviruses persist in a transcriptionally silent state that is refractory to both drugs and host immune responses. However, these memory T cells can be reactivated by an appropriate recall antigen, resulting in induction of a productive viral replication cycle (Chun et al., 1997; Wong et al., 1997). If this occurs after drug treatment has been stopped, HIV-1 will rapidly spread through the available CD4+ T cells and rekindle the same level of virus replication that was seen prior to antiviral drug treatment.
Efforts to purge the pool of latently infected cells have focused on two strategies. On the one hand, several groups have attempted to activate latent HIV-1 proviruses using drugs, including histone deacetylase inhibitors and PKC agonists (Xing and Siliciano, 2013). However, so far this strategy has not proven able to activate HIV-1 in a high percentage of latently infected cells. An alternative strategy would be to directly target and destroy latent proviruses using HIV-1-specific CRISPR/Cas combinations and indeed two papers have shown that this is feasible and that latent proviruses can in fact be excised from the host cell genome, and then destroyed, by cleavage in the HIV-1 long terminal repeat regions (Ebina et al., 2013; Hu et al., 2014a). In principle, the HIV-1 provirus is a perfect target for CRISPR/Cas as there is only a single proviral copy in the infected cell and, in the presence of antiviral drugs, no spread of the virus is possible. The problem, however, is that latently HIV-1 infected T cells are scattered throughout the body and infecting all of these seems currently to be an insurmountable problem, especially as T cells are poor targets for AAV infection. This contrasts with HBV, HSV and HPV, all of which are tightly localized in known tissues in the body that can be readily targeted by AAV. Therefore, in the absence of novel vector delivery systems that can target latently HIV-1 infected cells throughout the body, HIV-1 is likely to remain a technically challenging target for elimination by CRISPR/Cas in vivo.
Other DNA viruses
A number of other DNA viruses are associated with serious human diseases including Epstein-Barr virus (EBV), Kaposi's sarcoma-associated herpesvirus (KSHV) and Merkel cell polyomavirus (MCPyV). Of these, perhaps the most relevant is EBV, which is the etiologic agent of several cancers, including an epithelial cell tumor called nasopharyngeal carcinoma (NPC) that is highly prevalent in southern China and Southeast Asia (Rickinson and Kieff, 2007). In NPC cells, EBV is found in a form of viral latency that nevertheless involves the expression of several viral nonstructural proteins and microRNAs (Tsao et al., 2012). EBV+ NPCs share a number of characteristics with HPV-16+ H&N cancers and, as in the latter case, the continued presence and transcription of the viral, in this case EBV, genome is thought to be essential for tumor survival. It has already been demonstrated that EBV episomes are readily disrupted and destroyed by specific Spy Cas9/sgRNA combinations (Wang and Quake, 2014) and it seems likely that NPC cells would be excellent targets for transduction in vivo using Sau Cas9/sgRNA-based AAV vectors specific for the EBV genome.
Using CRISPR/Cas to define cellular factors that facilitate or restrict virus replication
Viruses have very compact genomes and therefore rely on the host cell for many activities required to support their replication cycle. Conversely, viruses are pathogens and put selective pressure on their host organisms, thus selecting for cellular restriction factors that can limit the level of viral replication. One of the best studied viruses in this context is HIV-1, which requires a wide range of human co-factors for replication in CD4+ T cells, several of which are lacking in other mammalian species, such as in mice. Conversely, HIV-1 is targeted by a wide range of human restriction factors and dedicates a substantial portion of its coding capacity to the neutralization of these restriction factors. For example, the HIV-1 Vif protein blocks the activity of the host APOBEC3 family of restriction factors, which otherwise interfere with the production of the HIV-1 provirus, while the HIV-1 Vpu protein neutralizes the cellular restriction factor tetherin, which otherwise blocks the release of progeny virions (Malim and Bieniasz, 2012).
The identity of cellular factors that either facilitate or restrict virus replication is of great interest, not least because these can provide targets for antiviral drug development. One approach to their identification has been to use RNA interference (RNAi) to knock down the expression of human genes in order to identify factors required for virus replication. However, while this approach has led to some interesting insights, it is also clear that RNAi screens for viral cofactors in different laboratories have often led to very different lists of cellular proteins with this potential activity. The reasons for this are unclear but probably relate to the use of different cell systems, different assays for viral replication and different RNAi reagents. As a result, most of the viral co-factors and restriction factors identified so far have in fact been defined using biochemical approaches, for example by identification of cellular factors that specifically bind to a viral protein, or genetic approaches, for example by complementation of a human and/or animal cell line that lacks a particular co-factor or restriction factor (Malim and Bieniasz, 2012).
CRISPR/Cas systems appear highly suitable for use in screens for non-essential viral co-factors or restriction factors, and indeed, several screens for cellular factors involved in cell transformation have been published (Shalem et al., 2014; Wang et al., 2014; Zhou et al., 2014). This could now easily be extended to analysis of the replication potential of, for example, gfp+ viruses in a 96-well or 384-well plate format. More generally, it seems likely that there will soon be comprehensive CRISPR/Cas generated libraries of cellular clones available for specific human cell lines that each lack a human gene that is dispensable for cell viability in a tissue culture setting. These clones would represent defined reagents, unlike the transient RNAi knockdown cells used previously, that should allow the reproducible and almost complete identification of cellular factors that either help or hinder virus replication in that human cell line in vitro. Of course, factors required for human cell viability would be missed but it seems likely that almost all host innate immune factors involved in restricting virus replication would not be essential and, while certainly factors required for host cell viability are, by definition, also essential for virus replication, such housekeeping functions are a priori less interesting, especially as they clearly would not provide potential targets for anti-viral drug development. In conclusion, we believe that the development of effective CRISPR/Cas-based gene editing tools has the potential to lead to the global identification of almost all cellular factors that regulate virus replication in culture, leading to a wealth of new insights into viral molecular biology and producing numerous potential targets for antiviral drug development. If such screens can indeed identify cellular factors that are required for virus replication but entirely dispensable for the health of the adult target organism, then it might be possible to also treat viral infections, including RNA virus infections, via the localized ablation of a cellular gene encoding such an essential viral co-factor using AAV vector delivered CAS9/sgRNA combinations, as described above for direct targeting of DNA virus genomes. Indeed, a similar approach has previously been reported using ZFNs to inactivate the HIV-1 co-receptor CCR5, to prevent the infection of CD4+ T cells, by transduction of hematopoietic stem cells followed by analysis of the production of HIV-1-resistant CD4+ human T cells in engrafted immunodeficient mice (Holt et al., 2010; Li et al., 2013). Whether this latter approach is feasible in human patients is, of course, currently unknown, but this possibility does open up a range of potential new approaches to targeted intervention in severe viral infections.
Acknowledgments
Research in the Cullen laboratory is supported by grants R01- DA030086, R01-AI067968, R21-AI113098 and R01-AI097376 from the National Institutes of Health and by a small grant from the Duke Center for AIDS Research funded by NIH P30-AI064518. EMK is supported by a postdoctoral fellowship funded by F. Hoffmann-LaRoche Ltd.
References
- Aubert M, Boyle NM, Stone D, Stensland L, Huang ML, Magaret AS, Galetto R, Rawlings DJ, Scharenberg AM, Jerome KR. In vitro inactivation of latent HSV by targeted mutagenesis using an HSV-specific homing endonuclease. Mol. Ther. Nucleic Acids. 2014;3:e146. doi: 10.1038/mtna.2013.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou R, Marraffini LA. CRISPR-Cas systems: prokaryotes upgrade to adaptive immunity. Mol. Cell. 2014;54:234–244. doi: 10.1016/j.molcel.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom K, Ely A, Mussolino C, Cathomen T, Arbuthnot P. Inactivation of hepatitis B virus replication in cultured cells and in vivo with engineered transcription activator-like effector nucleases. Mol. Ther. 2013;21:1889–1897. doi: 10.1038/mt.2013.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlin NL, Du B, de Lacalle S, Saper CB. Recombinant adeno-associated virus vector: use for transgene expression and anterograde tract tracing in the CNS. Brain. Res. 1998;793:169–175. doi: 10.1016/s0006-8993(98)00169-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Zhang W, Lin J, Wang F, Wu M, Chen C, Zheng Y, Peng X, Li J, Yuan Z. An efficient antiviral strategy for targeting hepatitis B virus genome using transcription activator-like effector nucleases. Mol. Ther. 2014;22:303–311. doi: 10.1038/mt.2013.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, Bogdanove AJ, Voytas DF. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics. 2010;186:757–761. doi: 10.1534/genetics.110.120717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JA, Baseler M, Lloyd AL, Nowak MA, Fauci AS. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA. 1997;94:13193–13197. doi: 10.1073/pnas.94.24.13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cradick TJ, Keck K, Bradshaw S, Jamieson AC, McCaffrey AP. Zinc-finger nucleases as a novel therapeutic strategy for targeting hepatitis B virus DNAs. Mol. Ther. 2010;18:947–954. doi: 10.1038/mt.2010.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dandri M, Lutgehetmann M. Mouse models of hepatitis B and delta virus infection. J. Immunol. Methods. 2014;410C:39–49. doi: 10.1016/j.jim.2014.03.002. [DOI] [PubMed] [Google Scholar]
- Ebina H, Misawa N, Kanemura Y, Koyanagi Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci. Rep. 2013;3:2510. doi: 10.1038/srep02510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esvelt KM, Mali P, Braff JL, Moosburner M, Yaung SJ, Church GM. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat. Methods. 2013;10:1116–1121. doi: 10.1038/nmeth.2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaj T, Gersbach CA, Barbas CF., 3rd ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau JE, Dupuis ME, Villion M, Romero DA, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadan AH, Moineau S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010;468:67–71. doi: 10.1038/nature09523. [DOI] [PubMed] [Google Scholar]
- Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleo-protein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA. 2012;109:E2579–E2586. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse S, Huot N, Mahiet C, Arnould S, Barradeau S, Clerre DL, Chion-Sotinel I, Jacqmarcq C, Chapellier B, Ergani A, Desseaux C, Cedrone F, Conseiller E, Paques F, Labetoulle M, Smith J. Meganuclease-mediated inhibition of HSV1 infection in cultured cells. Mol. Ther. 2011;19:694–702. doi: 10.1038/mt.2010.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt N, Wang J, Kim K, Friedman G, Wang X, Taupin V, Crooks GM, Kohn DB, Gregory PD, Holmes MC, Cannon PM. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat. Biotechnol. 2010;28:839–847. doi: 10.1038/nbt.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Z, Zhang Y, Propson NE, Howden SE, Chu LF, Sontheimer EJ, Thomson JA. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc. Natl. Acad. Sci. USA. 2013;110:15644–15649. doi: 10.1073/pnas.1313587110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howley PM, Lowry DR. Papillomaviruses. In: Knipe DM, Howley PM, editors. Fields Virology. Fifth ed. Philadelphia: Lippincott Williams & Wilkins; 2007. pp. 2299–2354. [Google Scholar]
- Hsu PD, Lander ES, Zhang F. Development and applications of CRISPRCas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, Cradick TJ, Marraffini LA, Bao G, Zhang F. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Kaminski R, Yang F, Zhang Y, Cosentino L, Li F, Luo B, Alvarez-Carbonell D, Garcia-Mesa Y, Karn J, Mo X, Khalili K. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc. Natl. Acad. Sci. USA. 2014a;111:11461–11466. doi: 10.1073/pnas.1405186111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Yu L, Zhu D, Ding W, Wang X, Zhang C, Wang L, Jiang X, Shen H, He D, Li K, Xi L, Ma D, Wang H. Disruption of HPV16-E7 by CRISPR/Cas system induces apoptosis and growth inhibition in HPV16 positive human cervical cancer cells. Biomed. Res. Int. 2014b;2014:612823. doi: 10.1155/2014/612823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy EM, Bassit LC, Mueller H, Kornepati AV, Bogerd HP, Nie T, Chatterjee P, Javanbakht H, Schinazi RF, Cullen BR. Suppression of hepatitis B virus DNA accumulation in chronically infected cells using a bacterial CRISPR/Cas RNA-guided DNA endonuclease. Virology. 2015 doi: 10.1016/j.virol.2014.12.001. (In press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy EM, Kornepati AV, Goldstein M, Bogerd HP, Poling BC, Whisnant AW, Kastan MB, Cullen BR. Inactivation of the human papillomavirus E6 or E7 gene in cervical carcinoma cells by using a bacterial CRISPR/Cas RNA-guided endonuclease. J. Virol. 2014;88:11965–11972. doi: 10.1128/JVI.01879-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klug A. The discovery of zinc fingers and their applications in gene regulation and genome manipulation. Annu. Rev. Biochem. 2010;79:213–231. doi: 10.1146/annurev-biochem-010909-095056. [DOI] [PubMed] [Google Scholar]
- Komatsu H. Hepatitis B virus: where do we stand and what is the next step for eradication? World J. Gastroenterol. 2014;20:8998–9016. doi: 10.3748/wjg.v20.i27.8998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotterman MA, Schaffer DV. Engineering adeno-associated viruses for clinical gene therapy. Nat. Rev. Genet. 2014;15:445–451. doi: 10.1038/nrg3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuscu C, Arslan S, Singh R, Thorpe J, Adli M. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat. Biotechnol. 2014;32:677–683. doi: 10.1038/nbt.2916. [DOI] [PubMed] [Google Scholar]
- Li L, Krymskaya L, Wang J, Henley J, Rao A, Cao LF, Tran CA, Torres-Coronado M, Gardner A, Gonzalez N, Kim K, Liu PQ, Hofer U, Lopez E, Gregory PD, Liu Q, Holmes MC, Cannon PM, Zaia JA, DiGiusto DL. Genomic editing of the HIV-1 coreceptor CCR5 in adult hematopoietic stem and progenitor cells using zinc finger nucleases. Mol. Ther. 2013;21:1259–1269. doi: 10.1038/mt.2013.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesegang TJ. Herpes simplex virus epidemiology and ocular importance. Cornea. 2001;20:1–13. doi: 10.1097/00003226-200101000-00001. [DOI] [PubMed] [Google Scholar]
- Lin SR, Yang HC, Kuo YT, Liu CJ, Yang TY, Sung KC, Lin YY, Wang HY, Wang CC, Shen YC, Wu FY, Kao JH, Chen DS, Chen PJ. The CRISPR/Cas9 system facilitates clearance of the intrahepatic HBV templates in vivo. Mol. Ther. Nucleic Acids. 2014;3:e186. doi: 10.1038/mtna.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisowski L, Dane AP, Chu K, Zhang Y, Cunningham SC, Wilson EM, Nygaard S, Grompe M, Alexander IE, Kay MA. Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature. 2014;506:382–386. doi: 10.1038/nature12875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malim MH, Bieniasz PD. HIV restriction factors and mechanisms of evasion. Cold Spring Harb. Perspect. Med. 2012;2:a006940. doi: 10.1101/cshperspect.a006940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mighty KK, Laimins LA. The role of human papillomaviruses in oncogenesis. Recent Results Cancer Res. 2014;193:135–148. doi: 10.1007/978-3-642-38965-8_8. [DOI] [PubMed] [Google Scholar]
- Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ, Dulay GP, Hua KL, Ankoudinova I, Cost GJ, Urnov FD, Zhang HS, Holmes MC, Zhang L, Gregory PD, Rebar EJ. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011;29:143–148. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- Mino T, Hatono T, Matsumoto N, Mori T, Mineta Y, Aoyama Y, Sera T. Inhibition of DNA replication of human papillomavirus by artificial zinc finger proteins. J. Virol. 2006;80:5405–5412. doi: 10.1128/JVI.01795-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pingoud A, Wilson GG, Wende W. Type II restriction endonucleases – a historical perspective and more. Nucleic Acids Res. 2014;42:7489–7527. doi: 10.1093/nar/gku447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, Zhang F. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickinson AB, Kieff E. Epstein-Barr virus. In: Knipe DM, Howley PM, editors. Fields Virology. Fifth ed. Philadelphia: Lippincott Williams & Wilkins; 2007. pp. 2655–2700. [Google Scholar]
- Roizman B, Knipe DM, Whitley RJ. Herpes simplex viruses. In: Knipe DM, Howley PM, editors. Fields Virology. Fifth ed. Philadelphia: Lippincott Williams & Wilkins; 2007. pp. 2501–2601. [Google Scholar]
- Sapranauskas R, Gasiunas G, Fremaux C, Barrangou R, Horvath P, Siksnys V. The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res. 2011;39:9275–9282. doi: 10.1093/nar/gkr606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- Schiffer JT, Aubert M, Weber ND, Mintzer E, Stone D, Jerome KR. Targeted DNA mutagenesis for the cure of chronic viral infections. J. Virol. 2012;86:8920–8936. doi: 10.1128/JVI.00052-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwank G, Koo BK, Sasselli V, Dekkers JF, Heo I, Demircan T, Sasaki N, Boymans S, Cuppen E, van der Ent CK, Nieuwenhuis EE, Beekman JM, Clevers H. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell. 2013;13:653–658. doi: 10.1016/j.stem.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiech L, Heidenreich M, Banerjee A, Habib N, Li Y, Trombetta J, Sur M, Zhang F. In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat. Biotechnol. 2014 doi: 10.1038/nbt.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thierry F, Howley PM. Functional analysis of E2-mediated repression of the HPV18 P105 promoter. New Biol. 1991;3:90–100. [PubMed] [Google Scholar]
- Tsao SW, Tsang CM, Pang PS, Zhang G, Chen H, Lo KW. The biology of EBV infection in human epithelial cells. Semin. Cancer Biol. 2012;22:137–143. doi: 10.1016/j.semcancer.2012.02.004. [DOI] [PubMed] [Google Scholar]
- Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature. 2008;454:780–783. doi: 10.1038/nature07103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Quake SR. RNA-guided endonuclease provides a therapeutic strategy to cure latent herpesviridae infection. Proc. Natl. Acad. Sci. USA. 2014;111:13157–13162. doi: 10.1073/pnas.1410785111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber ND, Stone D, Sedlak RH, Feelixge, De Silva, Roychoudhury HS, Schiffer P, Aubert JT, Jerome KRM. AAV-mediated delivery of zinc finger nucleases targeting hepatitis B virus inhibits active replication. PLoS One. 2014;9:e97579. doi: 10.1371/journal.pone.0097579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werle-Lapostolle B, Bowden S, Locarnini S, Wursthorn K, Petersen J, Lau G, Trepo C, Marcellin P, Goodman Z, Delaney WEt, Xiong S, Brosgart CL, Chen SS, Gibbs CS, Zoulim F. Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy. Gastroenterology. 2004;126:1750–1758. doi: 10.1053/j.gastro.2004.03.018. [DOI] [PubMed] [Google Scholar]
- Wong JK, Hezareh M, Gunthard HF, Havlir DV, Ignacio CC, Spina CA, Richman DD. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science. 1997;278:1291–1295. doi: 10.1126/science.278.5341.1291. [DOI] [PubMed] [Google Scholar]
- Wu X, Scott DA, Kriz AJ, Chiu AC, Hsu PD, Dadon DB, Cheng AW, Trevino AE, Konermann S, Chen S, Jaenisch R, Zhang F, Sharp PA. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat. Biotechnol. 2014;32:670–676. doi: 10.1038/nbt.2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Liang D, Wang Y, Bai M, Tang W, Bao S, Yan Z, Li D, Li J. Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell. 2013;13:659–662. doi: 10.1016/j.stem.2013.10.016. [DOI] [PubMed] [Google Scholar]
- Xing S, Siliciano RF. Targeting HIV latency: pharmacologic strategies toward eradication. Drug Discov. Today. 2013;18:541–551. doi: 10.1016/j.drudis.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Ran FA. Engineering and optimization of improved systems, methods and enzyme compositions for sequence manipulation. US20140242700 A1, U.S., US 14/222,930. Office. U.S.P. 2014
- Zhen S, Hua L, Takahashi Y, Narita S, Liu YH, Li Y. In vitro and in vivo growth suppression of human papillomavirus 16-positive cervical cancer cells by CRISPR/Cas9. Biochem. Biophys. Res. Commun. 2014;450:1422–1426. doi: 10.1016/j.bbrc.2014.07.014. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Zhu S, Cai C, Yuan P, Li C, Huang Y, Wei W. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature. 2014;509:487–491. doi: 10.1038/nature13166. [DOI] [PubMed] [Google Scholar]