

Summary
Background
In addition to their key role in hemostasis, platelets and megakaryocytes also regulate immune and inflammatory responses, in part through their expression of Toll-like receptors (TLRs). Among the TLRs, TLR3 recognizes double-stranded (ds) RNA associated with viral infection. Thrombocytopenia is a frequent complication of viral infection. However, the expression and functionality of TLR3 in megakaryocytes and platelets is not yet well understood.
Objective
To study the expression and functionality of TLR3 in the megakaryocytic lineage.
Methods and Results
RT-PCR, flow cytometric, and immunofluorescence assays showed that TLR3 is expressed in CD34+ cells, megakaryocytes, and platelets. Immunoblotting assays showed that stimulation of megakaryocytes with two synthetic agonists of TLR3, Poly(I:C) and Poly(A:U), activated the NF-κB, PI3K/Akt, ERK1/2, and p38 pathways. TLR3-megakaryocyte activation resulted in reduced platelet production in vitro and IFN-β release through the PI3K/Akt and NF-κB signaling pathways. TLR3 ligands potentiated the aggregation mediated by classical platelet agonists. This effect was also observed for ATP release, but not for P-selectin or CD40L membrane exposure, indicating that TLR3 activation was not involved in alpha granule release. In addition, TLR3 agonists induced activation of the NF-κB, PI3K/Akt, and ERK1/2 pathways in platelets. Reduction of platelet production and platelet fibrinogen binding mediated by Poly(I:C) or Poly(A:U) were prevented by the presence of an inhibitor of TLR3/dsRNA complex.
Conclusions
Our findings indicate that functional TLR3 is expressed in CD34+ cells, megakaryocytes, and platelets, and suggest a potential role for this receptor in the megakaryo/thrombopoiesis alterations that occur in viral infections.
Keywords: Toll-like receptor 3, interferon-beta, NF-kappa B, megakaryocytes, platelets
Introduction
Platelets play a key role in hemostasis and are important players in the immune and inflammatory responses [1]. Although the mechanisms involved in the immune-related functions mediated by platelets are not completely understood, it is known that the expression and/or release of cell adhesion molecules, cytokines, and immunomodulators are very important. Among the immunomodulatory molecules expressed by platelets and megakaryocytes are the Toll-like receptors (TLRs) an important family of receptors that detect the molecular patterns associated with a broad variety of pathogens [2]. Platelets express TLR1, TLR2, TLR4, TLR6, TLR7, TLR8 and TLR9 [3–7]. Pathogens can bind to platelets through TLRs, and present their products to neutrophils and cells in the reticuloendothelial system. Thus, the expression of TLRs on platelets links the innate and adaptive immune responses during infectious inflammation and atherosclerotic vascular disease [8, 9]. Most investigations of TLR functionality have been focused on TLR4 and the platelet response to its ligand, lipopolysaccharide (LPS), a component of the Gram-negative bacterial cell wall. Although it is generally accepted that platelets contain the molecular machinery required for signaling through TLR4 [10], the activation responses of platelets to LPS stimulation remains controversial [11–15]. Regarding platelet TLR2 functionality, it has been reported that stimulation of platelet TLR2 induces platelet aggregation, adhesion, and degranulation [15–17]. A subset of TLRs recognizes viral components and induces antiviral responses. TLR4 recognizes viral components at the cell surface, whereas TLR3, TLR7, TLR8, and TLR9 recognize viral nucleic acids on the endosomal membrane. In platelets, TLR9 is expressed internally [7], and unconventional TLR9 ligands promote platelet activation in vitro and thrombosis in vivo via the TLR9/MyD88 pathway [18]. TLR7 is also expressed in platelets, and its activation mediates platelet-viral immune responses through P-selectin exposure and increases platelet-neutrophil adhesion, without inducing thrombosis [6].
In contrast, only a few studies have examined the expression and function of TLRs in megakaryocytes and assessed whether or not these receptors have a role in platelet production. TLR4−/− mice were shown to have a lower platelet count than wild-type mice, suggesting a possible role for TLR4 in thrombopoiesis [19]. The megakaryocytic cell line Meg01 expresses TLR1, TLR6 and TLR2 [3, 20]. TLR2 stimulation of Meg01 promotes activation of pathways known to be downstream of TLRs and subsequent megakaryocyte maturation [20].
Thrombocytopenia is a frequent complication of viral infections. Several mechanisms are involved, depending on the nature of the infecting viruses, including immunological platelet destruction, increased clearance, inappropriate platelet activation and consumption, and impaired megakaryopoiesis [21].
We reported that Junin virus (JV), impairs in vitro thrombopoiesis by decreasing proplatelet formation and platelet release as well as the functionality of in vitro generated-platelets. The decrease in platelet release could be mimicked by Poly(I:C), a mimetic of double-stranded (ds) RNA replication products and ligand of TLR3, and by both type I interferons (IFN-I, IFN α/β) [22]. Moreover, both early progenitors and mature megakaryocytes express functional IFN-I receptors and synthetize/release IFN-β after stimulation with JV or Poly(I:C), indicating that megakaryocytes may play a role in antiviral defense as both IFN-I producers and responders [23].
TLR3 is the critical sensor of viral dsRNA, and its stimulation induces the activation of NF-κB and the production of IFN-I [24]. Although we previously demonstrated TLR3 expression in megakaryocytes by RT-PCR [23], its expression and functionality in the megakaryocytic linage has not yet been studied. Here, we report that TLR3 is expressed in hematopoietic progenitor cells, megakaryocytes, and platelets. Moreover, TLR3 activation decreases platelet production but increases their functionality. Our data further extend the novel role of TLRs in the megakaryocytic lineage and suggest a new mechanism through which virus interaction with megakaryocytes could result in selective alteration of thrombopoiesis.
Materials and Methods
Ethics statement
The study was approved by the Institutional Review Board of the National Academy of Medicine, Argentina. All individuals provided written informed consent for the collection of samples and their subsequent analysis.
Purification of CD34+ cells
Umbilical cord blood was collected following normal, full-term deliveries. After collection, CD34+ cells were purified as previously described [25]. The CD34+ cells typically ranged between 95% and 99%.
Megakaryocyte culture
CD34+ cells were cultured in IMDM supplemented with BIT9500 (STEMCELL Technologies, Vancouver, Canada) in the presence of thrombopoietin (TPO, 100 ng/mL) (Peprotech, Veracruz, Mexico). For PCR experiments, mature megakaryocytes were purified by immunomagnetic selection from cultures on days 12–14 using anti-CD61 magnetic beads (Miltenyi Biotec) according to the manufacturer’s instructions. The purity of the final cell suspension was > 97%, and cell viability was >80%.
Preparation of human platelets
Blood samples were obtained from healthy donors who had not taken non-steroidal anti-inflammatory drugs in the 10 days before sampling. Washed platelets (WPs) were prepared as previously described [26]. Highly purified platelets were obtained using a high-efficiency leukoreduction filter (Purecell PL; PALL Biomedical Products, East Hills, NY, USA), which decreased contamination to less than 1 leukocyte per 104 platelets.
Semi-quantitative RT-PCR
RNA was isolated from cell pellets by using TriReagent (Genbiotech, Buenos Aires, Argentina). cDNA was synthesized using random hexamers (Biodynamics, Buenos Aires, Argentina) and MMLV reverse transcriptase (Promega, Buenos Aires, Argentina). The PCR was conducted at an annealing temperature of 55°C. All reactions were confirmed to be within the linear range of amplification. The primer sequences used to amplify the target fragments were: TLR3 sense 5′-GATCTGTCTCATAATGGCTTG-3′ and antisense 5′-GACAGATTCCGAATGCTTGTG-3′; CD20 sense 5′-CGTGCTCCAGACCCAAATCTAACA-3′ and antisense 5′-GCGTGACAACACAAGCTGCTACAA-3′; and actin sense 5′- AACCCCAAGGCCAACCGCGAGAAGATGACC-3′ and antisense 5′-GGTGATGACCTGGCCGTCAGGCAGCTCGTA-3′. Results were analyzed with GELPRO software (Bethesda, MD, USA), and the values were normalized to actin.
Immunofluorescence
Cells were fixed with 1% paraformaldehyde, centrifuged, and permeabilized with 0.1% Triton X-100. After blocking, the cells were incubated with a goat polyclonal IgG anti-TLR3 antibody (Ab) or goat IgG and then with a FITC donkey anti-goat IgG (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Similar results were obtained with a primary anti-TLR3 mouse monoclonal antibody (clone TLR3.7; Abcam, Cambridge, UK), and then an Alexa-conjugated secondary Ab. In the case of megakaryocytes and platelets, PE-CD61 (BD Pharmingen) was used as a lineage marker. The nuclei were stained with DAPI. Slides were mounted with PolyMount and analyzed by fluorescence microscopy (BX60; Olympus, Tokyo, Japan).
Immunoblotting
Western blotting was performed as previously described [26]. Membranes were probed with phospho-ERK1/2, phospho-p38, phospho-NF-κB p65 (all from Santa Cruz Biotechnology), I-κB-α (BD Biosciences), and phospho-Akt (Abcam). Each membrane was reprobed with an Ab against β-actin (BD Biosciences) to ensure equal loading. Protein bands were visualized by using ECL. Immunoblotting results were semi-quantitated using Gel-Pro analyzer 3.1 software.
Determination of platelet production in culture
Platelets were counted using flow cytometry as previously described [25]. Briefly, after 17 days of culture, the cells were incubated with an FITC-CD61 antibody or an irrelevant isotype and fixed. Cells from each culture condition were distributed in an equal volume. For each sample, the acquisition rate was 1 μL/s for 100 s. Events were collected without gating by using a log scale for size (FSC) and intracellular granularity (SSC). An analytical gate was determined based on the scatter properties of normal blood platelets treated similar to the culture-derived platelets. Culture-derived platelets were counted as CD61+ events with the same scatter properties as human peripheral blood platelets. Anti-human IFN-β antibody (clone IFNb/A1) or its respective isotype (both from BioLegend, San Diego, CA, USA) were used in neutralizing experiments.
Determination of IFN-β level by ELISA
The concentration of IFN-β in the culture supernatant was determined using a commercial ELISA kit (PBL Interferon-Source, Piscataway, NJ, USA) according to the manufacturer’s instructions.
Platelet aggregation and ATP release
Aggregation and ATP release were measured simultaneously in a Lumi-aggregometer (Chrono-Log, Havertown, PA, USA). Aggregation was induced by alpha-thrombin (Enzyme Research Laboratories, Swansea, UK), arachidonic acid (AA), ADP (both from Sigma, San Diego, CA, USA), or collagen (Nycomed Pharma, Unterschleißheim, Germany). ATP levels were calculated at the end of the assay by adding a known amount of ATP (Sigma).
Flow cytometry studies
To evaluate intracellular staining, cells were fixed and permeabilized using Cytofix/Cytoperm™ solution (BD Biosciences) and incubated with a goat polyclonal anti-TLR3 IgG or with normal goat IgG. After washing, the cells were incubated with FITC-labeled donkey anti-goat IgG. To assess TLR3 expression in the megakaryocyte population, double staining was performed by labeling the cells with PE-CD61. WPs were stained and fixed with PE-CD62P (anti-P-selectin), Alexa-488-fibrinogen, FITC-CD40L or an equivalent amount of isotype-matched control antibody. The percentage of positive cells and the median fluorescence intensity (MFI) were analyzed using a FACSCalibur flow cytometer and FCS Express V3 software.
Statistical analysis
All results are expressed as mean ± standard error of the mean (SEM). According to the experiments, results were analyzed using Student’s paired t-test or ANOVA with normal distributions to determine the significance of differences between groups. P values less than 0.05 were considered statistically significant. All statistical analyses were performed by using Prism 5 software (GraphPad).
Results
TLR3 is expressed in human hematopoietic progenitor cells, megakaryocytes, and platelets
TLR3 mRNA expression in megakaryocytes was previously reported by our group [23]. To extend our understanding of TLR expression throughout the megakaryocytic linage, here, we first evaluated the expression of TLR3 in human CD34+ cells, cultured mature megakaryocytes, and peripheral blood platelets. Transcripts of the expected size for TLR3 were detected in all these cells. These transcripts were not due to leukocyte contamination as CD20, a marker very highly expressed by mononuclear cells (MNCs), was undetectable in these platelet and megakaryocyte samples (Fig. 1A). To verify that these mRNAs corresponded to TLR3 protein expression, TLR3 protein was visualized by immunofluorescence microscopy and flow cytometry. The immunofluorescence assays showed that TLR3 is expressed in CD34+ progenitor cells as well as in megakaryocytes and platelets (Fig. 1B). Taking into account that TLR3 expression has been reported to be cell-specific and could be localized to either the cell surface or the endosomal membrane [27], we analyzed the cellular localization of TLR3 by flow cytometry. Our analysis revealed that TLR3 was only slightly expressed in CD34+ cells, but not in megakaryocytes or platelet surface. However, intracellular staining showed that 95% ± 2% of CD34+ cells, 76.5% ± 5.2% of megakaryocytes, and 21.5% ± 1.8% of platelets expressed TLR3 (Fig. 1C).
Fig. 1. TLR3 is expressed in the megakaryocytic lineage.
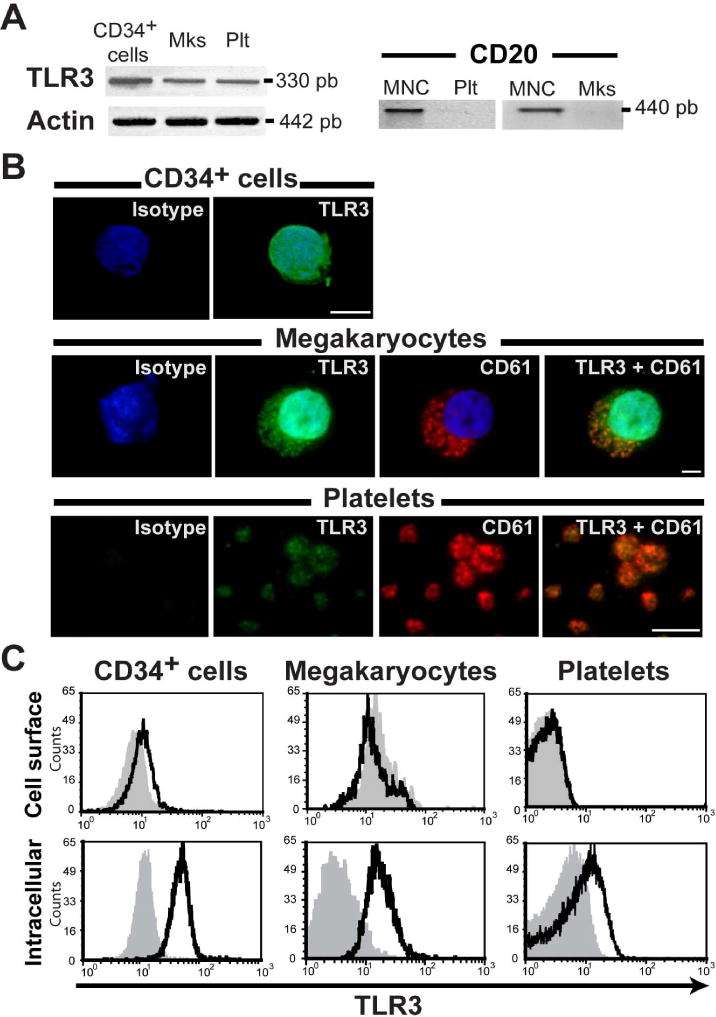
(A) mRNAs encoding TLR3 were detected in CD34+ cells, megakaryocytes (Mks), and platelets (Plt) by RT-PCR. No CD20 mRNA (a marker of leukocytes) was detected in platelets or megakaryocytes, indicating no leukocyte contamination, and mononuclear cells (MNC) were used as a positive control (n=3). (B) TLR3 (green) and CD61 (red) proteins were detected by immunofluorescence microscopy, and nuclei were visualized with DAPI (blue) (n=4) Original magnification, 60× (scale bar: 5 μm). (C) Flow cytometry analysis of TLR3 in cell surface and permeabilized cells throughout of the megakaryocyte lineage. Histograms showing TLR3 expression assayed in CD34+ purified cells (CD34+ >95%), in double stained mature megakaryocytes (CD61+/TLR3+ cells) and in washed platelets (WPs). Shown are representative histograms from one of three experiments. The gray histograms represent the isotype fluorescence.
Signaling transduction pathways activated by TLR3 stimulation in megakaryocytes
A recent report demonstrated that TLR2 activation in Meg01 cells promotes PI3K/Akt and MAPK activation [20]. To determine if these pathways are activated in megakaryocytes upon TLR3 activation, mature cultured megakaryocytes were stimulated with Poly(I:C), a synthetic dsRNA analogue that is extensively used as a TLR3 ligand. Megakaryocyte lysates were immunoblotted for phospho-AKT, phospho-ERK1/2, and p38 MAPK. Figs 2A and 2B show that Akt, ERK1/2, and p38 MAPK phosphorylation was triggered by Poly(I:C).
Fig. 2. TLR3 agonists activate the PI3K/Akt, ERK1/2, p38, and NF-κB pathways in megakaryocytes.
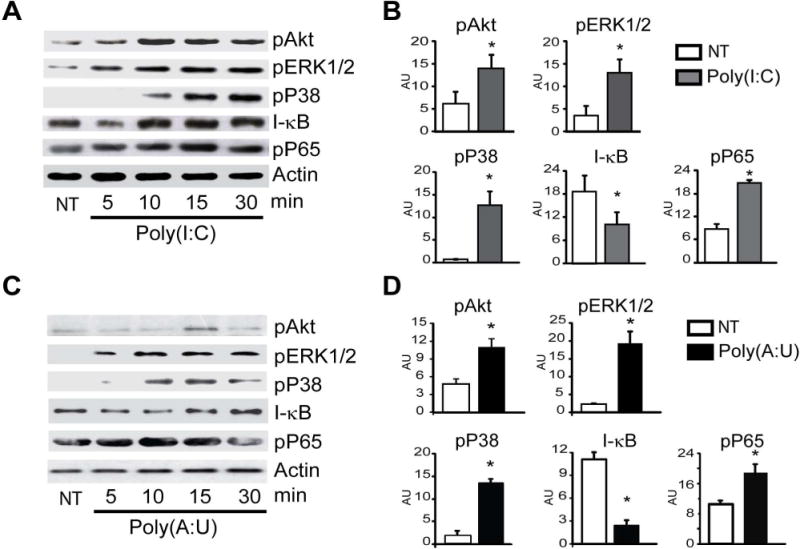
(A) Megakaryocytes were treated with 100 μg/mL Poly(I:C) for up to 30 min, and lysates were probed for proteins in each pathway and were compared to untreated cells (NT). (B) Each membrane was reprobed with an anti-β-actin antibody to calculate the relative OD. Mean ± SEM in arbitrary units (AU) is shown at the time point when the most significant differences were found for each pathway (*P<0.05 vs. NT, n=3). (C and D) The signaling pathways activated by 100 μg/mL Poly(A:U) were examined as described in (A).
In other cell types, activation of TLR3 leads to the activation of NF-κB, IRF3, and AP1 with subsequent production of inflammatory cytokines and IFN-I [27]. As shown in Figs 2A and 2B, Poly(I:C) induced the degradation of I-κB, the inhibitor of the NF-κB complex after 5 min of stimulation, which then returned to control levels. Accordingly, the levels of phospho-p65, a subunit of NF-κB were increased (Figs. 2A and B).
Poly(I:C) is recognized by both endosomal TLR3 and cytosolic receptors, including RNA helicases such as RIG-I and the melanoma differentiation-associated gene 5 (MDA5) [27]. To determine whether the described signaling cascade triggered by Poly(I:C) was due to TLR3-specific activation, megakaryocytes were stimulated with another synthetic dsRNA, polyadenylic-polyuridylic acid [Poly(A:U)], which only signals through TLR3 [28]. Figs 2C and 2D show a similar protein phosphorylation pattern after Poly(A:U) stimulation. Collectively, these data suggest that TLR3 activation induces not only ERK1/2, p38-MAPK, and PI3K/Akt signaling but also NF-κB signaling.
Megakaryocyte functional responses-mediated by TLR3 activation
We have previously demonstrated that infection of TPO-stimulated CD34+ cells with JV results in a reduction of platelet production due to IFN-β release [22]. We now show that both, Poly(I:C) and Poly(A:U), dampened platelet biogenesis in culture (Fig. 3A). This effect was not due to a reduced proliferation of CD34+ cells’ differentiation towards megakaryocyte or impaired megakaryocyte maturation since the percentage of CD61 and CD42b positive cells (maturation marker) was not modified in Poly(I:C) or Poly(A:U) treated cultures (Fig. 3B). The presence of an IFN-β blocking antibody in the cell culture completely prevented the decrease in platelet production (Fig. 3C). Moreover, a novel compound named 4a which acts as a specific inhibitor of the TLR3/dsRNA complex [29] significantly reduced the inhibitory effect mediated by Poly(I:C) or Poly(A:U) (Fig. 3C). To further confirm that IFN-β was the mediator involved in this effect, this cytokine was measured in the supernatant of megakaryocytes stimulated with the TLR3 ligands. As shown in Fig. 3D both dsRNAs induced significant levels of IFN-β compared to the control samples and the production of IFN-β was not observed in megakaryocytes treated with the TLR3 antagonist, 4a. Furthermore, the IFN-β production was completely abolished when megakaryocytes were pretreated with inhibitors of the PI3K/Akt (Ly294002) or NF-κB (Ro106-9920) pathways but not with inhibitors for ERK1/2 (U0126) or P38 MAPK (SB203580) pathways (Fig. 3D).
Fig. 3. Functional responses triggered by TLR3 agonists in megakaryocytes.
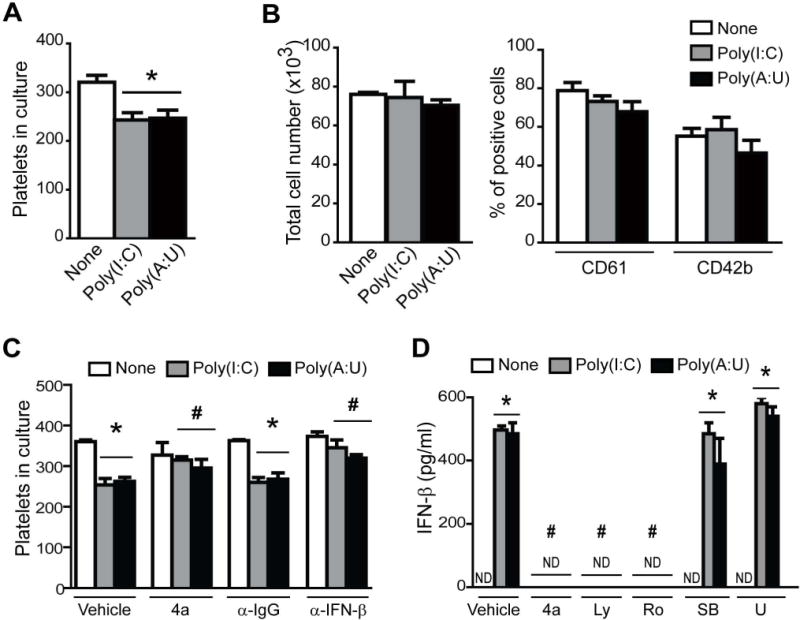
(A) Immature megakaryocytes (7 day culture) were treated with Poly(I:C) or Poly(A:U) (100 μg/mL), and platelets were counted on day 17 by flow cytometry (*P<0.05 vs. none, n=5). (B) CD34+ cells were cultured in presence of TPO and 100 μg/ml of Poly(I:C) or Poly(A:U) during 14 days. Total cells were counted with an hemocytometer and percentage of CD61+ cells as a differentiation marker, or CD42b+ cells as a maturation marker, were determined by flow cytometry (n=4). (C) The TLR3/dsRNA inhibitor (4a, 27μM), or the neutralizing anti-IFN-β or corresponding IgG (20 μg/ml) were added 1 h before megakaryocyte stimulation with Poly(I:C) or Poly(A:U), and platelet counts were determined at day 17 (*P<0.05 vs. none, #P<0.05 vs the same treatment with vehicle or IgG, n=4). (D) Mature megakaryocytes were preincubated with the TLR3/dsRNA inhibitor (4a, 27μM), the PI3K/Akt inhibitor Ly294002 (Ly, 25 μM), the NF-κB inhibitor Ro106-9920 (Ro, 12.5 μM), the p38 inhibitor SB203580 (SB, 25 μM) and the ERK1/2 inhibitor U0126 (U, 10 μM) 1 h before Poly(I:C) or Poly(A:U) (100 μg/mL) for 24h. IFN-β release in megakaryocyte supernatants was determined by ELISA. Values below 50 pg/mL cannot be detected by this ELISA assay, and are shown as not detectable (ND), (*P<0.05 vs. none, #P<0.05 vs vehicle, n=4).
TLR3 agonists differentially modulate platelet responses
Platelets were previously shown to express functional TLRs; however, the expression and functionality of TLR3 in platelets has not yet been studied. Having shown here that platelets express TLR3, in the next set of experiments, we tested whether Poly(I:C) and Poly(A:U) modulated platelet function. We observed that while neither Poly(I:C) or Poly(A:U) per se were able to induce platelet aggregation, both agonists potentiated aggregation mediated by suboptimal concentrations of classical platelet agonists such as ADP, collagen, arachidonic acid, and thrombin (Fig. 4A).
Fig. 4. TLR3 activation potentiates platelet aggregation and αIIbβ3 integrin conformational changes triggered by classical agonists.
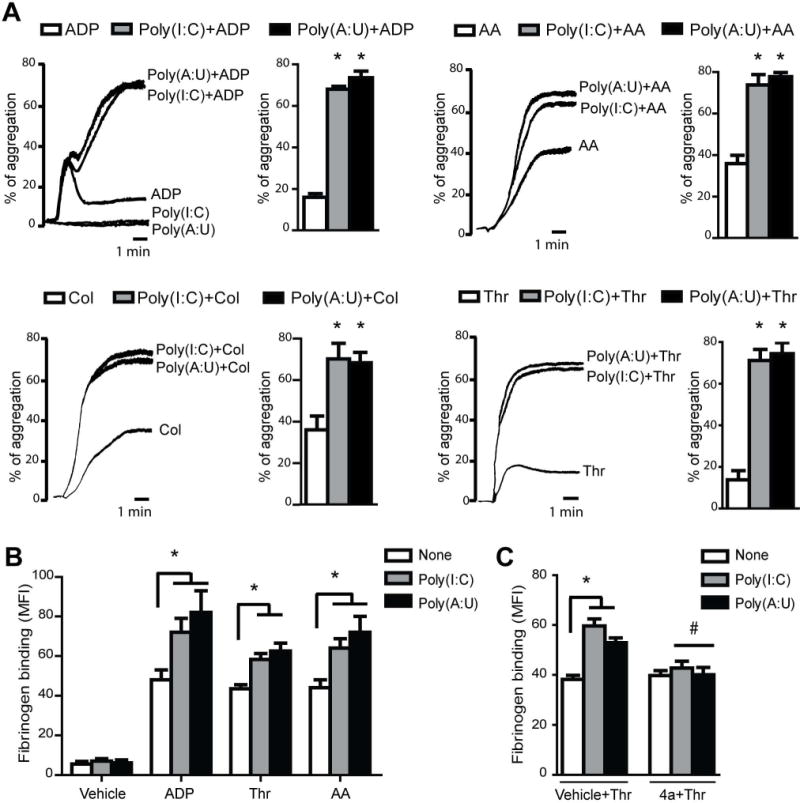
(A) WPs were incubated with Poly(I:C) or Poly(A:U) (50 μg/mL) for 1 min, and then aggregation was induced by a suboptimal concentration of ADP (1.25 μM), arachidonic acid (AA, 0.05 μM), collagen (Col, 1 μg/mL), or thrombin (Thr, 0.02 U/mL). Representative aggregation curves from 4 independent experiments are shown. (B) WPs were stimulated with the indicated dsRNA for 1 min, and then ADP (1.25 μM), Thr (0.02 U/mL), AA (0.05 μM) or vehicle was added and incubated for 5 min. Fibrinogen binding was analyzed by flow cytometry (*P<0.05 vs. each respective agonist alone, n=4). (C) WPs were preincubated with the TLR3/dsRNA inhibitor (4a) at the EC90 (27μM), 30 min before Poly(I:C) or Poly(A:U) (50 μg/mL) and then stimulated with Thr (0.02 U/mL) for 5 min (*P<0.05 vs. none, #P<0.05 vs vehicle+Thr, n=4).
The molecular mechanism of platelet-platelet interaction involves integrin αIIbβ3 binding to fibrinogen, which acts as a bridge between the two cells [30]. Our results show that the potentiation of aggregation was correlated with increased fibrinogen binding (Fig. 4B). The potentiating effect of dsRNAs in the fibrinogen binding induced by thrombin was abolished by the preincubation of platelets with the 4a compound (Fig. 4C). Similar results were observed with the others agonists used (data not shown). In addition, we found that both dsRNAs also potentiated ATP release (Fig. 5A).
Fig. 5. TLR3 activation potentiates ATP release but not alpha granule secretion.
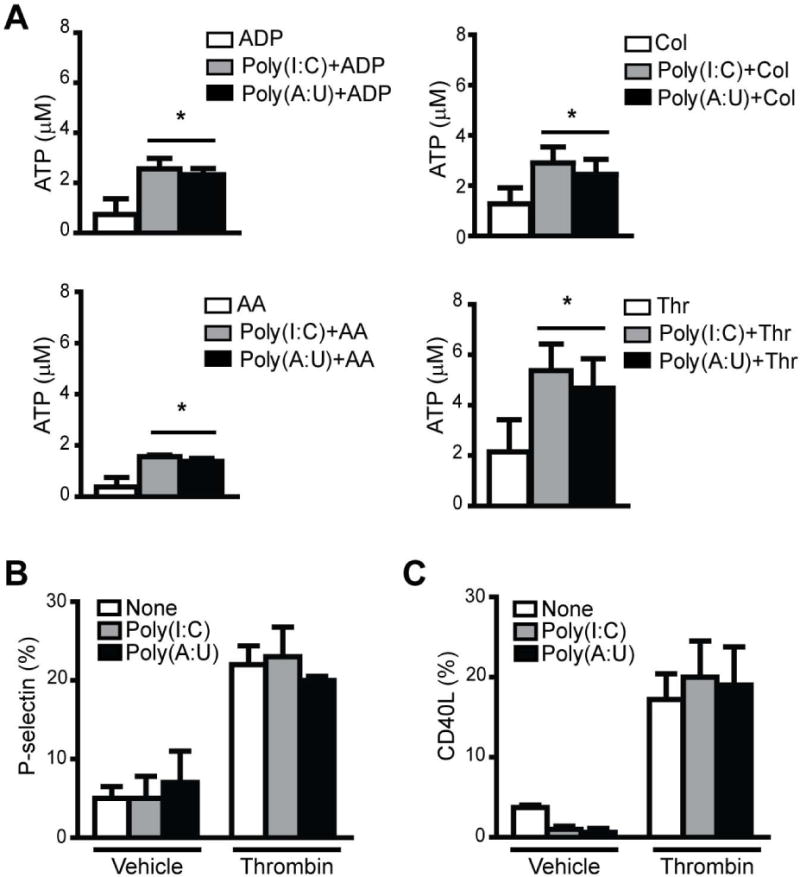
(A) WPs were incubated with Poly(I:C) or Poly(A:U) (50 μg/mL) for 1 min, and then ATP release was induced by suboptimal concentrations of ADP (1.25 μM), AA (0.05 μM), Col (1 μg/mL), or Thr (0.02 U/mL). (*P<0.05 vs. the respective agonist alone, n=4). (B) P-selectin and (C) CD40L expression was determined by flow cytometry in WPs treated with each TLR3 agonist (50 μg/mL) alone or with Thr 0.05 U/mL for 15 min (n=4).
We then determined whether TLR3 activation promoted alpha granule secretion of two mediators of platelet inflammatory responses, P-selectin and CD40 ligand (CD40L). Our results show that neither Poly(I:C) nor Poly(A:U)-mediated platelet stimulation resulted in P-selectin and CD40L surface exposure. In addition, TLR3 agonists were not able to potentiate alpha granule secretion caused by platelet agonists (Figs. 5B and 5C).
Poly(I:C) and Poly(A:U) triggered NF-κB, PI3K/Akt, and ERK1/2 activation in platelets
Similar to what was observed in megakaryocytes, in platelets, the PI3K/Akt, ERK1/2, and NF-κB signaling pathways were activated following stimulation by Poly(I:C) and Poly(A:U) (Figs. 6A and 6B). Interestingly, and in contrast to megakaryocytes, the p38 MAPK pathway was not activated by TLR3 agonists within the first five minutes of stimulation (data not shown). Notably, as was observed in the functionality assays, activation of these pathways by TLR3 agonists did not seem to be sufficient to induce platelet activation. However, these pathways were involved in the potentiating effect of platelet activation mediated by Poly(I:C) or Poly(A:U) and classical agonists, since inhibitors of the PI3K/Akt (Ly294002) and NF-κB (Ro106-9920) pathways restored integrin-αIIbβ3 activation to thrombin values (Fig. 6C).
Fig. 6. Signaling pathways triggered by TLR3 activation.
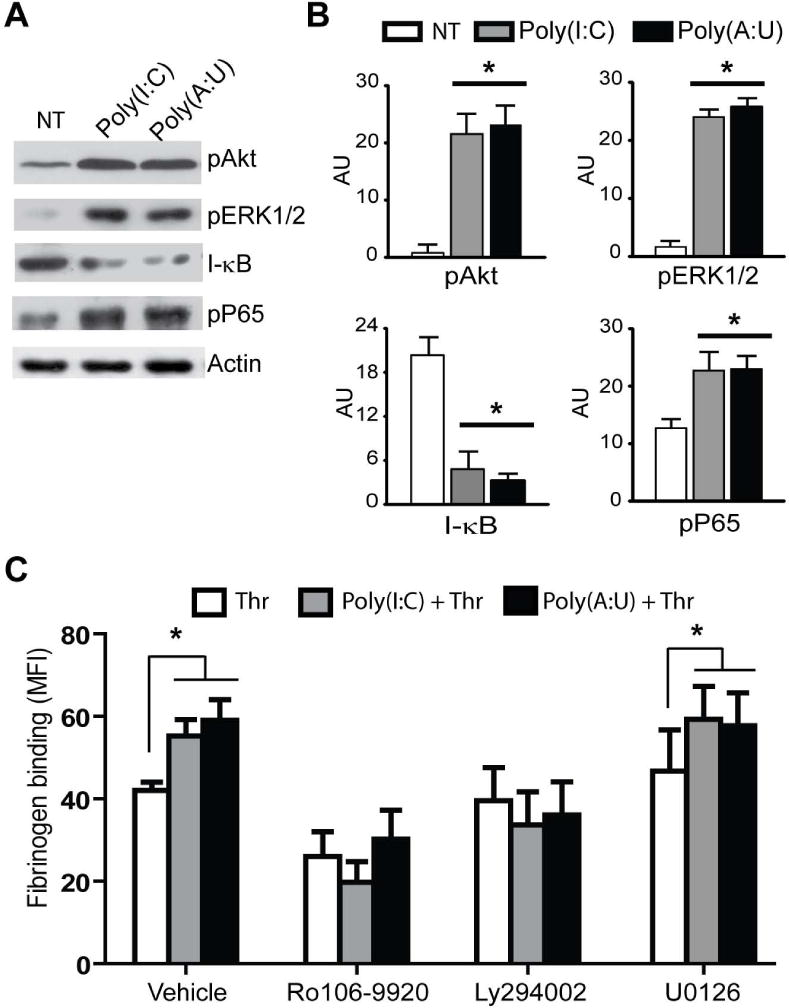
(A) WPs were stimulated or not (NT) for 5 min with Poly(I:C) or Poly(A:U) (50 μg/mL), and blots of cell lysates were probed for pAkt, pERK1/2, I-κB, and pP65 as indicators of NF-κB pathway activation. Actin was used as a loading control. The image shown is representative of three independent experiments. Mean ± SEM in arbitrary units (AU) are shown in (B) *P<0.05 vs. NT. (C) WPs were preincubated with the inhibitors of PI3K/Akt (Ly294002, 10 μM), NF-κB (Ro106-9920, 3 μM), or ERK1/2 (U0126, 20 μM) for 20 min, stimulated with 50 μg/mL Poly(I:C) or Poly(A:U) for 5 min, and then treated with 0.03 U/mL of Thr. The mean fluorescence intensity of fibrinogen binding was determined by flow cytometry (n=4, *P<0.05 vs. Thr).
Discussion
In the present study, we demonstrated that TLR3 mRNA and protein is present on CD34+ cells, megakaryocytes, and platelets. Moreover, stimulation of megakaryocytes with two TLR3 ligands, Poly(I:C) and Poly(A:U), impaired platelet biogenesis and synthesis, release IFN-I, and potentiated platelet activation triggered by classical agonists.
The expression and functionality of TLRs in murine and human CD34+ cells have been previously described [31]. Human CD34+ progenitors express TLR4, TLR7, TLR8, and TLR9, and stimulation of TLR7 and TLR8 in vitro induces myeloid differentiation of these progenitors [32]. TLR4 activation of HSCs also redirects lymphoid progenitors to produce dendritic cells, enter the cell cycle, and acquire lineage markers [33]. In addition, we previously showed that JV infection or Poly(I:C) stimulation of TPO-stimulated CD34+ cells reduces platelet generation in culture without affecting apoptosis or cell proliferation, suggesting the presence of dsRNA sensors in CD34+ cells [22]. Recently, it was reported that human peripheral blood CD34+ cells express TLR3 mRNA and that Poly(I:C) stimulation increases its expression as well as IFN-I synthesis and apoptosis [34]. Here, we confirmed these data, showing that TLR3 mRNA and protein are expressed in CD34+ cells derived from cord blood. We observed that the expression pattern of TLR3 decreased from CD34+ cells to megakaryocytes and platelets. Although the reason for this phenomenon is not clear, a similar observation was reported for TLR1 and TLR6 in Meg01 cells and platelets [3]. The higher expression of TLRs on CD34+ cells is in line with the suggestion that TLR expression on HSCs represents an early sensing mechanism for HSCs to directly detect infection and respond with immediate production of immune effector cells [35]. On the other hand, the expression of TLRs in HSC was shown to be regulated during their differentiation [36, 37]. Together, these data suggest that bacterial/viral products may influence hematopoiesis through activation of TLRs on HSCs.
In contrast to that with HSCs, few studies have examined the expression of TLRs on human megakaryocytes and their role in platelet biogenesis. Beaulieu et al. demonstrated that TLR2 activation promotes adhesion of Meg01 cells to extracellular matrix, increases polyploidization, and increases CD41 and GPIb mRNA and protein levels [20]. TLR9, a receptor for unmethylated CpG islands in bacterial and viral DNA, is expressed in murine megakaryocytes and is distributed to platelets during proplatelet production [7]. Mice with a deletion of TLR2 or TLR4 have lower platelet counts than wild type mice, suggesting a role for these TLRs in thrombopoiesis [19, 20].
Our study demonstrates that TLR3 is localized in the cytoplasm of megakaryocytes. Although TLR3 subcellular localization is associated mainly with the endosomal compartment in megakaryocytes, the staining pattern seems to be partially consistent with CD61, probably due to possible endoplasmic reticulum expression of both proteins [38–40]. To the best of our knowledge, we described here, for the first time, a functional intracellular TLR in human megakaryocytes that acts as a sensor of viral infection. Indeed, megakaryocytes are not only susceptible to viral infection [41–44] and replication [45, 46] but thrombocytopenia is also a common clinical feature in people infected with several viruses [21]. Increased platelet clearance, destruction, and activation are among the different causes of the reduced platelet count observed during viral infection. Our previous findings of IFN-β-mediated selective impairment of platelet production in JV infected hematopoietic progenitors and megakaryocytes [22], together with our current data showing a similar effect using two mimetics of viral replication products that are also TLR3 ligands, strongly indicate that activation of this pattern-recognition receptor in megakaryocytes may be another mechanism involved in virus-associated thrombocytopenia. The observation that in the presence of 4a, an inhibitor of TLR3/dsRNA complex, the reduction in platelet count was prevented and the IFN-β production was inhibited, gave further support to the functional role of TLR3 in megakaryocytes.
In an attempt to further characterize the molecular pathways involved in TLR3 activation, we demonstrated that TLR3 downstream signaling in megakaryocytes involves activation of NF-κB, ERK1/2, p38, and PI3K/Akt. Moreover, blockade of PI3K/Akt and NF-κB pathways abolished IFN-β synthesis triggered by TLR3 agonists, suggesting that, similar to inflammatory cells, in megakaryocytes, these pathways account for IFN-I production. Interestingly, Beaulieu et al. [20] showed that that signaling molecules activated downstream of TLR2 in Meg01 cells, also include NF-κB, PI3K/Akt, ERK1/2, and p38 MAPK. However, and in contrast to our data, the functional responses were cell polyploidization and platelet production. Unlike to that observed with other TLRs, TLR3 activation induces MyD88-independent pathways that activate IRF3 and IRF7, which induce the synthesis of IFN-I [47]. On the other hand, except for specific inflammatory cells and agonists [48], activation of TLR2 does not induce the production of IFN-I but rather inflammatory cytokines [49]. Therefore, the different signal transduction pathways and effector molecules triggered by activation of either TLR2 or TLR3 appear to determine the positive or negative fate of megakaryocyte development and platelet production, as well as the thrombocytosis or thrombocytopenia frequently observed in bacterial or viral infections, respectively.
Although there have been several studies of the expression and functionality of TLR2, TLR4, TLR7, and TLR9 in platelets, this is the first report of TLR3 expression at both the mRNA and protein levels. TLR3 was only expressed intracellularly in platelets and in contrast to platelet TLR9 [7], no upregulation or surface exposure of TLR3 was induced by dsRNAs or platelet agonists (data not shown). We observed that although stimulation of platelets with Poly(I:C) or Poly(A:U) failed to trigger platelet activation, it potentiated the αIIbβ3 activation, ATP release, and platelet aggregation induced by classical platelet agonists. P-selectin and CD40L expression were not altered, suggesting that alpha granule degranulation is not influenced by TLR3 activation. As in megakaryocytes, TLR3 agonists activated the PI3K/Akt, NF-κB, and ERK1/2 signaling pathways. Although activation of these pathways was not sufficient to trigger platelet effector responses, they were required to prime activation induced by platelet agonists as the synergistic effect exerted by dsRNA and thrombin on fibrinogen binding or αIIbβ3 activation was no longer observed in platelets pretreated with NF-κB or PI3K inhibitors.
Recently, very interesting data from Koupenova et al. in platelets revealed that stimulation of TLR7, another endosomal TLR that recognizes single-stranded (ss) RNA, did not induce platelet aggregation nor affect the ability of platelets to aggregate with a low concentration of platelet agonists but promoted P-selectin exposure and platelet-neutrophil aggregates without inducing thrombosis [6]. In contrast, priming of platelets with collagen and subsequent activation of TLR9 by viral oligodeoxynucleotides, results in increased P-selectin expression and platelet clumping [7]. Interestingly, activation of platelet TLR9 by carboxyalkylpyrrole protein adducts, the so-called altered-self molecules generated during oxidative stress, promotes platelet activation and aggregation in vitro and accelerates thrombosis in vivo in a TLR9/MyD88-dependent manner [18]. Together, these data indicate that activation of viral sensor TLRs in platelets could give rise to a plethora of effector responses that could influence immune and hemostatic responses.
In summary, our data indicate that CD34+ cells, megakaryocytes, and platelets, express functional TLR3. Activation of this receptor in megakaryocytes results in an IFN-β-mediated reduction in platelet generation, suggesting a potential role for TLR3 in the thrombocytopenia observed in viral infections. In addition, stimulation of TLR3 primes platelets for activation mediated by classical agonists. Whether this effect compensates for the decreased platelet number induced by activation of TLR3 on megakaryocytes or whether it has a thrombotic effect during viral infections require further investigation.
Acknowledgments
This work was supported by grants from Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT) PICT 0733-2011 and PICTO-Glaxo 009-2011 (M. Schattner), 0493-2011 (L. P. D’Atri), and from NIH R01GM101279 and R01GM103843 (H. Yin). L. P. D’Atri and M. Schattner are scientific researchers from CONICET; J. Etulain and L. Rivadeneyra hold fellowships from CONICET and M. J. Lapponi holds a fellowship from ANPCyT.
Footnotes
Addendum
L. P. D’Atri performed and designed experiments, interpreted results and wrote the manuscript; J. Etulain performed platelet functionality assays; L. Rivadeneyra performed RT-PCR studies, M. J. Lapponi performed immunofluorescence assays, M. Centurion contributed to obtain umbilical cord blood samples and revised the manuscript, K. Cheng and H. Yin contributed with the 4a compound. M. Schattner designed the study, interpreted results, formulated discussion, wrote the manuscript and assisted with manuscript preparation and editing. All authors read and approved the final manuscript.
Disclosure of Conflict of Interests
The authors state that they have no conflicts of interest.
References
- 1.Semple JW, Italiano JE, Jr, Freedman J. Platelets and the immune continuum. Nat Rev Immunol. 2011;11:264–74. doi: 10.1038/nri2956. [DOI] [PubMed] [Google Scholar]
- 2.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–84. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 3.Shiraki R, Inoue N, Kawasaki S, Takei A, Kadotani M, Ohnishi Y, Ejiri J, Kobayashi S, Hirata K, Kawashima S, Yokoyama M. Expression of Toll-like receptors on human platelets. Thromb Res. 2004;113:379–85. doi: 10.1016/j.thromres.2004.03.023. [DOI] [PubMed] [Google Scholar]
- 4.Cognasse F, Hamzeh H, Chavarin P, Acquart S, Genin C, Garraud O. Evidence of Toll-like receptor molecules on human platelets. Immunol Cell Biol. 2005;83:196–8. doi: 10.1111/j.1440-1711.2005.01314.x. [DOI] [PubMed] [Google Scholar]
- 5.Andonegui G, Kerfoot SM, McNagny K, Ebbert KV, Patel KD, Kubes P. Platelets express functional Toll-like receptor-4. Blood. 2005;106:2417–23. doi: 10.1182/blood-2005-03-0916. [DOI] [PubMed] [Google Scholar]
- 6.Koupenova M, Vitseva O, Mackay CR, Beaulieu LM, Benjamin EJ, Mick E, Kurt-Jones EA, Ravid K, Freedman JE. Platelet-TLR7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood. 2014;124:791–802. doi: 10.1182/blood-2013-11-536003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thon JN, Peters CG, Machlus KR, Aslam R, Rowley J, Macleod H, Devine MT, Fuchs TA, Weyrich AS, Semple JW, Flaumenhaft R, Italiano JE., Jr T granules in human platelets function in TLR9 organization and signaling. J Cell Biol. 2012;198:561–74. doi: 10.1083/jcb.201111136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cox D, Kerrigan SW, Watson SP. Platelets and the innate immune system: mechanisms of bacterial-induced platelet activation. J Thromb Haemost. 2011;9:1097–107. doi: 10.1111/j.1538-7836.2011.04264.x. [DOI] [PubMed] [Google Scholar]
- 9.Garraud O, Cognasse F. Platelet Toll-like receptor expression: the link between “danger” ligands and inflammation. Inflamm Allergy Drug Targets. 2010;9:322–33. doi: 10.2174/187152810793937991. [DOI] [PubMed] [Google Scholar]
- 10.Berthet J, Damien P, Hamzeh-Cognasse H, Pozzetto B, Garraud O, Cognasse F. Toll-like receptor 4 signal transduction in platelets: novel pathways. Br J Haematol. 2010;151:89–92. doi: 10.1111/j.1365-2141.2010.08292.x. [DOI] [PubMed] [Google Scholar]
- 11.Ward JR, Bingle L, Judge HM, Brown SB, Storey RF, Whyte MK, Dower SK, Buttle DJ, Sabroe I. Agonists of toll-like receptor (TLR)2 and TLR4 are unable to modulate platelet activation by adenosine diphosphate and platelet activating factor. Thromb Haemost. 2005;94:831–8. [PubMed] [Google Scholar]
- 12.Sheu JR, Hung WC, Su CH, Lin CH, Lee LW, Lee YM, Yen MH. The antiplatelet activity of Escherichia coli lipopolysaccharide is mediated through a nitric oxide/cyclic GMP pathway. Eur J Haematol. 1999;62:317–26. doi: 10.1111/j.1600-0609.1999.tb01909.x. [DOI] [PubMed] [Google Scholar]
- 13.Zhang G, Han J, Welch EJ, Ye RD, Voyno-Yasenetskaya TA, Malik AB, Du X, Li Z. Lipopolysaccharide stimulates platelet secretion and potentiates platelet aggregation via TLR4/MyD88 and the cGMP-dependent protein kinase pathway. J Immunol. 2009;182:7997–8004. doi: 10.4049/jimmunol.0802884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stahl AL, Svensson M, Morgelin M, Svanborg C, Tarr PI, Mooney JC, Watkins SL, Johnson R, Karpman D. Lipopolysaccharide from enterohemorrhagic Escherichia coli binds to platelets through TLR4 and CD62 and is detected on circulating platelets in patients with hemolytic uremic syndrome. Blood. 2006;108:167–76. doi: 10.1182/blood-2005-08-3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rivadeneyra L, Carestia A, Etulain J, Pozner RG, Fondevila C, Negrotto S, Schattner M. Regulation of platelet responses triggered by Toll-like receptor 2 and 4 ligands is another non-genomic role of nuclear factor-kappaB. Thromb Res. 2014;133:235–43. doi: 10.1016/j.thromres.2013.11.028. [DOI] [PubMed] [Google Scholar]
- 16.Keane C, Tilley D, Cunningham A, Smolenski A, Kadioglu A, Cox D, Jenkinson HF, Kerrigan SW. Invasive Streptococcus pneumoniae trigger platelet activation via Toll-like receptor 2. J Thromb Haemost. 2010;8:2757–65. doi: 10.1111/j.1538-7836.2010.04093.x. [DOI] [PubMed] [Google Scholar]
- 17.Blair P, Rex S, Vitseva O, Beaulieu L, Tanriverdi K, Chakrabarti S, Hayashi C, Genco CA, Iafrati M, Freedman JE. Stimulation of Toll-like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3-kinase. Circ Res. 2009;104:346–54. doi: 10.1161/CIRCRESAHA.108.185785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Panigrahi S, Ma Y, Hong L, Gao D, West XZ, Salomon RG, Byzova TV, Podrez EA. Engagement of Platelet Toll-Like Receptor 9 by Novel Endogenous Ligands Promotes Platelet Hyper-Reactivity and Thrombosis. Circ Res. 2012;112:103–12. doi: 10.1161/CIRCRESAHA.112.274241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jayachandran M, Brunn GJ, Karnicki K, Miller RS, Owen WG, Miller VM. In vivo effects of lipopolysaccharide and TLR4 on platelet production and activity: implications for thrombotic risk. J Appl Physiol. 2007;102:429–33. doi: 10.1152/japplphysiol.01576.2005. [DOI] [PubMed] [Google Scholar]
- 20.Beaulieu LM, Lin E, Morin KM, Tanriverdi K, Freedman JE. Regulatory effects of TLR2 on megakaryocytic cell function. Blood. 2011;117:5963–74. doi: 10.1182/blood-2010-09-304949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flaujac C, Boukour S, Cramer-Borde E. Platelets and viruses: an ambivalent relationship. Cell Mol Life Sci. 2010;67:545–56. doi: 10.1007/s00018-009-0209-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pozner RG, Ure AE, Jaquenod de Giusti C, D’Atri LP, Italiano JE, Torres O, Romanowski V, Schattner M, Gomez RM. Junin virus infection of human hematopoietic progenitors impairs in vitro proplatelet formation and platelet release via a bystander effect involving type I IFN signaling. PLoS Pathog. 2010;6:e1000847. doi: 10.1371/journal.ppat.1000847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Negrotto S, De Giusti CJ, Lapponi MJ, Etulain J, Rivadeneyra L, Pozner RG, Gomez RM, Schattner M. Expression and functionality of type I interferon receptor in the megakaryocytic lineage. J Thromb Haemost. 2011;9:2477–85. doi: 10.1111/j.1538-7836.2011.04530.x. [DOI] [PubMed] [Google Scholar]
- 24.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–8. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 25.D’Atri LP, Pozner RG, Nahmod KA, Landoni VI, Isturiz M, Negrotto S, Schattner M. Paracrine regulation of megakaryo/thrombopoiesis by macrophages. Exp Hematol. 2011;39:763–72. doi: 10.1016/j.exphem.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 26.Malaver E, Romaniuk MA, D’Atri LP, Pozner RG, Negrotto S, Benzadon R, Schattner M. NF-kappaB inhibitors impair platelet activation responses. J Thromb Haemost. 2009;7:1333–43. doi: 10.1111/j.1538-7836.2009.03492.x. [DOI] [PubMed] [Google Scholar]
- 27.Yu M, Levine SJ. Toll-like receptor 3, RIG-I-like receptors and the NLRP3 inflammasome: Key modulators of innate immune responses to double-stranded RNA viruses. Cytokine Growth Factor Rev. 2011;22:63–72. doi: 10.1016/j.cytogfr.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Conforti R, Ma Y, Morel Y, Paturel C, Terme M, Viaud S, Ryffel B, Ferrantini M, Uppaluri R, Schreiber R, Combadiere C, Chaput N, Andre F, Kroemer G, Zitvogel L. Opposing effects of toll-like receptor (TLR3) signaling in tumors can be therapeutically uncoupled to optimize the anticancer efficacy of TLR3 ligands. Cancer research. 2010;70:490–500. doi: 10.1158/0008-5472.CAN-09-1890. [DOI] [PubMed] [Google Scholar]
- 29.Cheng K, Wang X, Yin H. Small-molecule inhibitors of the TLR3/dsRNA complex. J Am Chem Soc. 2011;133:3764–7. doi: 10.1021/ja111312h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shattil SJ, Gao J, Kashiwagi H. Not just another pretty face: regulation of platelet function at the cytoplasmic face of integrin alpha IIb beta 3. Thromb Haemost. 1997;78:220–5. [PubMed] [Google Scholar]
- 31.Nagai Y, Garrett KP, Ohta S, Bahrun U, Kouro T, Akira S, Takatsu K, Kincade PW. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity. 2006;24:801–12. doi: 10.1016/j.immuni.2006.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sioud M, Floisand Y, Forfang L, Lund-Johansen F. Signaling through toll-like receptor 7/8 induces the differentiation of human bone marrow CD34+ progenitor cells along the myeloid lineage. J Mol Biol. 2006;364:945–54. doi: 10.1016/j.jmb.2006.09.054. [DOI] [PubMed] [Google Scholar]
- 33.Massberg S, Schaerli P, Knezevic-Maramica I, Kollnberger M, Tubo N, Moseman EA, Huff IV, Junt T, Wagers AJ, Mazo IB, von Andrian UH. Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell. 2007;131:994–1008. doi: 10.1016/j.cell.2007.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu J, Guo YM, Hirokawa M, Iwamoto K, Ubukawa K, Michishita Y, Fujishima N, Tagawa H, Takahashi N, Xiao W, Yamashita J, Ohteki T, Sawada K. A synthetic double-stranded RNA, poly I:C, induces a rapid apoptosis of human CD34(+) cells. Exp Hematol. 2012;40:330–41. doi: 10.1016/j.exphem.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 35.Baldridge MT, King KY, Goodell MA. Inflammatory signals regulate hematopoietic stem cells. Trends Immunol. 2011;32:57–65. doi: 10.1016/j.it.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dorner M, Brandt S, Tinguely M, Zucol F, Bourquin JP, Zauner L, Berger C, Bernasconi M, Speck RF, Nadal D. Plasma cell toll-like receptor (TLR) expression differs from that of B cells, and plasma cell TLR triggering enhances immunoglobulin production. Immunology. 2009;128:573–9. doi: 10.1111/j.1365-2567.2009.03143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takami M, Kim N, Rho J, Choi Y. Stimulation by toll-like receptors inhibits osteoclast differentiation. J Immunol. 2002;169:1516–23. doi: 10.4049/jimmunol.169.3.1516. [DOI] [PubMed] [Google Scholar]
- 38.Chen A, Yu H, Deng H, Mitchell WB. Mechanisms of αIIbβ3 Biogenesis in the Megakaryocyte: A Proteomics Approach. In: Lawrie CH, editor. Hematology – Science and Practice: InTech. pp. 171–94. [Google Scholar]
- 39.Blasius AL, Beutler B. Intracellular toll-like receptors. Immunity. 2010;32:305–15. doi: 10.1016/j.immuni.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 40.Duperray A, Troesch A, Berthier R, Chagnon E, Frachet P, Uzan G, Marguerie G. Biosynthesis and assembly of platelet GPIIb–IIIa in human megakaryocytes: evidence that assembly between pro-GPIIb and GPIIIa is a prerequisite for expression of the complex on the cell surface. Blood. 1989;74:1603–11. [PubMed] [Google Scholar]
- 41.Zucker-Franklin D, Seremetis S, Zheng ZY. Internalization of human immunodeficiency virus type I and other retroviruses by megakaryocytes and platelets. Blood. 1990;75:1920–3. [PubMed] [Google Scholar]
- 42.Boukour S, Masse JM, Benit L, Dubart-Kupperschmitt A, Cramer EM. Lentivirus degradation and DC-SIGN expression by human platelets and megakaryocytes. J Thromb Haemost. 2006;4:426–35. doi: 10.1111/j.1538-7836.2006.01749.x. [DOI] [PubMed] [Google Scholar]
- 43.Crapnell K, Zanjani ED, Chaudhuri A, Ascensao JL, St Jeor S, Maciejewski JP. In vitro infection of megakaryocytes and their precursors by human cytomegalovirus. Blood. 2000;95:487–93. [PubMed] [Google Scholar]
- 44.Gonelli A, Mirandola P, Grill V, Secchiero P, Zauli G. Human herpesvirus 7 infection impairs the survival/differentiation of megakaryocytic cells. Haematologica. 2002;87:1223–5. [PubMed] [Google Scholar]
- 45.Chelucci C, Federico M, Guerriero R, Mattia G, Casella I, Pelosi E, Testa U, Mariani G, Hassan HJ, Peschle C. Productive human immunodeficiency virus-1 infection of purified megakaryocytic progenitors/precursors and maturing megakaryocytes. Blood. 1998;91:1225–34. [PubMed] [Google Scholar]
- 46.Li X, Jeffers LJ, Garon C, Fischer ER, Scheffel J, Moore B, Reddy KR, Demedina M, Schiff ER. Persistence of hepatitis C virus in a human megakaryoblastic leukaemia cell line. J Viral Hepat. 1999;6:107–14. doi: 10.1046/j.1365-2893.1999.00140.x. [DOI] [PubMed] [Google Scholar]
- 47.Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity. 2006;25:349–60. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 48.Bauernfeind F, Hornung V. TLR2 joins the interferon gang. Nat Immunol. 2009;10:1139–41. doi: 10.1038/ni1109-1139. [DOI] [PubMed] [Google Scholar]
- 49.Takeda K, Akira S. Microbial recognition by Toll-like receptors. Journal of dermatological science. 2004;34:73–82. doi: 10.1016/j.jdermsci.2003.10.002. [DOI] [PubMed] [Google Scholar]