

Abstract
Current management of neuromyelitis optica (NMO) is noncurative and only partially effective. Immunosuppressive or immunomodulatory agents are the mainstays of maintenance treatment. Safer, better-tolerated, and proven effective treatments are needed. The perceived rarity of NMO has impeded clinical trials for this disease. However, a diagnostic biomarker and recognition of a wider spectrum of NMO presentations has expanded the patient population from which study candidates might be recruited. Emerging insights into the pathogenesis of NMO have provided rationale for exploring new therapeutic targets. Academic, pharmaceutical, and regulatory communities are increasingly interested in meeting the unmet needs of patients with NMO. Clinical trials powered to yield unambiguous outcomes and designed to facilitate rapid evaluation of an expanding pipeline of experimental agents are needed. NMO-related disability occurs incrementally as a result of attacks; thus, limiting attack frequency and severity are critical treatment goals. Yet, the severity of NMO and perception that currently available agents are effective pose challenges to study design. We propose strategies for NMO clinical trials to evaluate agents targeting recovery from acute attacks and prevention of relapses, the 2 primary goals of NMO treatment. Aligning the interests of all stakeholders is an essential step to this end.
Neuromyelitis optica (NMO) is a frequently relapsing inflammatory disease with a predilection for optic nerves and spinal cord. The specific biomarker aquaporin-4 (AQP4)–immunoglobulin G (IgG) is detected in most patients,1 but assays for AQP4-IgG vary in sensitivity and specificity.2 In vivo and in vitro studies and favorable response to plasma exchange (PLEX) suggest that AQP4-IgG is pathogenic.3–6 Some patients with NMO may develop symptoms implicating involvement of other CNS regions such as the area postrema and hypothalamus.7,8 Others have limited variants of NMO (e.g., recurrent optic neuritis or recurrent myelitis). Such patients are collectively referred to as having NMO spectrum disorders. In seropositive patients, a confident diagnosis can be made after a single clinical event. While AQP4-IgG detection facilitates diagnosis and predicts relapse,9 an important proportion of cases are seronegative.10,11
Acute NMO attacks are generally more severe than those of multiple sclerosis (MS) and may be fatal.12,13 Mortality estimates have improved from 30% at 5 years,12 to 9% at 6 years.13 Unlike MS, disability in NMO occurs as cumulative sequelae of attacks rather than during a secondary progressive phase.14 Thus, minimizing the frequency and severity of attacks is a primary therapeutic goal. IV corticosteroids and, in refractory cases, PLEX are currently used to treat acute attacks. Immunosuppressive agents (e.g., azathioprine, mycophenolate mofetil, rituximab, and corticosteroids) are prescribed to reduce attack frequency, based on small, uncontrolled studies.15–17 These agents are collectively referred to as “empiric” treatments herein, to avoid any suggestion that a standard of NMO therapy has been established.
Several obstacles have historically limited clinical trials in NMO. However, these barriers are now mitigated by important advances that increase the feasibility of informative trials (table 1). An increasing number of therapeutic candidates, including those repurposed for evaluation in NMO, and emerging candidates specific to AQP4 (e.g., aquaporumab),18 create an immediate need for definitive NMO clinical trials. Promising repurposed agents include the following:
Rituximab (anti-CD20 monoclonal antibody targeting B cells)19,20
Eculizumab (anti-C5 monoclonal antibody targeting complement)21
Tocilizumab (anti-IL-6R monoclonal antibody targeting T- and B-cell activation, Th17 differentiation, and plasmablast survival)22–24
Table 1.
Obstacles and advances influencing development of clinical trials for NMO
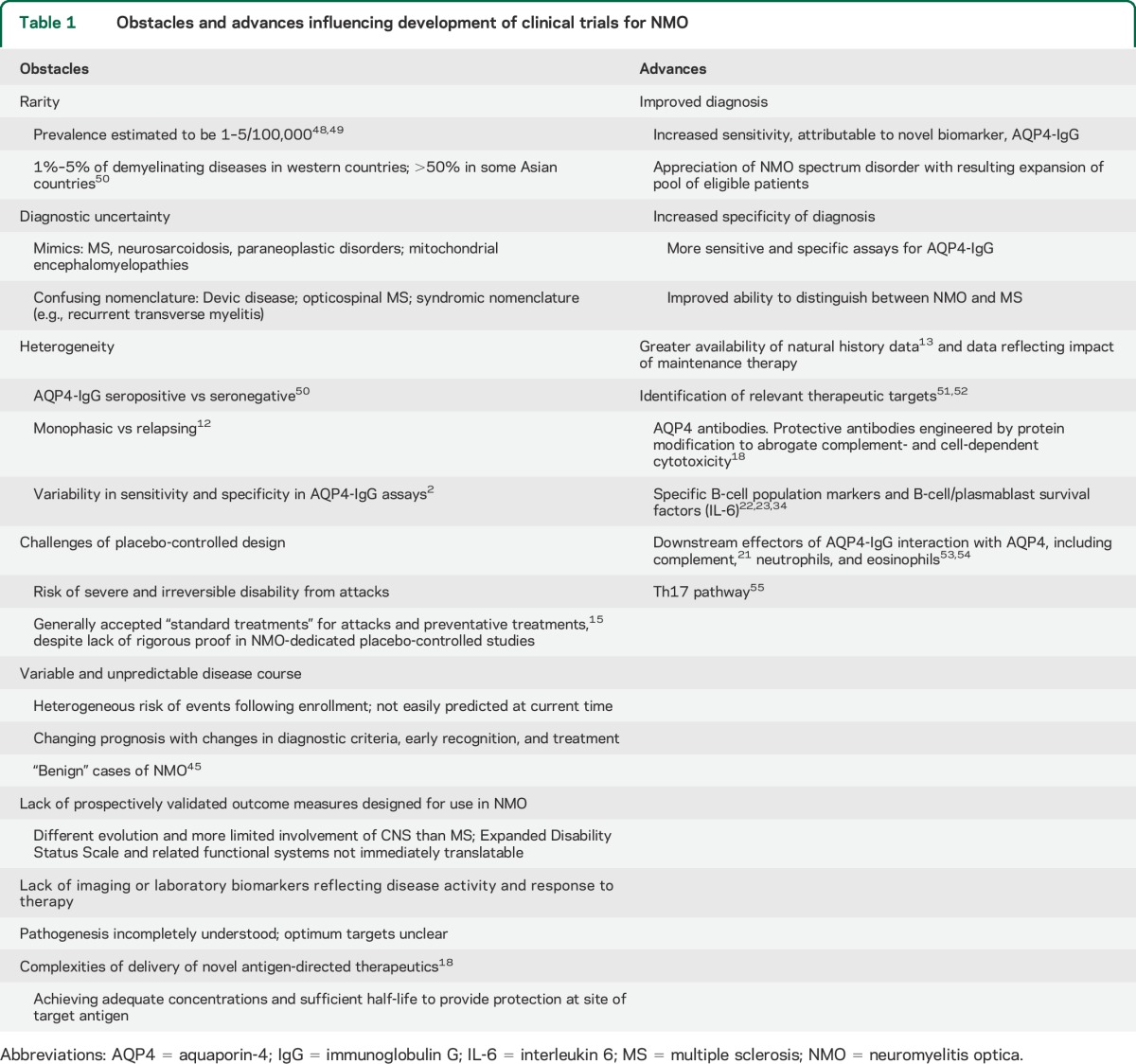
Clinical trial templates developed for MS are not directly applicable to NMO (table 2). Herein, we review issues pertinent to clinical trial development in NMO based on discussions among clinicians, researchers, industry partners, and patients at research-focused meetings of the Guthy-Jackson Charitable Foundation. Key concepts were further refined by all coauthors, including one serving in a regulatory capacity.
Table 2.
Selected features differentiating NMO from MS

OVERARCHING CONCEPTS FOR NMO CLINICAL TRIAL DESIGN
No drug or treatment has been proven to be safe and effective in NMO in randomized, controlled studies, and none has received regulatory approval.
Opinions vary widely among investigators regarding ethics of placebo-controlled studies for maintenance treatment of NMO. NMO-associated transverse myelitis and optic neuritis attacks can result in devastating neurologic consequences.12,13 Moreover, retrospective analyses suggest that current empiric therapies may be moderately effective.20,25,26 However, ascertainment bias and biases inherent in treatment discontinuation and reporting of attacks in retrospective studies preclude conclusions regarding efficacy of any currently used agent. Furthermore, rigorous studies frequently contradict theoretical benefit27 or reveal unexpected toxicity,28 especially when agents are tested in combination.29 Some investigators and regulatory agencies regard the evidence supporting effectiveness of current empiric treatments sufficiently inadequate that placebo-controlled studies in NMO are justified for several reasons, including:
Noninferiority studies require an established standard of treatment that currently does not exist.
NMO clinical trials must address safety as well as efficacy both for acute and chronic exposure, considering that long-term therapy is likely necessary.
Sensitive and meaningful NMO-customized outcomes measures are needed to inform treatment.
Attacks, rather than neurodegeneration, are the principal contributors to NMO-related disability.14 Thus, frequency and severity of attacks are the most suitable primary endpoints in NMO clinical trials. Validated criteria, including imaging evidence, that define specific signs or symptoms of an acute attack are necessary for optimal trial design. Whether nonattack-related effects of the disease (e.g., changes in cognitive function, behavior, or other neurologic functions) occur in NMO is uncertain. Likewise, laboratory-based biomarkers reflecting or predicting NMO attack frequency, severity, or therapeutic efficacy must be validated before consideration as surrogate outcomes.30–34 Poor cross-sectional correlation of AQP4 autoantibody titers with NMO disease activity and severity exemplifies this issue. Longitudinal assessment of titers within individuals may be more promising than between individuals, although this has not been adequately explored.35,36 Similar considerations apply to serum cytokine levels, immune cell profiles,34 or surrogates of tissue-specific injury (e.g., CSF glial fibrillary astrocytic protein37). CSF parameters are difficult to measure serially, and MRI signals are highly variable among individuals. Brain MRI abnormalities in NMO occur in selective regions, are difficult to quantify, and have inconsistent association with clinical disability. Changes in retinal nerve fiber layer thickness over time, as measured by optical coherence tomography, are more striking in NMO than in MS.38 However, some patients with NMO spectrum disorders do not have optic nerve events, and changes in optical coherence tomography are typically delayed after acute events.
SPECIFIC CONCEPTS FOR NMO CLINICAL TRIAL DESIGN
Two clinical situations represent major opportunities to mitigate worsening of the disease: (1) treatment of acute events, and (2) maintenance treatment (relapse prevention).
Acute event (onset or relapse).
Synopsis.
The key interacting elements in acute NMO lesions are AQP4-IgG, AQP4, complement, and granulocytes. Typically, acute events are treated with high-dose pulse corticosteroids, and with PLEX in refractory cases. The following considerations apply to clinical trials of treatment of acute NMO episodes.
Inclusion/exclusion criteria.
An issue common to all potential NMO clinical trials is whether participation should be confined to AQP4-IgG–seropositive individuals (table 3). Restricting enrollment to seropositive patients would enhance cohort homogeneity and diagnostic certainty, excluding NMO mimics, such as sarcoidosis or paraneoplastic disorders. The majority of potential study participants are seropositive. However, current serologic assays have imperfect sensitivity and may not predict treatment response.39 To address potential for differential efficacy, patients may need to be randomized based on serostatus.40
Table 3.
Pros and cons of design issues in NMO clinical trials
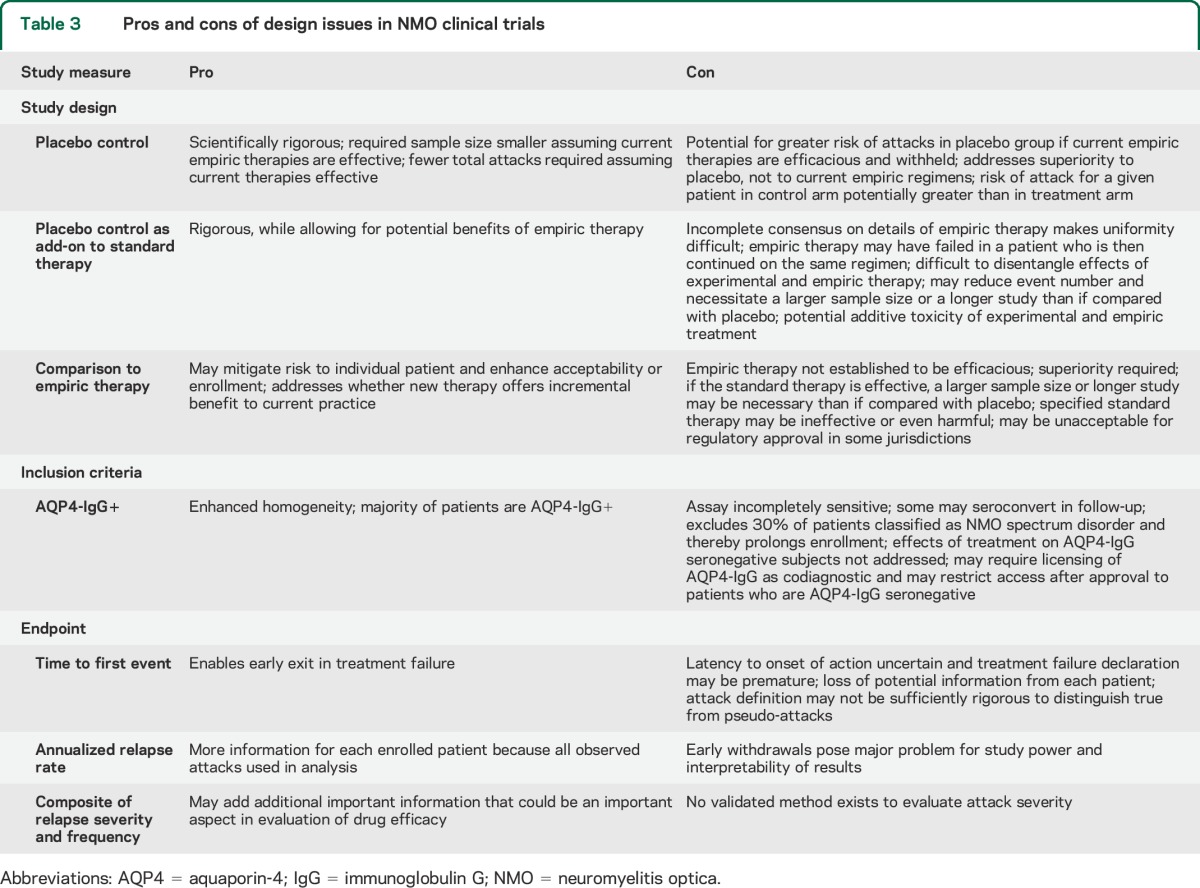
Clinical trial designs.
The potential for severe and irreversible morbidity during acute NMO attack is particularly relevant in evaluating the acceptability and ethical correctness of trial design (Table 3 and figure 1).
Figure 1. Study designs for treatment of attacks.

(A) An experimental treatment (Expt Tx) is compared with placebo. At enrollment while in relapse, a patient would either discontinue prior maintenance treatment (placebo-only design) or not (placebo-controlled add-on). The study design could allow patients whose initial treatment failed to cross over to the other treatment regimen. (B) An experimental treatment is added to a standard regimen for management of an acute attack such as pulse high-dose IV corticosteroids or plasma exchange (PLEX). The treatment may be used along with the other empiric treatment from the outset or after a predefined period of initial treatment in patients who have met failure criteria.
Single agent vs placebo.
Randomized placebo-controlled trials established that corticosteroids shorten recovery of acute optic neuritis and MS relapses (figure 1A).41,42 Although no comparable studies exist in NMO, widespread corticosteroid use and acceptable short-term adverse event profile make it difficult to prohibit their use. Following treatment with corticosteroids, subjects are randomized to an experimental drug vs placebo, either after or concurrent to their treatment with corticosteroid therapy. PLEX was superior to sham pheresis in a controlled study of acute, severe attacks of CNS demyelinating disease that failed corticosteroid treatment; that study included 2 patients with NMO.43 Subsequent uncontrolled prospective and retrospective clinical studies in patients with NMO also reported favorable outcomes. In the only sham-controlled study, patients failing the initial randomized treatment crossed over to the comparator, allowing both intra- and interindividual comparisons of PLEX efficacy with sham. The opportunity for favorable outcomes may decline as the interval from treatment initiation lengthens and neurologic injury becomes irreversible; therefore, it is necessary to consider the sequence of treatment in the analysis. Crossover of subjects who do not improve is also a risk mitigation and recruitment facilitation strategy by guaranteeing access to the active treatment in either the first or second treatment phase.
Add-on to corticosteroid treatment.
A randomized, placebo-controlled study of an add-on therapy may be appropriate for agents with theoretical additive or synergistic benefit (e.g., addition of a granulocyte inhibitor or complement inhibitor to pulse corticosteroid treatment) (figure 1B). If combination therapy proves efficacious, follow-up studies would be necessary to evaluate superiority of the experimental drug as a single agent.
Outcome measures.
Evaluation of attack-specific outcomes requires that neurologic deficits be assessed and recovery quantified with pertinent impairment scales. An integrated estimate of recovery (e.g., mild, moderate, marked) could be pooled regardless of the site of the attack. The proportion of patients who achieve these levels of recovery could be compared across treatments.43 Thus, patients having a variety of neurologic deficits (e.g., visual, motor, or sensory) could be included in a single study. Recovery of the patient-specific “targeted neurologic deficit” is a focused and relevant outcome43 and is typically assessed within 2 to 4 weeks after an event. Favorable treatment-related outcomes generally occur rapidly but delayed benefit is also of interest.
Attack prevention.
Synopsis.
Attack prevention studies are typically offered to patients in remission who may more favorably consider participation in a placebo-controlled trial. Current preventive regimens are nonspecific, and include low-dose oral corticosteroids and lymphocyte-directed immunosuppressants. Newer strategies may target upstream mechanisms involved in AQP4-IgG generation, such as specific plasmablast expansion,34 CNS trafficking, and autoreactive T-cell activation.44 The following considerations are relevant to clinical trials evaluating maintenance treatments.
Inclusion/exclusion criteria.
Restricting enrollment to seropositive individuals was discussed in the context of acute disease (table 3). Comorbidities, such as infections, immunodeficiencies, and agent-specific risk factors, typically exclude participation. A minimum level of recent disease activity is required to exclude subjects with quiescent disease. However, targeting patients with high levels of recent disease activity for enrollment to enhance study power may limit the ability to generalize results. Furthermore, the attack frequency may regress to a lower mean event rate on study.
Clinical trial designs.
Three potential clinical trial strategies are considered for phase IIb/III maintenance studies.
Experimental agent vs placebo.
Placebo-controlled studies are the standard for pivotal trials (figure 2A). We propose several mitigating strategies to reduce the risk of a disabling attack during placebo exposure, including the following:
Liberal “escape” criteria in the event of treatment failure (e.g., a single attack)
Ratio of subjects randomized to experimental treatment vs placebo >1:1, reducing the odds that a given patient will be assigned to the placebo group
Early recognition and uniform aggressive treatment of attacks
Limited duration of placebo-controlled period after which patients may receive active study drug or empiric treatment
Figure 2. Phase IIb/III study designs for maintenance (attack prevention) treatment.

(A) Patients are randomized to receive experimental treatment (Tx) or placebo. To minimize risk, liberal “escape” criteria are included in the protocol to address on-study disease activity, such as occurrence of first attack. Those whose experimental therapy failed could be reassigned to empiric treatment or an alternate treatment. (B) Patients are maintained on their existing regimen and randomized to receive experimental treatment or placebo as add-on therapy. Inclusion of an optional experimental treatment alone arm (group C) might allow evaluation of potential interaction(s) between empiric and experimental therapy. (C) Patients are randomized to receive either experimental or empiric treatment. (D) Patients are randomized to receive 1 of 2 existing empiric therapies. This design is similar to design C, but 2 empiric treatments are compared directly. For all studies, interim analysis for futility and for assessment of event rate and adjustment of sample size may be conducted with appropriate power correction.
Studies should be designed to observe the minimum number of events necessary for achieving interpretable and robust outcomes. Placebo-controlled studies require fewer observed attacks to achieve adequate statistical power than those comparing experimental and an efficacious empiric therapy; however, the risk of an attack for an individual patient may be greater in a placebo-controlled than in an active treatment comparator study. Placebo-controlled studies do not address superiority to current empiric treatments that may be perceived as moderately effective and relatively inexpensive. To address this concern, one or more empiric therapy arms could be added to a placebo-controlled study or subsequently evaluated against an experimental agent that is proven to be superior to placebo.
Add-on experimental agent vs placebo.
Subjects are randomized to experimental agent or placebo, while also receiving a protocol-defined empiric regimen (figure 2B). Investigators may terminate patient participation after a first on-study relapse. However, extended participation in the randomized phase beyond the first event is more acceptable to some stakeholders in an add-on than in a pure placebo-controlled trial. Extended participation increases power by allowing each patient to contribute more than a single event. Potential limitations include possible additive toxicity of multiple agents and unwarranted attribution of benefit to combined therapy when one agent might be solely responsible. Effective empiric therapy may also blur outcome specificity, thereby requiring recruitment of additional subjects or observation of more events than in a pure placebo trial. However, the per-subject risk of an event may be lower. As stated above, if a combination regimen proves efficacious, ensuing studies would be required to assess the experimental agent alone (figure 2B) vs the combination.
Experimental agent vs existing therapy.
Direct comparison of an investigational agent to an existing empiric therapy circumvents concerns about pure placebo exposure (figure 2C). Multiple options for empiric therapy during the trial may be necessary, as subjects may have failed one or more of comparator treatments. A priori consensus of empiric therapy mitigates, but does not eliminate, unwanted variability in the comparator. Active comparator studies require large sample sizes to detect modest differences in efficacy. Also, unrecognized deleterious effects of an unproven comparator treatment may convolute evaluation of efficacy of the experimental treatment. A variation of this approach is comparison of 2 empiric therapies to evaluate superiority (figure 2D).
Outcome measures.
Three study endpoints are potential measures of attack frequency or severity (table 3):
Time to first on-study event.
Ending the randomized phase at the first on-study attack mitigates the risk of placebo exposure, allowing subjects access to alternative therapies when treatment fails. Time to first attack is a continuous measure with inherent statistical advantages. Subjects experiencing an attack may be eligible for an open-label investigational drug or other empiric off-protocol treatment, but could no longer contribute to efficacy evaluation. However, withdrawal of patients after a first event sacrifices information about efficacy and safety as compared to assessment of the annualized relapse rate over the entire observational period. Early relapses could result in underestimation of efficacy for agents with delayed effectiveness. To minimize the consequences of delayed effectiveness, a protocol might define a period of initial treatment during which attacks do not qualify as endpoints, either for primary or secondary analysis. Alternatively, all study participants could receive corticosteroids for a defined period based on the pharmacodynamics of the experimental agent to minimize the risk of early relapse before the experimental agent becomes effective. While any combination of treatments could confound the interpretation of trial results, this approach equally distributes any beneficial signal attributable to corticosteroid treatment among subjects in all treatment limbs.
Annualized attack rate.
This metric, a common primary outcome in MS clinical trials, annualizes net frequency of all attacks. However, variation in the time over which relapse rate is calculated is problematic when dropout rates are high, and when they differ between treatment groups.
Attack frequency/severity composite.
Worsening in NMO reflects the cumulative result of attack-related disability. Although attacks are generally more severe than in MS, milder attacks may also occur.45 Heightened awareness of new or worsening symptoms in prospective clinical trials exaggerates the risk of recording events that are not true attacks. For example, in the recent open-label study of eculizumab, 2 on-study events (back pain alone in one subject and a one-line change of visual acuity in a second) were of equivocal significance.21 Although experience with stratification of attack severity is limited, a simple system for rating optic nerve and spinal cord attacks has been applied to retrospective studies.12 This strategy is similar to well-accepted clinical scales used in MS46 and focuses on visual, pyramidal, sensory, and sphincter functions that are most consistently affected in NMO. Theoretically, each NMO attack could be rated based on attack severity at nadir or after a fixed period of recovery (e.g., 1 month), and a composite developed to reflect attack frequency and severity (i.e., weighted composite relapse severity/frequency score = ∑ attacki × severityi/time, i = 1→n). Such an integrated attack frequency and severity metric would minimize the impact of mild equivocal events. Alternatively, the distribution of attacks using a categorical prespecified scale (mild, moderate, severe) is compared using nonparametric statistical tests to mitigate concerns surrounding variability of unvalidated severity scoring scales.
A masked adjudication panel empowered to evaluate all relevant data in real time, including clinical, laboratory, and imaging results, to independently confirm attacks and grade their severity might reduce study site–related variation in the application of an attack frequency endpoint. Furthermore, an independent adjudication panel would resolve potential conflicts of site investigators who may be inclined to interpret an equivocal event as an attack to facilitate treatment of the patient. The decisions of the adjudication panel would not influence treatment decisions for the trial subject, but could define the primary endpoint.
OTHER ISSUES
Phase of study.
The phase of clinical trial for an experimental NMO treatment relates to antecedent experience. Safety profiles of repurposed agents are better established than those of new therapies developed specifically for NMO, possibly eliminating the need for early-phase trials. Early-phase clinical trials would be required for de novo drug development; however, given the recent emphasis on drug repurposing, phase II and III clinical trial designs may be most relevant to planned NMO clinical trials:
Phase IIa exploratory studies.
Phase IIa or target engagement studies are conducted for promising agents for which adequate safety data are available (figure 3). Patients enrolled in these studies typically have active NMO that has failed one or more empiric treatments. Dramatic reductions in attack frequency compared with pretreatment in single-arm, open-label studies (e.g., as revealed in patients treated with rituximab [n = 8],19 eculizumab [n = 14],21 or tocilizumab [n = 7]24) may portend favorable results in controlled trials. However, “regression to the mean” may be an alternative explanation, and decline in event rates in placebo groups may exceed the difference between active and control groups.47 Nonetheless, phase IIa exploratory studies may be useful for screening novel NMO therapies in cohorts of 10 to 15 patients with recently active NMO whose other treatments have failed. If safety criteria are met and most subjects' treatment has not failed, the agent may be deemed “promising” and considered for phase IIb and III studies. Sensitive biomarkers, if validated, could accelerate drug development as has detection of new MRI lesions for MS.
Figure 3. Conceptual model of NMO clinical trial pipeline.

Patients are enrolled in a staggered schedule based on agent availability. Some agents have unacceptable adverse effects and are discontinued (agent A). Others fail prespecified futility criteria and are not considered further (agent C). Indeterminate results may be addressed by a longer period of observation and/or increased subject enrollment. Agents surviving the futility criteria and demonstrating acceptable safety are then eligible for further evaluation (agents B and D). If feasible, such a neuromyelitis optica (NMO) clinical trial “pipeline” might be a pathway by which multiple agents can be evaluated in a way that directs subjects most efficiently to potentially effective therapies.
Phase IIb and III studies.
Agents surviving the futility criteria of phase IIa may be examined in subsequent controlled studies to rigorously evaluate efficacy (figure 2, A–D). First-line treatments should be interrogated in relatively unselected populations representative of their intended use. However, second-line treatments, including potent agents with less favorable safety profiles, may be appropriate for severe or refractory NMO cohorts. Because no treatment is currently proven to be efficacious in NMO, superiority rather than noninferiority to the comparator is required.
Washout interval.
Washout periods enhance the likelihood that efficacy is attributed to the experimental agent rather than to residual effects of prior treatment. Patients with NMO receiving immunosuppressive drugs may exhibit long-lasting carryover effects. For example, rituximab-induced B-cell depletion may persist for more than 1 year. However, patients are potentially susceptible to relapse during washout periods, negatively impacting safety and enrollment. To address this concern, randomization may need to be stratified based on prior treatment.
Masking.
Definitive studies should be double-masked by ensuring that the placebo has similar characteristics (frequency and route of administration, color, etc.) and by providing appropriate concomitant medications to prevent infusion reactions. Where adverse effects result in unavoidable unmasking, an “evaluating” physician may be designated to assess the primary outcome while another physician evaluates safety, adverse effects, and decides whether termination based on safety is necessary, a frequently used strategy in MS trials.
Clinical trial pipeline.
Innovative strategies for coordinating NMO clinical trials to evaluate multiple agents concurrently are desirable for phase I or IIa studies (figure 3). In addition, if standardized inclusion criteria and study procedures are adopted, control subjects might be shared among multiple phase IIb and III controlled clinical trials. Theoretically, this strategy might provide a larger comparator, minimize exposure to placebo, and enhance efficient use of scarce patient resources. Moreover, subjects involved in trials of “failed” therapeutic candidates could be redirected into new studies through a streamlined crossover mechanism to facilitate enrollment and accrual. There are many hurdles to be overcome, however. Diagnostic criteria continue to evolve and have led to expansion of NMO disease spectrum. Differences in inclusion and exclusion criteria based on prior treatment and other subject antecedents vary among studies and could affect the feasibility of a common control group. Investigators, industry sponsors, and regulators would need to be willing to adopt standardized inclusion and exclusion criteria and common study procedures across trials to facilitate this initiative. However, it may be cost-effective to all parties to facilitate multiple phase IIa and possibly phase III studies proceeding simultaneously. Thus, an NMO clinical trial pipeline envisions a cooperative approach for reducing risk to patients, optimizing enrollment, offering patients greatest access to the most promising drug candidates, and promoting long-term resources (e.g., generation of clinical information and biological sample resources for investigational studies), ultimately advancing NMO care.
DISCUSSION
NMO presents unique opportunities and challenges for clinical trial design. Among the most difficult aspects of such trials is implementation of placebo-controlled studies, given the potential severity of NMO attacks. Preventing attacks and limiting their sequelae are highest priorities. Trial designs must be acceptable to all stakeholders and protect patient interests, while being informative and robust. Careful definition of attack frequency and severity, and discovery of surrogate efficacy biomarkers should facilitate these goals. Innovative approaches, such as shared placebo groups, may expand and speed the investigative pipeline.
Supplementary Material
ACKNOWLEDGMENT
The activities of the International Clinical Consortium and Neuromyelitis Optica Clinical Council were organized by the Guthy-Jackson Charitable Foundation.
GLOSSARY
- AQP4
aquaporin-4
- IgG
immunoglobulin G
- MS
multiple sclerosis
- NMO
neuromyelitis optica
- PLEX
plasma exchange
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Dr. B. Weinshenker participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Mr. G. Barron participated in the critical review of the manuscript for important intellectual content. Ms. J. Behne participated in the critical review of the manuscript for important intellectual content. Dr. J. Bennett participated in the critical review of the manuscript for important intellectual content. Dr. P. Chin participated in the critical review of the manuscript for important intellectual content. Dr. B. Cree participated in the critical review of the manuscript for important intellectual content. Dr. J. de Seze participated in the critical review of the manuscript for important intellectual content. Dr. A. Flor participated in the critical review of the manuscript for important intellectual content. Dr. K. Fujihara participated in the critical review of the manuscript for important intellectual content. Dr. B. Greenberg participated in the critical review of the manuscript for important intellectual content. Dr. S. Higashi participated in the critical review of the manuscript for important intellectual content. Dr. W. Holt participated in the critical review of the manuscript for important intellectual content. Dr. O. Khan participated in the critical review of the manuscript for important intellectual content. Dr. V. Knappertz participated in the critical review of the manuscript for important intellectual content. Dr. M. Levy participated in the critical review of the manuscript for important intellectual content. Ms. A. Melia participated in the critical review of the manuscript for important intellectual content. Dr. J. Palace participated in the critical review of the manuscript for important intellectual content. Dr. T. Smith participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. M.P. Sormani participated in the critical review of the manuscript for important intellectual content. Dr. K. Van Herle participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content. Dr. S. VanMeter participated in the critical review of the manuscript for important intellectual content. Dr. P. Villoslada participated in the critical review of the manuscript for important intellectual content. Dr. M. Walton participated in the critical review of the manuscript for important intellectual content. Dr. W. Wasiewski participated in the critical review of the manuscript for important intellectual content. Dr. D. Wingerchuk participated in the critical review of the manuscript for important intellectual content. Dr. M. Yeaman participated in the analysis, interpretation, writing, and critical review of the manuscript for important intellectual content.
STUDY FUNDING
Drs. Weinshenker, Van Herle, Smith, and Yeaman have received support for activities that led to this manuscript by the Guthy-Jackson Charitable Foundation.
DISCLOSURE
B. Weinshenker: member data safety monitoring boards: Novartis, Biogen Idec, and Mitsubishi; adjudication panel member: MedImmune; consultant: Elan, GlaxoSmithKline, Ono, CHORD Therapeutics, and Chugai; editorial board membership: Neurology®, the Canadian Journal of Neurological Sciences, and the Turkish Journal of Neurology; research support: Guthy-Jackson Charitable Foundation; royalties and patent: RSR Ltd. and Oxford University for a patent regarding AQP4-associated antibodies for diagnosis of neuromyelitis optica. G. Barron: employee: MedImmune Ltd. J. Behne reports no disclosures relevant to the manuscript. J. Bennett: consultant: Novartis, Alnylam Pharmaceuticals, MedImmune, Chugai, EMD Serono, Abbott, Genentech, Genzyme, and Questcor; founder/shareholder: Apsara Therapeutics; royalties and patent: Compositions and Methods for the Treatment of Neuromyelitis Optica; editorial board membership: Multiple Sclerosis Journal and Journal of Neuro-Ophthalmology. P. Chin: former employee of Novartis; holds stock in Novartis; after completion of manuscript, became employee of Genentech. B. Cree: consultant: AbbVie, Biogen Idec, EMD Serono, Genzyme/Sanofi Aventis, MedImmune, Novartis, Teva Neurosciences; research support: Acorda, Avanir, Biogen Idec, EMD Serono, Hoffmann-La Roche, and Novartis. J. de Seze: consultant: Alexion, Chugai. A. Flor: employee: MedImmune LLC. K. Fujihara: advisory board membership: Bayer Schering, Biogen Idec, Mitsubishi Tanabe Corporation, Novartis, Chugai, Ono, Nihon, Merck Serono, Alexion, MedImmune, and Medical Review; travel and speaker honoraria: Bayer Schering Pharma, Biogen Idec, Eisai, Mitsubishi Tanabe Corporation, Novartis, Astellas, Takeda, Asahi Kasei Medical Co., Daiichi Sankyo, Nihon Pharmaceutical, and Cosmic Corporation; editorial board member: Clinical and Experimental Neuroimmunology, Sri Lanka Journal of Neurology; research support: Bayer Schering Pharma, Biogen Idec Japan, Asahi Kasei Medical, The Chemo-Sero-Therapeutic Research Institute, Teva Pharmaceutical, Mitsubishi Tanabe Pharma, Teijin Pharma, Chugai Pharmaceutical, Ono Pharmaceutical, Nihon Pharmaceutical, and Genzyme Japan; primary (26293205, 2016–2018) and secondary (22229008, 2010–2015) investigator by the Grants-in-Aid for Scientific Research from the Ministry of Education, Science and Technology of Japan and as the secondary investigator by the Grants-in-Aid for Scientific Research from the Ministry of Health, Welfare and Labor of Japan (2010–present). B. Greenberg: consultant: Novartis, DioGenix, Amplimmune, Chugai; research support: NIH, Accelerated Cure Project, Guthy-Jackson Charitable Foundation, Biogen; equity: DioGenix and Amplimmune. S. Higashi: employee of Chugai Pharmaceuticals. W. Holt: employee of PPD, Inc. O. Khan: speakers bureau: Teva Neuroscience, Novartis, Biogen Idec; consultant: Genentech, Teva Neuroscience, Novartis, Biogen Idec; research funding: National Multiple Sclerosis Society, National Institute of Neurological Disorders and Stroke, and the Sastry Family Foundation. V. Knappertz: employee, Teva Pharmaceuticals. M. Levy: advisory board membership: Asterias; consultant: Chugai Pharmaceuticals, GlaxoSmithKline, MedImmune; research support: ApoPharma Inc., NIH, Guthy-Jackson Charitable Foundation, ViroPharma, Acorda, Sanofi, NeuralStem, Genentech; travel support: Asterias. A. Melia: employee of Chugai Pharmaceuticals. J. Palace: consultant: Merck Serono, Biogen Idec, Novartis, Teva, Chugai Pharma, Bayer Schering; research support: Merck Serono, Novartis, Biogen Idec, Bayer Schering, UK MS Society, Guthy-Jackson Charitable Foundation. T. Smith: consultant: Guthy-Jackson Charitable Foundation. M. Sormani: consultant: Biogen Idec, Novartis, Teva, Merck Serono, Genzyme, Synthon, Actelion. K. Van Herle: employee: Premier Health Care, Greater Good Foundation, All Greater Good Foundation. S. VanMeter: employee and stockholder: GlaxoSmithKline. P. Villoslada: board membership: Roche, Novartis, Neurotec Pharma, Bionure Farma; consultant: Novartis, Roche, TFS, Heidelberg Engineering, MedImmune, Digna Biotech, Neurotec Pharma; research support: European Commission, Instituto Salud Carlos III, Marato TV3, Novartis, Roche; patents: Digna Biotech, Bionure Farma; stock/stock options: Bionure Farma; travel expenses: Novartis. M. Walton: employee, Food and Drug Administration; the views presented in this article are those of the author and do not necessarily reflect those of the Food and Drug Administration; after completion of this manuscript, became an employee of Janssen Research and Development. W. Wasiewski: employee of Alexion Pharmaceuticals; after completion of the manuscript, became employee of Neurotrope BioScience Inc. D. Wingerchuk: research support: Alexion, Terumo BCT, Guthy-Jackson Charitable Foundation; consultant: Alexion, MedImmune, and Chugai; adjudication committee member: MedImmune; co-editor-in-chief, The Neurologist; editorial advisory boards: Current Medical Research & Opinion, Drugs in Context. M. Yeaman: research support: NIH, US Department of Defense; founder/shareholder: NovaDigm Therapeutics, Inc., Metacin, Inc.; consultant: Guthy-Jackson Charitable Foundation. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004;364:2106–2112. [DOI] [PubMed] [Google Scholar]
- 2.Jarius S, Wildemann B. Aquaporin-4 antibodies (NMO-IgG) as a serological marker of neuromyelitis optica: a critical review of the literature. Brain Pathol 2013;23:661–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bradl M, Misu T, Takahashi T, et al. Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann Neurol 2009;66:630–643. [DOI] [PubMed] [Google Scholar]
- 4.Saadoun S, Waters P, Bell BA, Vincent A, Verkman AS, Papadopoulos MC. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain 2010;133:349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang H, Bennett JL, Verkman AS. Ex vivo spinal cord slice model of neuromyelitis optica reveals novel immunopathogenic mechanisms. Ann Neurol 2011;70:943–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watanabe S, Nakashima I, Misu T, et al. Therapeutic efficacy of plasma exchange in NMO-IgG-positive patients with neuromyelitis optica. Mult Scler 2007;13:128–132. [DOI] [PubMed] [Google Scholar]
- 7.Misu T, Fujihara K, Nakashima I, Sato S, Itoyama Y. Intractable hiccup and nausea with periaqueductal lesions in neuromyelitis optica. Neurology 2005;65:1479–1482. [DOI] [PubMed] [Google Scholar]
- 8.Popescu BFG, Lennon VA, Parisi JE, et al. Neuromyelitis optica unique area postrema lesions: nausea, vomiting, and pathogenic implications. Neurology 2011;76:1229–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weinshenker B, Wingerchuk D, Vukusic S, et al. Neuromyelitis optica IgG predicts relapse following longitudinally extensive transverse myelitis. Ann Neurol 2006;59:566–569. [DOI] [PubMed] [Google Scholar]
- 10.Jiao Y, Fryer JP, Lennon VA, et al. Updated estimate of AQP4-IgG serostatus and disability outcome in neuromyelitis optica. Neurology 2013;81:1197–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Waters PJ, McKeon A, Leite MI, et al. Serologic diagnosis of NMO: a multicenter comparison of aquaporin-4-IgG assays. Neurology 2012;78:665–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wingerchuk DM, Hogancamp WF, O'Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic's syndrome). Neurology 1999;53:1107–1114. [DOI] [PubMed] [Google Scholar]
- 13.Kitley J, Leite MI, Nakashima I, et al. Prognostic factors and disease course in aquaporin-4 antibody-positive patients with neuromyelitis optica spectrum disorder from the United Kingdom and Japan. Brain 2012;135:1834–1849. [DOI] [PubMed] [Google Scholar]
- 14.Wingerchuk DM, Pittock SJ, Lucchinetti CF, Lennon VA, Weinshenker BG. A secondary progressive clinical course is uncommon in neuromyelitis optica. Neurology 2007;68:603–605. [DOI] [PubMed] [Google Scholar]
- 15.Kimbrough DJ, Fujihara K, Jacob A, et al. Treatment of neuromyelitis optica: review and recommendations. Mult Scler Relat Disord 2012;1:180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trebst C, Berthele A, Jarius S, et al. Diagnosis and treatment of neuromyelitis optica: consensus recommendations of the Neuromyelitis Optica Study Group [in German]. Nervenarzt 2011;82:768–777. [DOI] [PubMed] [Google Scholar]
- 17.Sellner J, Boggild M, Clanet M, et al. EFNS guidelines on diagnosis and management of neuromyelitis optica. Eur J Neurol 2010;17:1019–1032. [DOI] [PubMed] [Google Scholar]
- 18.Tradtrantip L, Zhang H, Saadoun S, et al. Anti-aquaporin-4 monoclonal antibody blocker therapy for neuromyelitis optica. Ann Neurol 2012;71:314–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cree BAC, Lamb S, Morgan K, Chen A, Waubant E, Genain C. An open label study of the effects of rituximab in neuromyelitis optica. Neurology 2005;64:1270–1272. [DOI] [PubMed] [Google Scholar]
- 20.Jacob A, Weinshenker BG, Violich I, et al. Treatment of neuromyelitis optica with rituximab: retrospective analysis of 25 patients. Arch Neurol 2008;65:1443–1448. [DOI] [PubMed] [Google Scholar]
- 21.Pittock SJ, Lennon VA, McKeon A, et al. Eculizumab in AQP4-IgG-positive relapsing neuromyelitis optica spectrum disorders: an open-label pilot study. Lancet Neurol 2013;12:554–562. [DOI] [PubMed] [Google Scholar]
- 22.Kieseier B, Stüve O, Dehmel T, et al. Disease amelioration with tocilizumab in a treatment-resistant patient with neuromyelitis optica: implication for cellular immune responses. JAMA Neurol 2013;70:390–393. [DOI] [PubMed] [Google Scholar]
- 23.Ayzenberg I, Kleiter I, Schröder A, et al. Interleukin 6 receptor blockade in patients with neuromyelitis optica nonresponsive to anti-CD20 therapy. JAMA Neurol 2013;70:394–397. [DOI] [PubMed] [Google Scholar]
- 24.Araki M, Matsuoka T, Miyamoto K, et al. Efficacy of the anti-IL-6 receptor antibody tocilizumab in neuromyelitis optica: a pilot study. Neurology 2014;82:1302–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Costanzi C, Matiello M, Lucchinetti CF, et al. Azathioprine: tolerability, efficacy, and predictors of benefit in neuromyelitis optica. Neurology 2011;77:659–666. [DOI] [PubMed] [Google Scholar]
- 26.Jacob A, Matiello M, Weinshenker BG, et al. Treatment of neuromyelitis optica with mycophenolate mofetil: retrospective analysis of 24 patients. Arch Neurol 2009;66:1128–1133. [DOI] [PubMed] [Google Scholar]
- 27.Echt DS, Liebson PR, Mitchell LB, et al. Mortality and morbidity in patients receiving encainide, flecainide, or placebo. N Engl J Med 1991;324:781–788. [DOI] [PubMed] [Google Scholar]
- 28.Hofmeyr GJ, Gulmezoglu AM, Novikova N, Lawrie TA. Postpartum misoprostol for preventing maternal mortality and morbidity. Cochrane Database Syst Rev 2013;7:CD008982. [DOI] [PubMed] [Google Scholar]
- 29.Engebraaten O, Edvardsen H, Løkkevik E, et al. Gefitinib in combination with weekly docetaxel in patients with metastatic breast cancer caused unexpected toxicity: results from a randomized phase II clinical trial. ISRN Oncol 2012;2012:176789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hinson SR, McKeon A, Fryer JP, Apiwattanakul M, Lennon VA, Pittock SJ. Prediction of neuromyelitis optica attack severity by quantitation of complement-mediated injury to aquaporin-4-expressing cells. Arch Neurol 2009;66:1164–1167. [DOI] [PubMed] [Google Scholar]
- 31.Ishizu T, Osoegawa M, Mei FJ, et al. Intrathecal activation of the IL-17/IL-8 axis in opticospinal multiple sclerosis. Brain 2005;128:988–1002. [DOI] [PubMed] [Google Scholar]
- 32.Uzawa A, Mori M, Arai K, et al. Cytokine and chemokine profiles in neuromyelitis optica: significance of interleukin-6. Mult Scler 2010;16:1443–1452. [DOI] [PubMed] [Google Scholar]
- 33.Uzawa A, Mori M, Sato Y, Masuda S, Kuwabara S. CSF interleukin-6 level predicts recovery from neuromyelitis optica relapse. J Neurol Neurosurg Psychiatry 2012;83:339–340. [DOI] [PubMed] [Google Scholar]
- 34.Chihara N, Aranami T, Sato W, et al. Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc Natl Acad Sci USA 2011;108:3701–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jarius S, Aboul-Enein F, Waters P, et al. Antibody to aquaporin-4 in the long-term course of neuromyelitis optica. Brain 2008;131:3072–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takahashi T, Fujihara K, Nakashima I, et al. Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain 2007;130:1235–1243. [DOI] [PubMed] [Google Scholar]
- 37.Takano R, Misu T, Takahashi T, Sato S, Fujihara K, Itoyama Y. Astrocytic damage is far more severe than demyelination in NMO: a clinical CSF biomarker study. Neurology 2010;75:208–216. [DOI] [PubMed] [Google Scholar]
- 38.Green AJ, Cree BA. Distinctive retinal nerve fibre layer and vascular changes in neuromyelitis optica following optic neuritis. J Neurol Neurosurg Psychiatry 2009;80:1002–1005. [DOI] [PubMed] [Google Scholar]
- 39.Magana SM, Keegan BM, Weinshenker BG, et al. Beneficial plasma exchange response in central nervous system inflammatory demyelination. Arch Neurol 2011;68:870–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Temple RJ; Center for Drug Evaluation and Research, US Food and Drug Administration. Enrichment strategies for clinical trials. 2013. Available at: http://www.fda.gov/downloads/Drugs/UCM345394.pdf. Accessed August 1, 2014.
- 41.Beck RW, Cleary PA. Optic neuritis treatment trial: one-year follow-up results [see comments]. Arch Ophthalmol 1993;111:773–775. [DOI] [PubMed] [Google Scholar]
- 42.Kinkel R. Methylprednisolone. In: Rudick R, Goodkin D, editors. Multiple Sclerosis Therapeutics. London: Martin Dunitz; 1999:349–370. [Google Scholar]
- 43.Weinshenker B, O'Brien P, Petterson T, et al. A randomized trial of plasma exchange in acute CNS inflammatory demyelinating disease. Ann Neurol 1999;46:878–886. [DOI] [PubMed] [Google Scholar]
- 44.Varrin-Doyer M, Spencer CM, Schulze-Topphoff U, et al. Aquaporin 4-specific T cells in neuromyelitis optica exhibit a Th17 bias and recognize clostridium ABC transporter. Ann Neurol 2012;72:53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Collongues N, Cabre P, Marignier R, et al. A benign form of neuromyelitis optica: does it exist? Arch Neurol 2011;68:918–924. [DOI] [PubMed] [Google Scholar]
- 46.Kurtzke JF. Rating neurological impairment in multiple sclerosis: an Expanded Disability Status Scale (EDSS). Neurology 1983;33:1444–1452. [DOI] [PubMed] [Google Scholar]
- 47.Spector R, Park GD. Regression to the mean: a potential source of error in clinical pharmacological studies. Drug Intell Clin Pharm 1985;19:916–919. [DOI] [PubMed] [Google Scholar]
- 48.Bizzoco E, Lolli F, Repice AM, et al. Prevalence of neuromyelitis optica spectrum disorder and phenotype distribution. J Neurol 2009;256:1891–1898. [DOI] [PubMed] [Google Scholar]
- 49.Asgari N, Lillevang ST, Skejoe HPB, Falah M, Stenager E, Kyvik KO. A population-based study of neuromyelitis optica in Caucasians. Neurology 2011;76:1589–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Siritho S, Nakashima I, Takahashi T, Fujihara K, Prayoonwiwat N. AQP4 antibody-positive Thai cases: clinical features and diagnostic problems. Neurology 2011;77:827–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Van Herle K, Behne JM, Van Herle A, Blaschke TF, Smith TJ, Yeaman MR. Integrative continuum: accelerating therapeutic advances in rare autoimmune diseases. Annu Rev Pharmacol Toxicol 2012;52:523–547. [DOI] [PubMed] [Google Scholar]
- 52.Verkman AS, Ratelade J, Rossi A, Zhang H, Tradtrantip L. Aquaporin-4: orthogonal array assembly, CNS functions, and role in neuromyelitis optica. Acta Pharmacol Sin 2011;32:702–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saadoun S, Waters P, MacDonald C, et al. Neutrophil protease inhibition reduces neuromyelitis optica-immunoglobulin G-induced damage in mouse brain. Ann Neurol 2012;71:323–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang H, Verkman AS. Eosinophil pathogenicity mechanisms and therapeutics in neuromyelitis optica. J Clin Invest 2013;123:2306–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Axtell RC, Raman C, Steinman L. Interferon-beta exacerbates Th17-mediated inflammatory disease. Trends Immunol 2011;32:272–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matthews L, Marasco R, Jenkinson M, et al. Distinction of seropositive NMO spectrum disorder and MS brain lesion distribution. Neurology 2013;80:1330–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.