

Abstract
AML is a diagnosis encompassing a diverse group of myeloid malignancies. Heterogeneous genetic etiology, together with the potential for oligoclonality within the individual patient, have made the identification of a single high-sensitivity marker of disease burden challenging. We developed a multiple gene measurable residual disease (MG-MRD) RQ–PCR array for the high-sensitivity detection of AML, retrospectively tested on 74 patients who underwent allo-SCT at the NHLBI in the period 1994–2012. MG-MRD testing on peripheral blood samples prior to transplantation demonstrated excellent concordance with traditional BM-based evaluation and improved risk stratification for post-transplant relapse and OS outcomes. Pre-SCT assessment by MG-MRD predicted all clinical relapses occurring in the first 100 days after allo-SCT compared with 57% sensitivity using WT1 RQ–PCR alone. Nine patients who were negative for WT1 prior to transplantation were correctly reclassified into a high-risk MG-MRD-positive group, associated with 100% post-transplant mortality. This study provides proof of principle that a multiple gene approach may be superior to the use of WT1 expression alone for AML residual disease detection.
Introduction
AML is a broad diagnostic category encompassing a genetically heterogeneous set of myeloid malignancies,1, 2, 3, 4 with molecularly distinct subgroups displaying different outcomes to treatment5, 6 and the potential for intrapatient oligoclonality such that the predominant clone at presentation is not necessarily the leukemic clone ultimately responsible for clinical relapse and death.7 Relapse of AML after allo-SCT has a dismal prognosis and remains the most common form of treatment failure.8, 9, 10, 11, 12
For AML patients in CR after initial treatment the use of high-sensitivity tests to detect measurable residual disease (MRD,13, 14, 15, 16, 17, 18, 19, 20) prior to allo-SCT can identify patients with higher rates of relapse and death compared with MRD-negative patients and may supersede stratification based on clinical discriminators, such as CR1 vs CR2.21 In situations where highly specific assays are available, such as core-binding factor AML, MRD is the most important prognostic factor for relapse prediction even when high-risk clinical features and pre-treatment molecular risk stratification markers are considered,22, 23 and can be used to optimize subsequent treatment.24 Unfortunately, due in part to the heterogeneity of AML, no single universal AML MRD assay has yet been developed.25, 26
WT1 is a tumor suppressor gene that encodes for a zinc-finger transcription factor and is expressed in ~85–90% of AML cases, and is aberrantly overexpressed to the extent that it can be used as a marker for MRD in AML in between 46 and 74% of cases.27, 28, 29, 30 WT1 expression by RQ–PCR27, 28, 31, 32, 33, 34, 35, 36, 37, 38, 39 has been tested extensively for AML MRD, as have flow cytometry approaches,40, 41, 42, 43, 44, 45, 46 but unfortunately neither method is applicable to all AML patients. We therefore sought to test whether a novel AML MRD approach incorporating detection of multiple potentially overexpressed genes could offer superior pre-SCT relapse risk stratification compared with the use of WT1 alone.
Patients and methods
Patient eligibility
Laboratory analysis was performed on samples previously collected from AML (excluding APL) patients who had received allo-SCT as part of clinical protocols (MB, AJB) performed between 1994 and 2012. These were predominately myeloablative, T-cell depleted, PBSC sibling-matched allogeneic transplants with cyclosporine-based GVHD prophylaxis (Supplementary Table S1). Eligibility criteria for MRD testing were a stored pre-SCT peripheral blood sample, pathological evaluation of disease status within 2 months prior to transplant date and at least 12 months of post-SCT clinical outcome data or until date of death if this occurred before 12 months. All clinical protocols were conducted in accordance with Declaration of Helsinki principles and were approved by the institutional review board. Written informed consent was obtained from all subjects.
Clinical samples
Peripheral blood samples from fifty healthy adult donors were collected as baseline controls. Patient samples immediately prior to transplantation were taken from aliquots of a research leukapheresis product processed using ficoll hypaque isolation, followed by freezing and storage in the vapor phase of liquid nitrogen. RNA was isolated from peripheral blood samples of both patient and healthy donors using AllPrep Mini Kits (Qiagen, Valencia, CA, USA), and assessed using a Nanodrop 1000 Spectrophotometer (Wilmington, DE, USA).
Real-time PCR array
One μg of total RNA was reverse-transcribed into cDNA using the RT2 First Strand Kit (Qiagen). Custom RT2 Profiler PCR array plates including controls for human genomic DNA contamination, reverse transcription and PCR efficacy (SABiosciences, Qiagen) were used for RQ–PCR reactions performed using RT2 SYBR Green ROX qPCR Mastermix (SABiosciences, Valencia, CA, USA) on an ABI 7900 thermal cycler (Applied Biosystems, Foster City, CA, USA) as described previously.47
Data handling and statistical analysis
During the study period, the format and content of pathologist reports at our institution varied; therefore the term ‘active disease' (relapsed or refractory) has been used here to refer to the current consensus AML response criteria48 of at least 5% blasts on BM aspirate differential and/or an AML-defining chromosomal abnormality. Clinical annotation of samples for pathological diagnosis and clinical outcome was performed independently by two physicians (KL, NJ) blinded to research laboratory testing results. Statistical analysis was performed using GraphPad Prism (La Jolla, CA, USA) with comparison between survival and relapse curves using the log-rank (Mantel–Cox) test, McNemar's test as described,49 and receiver-operating characteristic curves created using StAR.50
Results
Patients
Eligible samples were identified from 74 of the 111 AML (excluding APL) patients transplanted using these protocols during the study time period (Table 1; Supplementary Table S2; Table 2). Patients studied were transplanted a median of 9.9 years (range: 1.75–20.2 years) before this laboratory analysis.
Table 1. Patient characteristics.
|
All AML patients with pre-SCT samples available (n=74) | |
| Age: median | 38 (range 12–72) |
| Sex | 54% female |
| Pathological diagnosis at time of allo-SCT | |
| CR1 | 32% |
| CR2 | 30% |
| ⩾CR3 | 3% |
| Active | 35% |
| Three-year outcomes following allo-SCT | |
| Alive | 36% |
| Dead—relapse | 34% |
| Dead—non-relapse | 30% |
| Median survival post allo-SCT | 343 days (range: 6–7000 days) |
Table 2. Individual patient characteristics.
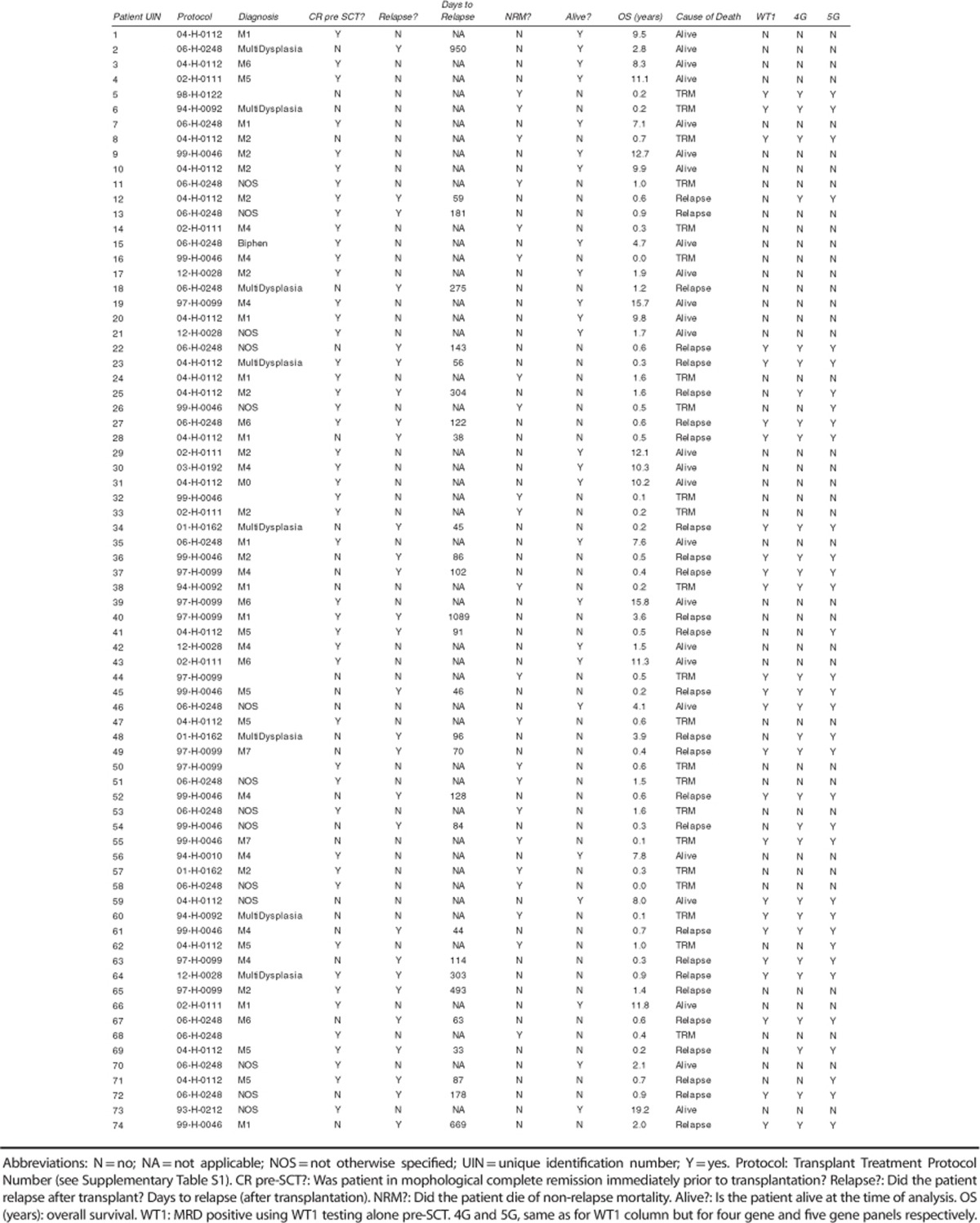
Twenty-two patients suffered non-relapse mortality (30%), 18 within the first year after allo-SCT (24%). Twenty-eight (38%) patients relapsed, a median of 99 days after allo-SCT (range 33–1089 days); relapse was fatal in 96% of cases, with one patient still alive 81 days after relapse at the time of analysis. The median survival after relapse was 85 days (range 0–1328). Twenty-six patients (35%) had active disease immediately prior to allo-SCT; only three were alive at time of analysis (12%) including one with ongoing relapse.
Design and validation of a multigene (MG-MRD) RQ–PCR array for AML
We previously reported expression of potential leukemia-associated antigens51 using a highly reproducible RQ–PCR array in primary untreated human AML samples.47 We reanalyzed this data set to focus on 13 out of 30 peripheral blood samples that lacked WT1 expression of at least 50-fold higher than the median seen in normal individuals (Figure 1). We identified that a combination of cyclin A1 (CCNA1, gene ID: 8900), proteinase 3 (PRTN3, gene ID: 5657), preferentially expressed antigen in melanoma (PRAME, Gene ID: 23532) and mesothelin (MSLN, Gene ID: 10232) (Supplementary Table S3) could provide at least one gene overexpressed at least 50-fold in 9 of these 13 patients, which together with WT1 would provide at least one gene highly overexpressed in 26 of all 30 newly diagnosed untreated patients (that is, 87% sensitivity for at least one 50-fold overexpressed gene).
Figure 1.

Overexpressed genes in AML patients with low levels of WT1 expression. Samples of peripheral blood from 30 newly diagnosed, untreated, AML patients were assayed for gene overexpression by quantitative real-time PCR as previously described.47 Data were reanalyzed to identify genes overexpressed at least 50-fold in those patients with less than 50-fold WT1 overexpression compared with the normal donors. Red: 50–99-fold overexpression; bright red: 100-fold or greater overexpression; black: not detected; white: transcript detected but less than 50-fold overexpressed compared with normal donors. CCNA1, cyclin A1; MSLN, mesothelin; PRAME, preferentially expressed antigen in melanoma; PRTN3, proteinase 3; WT1, Wilms tumor 1.
A custom RQ–PCR array containing these genes together with the housekeeping gene c-abl (ABL, gene ID: 25) was then tested using peripheral blood from 50 reportedly cancer-free healthy donors (Figure 2; Supplementary Figure S2). For each gene tested, a single high-expression outlier was removed. For WT1, PRAME, CCNA1 and PRTN3, a threshold value for MRD was then established as this 98th percentile expression level seen in healthy donors plus the addition of a ‘cushion' of one half of s.d. (Figure 2). This is referred to as the 4-gene panel (4G-MRD), with elevation of any gene above that threshold classified as 4G-MRD positive. An exception to this method was used for MSLN, as a large s.d. was observed in expression levels in normal donors; the threshold value was therefore determined based on the maximum value seen in pre-transplant samples of those who did not relapse post transplant. Accordingly, as the set used for MSLN threshold discovery is part of the cohort under investigation, further validation will be required, and results from this ‘5G-MRD' panel are therefore reported separately. As before, elevation of any one of the five genes above the threshold was considered as MRD.
Figure 2.

Patterns of MG-MRD expression. Level of expression of the constituent gene transcripts of the MG-MRD array in normal healthy donor control and pre-transplant AML patient peripheral blood samples. Gene expression normalized to expression of the ABL control gene. Selected threshold values are indicated by a horizontal dotted line. Two non-relapsing patients with pathologist-detectable disease pre-SCT also had 4G-MRD values above the threshold, one with both WT1 and CCNA1, the other with WT1 and PRTN3. Several patients in this historical self-described healthy donor cohort had MSLN expression above the selected threshold; MSLN is not a specific AML antigen and has also been described as being overexpressed in a variety of solid tumors.62 CCNA1, cyclin A1; MSLN, mesothelin; PRAME, preferentially expressed antigen in melanoma; PRTN3, proteinase 3; WT1, Wilms tumor 1.
Comparison between traditional BM pathological diagnosis and peripheral blood molecular testing
Pathological examination of the BM for assessment of both treatment response and relapse is highly integrated into our care of patients with AML.48 We wished to test how our peripheral blood-based molecular assay compared with traditional BM testing in the assessment of clinically evident AML disease burden prior to allo-SCT. Twenty-six patients (35%) had a clinical diagnosis of active disease prior to transplant based on BM examination (Table 1). Molecular testing of peripheral blood from the same pre-transplantation time period identified 22 patients using WT1 testing (85% concordance) or 24 using multiple gene MRD (MG-MRD) testing (92% concordance) (Figure 3a). The two patients identified by both morphological criteria and MG-MRD, but not WT1 testing alone, suffered from very early relapse (84 and 96 days) and are therefore likely to represent active (but WT1 negative) disease at the time of allo-SCT.
Figure 3.

MG-MRD correlates with pathological diagnosis and relapse risk. (a) Peripheral blood-based MG-MRD testing has good concordance with pathologist BM diagnosis pre-SCT. Blue: pathologist determination of active disease based on clinical examination of BM (Path+), but negative for peripheral blood MRD testing. Purple: both BM pathological diagnosis and MRD positive. Red: negative for active AML by pathologist examination (‘remission marrow'), but positive for residual disease by peripheral blood-based MRD testing. (b) MG-MRD can identify patients mistakenly classified as low risk by WT1 MRD. Patients with pre-SCT positivity for WT1 represent the high-risk group. MG-MRD testing can identify additional high-risk individuals from the ‘low risk' WT1-negative group. The mortality in the additional nine patients (12%) reclassified as high risk was 100%. Six of those nine patients reclassified as high risk by MG-MRD were in a CR pre-transplant. (c) Pre-SCT MG-MRD testing improves prediction of early clinical relapses post SCT. All 28 relapses post SCT are plotted by day of clinical relapse aligned with the result of pre-SCT MRD testing. MG-MRD prior to transplantation correctly predicted all relapses in the first 180 days after SCT and was particularly useful in correctly identifying those at risk of early (that is, before median relapse of 99 days) relapse but not identified by WT1 testing. Patients relapsing at 33, 56, 59, 87, 91, 122, 181, 303, 304, 493 and 1089 days post SCT were in a CR prior to SCT (bold).
One of the two patients previously categorized as having active disease by BM examination but not identified by MG-MRD molecular testing of peripheral blood was an atypical case with only 5% blasts prior to transplant, but with cytogenetics showing a leukemia-defining 8,21 translocation in 4 of 20 metaphases. This patient suffered relapse 275 days after transplantation.
Risk stratification based on BM examination or WT1 testing
Our hypothesis was that highly sensitive detection of disease burden prior to allo-SCT could offer improved prediction of both relapse and survival. Traditional response criteria based on BM examination could risk-stratify these 74 patients into two groups: the ‘CR' group (n=48) with a 3-year OS of 48%, and a group of 26 patients with active disease at the time of allo-SCT with a 3-year OS of only 15%.
Peripheral blood testing for WT1 overexpression identified 25 patients as positive who experienced a post-SCT 3-year OS of only 8%. WT1 testing, however, failed to predict 9 of 24 patients who relapsed in the first year post allo-SCT (63% sensitivity) (Figure 3c; Table 3). For patients in CR before SCT, WT1 correctly predicted just three of nine relapses in the year after SCT. Excluding patients who did not have the opportunity to relapse due to non-relapse mortality, WT1 has an excellent positive predictive power of 83–100% for occurrence of relapse in the year following transplantation, but failed to identify the majority who relapsed in this time period (Figure 4b; Table 3).
Table 3. Summary of MRD test performance characteristics (relapse 1 year post SCT).
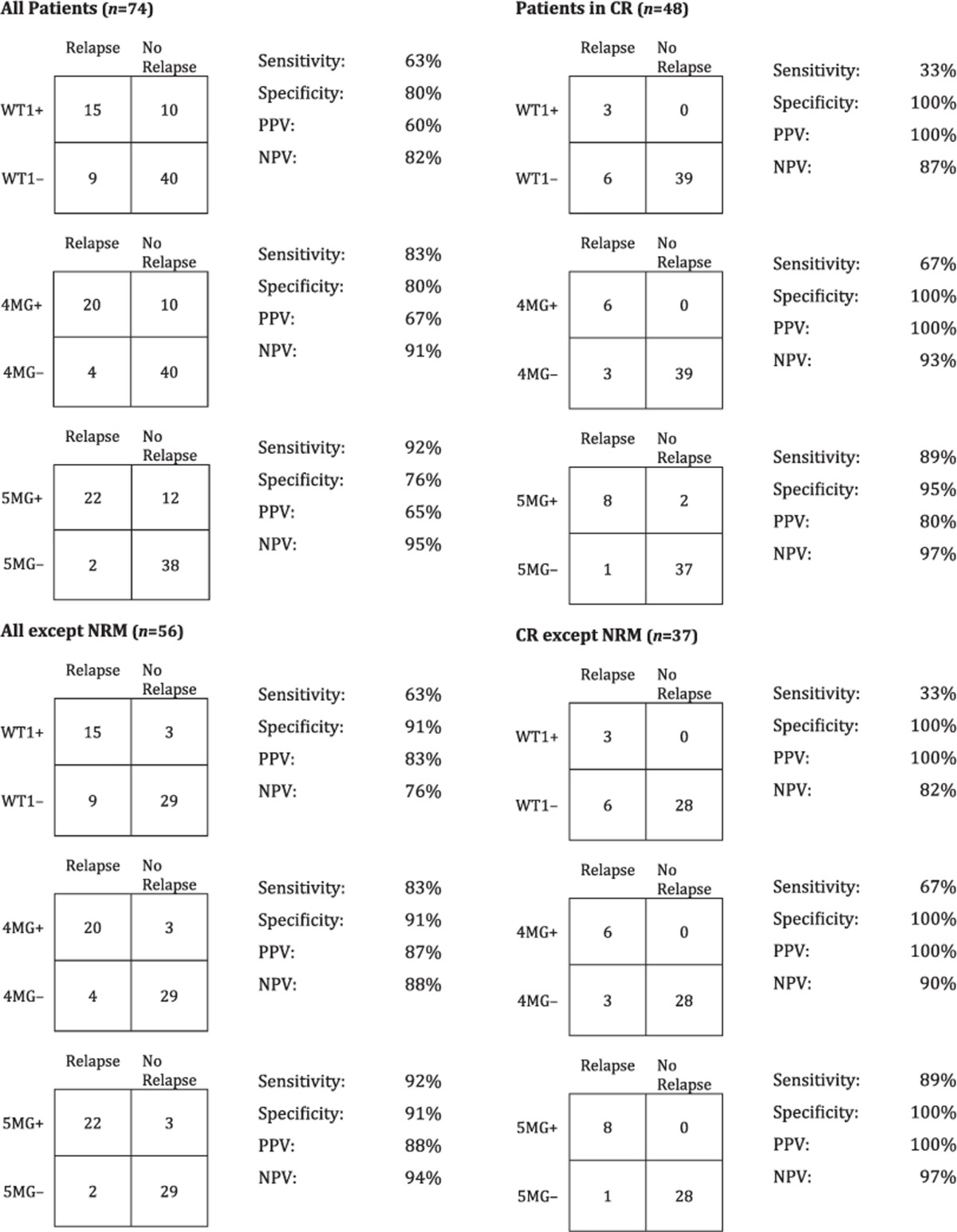
Figure 4.

MG-MRD allows stratification into highly polarized groups for survival and relapse risk. (a) Three-year OS. MG-MRD can effectively segregate patients based on pre-SCT peripheral blood gene expression profile into groups with high and low risk of survival following transplantation. Color pie chart below survival curve illustrates the fraction of the entire cohort triaged to the high-risk category (red) based on the MRD method used. (b) Relapses in the first year after transplantation. Including only patients in pathologist-confirmed CR prior to transplantation and excluding patients dying of non-relapse causes. The green pie chart illustrates sensitivity of each pre-SCT MRD test to predict relapse in the year following transplantation. Statistical analysis was performed using GraphPad Prism with comparison between survival and relapse curves performed using the log-rank (Mantel–Cox) test.
Pre-SCT multigene MRD testing for prediction of post-SCT outcomes
On the basis of the premise that the addition of individually suboptimal secondary MRD assays to the backbone of WT1 testing would increase the overall sensitivity of MRD assessment, we applied the MG-MRD array to peripheral blood samples from this same pre-transplant time point using either WT1 plus three other genes (4G-MRD) or 4G-MRD plus a fifth gene (5G-MRD). MG-MRD risk stratification was superior to the use of WT1 RQ–PCR-based MRD alone. Use of additional genes in the MRD assay correctly reclassified patients assigned by WT1 testing to a ‘low risk' MRD-negative category into a ‘high risk' MRD-positive category (Figure 3b). In total, all nine patients additionally identified using MG-MRD from the cohort designated as ‘low risk' based on testing negative for WT1 alone relapsed and/or died in the first year after transplantation (Figure 3b). Seven of these nine patients had been determined to be in pathological CR prior to allo-SCT. Six relapsed within 100 days of allo-SCT (four had been determined clinically to be in CR at time of allo-SCT), another relapsed 304 days after transplant and two died of non-relapse mortality in the first year after SCT. The sensitivity in predicting clinical relapse in the year after transplant was improved (92%) compared with using WT1 alone (63%). Importantly pre-SCT MG-MRD testing correctly predicted every case of early relapse within 100 days after allo-SCT compared with just 57% using WT1 alone (Figure 3c). The 46% of patients who were 5G-MRD positive prior to transplantation had a 3-year OS of just 9% and experienced 22 out of the total of 24 relapses observed in the first year following transplantation.
In patients entering transplant in CR, the improvement in sensitivity of pre-allo-SCT MRD assessment using MG-MRD (89%) versus WT1 alone (33%) to predict 1-year post-allo-SCT relapse was pronounced (Figure 4b; Table 3) and statistically significant (P=0.031, 95% confidence interval: 0.0–0.821, NcMemar's exact test, Supplementary Figure S3). There was no statistically significant difference in specificity between these tests. MG-MRD prior to allo-SCT identified eight of the nine CR patients who relapsed in the first year (Figures 3c and 4b). MG-MRD in patients in CR prior to transplant was associated with a positive predictive value of 100% for death following transplant (all 4G-MRD-positive patients relapsed and died; all 5G-MRD-positive patients died, 80% from relapse, Figure 3a).
Discussion
WT1 remains the best single universal molecular marker for detection of AML minimal residual disease.27, 29, 31, 34, 35, 39, 52 It is, however, an imperfect test suitable for use in MRD detection in only ~50–75% cases of AML27, 28, 47 whilst also exhibiting significantly different patterns of expression in different AML subtypes.27, 28, 32 Rather than searching for a single superior alternative marker to WT1, we attempted to mitigate the limitations of this good, but suboptimal, test of AML MRD by the creation of a panel that allowed detection of not only WT1 but also of other genes that may be overexpressed in those AML samples with lower levels of WT1 expression (Figure 1). In prior studies, those AML patients in CR without detectable WT1 expression prior to allo-SCT were assigned to a ‘low risk' group with a reported relapse rate between 20%34 and 27%,31 which compares well with the 24% 3-year relapse rate observed in the WT1-negative group in this large retrospective study. By incorporating other genes in addition to WT1 for the detection of MRD, our MG-MRD assay correctly reclassified, even when assessed prior to allo-SCT, between 10 and 20% of patients from the WT1 MRD-negative group into a high-risk group with 100% mortality (Figures 3 and 4). In this historical single-institution cohort, the 49% of patients (36/74) with detectable disease based on either traditional BM morphological examination and/or peripheral blood 5G-MRD positivity prior to allo-SCT experienced a 3-year relapse-free survival rate of only 6%.
Active disease identified by BM examination prior to allo-SCT is a well-known risk factor for relapse and death post allo-SCT,53 and in our series was associated with a mortality rate of 88% (23 of 26 patients). Notably, even for those patients in CR prior to allo-SCT, the presence of pre-transplantation 5G-MRD positivity in peripheral blood testing (seen in 10 patients, Figures 3a and 4b) was associated with survival statistically indistinguishable from those with pathologist BM-based diagnosis of active disease (P=0.78, curve comparison by a log-rank Mantel–Cox test, Supplementary Figure 1) and with a 100% mortality rate (at least 80% of which was attributable to relapse).
Testing peripheral blood prior to transplantation, 4G- and 5G-MRD assays identified 20 or 22, respectively, of the 24 patients who would suffer hematological relapse in the first year after allo-SCT (Figure 3c). Excluding those who died of the competing risk of non-relapse mortality, all three ‘false positive' tests were also seen with WT1 testing, and no additional false-positive results were created by MG-MRD testing. Notably, one of these three ‘false positives' did relapse in the second year after transplant. The two others were also positive for active disease prior to transplantation by marrow morphology, and likely represent successful cures by allo-SCT.
Although this multigene MRD assay represents an advance in high-sensitivity RQ–PCR-based detection of AML, several caveats should be noted. (1) Sensitivity to predict post-allo-SCT relapse was improved compared with use of WT1 alone, but remained less than 100% (67–92%, Table 3) due to the inability to detect late relapses based solely on pre-SCT assessment (Figure 3c). Surveillance monitoring for MRD in the post-SCT period may mitigate this. (2) Although prediction based on detectable WT1 expression in peripheral blood (that is, above the background threshold level seen in normal healthy donors) was in good agreement with that reported in prior studies,27, 28, 47 we did not use the ELN-validated WT1 PCR test27 as a direct comparator, and there is a possibility that the performance characteristics of the WT1 test could be further improved. Nevertheless, WT1 itself was also a core constituent of the multigene panel, and further optimization of the primer combinations used to detect other genes assayed by our multigene MRD panel is possible. Future work will evaluate use of more sophisticated methods for threshold setting, including machine-learning approaches.54 (3) Thresholds most predictive of relapse risk may vary between transplants with different conditioning and immunosuppression regimens and different AML subtypes. The transplant protocols in this series (Supplementary Table S1) were broadly similar. Future work will also collect detailed information on AML cytogenetic and molecular subtypes, which was not available here, and test a wider variety of transplant approaches, including reduced intensity conditioning and alternative donor transplants. (4) Finally, it should be emphasized that findings from this proof-of-concept study require validation in an independent cohort to determine the optimum sequence and specificity of the panel of primers that give maximum breadth of coverage and reliably detect AML in low-disease burden states. For example, although only two (of 24) relapses in the year following allo-SCT were not predicted by the 5G-MRD panel tested on pre-allo-SCT peripheral blood samples, it is notable that one of these had a diagnosis of AML based on a karyotype that included the leukemia-defining 8,21 translocation in the context of a low (~5%) level of CD34-positive cells on immunohistochemistry of BM. Other reports have previously commented on the trend for lower WT1 expression in AML containing the t(8;21) fusion protein AML1-ETO.27, 28 Subsequent iterations of this MG-MRD array will now also contain primers specific for t(8;21), Inv16,55 mutated NPM156 and potentially also other recurrent1, 57, 58 or personalized somatic mutations seen in AML. The other patient falsely classified as negative, based on pre-SCT MG-MRD testing, had a confirmed CR by BM examination but had a history of CNS involvement by AML.
The ability, using MG-MRD, to identify a group with exceptionally high post-transplant mortality primarily from disease relapse sets the stage for MRD-based clinical interventions based on relapse risk. This could include maintenance therapy with additional agents post allo-SCT to prevent relapse in MRD-positive patients,59, 60, 61 and de-escalation of conditioning in patients that are MRD negative prior to allo-SCT to avoid excessive toxicity. In our series, 13 of the 15 deaths seen in the 3 years following transplantation in the 38 pathological CR, MRD-negative patients were due to non-relapse causes. Direct assessment of tumor burden, and hence relapse risk, prior to transplantation could potentially help ‘tune' immunosuppression and T-cell dose strategy, and given that some MRD-negative patients may already be cured prior to transplant may be able to identify a subset of individuals who could be spared allo-SCT.
Acknowledgments
We thank Alan Hoofring and Ethan Tyler of the NIH Medical Arts Service for assistance. This work was supported by the Intramural Research Program of the National Heart, Lung, Blood Institute of the National Institutes of Health. JC was supported by NIH Award Number T32CA154274 from the National Cancer Institute.
Author Contributions
CSH designed and performed research, analyzed and interpreted data, and wrote the manuscript. MG performed research, analyzed and interpreted data. NFH, JT and KSM performed research. JC analyzed and interpreted data. KRC, KL and NJ collected and interpreted data. MB and AJB conducted the clinical protocols and interpreted data.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on Bone Marrow Transplantation website (http://www.nature.com/bmt)
Supplementary Material
References
- Cancer Genome Atlas Research N Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valk PJ, Verhaak RG, Beijen MA, Erpelinck CA, Barjesteh van Waalwijk van Doorn-Khosrovani S, Boer JM, et al. Prognostically useful gene-expression profiles in acute myeloid leukemia. N Engl J Med. 2004;350:1617–1628. doi: 10.1056/NEJMoa040465. [DOI] [PubMed] [Google Scholar]
- Bullinger L, Dohner K, Bair E, Frohling S, Schlenk RF, Tibshirani R, et al. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N Engl J Med. 2004;350:1605–1616. doi: 10.1056/NEJMoa031046. [DOI] [PubMed] [Google Scholar]
- Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366:1079–1089. doi: 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimwade D, Walker H, Oliver F, Wheatley K, Harrison C, Harrison G, et al. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children's Leukaemia Working Parties. Blood. 1998;92:2322–2333. [PubMed] [Google Scholar]
- Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–510. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroger N, Bacher U, Bader P, Bottcher S, Borowitz MJ, Dreger P, et al. NCI First International Workshop on the Biology, Prevention, and Treatment of Relapse after Allogeneic Hematopoietic Stem Cell Transplantation: report from the Committee on Disease-Specific Methods and Strategies for Monitoring Relapse following Allogeneic Stem Cell Transplantation. Part I: Methods, acute leukemias, and myelodysplastic syndromes. Biol Blood Marrow Transplant. 2010;16:1187–1211. doi: 10.1016/j.bbmt.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne AS, Giralt S, Kroger N, Bishop MR. Proceedings from the National Cancer Institute's Second International Workshop on the biology, prevention, and treatment of relapse after hematopoietic stem cell transplantation: introduction. Biol Blood Marrow Transplant. 2013;19:1534–1536. doi: 10.1016/j.bbmt.2013.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder T, Czibere A, Platzbecker U, Bug G, Uharek L, Luft T, et al. Azacitidine and donor lymphocyte infusions as first salvage therapy for relapse of AML or MDS after allogeneic stem cell transplantation. Leukemia. 2013;27:1229–1235. doi: 10.1038/leu.2013.7. [DOI] [PubMed] [Google Scholar]
- Bacher U, Talano JA, Bishop MR. Monitoring and prevention of relapse after allogeneic hematopoietic cell transplantation for myeloid malignancies. Biol Blood Marrow Transplant. 2012;18:S62–S73. doi: 10.1016/j.bbmt.2011.10.028. [DOI] [PubMed] [Google Scholar]
- Barrett AJ, Battiwalla M. Relapse after allogeneic stem cell transplantation. Expert Rev Hematol. 2010;3:429–441. doi: 10.1586/ehm.10.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman JM, Gale RP. What does MRD in leukemia really mean. Leukemia. 2014;28:1131. doi: 10.1038/leu.2013.318. [DOI] [PubMed] [Google Scholar]
- Appelbaum FR. Measurement of minimal residual disease before and after myeloablative hematopoietic cell transplantation for acute leukemia. Best Pract Res Clin Haematol. 2013;26:279–284. doi: 10.1016/j.beha.2013.10.008. [DOI] [PubMed] [Google Scholar]
- Carlson KS, Guzman ML. Is minimal residual disease monitoring clinically relevant in adults with acute myelogenous leukemia. Curr Hematol Malig Rep. 2013;8:109–115. doi: 10.1007/s11899-013-0157-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossenkoppele G, Schuurhuis GJ. MRD in AML: time for redefinition of CR. Blood. 2013;121:2166–2168. doi: 10.1182/blood-2013-01-480590. [DOI] [PubMed] [Google Scholar]
- Paietta E. Minimal residual disease in acute myeloid leukemia: coming of age. Hematology Am Soc Hematol Educ Program. 2012;2012:35–42. doi: 10.1182/asheducation-2012.1.35. [DOI] [PubMed] [Google Scholar]
- Hokland P, Ommen HB. Towards individualized follow-up in adult acute myeloid leukemia in remission. Blood. 2011;117:2577–2584. doi: 10.1182/blood-2010-09-303685. [DOI] [PubMed] [Google Scholar]
- Hokland P, Ommen HB, Nyvold CG, Roug AS. Sensitivity of minimal residual disease in acute myeloid leukaemia in first remission—methodologies in relation to their clinical situation. Br J Haematol. 2012;158:569–580. doi: 10.1111/j.1365-2141.2012.09203.x. [DOI] [PubMed] [Google Scholar]
- Buckley SA, Appelbaum FR, Walter RB. Prognostic and therapeutic implications of minimal residual disease at the time of transplantation in acute leukemia. Bone Marrow Transplant. 2013;48:630–641. doi: 10.1038/bmt.2012.139. [DOI] [PubMed] [Google Scholar]
- Walter RB, Buckley SA, Pagel JM, Wood BL, Storer BE, Sandmaier BM, et al. Significance of minimal residual disease before myeloablative allogeneic hematopoietic cell transplantation for AML in first and second complete remission. Blood. 2013;122:1813–1821. doi: 10.1182/blood-2013-06-506725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdan E, Boissel N, Chevret S, Delabesse E, Renneville A, Cornillet P, et al. Prospective evaluation of gene mutations and minimal residual disease in patients with core binding factor acute myeloid leukemia. Blood. 2013;121:2213–2223. doi: 10.1182/blood-2012-10-462879. [DOI] [PubMed] [Google Scholar]
- Yin JA, O'Brien MA, Hills RK, Daly SB, Wheatley K, Burnett AK. Minimal residual disease monitoring by quantitative RT-PCR in core binding factor AML allows risk stratification and predicts relapse: results of the United Kingdom MRC AML-15 trial. Blood. 2012;120:2826–2835. doi: 10.1182/blood-2012-06-435669. [DOI] [PubMed] [Google Scholar]
- Zhu HH, Zhang XH, Qin YZ, Liu DH, Jiang H, Chen H, et al. MRD-directed risk stratification treatment may improve outcomes of t(8;21) AML in the first complete remission: results from the AML05 multicenter trial. Blood. 2013;121:4056–4062. doi: 10.1182/blood-2012-11-468348. [DOI] [PubMed] [Google Scholar]
- Hourigan CS, Karp JE. Minimal residual disease in acute myeloid leukaemia. Nat Rev Clin Oncol. 2013;10:460–471. doi: 10.1038/nrclinonc.2013.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeijlemaker W, Gratama JW, Schuurhuis GJ. Tumor heterogeneity makes AML a "moving target" for detection of residual disease. Cytometry. 2014;86:3–14. doi: 10.1002/cyto.b.21134. [DOI] [PubMed] [Google Scholar]
- Cilloni D, Renneville A, Hermitte F, Hills RK, Daly S, Jovanovic JV, et al. Real-time quantitative polymerase chain reaction detection of minimal residual disease by standardized WT1 assay to enhance risk stratification in acute myeloid leukemia: a European LeukemiaNet study. J Clin Oncol. 2009;27:5195–5201. doi: 10.1200/JCO.2009.22.4865. [DOI] [PubMed] [Google Scholar]
- Ostergaard M, Olesen LH, Hasle H, Kjeldsen E, Hokland P. WT1 gene expression: an excellent tool for monitoring minimal residual disease in 70% of acute myeloid leukaemia patients - results from a single-centre study. Br J Haematol. 2004;125:590–600. doi: 10.1111/j.1365-2141.2004.04952.x. [DOI] [PubMed] [Google Scholar]
- Inoue K, Ogawa H, Sonoda Y, Kimura T, Sakabe H, Oka Y, et al. Aberrant overexpression of the Wilms tumor gene (WT1) in human leukemia. Blood. 1997;89:1405–1412. [PubMed] [Google Scholar]
- Miwa H, Beran M, Saunders GF. Expression of the Wilms' tumor gene (WT1) in human leukemias. Leukemia. 1992;6:405–409. [PubMed] [Google Scholar]
- Pozzi S, Geroldi S, Tedone E, Luchetti S, Grasso R, Colombo N, et al. Leukaemia relapse after allogeneic transplants for acute myeloid leukaemia: predictive role of WT1 expression. Br J Haematol. 2013;160:503–509. doi: 10.1111/bjh.12181. [DOI] [PubMed] [Google Scholar]
- Ommen HB, Nyvold CG, Braendstrup K, Andersen BL, Ommen IB, Hasle H, et al. Relapse prediction in acute myeloid leukaemia patients in complete remission using WT1 as a molecular marker: development of a mathematical model to predict time from molecular to clinical relapse and define optimal sampling intervals. Br J Haematol. 2008;141:782–791. doi: 10.1111/j.1365-2141.2008.07132.x. [DOI] [PubMed] [Google Scholar]
- Rossi G, Carella AM, Minervini MM, Savino L, Fontana A, Pellegrini F, et al. Minimal residual disease after allogeneic stem cell transplantation: a comparison among multiparametric flow cytometry, Wilms tumor 1 expression and chimerism status (complete chimerism versus low level mixed chimerism) in acute leukemia. Leuk Lymphoma. 2013;54:2660–2666. doi: 10.3109/10428194.2013.789508. [DOI] [PubMed] [Google Scholar]
- Kwon M, Martinez-Laperche C, Infante M, Carretero F, Balsalobre P, Serrano D, et al. Evaluation of minimal residual disease by real-time quantitative PCR of Wilms' tumor 1 expression in patients with acute myelogenous leukemia after allogeneic stem cell transplantation: correlation with flow cytometry and chimerism. Biol Blood Marrow Transplant. 2012;18:1235–1242. doi: 10.1016/j.bbmt.2012.01.012. [DOI] [PubMed] [Google Scholar]
- Ogawa H, Tamaki H, Ikegame K, Soma T, Kawakami M, Tsuboi A, et al. The usefulness of monitoring WT1 gene transcripts for the prediction and management of relapse following allogeneic stem cell transplantation in acute type leukemia. Blood. 2003;101:1698–1704. doi: 10.1182/blood-2002-06-1831. [DOI] [PubMed] [Google Scholar]
- Zhao XS, Yan CH, Liu DH, Xu LP, Liu YR, Liu KY, et al. Combined use of WT1 and flow cytometry monitoring can promote sensitivity of predicting relapse after allogeneic HSCT without affecting specificity. Ann Hematol. 2013;92:1111–1119. doi: 10.1007/s00277-013-1733-1. [DOI] [PubMed] [Google Scholar]
- Cilloni D, Gottardi E, De Micheli D, Serra A, Volpe G, Messa F, et al. Quantitative assessment of WT1 expression by real time quantitative PCR may be a useful tool for monitoring minimal residual disease in acute leukemia patients. Leukemia. 2002;16:2115–2121. doi: 10.1038/sj.leu.2402675. [DOI] [PubMed] [Google Scholar]
- Lange T, Hubmann M, Burkhardt R, Franke GN, Cross M, Scholz M, et al. Monitoring of WT1 expression in PB and CD34(+) donor chimerism of BM predicts early relapse in AML and MDS patients after hematopoietic cell transplantation with reduced-intensity conditioning. Leukemia. 2011;25:498–505. doi: 10.1038/leu.2010.283. [DOI] [PubMed] [Google Scholar]
- Zhao XS, Jin S, Zhu HH, Xu LP, Liu DH, Chen H, et al. Wilms' tumor gene 1 expression: an independent acute leukemia prognostic indicator following allogeneic hematopoietic SCT. Bone Marrow Transplant. 2012;47:499–507. doi: 10.1038/bmt.2011.121. [DOI] [PubMed] [Google Scholar]
- Terwijn M, van Putten WL, Kelder A, van der Velden VH, Brooimans RA, Pabst T, et al. High prognostic impact of flow cytometric minimal residual disease detection in acute myeloid leukemia: data from the HOVON/SAKK AML 42A study. J Clin Oncol. 2013;31:3889–3897. doi: 10.1200/JCO.2012.45.9628. [DOI] [PubMed] [Google Scholar]
- Walter RB, Gyurkocza B, Storer BE, Godwin CD, Pagel JM, Buckley SA, et al. Comparison of minimal residual disease as outcome predictor for AML patients in first complete remission undergoing myeloablative or nonmyeloablative allogeneic hematopoietic cell transplantation. Leukemia. 2014;29:137–144. doi: 10.1038/leu.2014.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki T, Fujita H, Fujimaki K, Hosoyama T, Watanabe R, Tachibana T, et al. Clinical significance of minimal residual disease detected by multidimensional flow cytometry: serial monitoring after allogeneic stem cell transplantation for acute leukemia. Leuk Res. 2012;36:998–1003. doi: 10.1016/j.leukres.2012.04.005. [DOI] [PubMed] [Google Scholar]
- Leung W, Pui CH, Coustan-Smith E, Yang J, Pei D, Gan K, et al. Detectable minimal residual disease before hematopoietic cell transplantation is prognostic but does not preclude cure for children with very-high-risk leukemia. Blood. 2012;120:468–472. doi: 10.1182/blood-2012-02-409813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba H, Coustan-Smith E, Cao X, Pounds SB, Shurtleff SA, Wang KY, et al. Comparative analysis of different approaches to measure treatment response in acute myeloid leukemia. J Clin Oncol. 2012;30:3625–3632. doi: 10.1200/JCO.2011.41.5323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubnitz JE, Inaba H, Dahl G, Ribeiro RC, Bowman WP, Taub J, et al. Minimal residual disease-directed therapy for childhood acute myeloid leukaemia: results of the AML02 multicentre trial. Lancet Oncol. 2010;11:543–552. doi: 10.1016/S1470-2045(10)70090-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaso JM, Wang SA, Jorgensen JL, Lin P. Multi-color flow cytometric immunophenotyping for detection of minimal residual disease in AML: past, present and future. Bone Marrow Transplant. 2014;49:1129–1138. doi: 10.1038/bmt.2014.99. [DOI] [PubMed] [Google Scholar]
- Goswami M, Hensel N, Smith BD, Prince GT, Qin L, Levitsky HI, et al. Expression of putative targets of immunotherapy in acute myeloid leukemia and healthy tissues. Leukemia. 2014;28:1167–1170. doi: 10.1038/leu.2014.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell MR, Abboud CN, Altman J, Appelbaum FR, Arber DA, Attar E, et al. Acute myeloid leukemia. J Natl Compr Canc Netw. 2012;10:984–1021. doi: 10.6004/jnccn.2012.0103. [DOI] [PubMed] [Google Scholar]
- McNemar Q. Note on the sampling error of the difference between correlated proportions or percentages. Psychometrika. 1947;12:153–157. doi: 10.1007/BF02295996. [DOI] [PubMed] [Google Scholar]
- Vergara IA, Norambuena T, Ferrada E, Slater AW, Melo F. StAR: a simple tool for the statistical comparison of ROC curves. BMC Bioinformatics. 2008;9:265. doi: 10.1186/1471-2105-9-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hourigan CS, Levitsky HI. Evaluation of current cancer immunotherapy: hemato-oncology. Cancer J. 2011;17:309–324. doi: 10.1097/PPO.0b013e3182341fde. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi G, Carella AM, Minervini MM, Savino L, Fontana A, Pellegrini F, et al. Minimal residual disease after allogeneic stem cell transplant: a comparison among multiparametric flow cytometry, Wilms tumor 1 expression and chimerism status (Complete chimerism versus Low Level Mixed Chimerism) in acute leukemia. Leuk Lymphoma. 2013;54:2660–2666. doi: 10.3109/10428194.2013.789508. [DOI] [PubMed] [Google Scholar]
- Duval M, Klein JP, He W, Cahn JY, Cairo M, Camitta BM, et al. Hematopoietic stem-cell transplantation for acute leukemia in relapse or primary induction failure. J Clin Oncol. 2010;28:3730–3738. doi: 10.1200/JCO.2010.28.8852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candia J, Maunu R, Driscoll M, Biancotto A, Dagur P, McCoy JP, Jr, et al. From cellular characteristics to disease diagnosis: uncovering phenotypes with supercells. PLoS Comput Biol. 2013;9:e1003215. doi: 10.1371/journal.pcbi.1003215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabert J, Beillard E, van der Velden VH, Bi W, Grimwade D, Pallisgaard N, et al. Standardization and quality control studies of 'real-time' quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia - a Europe Against Cancer program. Leukemia. 2003;17:2318–2357. doi: 10.1038/sj.leu.2403135. [DOI] [PubMed] [Google Scholar]
- Kronke J, Schlenk RF, Jensen KO, Tschurtz F, Corbacioglu A, Gaidzik VI, et al. Monitoring of minimal residual disease in NPM1-mutated acute myeloid leukemia: a study from the German-Austrian acute myeloid leukemia study group. J Clin Oncol. 2011;29:2709–2716. doi: 10.1200/JCO.2011.35.0371. [DOI] [PubMed] [Google Scholar]
- Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506:328–333. doi: 10.1038/nature13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hourigan CS. Next Generation MRD. Biol Blood Marrow Transplant. 2014;20:1259–1260. doi: 10.1016/j.bbmt.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan CH, Liu DH, Liu KY, Xu LP, Liu YR, Chen H, et al. Risk stratification-directed donor lymphocyte infusion could reduce relapse of standard-risk acute leukemia patients after allogeneic hematopoietic stem cell transplantation. Blood. 2012;119:3256–3262. doi: 10.1182/blood-2011-09-380386. [DOI] [PubMed] [Google Scholar]
- Hourigan CS, McCarthy P, de Lima M. Back to the future! The evolving role of maintenance therapy after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2014;20:154–163. doi: 10.1016/j.bbmt.2013.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platzbecker U, Wermke M, Radke J, Oelschlaegel U, Seltmann F, Kiani A, et al. Azacitidine for treatment of imminent relapse in MDS or AML patients after allogeneic HSCT: results of the RELAZA trial. Leukemia. 2012;26:381–389. doi: 10.1038/leu.2011.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hucl T, Brody JR, Gallmeier E, Iacobuzio-Donahue CA, Farrance IK, Kern SE. High cancer-specific expression of mesothelin (MSLN) is attributable to an upstream enhancer containing a transcription enhancer factor dependent MCAT motif. Cancer Res. 2007;67:9055–9065. doi: 10.1158/0008-5472.CAN-07-0474. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.