

Abstract
Scavenger Receptor Associated with Endothelial Cells (SREC-I) was previously shown to be expressed by immune cells and to play a role in CD8+-mediated T cell immunity. SREC-I was also shown to modulate the function of Toll like receptors with essential roles in innate immunity. Here we have shown that SREC-I enhanced double stranded RNA (dsRNA)-mediated Toll like receptor-3 (TLR3) activation. Viral double stranded RNA (dsRNA) was demonstrated to be a pathogen associated molecular pattern (PAMP) signaling viral infection. We found that in human monocyte/macrophage THP1 cells as well as murine bone marrow derived macrophages SREC-I led to elevated responses to the dsRNA-like molecule polyinosine-polycytidylic acid (Poly I:C) and enhanced production of inflammatory cytokines. Our data also showed that intracellular/endocytic TLR3 could directly interact with SREC-I in the presence of Poly I: C. The internalized ligand, along with TLR3 and SREC-I localized in endosomes within macrophages and in HEK293 cells engineered to express TLR3 and SREC-I. SREC-I also stimulated dsRNA-mediated TLR3 activation of signaling through the NFκβ, MAP kinase and interferon regulatory factor 3 (IRF3) pathways leading to expression of cytokines, most notably interleukin-8 and interferon-β. We therefore hypothesized that SREC-I could be a receptor capable of internalizing Poly I: C, boosting TLR3 mediated inflammatory signaling and stimulating cytokine production in macrophages.
Keywords: TLR3, SREC-I, THP1, Poly I:C, double stranded RNA, Cytokines
Introduction
Toll like receptors (TLRs) and pattern recognition receptors have been shown to be responsible for activating immune responses in the presence of pathogens or pathogen-associated molecules (Akira and Takeda, 2004). These receptors were also shown to transduce signals for innate immunity in the presence of pathogen associated molecular pattern (PAMP) molecules (Akira and Takeda, 2004, Beutler 2004). The TLRs are members of a protein family the individual members of which differ in terms of their ligand specificity, cellular localization and signaling pathways. TLRs 1, 2, 4, 5 and 6 were detected on the plasma membrane while others such as TLR 3, 7 and 9 were characterized as “endosomal TLRs” (Akira and Takeda, 2004; McGettrick et al., 2010). Among endosomal TLRs, TLR3 was unique in being activated by binding viral double stranded RNA (dsRNA) (Kumar et al., 2006 and Takashi et al., 2006). Viral double stranded RNA, produced during the replication of many viruses has been shown to be a PAMP, indicating viral infection (Pirher et al., 2008; Alexopoulou et al., 2001). The other commonly known nucleic acid PAMP, CpG DNA (unmethylated bacterial DNA) was found to be recognized by another member of the endosomal TLR family, TLR9 (Chuang et al., 2002). These endosomal receptors were shown to translocate to from their storage site in the endoplasmic reticulum (ER) to endosomal sites upon arrival of internalized PAMP ligands at the endosomes. It has been shown that immune cells possess several systems for response to dsRNA including TLR3 present in endosomes, as well as number of cytoplasmic sensors including protein kinase R, RIG-1 (retinoic acid-inducible gene I) and the recently discovered surface relocated TLR3 (Pohar et al., 2013). These molecules were established as sensors of dsRNA in several cellular compartments and were shown to activate the innate immune response by triggering a number of signaling cascades, including the NFkB, IRF3 and MAP kinase pathways Liu et al., 2008. Such dsRNA sensors were thus implicated in transcription of cytokine genes. Recently it was shown that TLR3 could localize to the cell surface in the presence of dsRNA and UNC93B1 (an accessory protein shown to interact with TLRs 3, 7, 9, 11 and 14) in some endothelial, epithelial and fibroblastic cells (such as human lung fibroblast cells) (Andrade et al., 2013; Brinkmann et al., 2007; Pifer et al., 2011). Recently there was a report on the role of the ectodomain of TLR3 in trafficking to the plasma membrane (Pohar et al., 2014).
We have examined another class of potential sensors for dsRNA - scavenger receptors which have been shown to bind polyanionic ligands. We detected SREC-I both on the cell surface and in endosomes along with TLR3, on stimulation with Poly I:C. SREC-I could play roles in detecting dsRNA in alternative cellular compartments and could potentially trigger the trafficking of TLR3 from the surface to endosomes where actual signaling could likely occur. We have therefore examined the ability of SREC-I to modulate the response of immune cells to the model dsRNA-Poly I:C.
Materials and Methods
Reagents and Abs
Rabbit polyclonal human anti–SREC-I Ab was rabbit monoclonal was custom synthesized by GenScript (Piscataway, NJ) against the specific peptide sequence (TQGTQGSTLDPAGQC). Commercially available anti-SREC-I abs was also purchased from Atlas Antibodies. Anti–TLR3 ab was from Abcam. All secondary fluorescent Abs were from Jackson Immunoresearch Laboratories. Rhodamine-labeled TLR3 was purchased from Invivogen. Poly I: C HMW was purchased from Invivogen. TLR3-CFP plasmid was from Addgene. Anti-Phospho-p38, anti-p38, anti phospho-JNK, anti JNK, anti phospho-p65, anti p65, anti Src anti phosphor-Src (416), IRF3 antibodies were purchased from Cell Signaling Technology. Anti LAMP1 and anti GFP antibodies were from Abcam. Anti-phospho-IRF3 antibodies were from Abcam. PP2 and Bafilomycin were purchased from Sigma-Aldrich. Normocin was from Invivogen.
Cells and culture conditions
THP1 and HEK293 cells were transfected with human full length SREC-I in pcDNA3 for stable expression of SREC-I. HEK293 cells stably TLR3 was purchased from Invivogen and cultured according to manufacturer’s instruction. THP1 and HEK293 cells were maintained in RPMI 1640 and DMEM respectively with 10% heat inactivated FBS and penicillin-streptomycin. For generation of stable SREC-I-expressing cell lines, cells were selected and maintained in the same medium plus 400 mg/ml G418. Hek293-TLR3 stable cell line was also maintained with 100 μg/ml Normocin. Differentiation of THP1 macrophages from undifferentiated ones was performed by treating the cells with 5–10 ng/ml of phorbol 12-myristate 13-acetate (PMA) for 72 hours or 50 ng/ml for overnight. SREC-I expression in differentiated THP1 macrophages was induced by incubating cells with 1–5 ng/ml of LPS for 12 hours. Bone marrow derived cells were isolated from C57/BL6J mice. The cells were then grown in RPMI supplemented with 10% heat inactivated serum and penicillin and streptomycin, along with L929 supernatant (media) to differentiate into macrophages. For inducing SREC-I expression in these macrophages, cells were incubated with 1–5 ng/ml of LPS for 12 hours.
Plasmids
The pcDNA3.1-SREC-I (human) was a generous gift from Dr. H. Adachi. FLAG–SREC-I construct was made in 3xFLAG-CMV vector as described in Murshid et al., 2010. TLR3-CFP was from Addgene. Human SREC-I-GFP was constructed in EGFP-N1 vector (Clontech). Both human and mice SREC-I (siRNA) and TLR3 (siRNA) constructs were purchased from Santa Cruz Biotech.
Immunofluorescence and microscopic analysis of Poly I: C internalization
HEK293 and bone marrow derived macrophages cells were incubated with Poly I: C for 20–30 min on ice. The ice-cold medium was then replaced by warm medium and incubated at 37°C for different periods. Cells were later washed with ice-cold stripping buffer (50 mM sodium citrate and 280 mM sucrose [pH 4.6]) to remove unbound ligand. Later, the cells were fixed with 4% para-formaldehyde and permeabilized with 0.1% Triton X-100. Cells were stained with different primary (anti FLAG m2, anti LAMP1, anti TLR3 antibodies) and secondary antibodies (Goat anti mouse Alexa 488, Goat anti rabbit/mouse Cy3, Goat anti mouse/rabbit Cy5) and later analyzed using a Zeiss 510 confocal microscope (Carl Zeiss, Jena, Germany). Fluorophores were visualized using the following filter sets: 488 nm excitation and band pass 505–530 emission filter for Alexa 488; 543 nm excitation and band pass 560–615 for Cy3; and 633 excitation and long pass 650 for Cy5. Images were taken using a 633 numerical aperture 1.4 oil immersion objective lens (Carl Zeiss, Jena, Germany). Figures were made using Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA) with little or no contrast adjustments without altering original images.
SEAP reporter assay
The secreted form of embryonic alkaline phosphatase (SEAP)–NFκβ promoter-reporter assay kit was utilized as a convenient and sensitive method to determine promoter activity in cells transfected with the SEAP expression plasmid. HEK293 cells were transfected with plasmids encoding FLAG-SREC-I, TLR3 and the reporter constructs NFkβ-SEAP and CMV-(control expression vector). NFkβ activity was measured indirectly by catalytic hydrolysis reaction of p-nitrophenyl phosphate producing a yellow end product that was read spectrophotometrically at 405 nm.
Western Blotting and Immunoprecipitation
HEK293 cells stably expressing TLR3 were transfected with SREC-I-GFP and then incubated with or without Poly I:C (10μg/ml) for 30 minutes. Cells were then washed with ice-cold Dulbecco’s phosphate-buffered saline (PBS) and lysed in NP-40 lysis buffer (containing 1% Nonidet P-40, 150mM NaCl, 1mM EDTA, 1mM PMSF, 1 x HALT protease and phosphatase inhibitor cocktail (Thermo Scientific). For Western blotting, 15–30 μg of protein were resolved by 4–15% gradient SDS-PAGE and transferred to PVDF (polyvinylidene fluoride) membranes. Membranes were immunoblotted with primary antibodies and later secondary antibodies that are HRP-conjugated. The membrane reactions were visualized by Perkin Elmer enhanced chemiluminescence reagents. For immunoprecipitation, 1 mg of cell extract was incubated with 5 ug of anti-rabbit-GFP antibody or IgG (control) for 2 hours at 4°C followed by incubation with 20μl of protein G (50% slurry, GE healthcare) plus-sepharose beads for either 2 hours at room temperature or overnight at 4°C. The beads were then washed with NP40-lysis buffer and complexes were eluted by boiling in Laemmli sample buffer.
ELISA
A human microanylate ELISA array was used to measure cytokine production by human THP1 cells treated with TLR3 ligand Poly I:C. Values represent the optical density. The kit was purchased from Qiagen (SA Biosciences) according to manufacturer’s protocol. THP1 cells and BMDM (bone marrow derived macrophages) were treated transfected with siRNA of SREC-I for 72 hours and cells were then incubated with Poly I:C (10μg/ml) or not. The secreted cytokines were measured using ELISA microanylate array kit or other kits from BD Biosciences (IL-6, IL-8, IFNβ). The optical density was measured either using BioRad plate and BioTec reader using appropriate software and wavelength.
Results
We first investigated the relative cellular locations of SREC-I and TLR3 without and with exposure to ligand. Experiments were carried out in HEK 293 initially for ease in gene transfection. Prior to addition of Poly I:C, FLAG-SREC-I was found largely in intracellular organelles resembling endosomes or endoplasmic reticulum (ER), while TLR3 (TLR3-CFP) appeared to reside largely in cytoplasmic membrane structures (Fig. 1A). Upon Poly I:C exposure in HEK293 cells expressing FLAG-SREC-I, the TLR3-CFP appeared to become co-localized along with the SREC-I at plasma membrane-endosomal sites (Fig. 1B). Incubation of the cells expressing FLAG-SREC-I and TLR3-CFP with Poly I:C at 37°C, resulted in relocalization of the receptors to an intracellular compartment (Fig. 1C). On the other hand, in control experiments, we did not observe Poly I:C induced translocation of FLAG-SREC-I to the plasma membrane from cytosolic compartments in HEK293 cells lacking TLR3 (Fig. 1D). In addition, TLR3-CFP was not re-localized to the plasma membrane in the presence of Poly I:C in HEK293 cells that did not express SREC-I (Fig. 1D) suggesting a role for SREC-I for relocation of TLR3.
Figure 1. TLR3 was colocalized with SREC-I at the cell surface and in intracellular compartments in the presence of Poly I:C.

A and B, TLR3 colocalized with SREC-I in HEK 293-TLR3-SREC-I overexpressing cells. HEK293 cells were transfected with FLAG-SREC-I and TLR3-CFP for 22 hours. Cells were then incubated without (A) or with (B) Poly I:C (10ug/ml) on ice for 30 min. Cells (A) and (B) were then fixed with 4% para formaldehyde and permeabilized with 0.1% Triton X100 (A) or not (B). Cells were then stained for FLAG with anti-FLAG M2 antibody (green) C, HEK293 cells were transfected with FLAG-SREC-I and TLR3-CFP for 22 hr. Cells were then incubated Poly I:C (10ug/ml) on ice for 30 min followed by incubation with warm media at 37°C for 20 min. Cells were then fixed with 4% PFA and permeabilized with 0.1% Triton X100. Cells were stained with anti-FLAG antibody (green). D, HEK293 cells were transfected with FLAG-SREC-1 or TLR3-CFP for 18 hours. Cells were incubated with Poly I:C (10μg/ml) for 1 hour and then fixed and stained with anti-FLAG M2 ab (green). E, Differentiated THP1 cells were transfected with FLAG-SREC-I and TLR3-CFP for 22 hr. Cells were then incubated with Poly I:C (10μg/ml) for 1 hr and then fixed and permeabilized as in D. Cells were stained for SREC-I (green), with anti FLAG ab, LAMP1 with anti LAMP1 (red). TLR3-CFP is in blue. Experiments were repeated twice with reproducible findings. Scale bar 2 micron.
We also carried out experiments in cells of myeloid lineage (Fig. 1E). These were THP1 human monocytes differentiated to macrophages by exposure to PMA. In common with the HEK293 experiments (Fig. 1C), treatment with Poly I:C led to localization of both SREC-I and TLR3 in intracellular vesicles (Fig. 1E). Interestingly, many of these structures were stained with antibodies to the lysosomal protein LAMP1 suggesting co-locatization of SREC-I and TLR3 to a lysosomal/endolysosomal compartment after Poly I:C treatment.
It was suggested in earlier reports that TLR3 trafficking to the endosomes and plasma membrane required UNC93B1 activity (Pohar et al., 2013). However we observed trafficking of TLR3 to the plasma membrane in HEK293 cells that do not express UNC93B1cells but engineered to express SREC-I. It was shown recently by Poher et al., that even though UNC93B1 could activate TLR3 and be responsible for TLR3 trafficking from ER to endo/lysosomal sites, this adaptor itself did not traffic to the plasma membrane along with TLR3 (Kim et al., 2008). This group later reported that the ectodomain was responsible for plasma membrane translocation of TLR3 in the presence of UNC93B1 (Pohar et al., 2013, 2014). Here we observed that TLR3 could traffic to the plasma membrane in the absence of UNC93B1 expression. We also observed colocalization of TLR3 and SREC-I in the endo/lysosomal compartment in the presence of Poly I:C in bone marrow derived macrophages (data not shown). As these two receptors are localized in ER or endosomal membrane compartments without or with Poly I:C respectively, it could be predicted that activated SREC-I might be an ER to cell surface trafficking/translocating receptor for TLR3. Thus SREC-I could potentially substitute for UNC93B1 in cells lacking or deficient in UNC93B1 expression.
We next asked whether SREC-I could interact physically with TLR3 using co-immunoprecipitation analysis. Indeed, SREC-I was minimally co-immunoprecipitated with TLR3 by exposure of Hek293-TLR3 stable cells overexpressing SREC-I-GFP to Poly I:C (Fig. 2). We did not see any sort of interaction between these two receptors in the absence of TLR3 ligand. Thus these two receptors might interact directly in response to Poly I:C.
Figure 2. TLR3 was associated with SREC-I in the presence of Poly I:C in HEK293 cells.
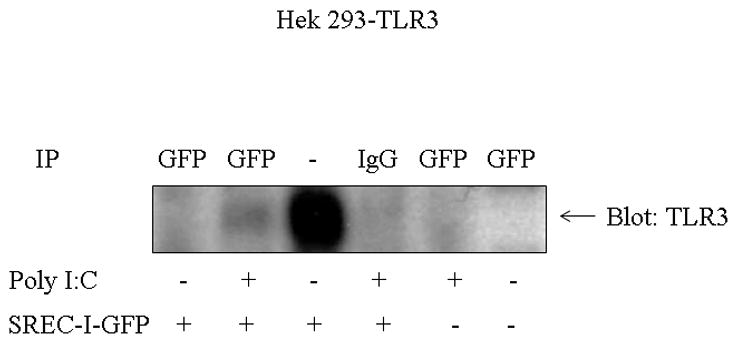
HEK293 cells stably expressing TLR3 was transfected with SREC-I-GFP for 22 hr. Cells were then treated with or without 10ug/ml Poly I:C for 30 min. Cell lysates were collected and SREC-I-GFP was immunoprecipitated using anti-GFP antibody and anti GFP ab and then the precipitated complexes were subjected to SDS-PAGE analysis followed by blotting for TLR3 using anti-TLR3 antibody. Experiments were carried out twice, reproducibly.
We next examined signaling pathways that could potentially transmit the effects of poly I:C-induced SREC-I-TLR3 interactions into the cytoplasm. We investigated activity of the NFkβ pathway and of the MAP kinases, p38 and c-jun kinase (JNK) using phospho-specific antibodies that detect the phosphorylated, active forms of p65, p38 and JNK (Fig. 3A, B, C) and NFkβ activity was also monitored by the SEAP promoter-reporter assay (Fig. 3F). Experiments were initially carried out in HEK293 cells with forced expression of: (a) neither receptor, (b) TLR3 alone or (c) TLR3 plus SREC-I. We found that maximal activation of p38, JNK and NFkB required both TLR3 and SREC-I (Fig. 3A, B, C). Likewise with NFkB reporter assays, maximal NFkB activation required expression of both TLR3 and SREC-I (Fig. 3E).
Figure 3. Poly I:C enhanced MAP Kinase and NFkβ activity in the presence of both TLR3 and SREC-I.

A, B and C, HEK293-TLR3 cells were transfected with SREC-I or untransfected HEK293-TLR3 cells were then incubated with or without Poly I:C (10μg/ml) for 2 hr. Cell lysates were collected and then subjected to SDS-PAGE and western blotting with appropriate antibodies. D, Bone marrow derived macrophages (BMDM) were transfected with siRNA of SREC-I for 72 hr. Cells were then incubated with Poly I:C (or not) as in A. Cell were lysed and equal amount of protein were subjected to SDS-PAGE and western blotting using anti phospho-p65 antibody and anti p65 antibody. E, BMDM cells were treated as in D and then cell lysates were subjected to SDS-PAGE and western blotting using antibodies shown in E. F, HEK293-TLR3 cells were transfected with SREC-I or not and also NFkβ-SEAP/CMV-SEAP constructs. Cells were incubated with Poly I:C (10μg/ml)/ODN2395 (10μg/ml), a non TLR3 ligand. NFkβ activity was measured as instructed by NFkβ-SEAporter assay kit. Similar results were observed in two separate experiments.
We also carried out parallel experiments on NFkB activity in BMDM and observed an elevated level of phospho-p65 in BMDM expressing both TLR3 and SREC-I (Fig. 3D). As BMDM express both receptors constitutively, levels of SREC-I were modulated by RNA interference using siRNA. Indeed, BMDM cells knocked down for SREC-I showed decreased levels of phosphorylated p65 comparing to the level seen in cells expressing both receptors (Fig. 3D).
We next measured activity of IFR3 (Interferon Regulatory factor 3) in BMDM cells expressing both TLR3 and SREC-I or cells expressing TLR3 alone (SREC-I was knocked down using siRNA). IRF3 plays a critical role in immunity to DNA and RNA viruses and becomes phosphorylated in the presence of dsRNA (Gu et al., 2014). We observed the level of phosphorylated IFR3 in BMDM to be increased in cells expressing both SREC-I and TLR3, in the presence of Poly I:C, compared to TLR3 only where activation was minimal, (Fig. 3F).
We next studied the role of SREC-I in Poly I:C-mediated cytokine release. We used an array approach to examine a range of cytokines. Assays were carried out on wild type, undifferentiated THP1 monocytic cells overexpressing TLR3 with or without SREC-I (Fig. 4). Secretion of IL-8 (Interleukin-8) from THP1 cells was minimal in the absence of ligand when either receptor was absent (Fig. 4). However, ligand-dependent secretion of IL-8 was strongly enhanced when SREC-I was overexpressed along with TLR3 (compared to TLR3 alone). There were minimal SREC-I mediated changes in secretion of other cytokines including IFNγ and TNFα. As IL-8 seemed a major target for Poly I:C-SREC-I-TLR3, we then examined a range of other chemokines (Fig. 4B). As in Fig. 4A, IL-8 was released at maximum levels by THP1 cells expressing both receptors in the presence of Poly I:C compared to control cells, while the release of MCP-1, RANTES, IP-10 and MIG was strongly induced in the presence of TLR3 with or without SREC-I overexpression (Fig. 4B). We observed an approximate 2.5 fold increase in IL-8 release in cells expressing both SREC-I and TLR3 compared with those expressing TLR3 alone (Fig. 4B). Again these effects were specific for IL-8 and were not duplicated for the other chemokines.
Figure 4. SREC-I enhanced IL-8 release from THP1 cells in the presence of TLR3 ligand.

A, THP1 cells were transfected with TLR3 or TLR3 and SREC-I expression plasmids for 22 hr. Cells were incubated with 10ug Poly I:C for 12 hr and then assayed for cytokine production using a human cytokine multianylate ELISA array kit according to manufacturer’s protocol. B, THP1 cells treated as in (A) and then assayed for chemokines using multianylate ELISA array kit according to Manufacturer’s protocol. Data represent the mean of two independent experiments.
We then investigated the role of MAPK phosphorylation in IL-8 and IL-6 secretion by THP1 cells. THP1 cells engineered to overexpress both SREC-I and TLR3 released significantly more IL-8 in the presence of 10μg of Poly I:C in comparison to the cells expressing SREC-I or TLR3 only (Fig. 5A). SREC-I alone did not mediate a detectable response to Poly I:C. Release of IL-6 after Poly I:C exposure was similarly regulated by SREC-I and TLR3 (Fig. 5C). Earlier we had observed an increase in phosphorylation of MAP kinases in cells expressing both TLR3 and SREC-I in the presence of Poly I:C (Fig. 2A,B). We now examined whether activation of these upstream MAP kinase pathways was necessary for IL-8 and IL-6 release by these cells. THP1 cells were incubated with PD98059 a specific ERK inhibitor, SB203580 a p38 inhibitor or the JNK inhibitor II for 1 hour before incubation with Poly I:C (Fig. 5B, D). We observed sharp decreases in both Poly I:C –induced IL-8 and IL-6 release when the cells were exposed to each of these inhibitors prior to incubation with Poly I:C. It therefore seemed that, cytokine release involved activation of each of the three MAP kinase pathways. We also observed increased secretion of IL-6 in an alternative cell type, BMDM expressing both TLR3 and SREC-I (Fig. 5F). Knocking down either of these receptors by siRNA decreased secretion of this proinflammatory cytokine, Il-6 by these cells (Fig. 5F).
Figure 5. Poly I:C-SREC-I-TLR3-induced IL-8 release required MAP Kinase activity.

A, THP1 cells were transfected with TLR3/SREC-I or TLR3 and SREC-I expression plasmids for 22 hr. Cells were incubated with Poly I:C (10ug/ml) for 12 hr and then assayed for IL-8 release. B, THP1 cells were transfected with SREC-I and TLR3 for 22 hr and then incubated with the ERK inhibitor (PD98059), p38 inhibitor (SB203580) or the JNK inhibitor (JNK inhibitor II) (10uM) for 1 hr right before incubation with incubation with Poly I:C for 12 hr. IL-8 secretion was assayed according to manufacturer’s instructions. C, THP1 cells were treated as in A and then IL-6 release was assayed according to manufacturer’s instruction. D, THP1 cells were treated as in B and IL-6 release was assayed. E, THP1 cells were treated with PMA (5–10ng/ml) for 72 hours. Cells were then transfected with siRNA SREC-I or siRNA TLR3. Cells were incubated with Poly I:C for 12 hr and then Ifnβ release was measured according to manufacturer’s protocol. F. BMDM cells were transfected with siRNA SREC-I/TLR3 or not. Wild type or TLR3 knocked down (siRNA) cells were also treated with 1–5 ng/ml of LPS for 12hrs. Cells were then incubated with Poly I:C for 12 hr. Secreted IL-6 in media was measured according to manufacturer’s protocol. Experiment was repeated twice. Data shown are the mean ±SD of results from those two experiments.
We next examined the release of the IRF3 target cytokine Ifnβ in THP1 after differentiation of these cells to macrophages with PMA for a prolonged period, 72 hrs. (Fig. 5E) (Differentiation upregulates TLR3 to detectable levels – data not shown). Wild type differentiated THP1 macrophages were also treated with LPS (1–5ng/ml) for 12 hours for inducing SREC-I expression prior to incubation with Poly I:C (Fig. 5E). It has been shown earlier that LPS incubation can induce expression of SREC-I in macrophages (Tamura et al., 2004). These cells were then transfected with either siRNA species targeting SREC-I or TLR3. Cells expressing TLR3 only (SREC-I knockdown) responded to Poly I:C to release Ifnβ while secretion was increased when cells expressed both the receptors (Fig. 5E). A limited number of parallel experiments were carried out on BMDM. We observed sharp increases in IL-6 release by BMDM expressing IRF3 treated with Poly I:C and these were amplified by SREC-I expression.
Since IL-8 release in the presence of Poly I:C and SREC-I was significant in the experiments shown earlier, we next focused on this cytokine. In kinetic studies, we found IL-8 secretion to be significantly increased after one hour incubation with Poly I:C when THP1 cells overexpressed both receptors (Fig. 6A). We also noticed that the amount of IL-8 release was decreased with 4 hr incubation with Poly I:C (Fig. 6A). Later we also observed a second wave of IL-8 release after 6 hr and 12hr incubation with Poly I:C in the same THP1 cells overexpressing both SREC-I and TLR3. Although we saw earlier that both TLR3 and SREC-I could be colocalized on the surface in the presence of Poly I:C, it was not clear whether signaling was initialized at the cell surface or at the endosomal site.
Figure 6. Poly I:C-TLR3-mediated IL-8 release required SREC-I mediated internalization and acidification of endosomes.

A, THP1-TLR3 cells were transfected with or without SREC-I for 22 hr and then incubated with 10ug Poly I:C for indicated time. IL-8 release from cells was then assayed. B, THP1-TLR3 cells were transfected as in A and then incubated with or without Bafilomycin A (0.2 uM for 20 min) or PP2 (10uM for 12 hr). Cells were incubated with Poly I:C for 12 hr and then IL-8 release from THP1 cells were assayed. C, Differentiated THP1 cells were transfected with siRNA SREC-I for 72 hr and then cells were treated with Poly I:C (10μg/ml) for 2 hr. Cells were then lysed and equal amount of protein in lysate were subjected to SDS-PAGE and western blotting using appropriate antibodies. Experiments were carried out twice reproducibly.
In previous studies we had observed that, in case of LPS-TLR4-SREC-I signaling, the kinase c-Src was necessary for cytokine release and internalization of ligand-receptor complexes (Murshid et al., submitted). In addition, it was not clear whether MAP kinase signaling was initiated at the cell surface upon Poly I:C-TLR3 interaction or in the endosomes after internalization of Poly I:C-TLR3-SREC-I complexes. TLR3 activation and ligand binding affinity have been shown to be dependent on both dsRNA length and pH (Cain et al., 1989; De Bouteiller et al., 2005). It was additionally shown that slightly acidic pH (pH 4.5–6.7) was optimum for dsRNA-TLR3 response (De Bouteiller, et al., 2005). Even though plasma membrane association of both the receptors SREC-I and TLR3 with Poly I:C was detected above, it was not clear whether this ligand-receptor interaction could initiate signaling from the plasma membrane. We therefore examined the effects of c-Src inhibition as well as blockade of endosomal acidification on IL-8 release. We observed inhibition of Poly I:C - induced cytokine release after incubation with bafilomycin A (inhibitor of endosomal acidification) treatment as well as PP2 (the Src kinase inhibitor) (Fig. 6B) in THP1 expressing both TLR3 and SREC-I. We knocked down SREC-I and found a sharp decrease in IL-8 release compared to that of cells expressing both receptors (Fig. 6B, Lane 3 and 6). Our findings suggested that both c-Src activity and endosomal acidification were required for IL-8 secretion induced by formation of an activated Poly I:C-TLR3-SREC-I complex. In control experiments, we also observed increased phosphorylated cSrc (Y416) in differentiated THP1 macrophage cells expressing TLR3 and SREC-I compared to cells not treated with Poly I:C (Fig. 6C).
Discussion
Our experiments demonstrated that SREC-I could influence the intracellular location of TLR3, the rate of MAPK and NFkβ signaling and the degree of cytokine expression in monocytes and macrophages exposed to Poly I:C (Fig. 7). SREC-I thus could be important in antiviral responses in monocytes and macrophages. Our data suggested that SREC-I could mediate Poly I:C induced trafficking of TLR3 from the ER to the plasma membrane and to endosomes at which site activated TLR3 likely mediated signaling to NFkβ, IRF3 and the MAPK family (Liu et al., 2008). Localization of SREC-I to endosome/lysosome organelles may be key, as signaling through TLR3 was shown previously to require the low pH environment offered by these compartments (De Bouteiller et al., Cain et al.). Interestingly SREC-I appeared to have a similar effect on MHC class II signaling in DC in which, in previous studies, we saw trafficking of MHC class II molecules through an ER>plasma membrane> endosome/lysosome pathway in a SREC-I dependent manner. In this case the end result was increased antigen presentation by the MHC class II molecules and triggering of adaptive immunity (Murshid, et al, 2014) rather than the effects on innate immunity envisaged here. Among pro-inflammatory cytokines, SREC-I appeared to strongly bias expression towards IL-8 (Fig. 4). The IL-8 promoter has been shown to interact with the factors NFkB, AP-1 and C/EBP β. This is consistent with the cell signaling experiments carried out here as AP-1 and C/EBP β activated downstream of MAPK signaling (Fig. 3). However, these factors are also involved in activation of many other cytokines, suggesting a novel input from SREC-I signaling that is selective for IL-8. The large intracellular domain of SREC-I is largely uncharacterized and future studies of this structure may clarify such regulation (A. Murshid & SK Calderwood, unpublished).
Figure 7. Role of SREC-I in response to double stranded RNA.
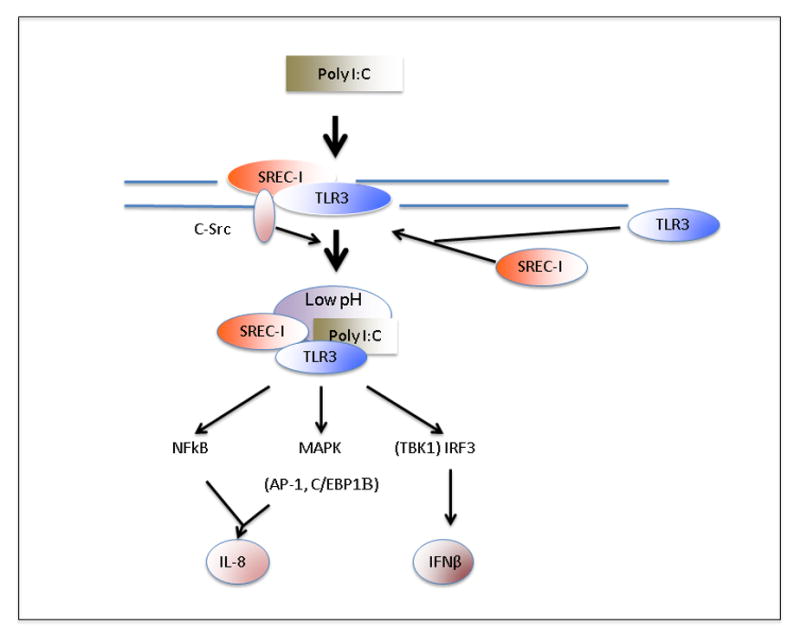
Double stranded RNA species Poly I:C interacts with macrophages leading to recruitment of SREC-I and TLR3 to the cell surface where they form a membrane complex that interacts with c-Src. The latter kinase then regulates endocytosis of the SREC-I/TLR3/Poly I:C complexes in endosomes. The complex finally resides in endosomes with low intravesicular pH, marked with lysosomal protein LAMP1. TLR3 is able to signal from such complexes and launches NFkB, MAPK and IRF3 signaling. (IRF3 is activated by the kinase TBK1). Activated NFkB, AP-1 and C/EBPβ are known to interact with the IL-8 gene while activated IRF3 leads to synthesis of Ifn-β.
TLR3 has been shown to participate in adaptive immunity by triggering maturation of CD8α+ DC and T cell activation in response to virally infected cells. SREC-I, which was shown previously to be present in DC, could play a significant role in this regard as it appeared to be involved in engulfment of cell corpses (Ramirez-Ortiz et al., 2013). SREC-I was identified as a paralog of the C. elegans engulfment factor CED-1 (Zhou et al., 2001). SREC-I could thus contribute to cell corpse engulfment as well as assisting in the TLR3 response to viral PAMPs such as ds RNA in the dead cells, leading to inflammatory signaling cascades including the NFkβ pathway upstream of DC maturation (Ramirez-Ortiz et al., 2013). Our current studies showed sturdy activation of NFkB by Poly I:C downstream of SREC-I-TLR3 interactions (Fig. 3). Mature CD8α+ DC could then signal to CD4+ and CD8+ T cells and trigger an antigen-specific immune response to virus. The role of IL-8 in the antiviral response is less clear. However IL-8 was shown to induce chemotaxis of target cells as well as endocytic activity in respiratory tract infection with Respiratory Syncytial Virus and was thus associated with extravasation and trafficking of leukocytes to infected regions (Fiedler et al., 1995; Hacking et al., 2004). Recent studies have shown that Poly I:C induced proinflammatory cytokine release required activation of MAP kinase, ERK, p38 and JNK and phosphorylation of p65 subunit of NFkβ (Liu et al., 2008). We found that the activation of MAP kinase and NFkβ in the presence of Poly I:C and TLR3 only was relatively minor compared to cells expressing both TLR3 and SREC-I, suggesting that this scavenger receptor participated in Poly I:C recognition and internalization as well as immune signaling.
Acknowledgments
This work was supported by US National Institutes of Health research grants RO-1CA047407, RO-1CA119045 and RO-1CA094397. AM is a recipient of JCRT and TJB is a recipient of CAPES fellowship. The authors alone are responsible for the content and writing of the paper.
Footnotes
Conflict of interest statement
There are no conflicts of interest for any of the authors regarding this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 2.Beutler B. Innate immunity: an overview. Mol Immunol. 2004;40:845–59. doi: 10.1016/j.molimm.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 3.McGettrick AF, O’Neill LA. Curr Opin Immunol. 2010;22:20–7. doi: 10.1016/j.coi.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 4.Kumar A, Zhang J, Yu FS. Toll-like receptor 3 agonist poly(I:C)-induced antiviral response in human corneal epithelial cells. Immunology. 2006;117:11–21. doi: 10.1111/j.1365-2567.2005.02258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takahashi N, Yamada T, Narita N, Fujieda S. Double-stranded RNA induces production of RANTES and IL-8 by human nasal fibroblasts. Clin Immunol. 2006;118:51–8. doi: 10.1016/j.clim.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 6.Pirher N, Ivicak K, Pohar J, Bencina M, Jerala R. A second binding site for double-stranded RNA in TLR3 and consequences for interferon activation. Nat Struct Mol Biol. 2008;15:761–3. doi: 10.1038/nsmb.1453. [DOI] [PubMed] [Google Scholar]
- 7.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–8. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 8.Chuang TH, Lee J, Kline L, Mathison JC, Ulevitch RJ. Toll-like receptor 9 mediates CpG-DNA signaling. J Leukoc Biol. 2002;71:538–44. [PubMed] [Google Scholar]
- 9.Pohar J, Pirher N, Benčina M, Manček-Keber M, Jerala R. The ectodomain of TLR3 receptor is required for its plasma membrane translocation. PLoS One. 2014;9:e92391. doi: 10.1371/journal.pone.0092391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Y, Kimura K, Yanai R, Chikama T, Nishida T. Cytokine, chemokine, and adhesion molecule expression mediated by MAPKs in human corneal fibroblasts exposed to poly(I:C) Invest Ophthalmol Vis Sci. 2008;49:3336–44. doi: 10.1167/iovs.07-0972. [DOI] [PubMed] [Google Scholar]
- 11.Andrade WA, Souza M, do C, Ramos-Martinez E, Nagpal K, Dutra MS, Melo MB, Bartholomeu DC, Ghosh S, Golenbock DT, Gazzinelli RT. Combined action of nucleic acid-sensing Toll-like receptors and TLR11/TLR12 heterodimers imparts resistance to Toxoplasma gondii in mice. Cell Host Microbe. 2013;13:42–53. doi: 10.1016/j.chom.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brinkmann MM, Spooner E, Hoebe K, Beutler B, Ploegh HL, Kim YM. The interaction between the ER membrane protein UNC93B and TLR3, 7, and 9 is crucial for TLR signaling. J Cell Biol. 2007;177:265–75. doi: 10.1083/jcb.200612056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pifer R, Benson A, Sturge CR, Yarovinsky F. UNC93B1 is essential for TLR11 activation and IL-12-dependent host resistance to Toxoplasma gondii. J Biol Chem. 2011;286:3307–14. doi: 10.1074/jbc.M110.171025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murshid A, Gong J, Calderwood SK. Heat shock protein 90 mediates efficient antigen cross presentation through the scavenger receptor expressed by endothelial cells-I. J Immunol. 2010;185:2903–17. doi: 10.4049/jimmunol.0903635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim YM, Brinkmann MM, Paquet M, Ploegh HL. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature. 2008;452:234–8. doi: 10.1038/nature06726. [DOI] [PubMed] [Google Scholar]
- 16.Pohar J, Pirher N, Benčina M, Manček-Keber M, Jerala R. The role of UNC93B1 protein in surface localization of TLR3 receptor and in cell priming to nucleic acid agonists. J Biol Chem. 2013;288:442–54. doi: 10.1074/jbc.M112.413922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cain CC, Sipe DM, Murphy RF. Regulation of endocytic pH by the Na+,K+-ATPase in living cells. Proc Natl Acad Sci U S A. 1989;86:544–8. doi: 10.1073/pnas.86.2.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Bouteiller O, Merck E, Hasan UA, Hubac S, Benguigui B, Trinchieri G, Bates EE, Caux C. Recognition of double-stranded RNA by human toll-like receptor 3 and downstream receptor signaling requires multimerization and an acidic pH. J Biol Chem. 2005;280:38133–45. doi: 10.1074/jbc.M507163200. [DOI] [PubMed] [Google Scholar]
- 19.Ramirez-Ortiz ZG, Pendergraft WF, 3rd, Prasad A, Byrne MH, Iram T, Blanchette CJ, Luster AD, Hacohen N, El Khoury J, Means TK. The scavenger receptor SCARF1 mediates the clearance of apoptotic cells and prevents autoimmunity. Nat Immunol. 2013;14:917–26. doi: 10.1038/ni.2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou Z, Hartwieg E, Horvitz HR. CED-1 is a transmembrane receptor that mediates cell corpse engulfment in C. elegans. Cell. 2001;104:43–56. doi: 10.1016/s0092-8674(01)00190-8. [DOI] [PubMed] [Google Scholar]
- 21.Fiedler MA, Wernke-Dollries K, Stark JM. Respiratory syncytial virus increases IL-8 gene expression and protein release in A549 cells. Am J Physiol. 269:L865–72. doi: 10.1152/ajplung.1995.269.6.L865. [DOI] [PubMed] [Google Scholar]
- 22.Hacking D, Knight JC, Rockett K, Brown H, Frampton J, Kwiatkowski DP, Hull J, Udalova IA. Increased in vivo transcription of an IL-8 haplotype associated with respiratory syncytial virus disease-susceptibility. Genes Immun. 2004;5:274–82. doi: 10.1038/sj.gene.6364067. [DOI] [PubMed] [Google Scholar]
- 23.Tamura Y, Osuga J, Adachi H, Tozawa R, Takanezawa Y, et al. Scavenger receptor expressed by endothelial cells I (SREC-I) mediates the uptake of acetylated low density lipoproteins by macrophages stimulated with lipopolysaccharide. J Biol Chem. 2004;279(30):30938–44. doi: 10.1074/jbc.M313088200. [DOI] [PubMed] [Google Scholar]
- 24.Gu M, Zhang T, Lin W, Liu Z, Lai R, Xia D, Huang H, Wang X. Protein phosphatase PP1 negatively regulates the Toll-like receptor- and RIG-I-like receptor-triggered production of type I interferon by inhibiting IRF3 phosphorylation at serines 396 and 385 in macrophage. Cell Signal. 2014;26(12):2930–2939. doi: 10.1016/j.cellsig.2014.09.007. [DOI] [PubMed] [Google Scholar]