

Abstract
Sudden unexpected death in epilepsy (SUDEP) is a fatal epileptic event. The DBA/1 mouse is a relevant animal model for study of SUDEP, as these mice exhibit seizure-induced respiratory arrest (S-IRA) leading to death, which has been observed in witnessed SUDEP patients. Fluoxetine, a selective serotonin (5-hydroxytryptamine or 5-HT) reuptake inhibitor (SSRI), reduces S-IRA in DBA/1 mice. Given that DBA/1 mice with S-IRA can be resuscitated using a ventilator, we hypothesized that breathing stimulants can prevent S-IRA and that fluoxetine prevents S-IRA by enhancing ventilation in these mice. Spontaneous respiratory function in anesthetized or awake DBA/1 mice was examined using non-invasive plethysmography before and after administering fluoxetine or breathing stimulants, doxapram and 5,6,7,8-tetrahydropyrido[4,3-d]pyrimidine (PK-THPP). The effects of these drugs on S-IRA in DBA/1 mice were tested. As reported previously, systemic administration of fluoxetine reduced S-IRA in awake DBA/1 mice, but fluoxetine in anesthetized and awake DBA/1 mice did not increase basal ventilation or the ventilatory response to 7% CO2. Both doxapram and PK-THPP increased ventilation in room air and in air + 7% CO2 in anesthetized DBA/1 mice. However, neither of the breathing stimulants reduced the incidence of S-IRA. Our studies confirm that fluoxetine reduces S-IRA in DBA/1 mice, but without enhancing basal ventilation in the absence of seizures. Although breathing stimulants increased ventilation in the absence of seizures, they were ineffective in reducing S-IRA, indicating that drug-induced increases in ventilation are insufficient to compensate for S-IRA in DBA/1 mice.
Keywords: ventilation, seizure-induced respiratory arrest, SUDEP, SSRI, breathing stimulants
1. Introduction
Sudden unexpected death in epilepsy (SUDEP) is a devastating event for patients and their families [1], accounting for up to 17% of deaths in patients with epilepsy [2] and ranking second to stroke in public health burden among common neurological diseases [3]. In most cases of witnessed SUDEP, patients exhibit generalized convulsive seizures, especially tonic–clonic seizures, prior to death [1, 4]. Respiratory dysfunction and cardiac arrhythmias are widely accepted as two major pathophysiological mechanisms for SUDEP [4–6]. The DBA/1 mouse is a relevant SUDEP animal model, as these mice often die from seizure-induced respiratory arrest (S-IRA) after generalized audiogenic seizures (AGS), consistent with the observation that the majority of witnessed SUDEP patients exhibit respiratory dysfunction prior to death [7–9]. Studies also indicate that changes in cardiac function occur subsequent to S-IRA in DBA/1 mice, which further suggests that S-IRA is the primary cause of death in this animal model [7].
A previous study has reported that both acute and chronic systemic administration of fluoxetine, a selective serotonin (5-hydroxytryptamine or 5-HT) reuptake inhibitor (SSRI), prevents S-IRA in DBA/1 mice [8]. Another SSRI, citalopram, also reduces mortality of Lmx1bf/f mice after acute seizure induction by maximal electroshock [10]. In addition, a human retrospective study observed that partial seizure-associated respiratory depression in SSRI-treated patients is lower than that in patients not taking these agents [11]. These studies suggest that 5-HT neurotransmission may play a critical role in prevention of S-IRA. 5-HT is an important modulator for normal respiration [12]. It also modulates the ventilatory response to hypercapnia [13], and hypercapnia commonly occurs during seizures [14, 15]. In addition, DBA/1 mice with S-IRA can be resuscitated using a rodent ventilator [7, 8]. Thus, we hypothesized that fluoxetine reduces S-IRA in DBA/1 mice by enhancing basal ventilation and/or the ventilatory response to CO2.
We further hypothesized that two known breathing stimulants, doxapram and 5,6,7,8-tetrahydropyrido[4,3-d]pyrimidine (PK-THPP), could reduce S-IRA in awake DBA/1 mice. Doxapram, a TWIK(tandem of pore domains in a weak inward rectifying potassium channel)-related acid-sensitive potassium (TASK) channel antagonist, has been used to treat patients with respiratory dysfunction, especially when they develop symptomatic acidosis [16, 17]. Recently, a novel TASK antagonist PK-THPP has been shown to be a potent breathing stimulant in rats [18]. Therefore in the present study, we investigated the effect of fluoxetine on basal breathing and ventilatory CO2 sensitivity at a dosage known to reduce S-IRA in awake DBA/1 mice. We also examined the effect of pre-seizural administration of breathing stimulants on the incidence of S-IRA in this model of SUDEP at dosages that enhance basal ventilation and ventilatory CO2 sensitivity in the absence of seizures.
2. Materials and methods
2.1. Animals
DBA/1 mice were obtained from Harlan Laboratories. Mice were housed and bred in the animal facility at Massachusetts General Hospital with food pellets and water available ad libitum. From postnatal day 26–28, AGS were induced daily for 3 days to develop S-IRA susceptibility in DBA/1 mice, as described below. Only those mice that exhibited consistent S-IRA were used in experiments. Mice were randomly divided into three groups, each with drug and vehicle control subgroups: Group A, fluoxetine in 10% DMSO; Group B, doxapram in saline; Group C, PK-THPP in 4% DMSO + 10% cremophor. Animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Massachusetts General Hospital.
2.2. Drugs
Fluoxetine, doxapram, PK-THPP and vehicles were administrated intraperitoneally (i.p.) at 30 mg/kg, 50 mg/kg, 10 mg/kg and 10 ml/kg, respectively. The volume injected was 0.1–0.3 ml for each mouse. Fluoxetine was obtained from Sigma-Aldrich (St. Louis, MO), and Doxapram (Respiram) was obtained from Modern Veterinary Therapeutics, LLC (Coral Gables, FL). PK-THPP was synthesized by Aberjona Laboratories (Beverly, MA) using published methods [19, 20].
2.3. Seizure induction and resuscitation
Each mouse was placed in a cylindrical plexiglass chamber (14″ diameter) mounted with a video camera. AGS were induced using an electric bell (96 dB SPL). The stimulus was given for a maximum duration of 60 s or until the mouse exhibited a tonic seizure, which in most cases ended in S-IRA. Mice exhibiting S-IRA were resuscitated within 5 s after the final respiratory gasp using a polyethylene tube connected to the outflow of a rodent respirator (Harvard Apparatus 680, Holliston, MA), operating at 180 strokes/min at a volume of 1 ml in room air. The S-IRA susceptibility was always re-confirmed 24 hr prior to drug or vehicle treatment. Fluoxetine or vehicle was administered 30 min prior to S-IRA induction. Doxapram and PK-THPP reached their peak effect on ventilatory stimulation ~ 15 min after administration (see experimental results below), and thus S-IRA was examined 15 min after treatment with each of these drugs or vehicles. AGS pattern/duration and S-IRA were digitally recorded for offline analysis. When a mouse was reused in experiments, the animal was recovered in the animal facility for at least one week allowing for clearance of the administered drug. Twenty-four hrs prior to testing of the next drug/vehicle, the mouse’s susceptibility to S-IRA was re-confirmed.
2.4. Anesthetized mouse breathing studies
DBA/1 mouse breathing was quantified using a nose-only plethysmography chamber. Studies were conducted in mice under 1.5% inhaled isoflurane (Baxter Healthcare, Deerfield, IL) anesthesia. The custom-built, acrylic nose chamber was crafted from a 1-inch length of 1.5-inch diameter clear acrylic pipe. The nose of the mouse was inserted into the chamber through a 0.25- inch hole in a 0.125-inch thick latex diaphragm (McMaster Carr, Robbinsville, NJ) covering one chamber wall. The chamber was continuously flushed (1 l/min) with fresh room air via luer ports using mass flow controller valves (Model GE50A, MKS Instruments Inc., Andover, MA), and chamber gas composition was monitored with a Capnomac Ultima medical gas analyzer (GE Healthcare, Buckinghamshire, U.K.). Changes in gas flow through the chamber induced by mouse breathing were detected using a heated pneumotachometer (Model 8420; Hans Rudolph Inc., Shawnee, KS). Mouse breathing was converted to an analog signal with a differential pressure transducer and a demodulator (Models CD15 and MP45–14-871; Validyne Engineering, Northridge, CA). The system was calibrated using a 1-ml syringe to inject air with the 0.25-inch hole in the latex diaphram occluded. Plethysmography data acquisition, analysis, and gas flows were controlled using LabView 2013 software (National Instruments, Austin, TX) run on an Apple computer interfaced with two USB-6009 data acquisition boards (National Instruments). Data were analyzed in 4-s time epochs to determine minute ventilation (VE), tidal volume (VT) and respiratory frequency (fR) (VE = VT x fR). The mouse body temperature was continuously measured by a rectal thermistor and maintained at 37°C by a microcontroller automated heat lamp. Following placement in the chamber, the anesthetized DBA/1 mouse was allowed to breathe room air for 30 min in the absence of drug treatment. The recordings of VE, VT and fR for the last 5 min were used as baseline measurements for normalization. After 30 min baseline recording, drugs or vehicle were then administered through an i.p. 24-gauge angiocatheter, which was placed prior to initiation of breathing measurements. Ventilatory response to CO2 was performed by switching to a CO2 gas mixture (7% CO2) for 10 min. Drug- and 7% CO2-induced changes in VE, fR and VT were recorded and normalized to the corresponding baseline. The peak response of each parameter was compared between drug group and its corresponding vehicle control.
2.5. Conscious mouse breathing studies
Conscious DBA/1 mouse breathing was studied in a custom-built, whole body plethysmography chamber constructed from 1.25-inch diameter, clear PVC pipe (U.S. Plastics Corporation, Lima, OH) (Fig. 1). The closed chamber was intermittently flushed with 1 l/min air using mass flow controllers (Model GE50A, MKS Instruments Inc.) via microcontroller operated solenoid valves (Cole Parmer, Vernon Hills, IL). Gas flushed from the chamber was continuously monitored for CO2 levels with a Capnomac Ultima medical gas analyzer (GE Healthcare). Pressure in the chamber was monitored with a pressure transducer and demodulator (Models CD15 and MP45-14-871; Validyne Engineering). The analog pressure signal was high-pass filtered at 15 s, digitized at 128 Hz, and analyzed in 8-s epochs. The chamber was calibrated by injecting 0.1 ml of air through a Luer port with a mouse-sized object occupying the chamber, and tidal volumes during intermittent chamber closure were estimated using methods described by Drorbaugh and Fenn [21]. LabView 2013 (National Instruments) run on an Apple computer interfaced with two USB-6009 data acquisition boards (National Instruments) was used for data acquisition and analysis and gas flow control. Chamber temperature and humidity used in tidal volume estimates were recorded continuously (DHT22, Aosong Electronics, Guangzhou, China). Mouse temperature was assumed to be 37°C in our analysis.
Fig. 1. Whole body plethysmography chamber used in this study.

Major components are labeled. The temperature and humidity sensor is inside the chamber.
2.6. Statistical analysis
Data are reported as mean ± SEM. Statistical analysis was performed using Prism 6 software (GraphPad Software Inc., La Jolla, CA). The effects of each drug on VE, fR and VT were compared with its vehicle control using unpaired Student t test. The incidence of S-IRA between drug and control groups was compared using Chi-square test. Statistical significance was inferred if p<0.05.
3. Results
3.1. Doxapram, PK-THPP but not fluoxetine increased basal ventilation in anesthetized DBA/1 mice
We first examined the effect of drugs on ventilation in anesthetized DBA/1 mice in room air. After injection of doxapram (50 mg/kg, i.p.) or PK-THPP (10 mg/kg, i.p.), the VE of anesthetized DBA/1 mice gradually increased within 5 min and reached a peak value at approximately 15 min (Fig. 2A). Administration of doxapram (n = 5) significantly increased VE (p<0.01), VT (p<0.05) and fR (p<0.01) as compared with vehicle treatment (Fig. 2B). PK-THPP (n = 3) produced a significant enhancement of VE (p<0.01) and fR (p<0.05), although it did not significantly increase VT as compared with vehicle treatment (Fig. 2B). Fluoxetine (30 mg/kg, i.p., n = 6) did not significantly alter VE, fR or VT as compared with vehicle treatment (n = 3) in anesthetized DBA/1 mice (Fig. 3A, B).
Fig. 2. Doxapram and PK-THPP stimulate basal breathing and increase ventilatory response to CO2 in anesthetized DBA/1 mice.

A, representative traces of minute ventilation (VE), respiratory frequency (fR) and tidal volume (VT) from anesthetized DBA/1 mice treated with doxapram (50 mg/kg), PK-THPP (10 mg/kg) or their corresponding vehicles, in room air or exposure to air + 7% CO2. Data were normalized to the average VE, fR or VT baseline value. Baseline VE, fR and VT were 13.15 ± 0.66 ml/20g/min, 95.37 ± 2.22 breaths/min and 0.14 ± 0.01 ml/20g/min, respectively (n = 25). Traces between the two dotted lines indicate the exposure time to 7% CO2 gas mixture. B, effects of doxapram and PK-THPP on the normalized VE, fR and VT in room air and in air + 7% CO2 in anesthetized DBA/1 mice. The error bars represent SEMs. * p<0.05; ** p<0.01: significantly different from corresponding controls.
Fig. 3. Fluoxetine did not increase basal breathing and ventilatory response to CO2 in anesthetized and conscious DBA/1 mice.

A, representative traces of minute ventilation (VE), respiratory frequency (fR) and tidal volume (VT) from anesthetized DBA/1 mice treated with fluoxetine (30 mg/kg) or vehicle, in room air or exposure to air + 7% CO2. Data were normalized to the average VE, fR or VT baseline value. Traces between the two dotted lines indicate the exposure time to 7% CO2 gas mixture. B, effects of fluoxetine on the normalized VE, fR and VT in room air and in air + 7% CO2 in anesthetized DBA/1 mice. C, effects of fluoxetine on the normalized VE, fR and VT in room air and in air + 7% CO2 in conscious DBA/1 mice. The error bars represent SEMs. * p<0.05; ** p<0.01: significantly different from corresponding vehicle control.
3.2. Doxapram and PK-THPP but not fluoxetine increased ventilatory response to CO2 in anesthetized DBA/1 mice
The effect of doxapram, PK-THPP and fluoxetine on the ventilatory response to 7% CO2 was tested in anesthetized DBA/1 mice following the recording in room air in the presence of drugs (Fig. 2A, 3A). After treatment with doxapram, 7% CO2 caused significant increases in VE (p<0.01) and fR (p<0.05), but not in VT as compared with vehicle treatment (Fig. 2B). After treatment with PK-THPP, 7% CO2 also induced a significant increase in VE (p<0.05), but not in fR and VT (Fig. 2B). In contrast, after treatment with fluoxetine in anesthetized DBA/1 mice, 7% CO2 did not cause significant changes in VE and fR, and actually decreased VT (p<0.05) as compared with vehicle control (Fig. 3B).
Isoflurane is a known ventilatory depressant and suppresses the hypercapnic breathing response [22]. Since isoflurane might interfere with fluoxetine effects on breathing, we also performed plethysmography in awake DBA/1 mice before and after administering fluoxetine. Fluoxetine (30 mg/kg, i.p., n = 5) significantly decreased VE (p<0.01) and fR (p<0.01) but not VT as compared with vehicle control (n = 4); and, after fluoxetine treatment, the ventilatory response to 7% CO2 was unchanged (Fig. 3C). Doxapram and PK-THPP exert similar effects on breathing in anesthetized and conscious rats (J.F. Cotten, unpublished observations). Thus, we did not test effects of these drugs on breathing in conscious DBA/1 mice.
3.3. Fluoxetine but not doxapram or PK-THPP decreased S-IRA susceptibility in awake DBA/1 mice
Consistent with a prior report [8], we determined that fluoxetine (30 mg/kg, i.p.) significantly reduced the incidence of S-IRA in DBA/1 mice (p<0.01, n = 10) (Fig. 4). Fluoxetine blocked only S-IRA but not seizure behavior in 40% of DBA/1 mice tested. In 30% of mice, fluoxetine blocked both S-IRA and tonic seizures, but these mice still exhibited wild running and/or clonic seizures. Interestingly, administration of doxapram (50 mg/kg, i.p., n = 8) or PK-THPP (10 mg/kg, i.p., n = 8) exerted no effect on the incidence of S-IRA in DBA/1 mice (Fig. 4). These breathing stimulants did not alter the seizure pattern in these animals.
Fig. 4. Fluoxetine but not doxapram or PK-THPP reduced the incidence of S-IRA in awake DBA/1 mice.
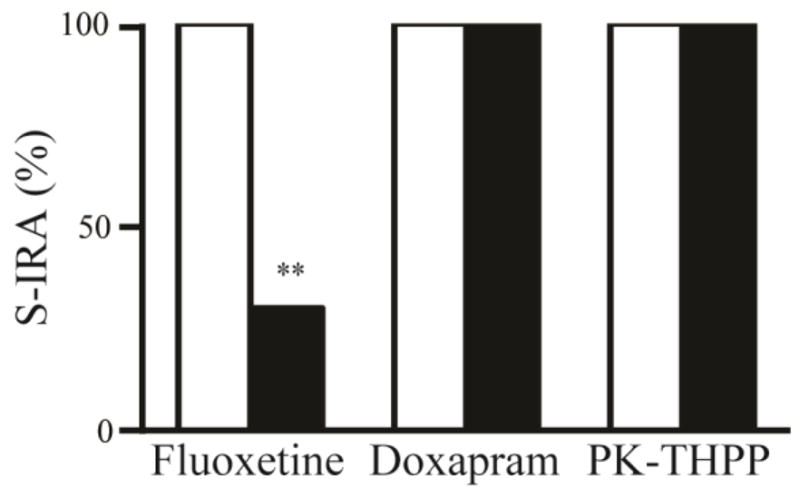
Effect of systemic administration (i.p.) of fluoxetine (30 mg/kg), doxapram (50 mg/kg) and PK-THPP (10 mg/kg) on S-IRA in DBA/1 mice. Fluoxetine significantly decreased the incidence of S-IRA 30 min after injection (p<0.01, n = 10) as compared with vehicle control (n = 9). Doxapram (n = 8) and PK-THPP (n = 8) did not show any effect on the incidence of S-IRA 15 min after injection as compared with corresponding vehicle controls (n = 6–7). The blank bars indicate the incidence of S-IRA with vehicle injections, and the filled bars indicate that with drug injections. ** p<0.01: significantly different from vehicle control.
4. Discussion
4.1. Breathing stimulants do not prevent S-IRA in DBA/1 mice
DBA/1 mice with S-IRA can be mechanically resuscitated using a rodent respirator [8], and oxygenation prevents sudden death in AGS-susceptible mice [23]. Thus, we postulated that breathing stimulants might be useful as a pharmacological approach to preventing S-IRA. We investigated the effects of two known breathing stimulants, doxapram and PK-THPP on breathing and on S-IRA in DBA/1 mice. Consistent with prior studies in rats [18], both drugs increased ventilation and ventilatory response to CO2 in anesthetized DBA/1 mice. However, neither breathing stimulant reduced the incidence of S-IRA in awake DBA/1 mice. These data indicate that drug-induced increases in ventilation are insufficient to compensate for S-IRA in DBA/1 mice. Although respiratory dysfunction and elevated arterial CO2 are observed in patients following seizures [14, 15], no attempts have been made to treat respiratory failure using breathing stimulants. A previous study reported that treatment with doxapram elevated the seizure threshold in amygdaloid kindled rats [24]. However, in our current study, doxapram and PK-THPP did not alter the threshold and/or the severity of AGS at doses that exert substantial effect on breathing.
4.2. Fluoxetine reduces S-IRA in DBA/1 mice without enhancing ventilation
Since 5-HT modulates normal breathing, we hypothesized that fluoxetine prevents S-IRA in DBA/1 mice by enhancing ventilation and/or the ventilatory response to CO2. Several, but not all tested SSRIs, are effective in blocking S-IRA in DBA/1 mice, and a 5-HT antagonist increases S-IRA susceptibility [25, 26]. It is not known why some SSRIs such as fluoxetine are more effective than others such as paroxetine in reducing S-IRA in DBA mice [26]. In addition to 5-HT, SSRIs also inhibit the reuptake of other neurotransmitters such as norepinephrine [27]. The differential effect of SSRIs on S-IRA may be related to their actions on non-serotonergic neurotransmission, as well. In the present study, we observed that acute systemic treatment with fluoxetine suppressed ventilation in room air without affecting that in air + 7% CO2 in conscious DBA/1 mice, whereas fluoxetine did not alter ventilation in either condition in anesthetized DBA/1 mice. These data suggest that although fluoxetine reduces S-IRA in DBA/1 mice, this protective effect is not due to enhanced basal ventilation and sensitivity to CO2 in the absence of seizures. The mechanisms underlying fluoxetine prevention of S-IRA in DBA/1 mice remain elusive but may be related to increase in 5-HT release during seizures [28]. The pre-Bötzinger complex in the rostral ventrolateral medulla is a key structure for respiratory rhythm generation [12]. The midline raphe nuclei contain spontaneously active 5-HT neurons, some of which have synaptic connections with the pre-Bötzinger complex, and play a potential role in promoting rhythm generation [29]. If S-IRA involves disruption of respiratory rhythm, it is possible that fluoxetine may prevent S-IRA by promoting this rhythm. Additionally, 5-HT neurons located in the rostral midbrain are believed to be central chemoreceptors inducing arousal in response to increased arterial pCO2 during sleep. A defect in the CO2-evoked arousal response was observed in Lmx1bf/f/p mice [13], in which 5-HT neurons were genetically deleted. There is also a reduced hypercapnic ventilatory response in these mice [30], and in Brown Norway rats [31], whose levels of 5-HT and its metabolite 5-hydroxyindolacetic acid (5-HIAA) were low in the brain [32]. Administration of fluoxetine can reverse the arousal deficit in Brown Norway rats [33]. If this defect in arousal contributes to S-IRA in DBA/1 mice, fluoxetine may prevent S-IRA by enhancing arousal mechanisms.
Previous studies on the effect of fluoxetine on breathing have yielded conflicting results [33–35]. For example, centrally administered fluoxetine increased basal breathing and the ventilatory response to CO2 in one study [35]. Another study observed that acute treatment with fluoxetine decreased VE and fR in a dose-dependent manner, although subchronic administration of fluoxetine exerted an opposite effect [34]. Consistent with this latter study, our results show that acute administration of fluoxetine suppresses ventilation and leaves the ventilatory response to CO2 unaffected in conscious DBA/1 mice. Some SSRIs evoke a depressant effect on serotonergic neuron firing through enhanced 5HT1A receptor activation [36]. It is possible that fluoxetine reduces ventilation by suppressing 5-HT neuronal activity. DBA/1 mice express reduced 5-HT2B/2C and 5-HT3B receptors in the brainstem [37]. Among these receptors, the 5-HT2B receptor exerts a stronger excitatory effect than other receptors on respiration [38]. Furthermore, a polymorphism (C1473G) of tryptophan hydroxylase-2 (TPH2), the key enzyme for synthesis of 5-HT in the brain, is found in DBA mice [39]. This polymorphism compromises the function of TPH2, leading to reduced synthesis of 5-HT [40]. It is not known if these deficits in 5-HT neurotransmission contribute to the observed respiratory suppression by fluoxetine. It is technically difficult to study respiratory function during convulsive seizures with the methods used here. Future studies are needed to explore if fluoxetine produces different effects on breathing in DBA/1 mice during seizures from those that are awake or under anesthesia, and if there are differences between acute and chronic treatments.
5. Conclusion
Our study demonstrates that, although breathing stimulants like TASK channel antagonists increase ventilation in spontaneously breathing DBA/1 mice, they are ineffective in reducing the incidence of S-IRA. Thus, drug-induced increases in ventilatory drive are insufficient to prevent S-IRA in DBA/1 mice. Fluoxetine, which did protect DBA/1 mice from S-IRA, had a slightly depressant effect on breathing. Therefore, suppression of S-IRA by fluoxetine in DBA/1 mice is unlikely to be achieved by enhancing basal ventilation and may involve other neuronal mechanisms/processes such as promotion of respiratory rhythmogenesis and/or arousal response.
Highlights.
Fluoxetine reduces S-IRA, but without enhancing the ventilation.
Breathing stimulants enhance ventilation but have no effect on S-IRA.
Fluoxetine prevents S-IRA via mechanisms other than enhancement of ventilation.
Acknowledgments
This work was supported by R03NS078591 and CURE (Citizens United for Research in Epilepsy) foundation to HJF, and R01HL117871 to JC. We thank the support from the Massachusetts General Hospital Department of Anesthesia, Critical Care and Pain Medicine. We thank Zuohao Li for help with data analysis and figure preparation.
Footnotes
Conflict of interest
The authors have no conflict of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tomson T, Nashef L, Ryvlin P. Sudden unexpected death in epilepsy: current knowledge and future directions. Lancet Neurol. 2008;7:1021–31. doi: 10.1016/S1474-4422(08)70202-3. [DOI] [PubMed] [Google Scholar]
- 2.Hughes JR. A review of sudden unexpected death in epilepsy: prediction of patients at risk. Epilepsy Behav. 2009;14:280–7. doi: 10.1016/j.yebeh.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 3.Thurman DJ, Hesdorffer DC, French JA. Sudden unexpected death in epilepsy: assessing the public health burden. Epilepsia. 2014;55:1479–85. doi: 10.1111/epi.12666. [DOI] [PubMed] [Google Scholar]
- 4.Opeskin K, Berkovic SF. Risk factors for sudden unexpected death in epilepsy: a controlled prospective study based on coroners cases. Seizure. 2003;12:456–64. doi: 10.1016/s1059-1311(02)00352-7. [DOI] [PubMed] [Google Scholar]
- 5.Langan Y, Nashef L, Sander JW. Sudden unexpected death in epilepsy: a series of witnessed deaths. J Neurol Neurosurg Psychiatry. 2000;68:211–3. doi: 10.1136/jnnp.68.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schuele SU, Widdess-Walsh P, Bermeo A, Luders HO. Sudden unexplained death in epilepsy: the role of the heart. Cleve Clin J Med. 2007;74 (Suppl 1):S121–7. doi: 10.3949/ccjm.74.suppl_1.s121. [DOI] [PubMed] [Google Scholar]
- 7.Faingold CL, Randall M, Tupal S. DBA/1 mice exhibit chronic susceptibility to audiogenic seizures followed by sudden death associated with respiratory arrest. Epilepsy Behav. 2010;17:436–40. doi: 10.1016/j.yebeh.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 8.Faingold CL, Tupal S, Randall M. Prevention of seizure-induced sudden death in a chronic SUDEP model by semichronic administration of a selective serotonin reuptake inhibitor. Epilepsy Behav. 2011;22:186–90. doi: 10.1016/j.yebeh.2011.06.015. [DOI] [PubMed] [Google Scholar]
- 9.So EL, Sam MC, Lagerlund TL. Postictal central apnea as a cause of SUDEP: evidence from near-SUDEP incident. Epilepsia. 2000;41:1494–7. doi: 10.1111/j.1528-1157.2000.tb00128.x. [DOI] [PubMed] [Google Scholar]
- 10.Buchanan GF, Murray NM, Hajek MA, Richerson GB. Serotonin neurones have anticonvulsant effects and reduce seizure-induced mortality. J Physiol. 2014;592:4395–410. doi: 10.1113/jphysiol.2014.277574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bateman LM, Li CS, Lin TC, Seyal M. Serotonin reuptake inhibitors are associated with reduced severity of ictal hypoxemia in medically refractory partial epilepsy. Epilepsia. 2010;51:2211–4. doi: 10.1111/j.1528-1167.2010.02594.x. [DOI] [PubMed] [Google Scholar]
- 12.Feldman JL, Mitchell GS, Nattie EE. Breathing: rhythmicity, plasticity, chemosensitivity. Annu Rev Neurosci. 2003;26:239–66. doi: 10.1146/annurev.neuro.26.041002.131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buchanan GF, Richerson GB. Central serotonin neurons are required for arousal to CO2. Proc Natl Acad Sci U S A. 2010;107:16354–9. doi: 10.1073/pnas.1004587107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bateman LM, Li CS, Seyal M. Ictal hypoxemia in localization-related epilepsy: analysis of incidence, severity and risk factors. Brain. 2008;131:3239–45. doi: 10.1093/brain/awn277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moseley BD, Nickels K, Britton J, Wirrell E. How common is ictal hypoxemia and bradycardia in children with partial complex and generalized convulsive seizures? Epilepsia. 2010;51:1219–24. doi: 10.1111/j.1528-1167.2009.02490.x. [DOI] [PubMed] [Google Scholar]
- 16.Bjork L, Arborelius M, Jr, Renck H, Rosberg B. Doxapram improves pulmonary function after upper abdominal surgery. Acta Anaesthesiol Scand. 1993;37:181–8. doi: 10.1111/j.1399-6576.1993.tb03697.x. [DOI] [PubMed] [Google Scholar]
- 17.Gomersall CD, Joynt GM, Freebairn RC, Lai CK, Oh TE. Oxygen therapy for hypercapnic patients with chronic obstructive pulmonary disease and acute respiratory failure: a randomized, controlled pilot study. Crit Care Med. 2002;30:113–6. doi: 10.1097/00003246-200201000-00018. [DOI] [PubMed] [Google Scholar]
- 18.Cotten JF. TASK-1 (KCNK3) and TASK-3 (KCNK9) tandem pore potassium channel antagonists stimulate breathing in isoflurane-anesthetized rats. Anesth Analg. 2013;116:810–6. doi: 10.1213/ANE.0b013e318284469d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coburn CA, Luo Y, Cui M, Wang J, Soll R, Dong J, Hu B, Lyon MA, Santarelli VP, Kraus RL, Gregan Y, Wang Y, Fox SV, Binns J, Doran SM, Reiss DR, Tannenbaum PL, Gotter AL, Meinke PT, Renger JJ. Discovery of a pharmacologically active antagonist of the two-pore-domain potassium channel K2P9. 1 (TASK-3) Chem Med Chem. 2012;7:123–33. doi: 10.1002/cmdc.201100351. [DOI] [PubMed] [Google Scholar]
- 20.Peukert S, Brendel J, Pirard B, Bruggemann A, Below P, Kleemann HW, Hemmerle H, Schmidt W. Identification, synthesis, and activity of novel blockers of the voltage-gated potassium channel Kv1. 5. J Med Chem. 2003;46:486–98. doi: 10.1021/jm0210461. [DOI] [PubMed] [Google Scholar]
- 21.Drorbaugh JE, Fenn WO. A barometric method for measuring ventilation in newborn infants. Pediatrics. 1955;16:81–7. [PubMed] [Google Scholar]
- 22.Teran FA, Massey CA, Richerson GB. Serotonin neurons and central respiratory chemoreception: where are we now? Prog Brain Res. 2014;209:207–33. doi: 10.1016/B978-0-444-63274-6.00011-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Venit EL, Shepard BD, Seyfried TN. Oxygenation prevents sudden death in seizure-prone mice. Epilepsia. 2004;45:993–6. doi: 10.1111/j.0013-9580.2004.02304.x. [DOI] [PubMed] [Google Scholar]
- 24.Albertson TE, Stark LG, Joy RM. The effects of doxapram, diazepam, phenobarbital and pentylenetetrazol on suprathreshold and threshold stimulations in amygdaloid kindled rats. Neuropharmacology. 1983;22:245–8. doi: 10.1016/0028-3908(83)90016-3. [DOI] [PubMed] [Google Scholar]
- 25.Faingold CL, Randall M. Effects of age, sex, and sertraline administration on seizure-induced respiratory arrest in the DBA/1 mouse model of sudden unexpected death in epilepsy (SUDEP) Epilepsy Behav. 2013;28:78–82. doi: 10.1016/j.yebeh.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 26.Faingold CL, Kommajosyula SP, Long X, Plath K, Randall M. Serotonin and sudden death: Differential effects of serotonergic drugs on seizure-induced respiratory arrest in DBA/1 mice. Epilepsy Behav. 2014;37:198–203. doi: 10.1016/j.yebeh.2014.06.028. [DOI] [PubMed] [Google Scholar]
- 27.Shank RP, Vaught JL, Pelley KA, Setler PE, McComsey DF, Maryanoff BE. McN-5652: a highly potent inhibitor of serotonin uptake. J Pharmacol Exp Ther. 1988;247:1032–8. [PubMed] [Google Scholar]
- 28.Fisher RS, Schachter SC. The Postictal State: A Neglected Entity in the Management of Epilepsy. Epilepsy Behav. 2000;1:52–59. doi: 10.1006/ebeh.2000.0023. [DOI] [PubMed] [Google Scholar]
- 29.Ptak K, Yamanishi T, Aungst J, Milescu LS, Zhang R, Richerson GB, Smith JC. Raphe neurons stimulate respiratory circuit activity by multiple mechanisms via endogenously released serotonin and substance P. J Neurosci. 2009;29:3720–37. doi: 10.1523/JNEUROSCI.5271-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hodges MR, Tattersall GJ, Harris MB, McEvoy SD, Richerson DN, Deneris ES, Johnson RL, Chen ZF, Richerson GB. Defects in breathing and thermoregulation in mice with near-complete absence of central serotonin neurons. J Neurosci. 2008;28:2495–505. doi: 10.1523/JNEUROSCI.4729-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hodges MR, Forster HV, Papanek PE, Dwinell MR, Hogan GE. Ventilatory phenotypes among four strains of adult rats. J Appl Physiol (1985) 2002;93:974–83. doi: 10.1152/japplphysiol.00019.2002. [DOI] [PubMed] [Google Scholar]
- 32.Hodges MRM, GC, Miller J, Muere C, Forster HV. Altered brainstem neuromodulators relevant to central chemoreceptor function in Brown Norway (BN) rats. FASEB J. 2012;26:894–895. [Google Scholar]
- 33.Hodges MR, Echert AE, Puissant MM, Mouradian GC., Jr Fluoxetine augments ventilatory CO2 sensitivity in Brown Norway but not Sprague Dawley rats. Respir Physiol Neurobiol. 2013;186:221–8. doi: 10.1016/j.resp.2013.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Annerbrink K, Olsson M, Hedner J, Eriksson E. Acute and chronic treatment with serotonin reuptake inhibitors exert opposite effects on respiration in rats: possible implications for panic disorder. J Psychopharmacol. 2010;24:1793–801. doi: 10.1177/0269881109106908. [DOI] [PubMed] [Google Scholar]
- 35.Sahin G, Guner I, Yelmen N, Yaman O, Mengi M, Simsek G, Sipahi S. Alterations of central hypercapnic respiratory response induced by acute central administration of serotonin re-uptake inhibitor, fluoxetine. Chin J Physiol. 2011;54:356–66. [PubMed] [Google Scholar]
- 36.Blier P, de Montigny C. Electrophysiological investigation of the adaptive response of the 5-HT system to the administration of 5-HT1A receptor agonists. J Cardiovasc Pharmacol. 1990;15 (Suppl 7):S42–8. [PubMed] [Google Scholar]
- 37.Faingold CL, Randall M, Mhaskar Y, Uteshev VV. Differences in serotonin receptor expression in the brainstem may explain the differential ability of a serotonin agonist to block seizure-induced sudden death in DBA/2 vs. DBA/1 mice. Brain Res. 2011;1418:104–10. doi: 10.1016/j.brainres.2011.08.043. [DOI] [PubMed] [Google Scholar]
- 38.Niebert M, Vogelgesang S, Koch UR, Bischoff AM, Kron M, Bock N, Manzke T. Expression and function of serotonin 2A and 2B receptors in the mammalian respiratory network. PLoS One. 2011;6:e21395. doi: 10.1371/journal.pone.0021395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Osipova DV, Kulikov AV, Mekada K, Yoshiki A, Moshkin MP, Kotenkova EV, Popova NK. Distribution of the C1473G polymorphism in tryptophan hydroxylase 2 gene in laboratory and wild mice. Genes Brain Behav. 2010;9:537–43. doi: 10.1111/j.1601-183X.2010.00586.x. [DOI] [PubMed] [Google Scholar]
- 40.Zhang X, Beaulieu JM, Sotnikova TD, Gainetdinov RR, Caron MG. Tryptophan hydroxylase-2 controls brain serotonin synthesis. Science. 2004;305:217. doi: 10.1126/science.1097540. [DOI] [PubMed] [Google Scholar]