

Abstract
Chronic hepatitis B virus (HBV) infection is a leading cause of hepatitis, liver failure, and hepatocellular carcinoma. An outstanding vaccine is available; however the number of infections remains high. Current anti-HBV treatments with interferon α and nucleos(t)ide analogs clear the infection in only a small minority of patients, and either induce serious side-effects or are of very long duration. HBV is a small, enveloped DNA virus that replicates by reverse transcription via an RNA intermediate. The HBV ribonuclease H (RNaseH) is essential for viral replication, but it has not been exploited as a drug target. Recent low-throughput screening of compound classes with anti-Human Immunodeficiency Virus RNaseH activity led to identification of HBV RNaseH inhibitors in three different chemical families that block HBV replication. These inhibitors are promising candidates for development into new anti-HBV drugs. The RNaseH inhibitors may help improve treatment efficacy enough to clear the virus from the liver when used in combination with existing anti-HBV drugs and/or with other novel inhibitors under development. This article forms part of a symposium in Antiviral Research on “An unfinished story: from the discovery of the Australia antigen to the development of new curative therapies for hepatitis B.”
Keywords: Hepatitis B Virus, Ribonuclease H, Reverse transcription, Inhibitors
1. HBV: Disease and genomic replication
Hepatitis B Virus (HBV) is an enveloped DNA virus that replicates in the liver. It is a member of the Hepadnaviridae family that includes the animal viruses Duck Hepatitis B Virus (DHBV) and Woodchuck Hepatitis Virus (WHV) (Dandri et al., 2005). HBV chronically infects up to 350 million people world-wide (Seeger et al., 2013). The infection causes hepatitis, fibrosis, cirrhosis, liver failure and over half of all cases of hepatocellular carcinoma (Lavanchy, 2005). Together, this leads to an annual death toll of over 500,000 (Sorrell et al., 2009).
HBV replicates its genome by reverse transcription of a viral pregenomic RNA within cytoplasmic capsid particles (Seeger et al., 2007; Summers and Mason, 1982; Tavis and Badtke, 2009). Reverse transcription is catalyzed by two enzymatic activities located on different domains of the viral polymerase protein (Chang et al., 1990; Radziwill et al., 1990). The reverse transcriptase copies the pregenomic RNA into minus-polarity DNA, and the ribonuclease H (RNaseH) destroys the viral RNA after it has been copied so that the plus-polarity DNA strand can be made. The direct product of HBV replication is a partially double-stranded DNA molecule within cytoplasmic capsid particles. These capsids may be enveloped and secreted from the cell as mature virions, or they may be transported to the nucleus where the DNA is converted to an episomal covalently-closed circular molecule (cccDNA) (Fig. 1). The cccDNA is key to HBV biology because it is the transcriptional template for all HBV RNAs (it is functionally equivalent to an integrated retroviral provirus).
Fig. 1. HBV replication cycle.
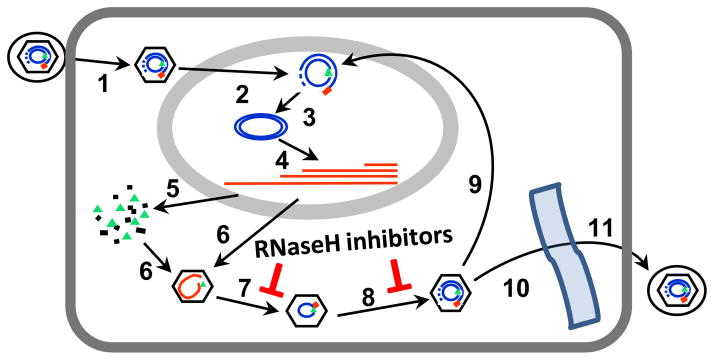
Binding of HBV virions to hepatocytes followed by fusion of the viral envelope with the plasma membrane releases core particles into the cytoplasm (1). Core particles are transported to the nucleus, where they release the partially double-stranded viral DNA (2), and the DNA is converted into cccDNA inside the nucleus (3). Viral RNAs are transcribed (4) and translated to produce the viral proteins (5). The viral pregenomic RNA is encapsidated into core particles as a complex with viral polymerase protein (6). The minus-polarity DNA strand is synthesized by the reverse transcriptase activity of the polymerase with concomitant degradation of the pregenomic RNA by the RNaseH activity (7). The plus-polarity DNA strand is synthesized by the reverse transcriptase (8). Mature core particles are then either transported back into the nucleus to maintain the cccDNA pool (9) or are enveloped by budding into the endoplasmic reticulum (10) and are non-cytolytically secreted as mature virions (11). RNaseH inhibitors block steps 7 and 8. Modified from (Hu et al., 2013).
2. Limitations to current anti-HBV therapy
The nucleos(t)ide analog drugs that dominate HBV therapy have transformed management of HBV chronic infections. The best drugs, tenofovir and entecavir, suppress HBV replication by 4–5 log10 or more in up to 70–90% of patients, often to below the common detection limit of ~200–400 copies/ml (Cox and Tillmann, 2011; Kwon and Lok, 2011; van Bommel et al., 2010; Woo et al., 2010) with little to no drug resistance even after prolonged treatment (Zoulim, 2011). This remarkable success for a monotherapy has made HBV infection controllable for those able to afford its high costs (Block et al., 2013; Lui et al., 2010), with major health benefits for the treated individuals (Dienstag, 2009; Liaw, 2013; Marcellin and Asselah, 2014).
Despite the profound suppression of HBV titers induced by nucleos(t)ide analogs, treatment reduces cccDNA levels by only about 1 log10 even after years of continuous drug exposure (Cheng et al., 2011; Werle-Lapostolle et al., 2004; Wong et al., 2006). Consequently, HBV infections are cleared in only 2–8% of patients after many years of treatment (Liaw, 2013). This persistence of the cccDNA causes viral titers to resurge if the nucleos(t)ide analogs are withdrawn, and hence treatment is essentially life-long. cccDNA persistence is in part due to its long apparent halflife, which is estimated to be 10 days in non-dividing tissue culture cells (Cai et al., 2012) and may be up to 30–60 days in the liver based on inhibitor studies in animal models (Addison et al., 2002; Moraleda et al., 1997; Zhu et al., 2001). However, maintenance of the cccDNA is also due to ongoing viral HBV replication during nucleos(t)ide analog therapy. This is revealed by sequential accumulation of resistance mutations to the nuceos(t)ide analogs (Ghany and Liang, 2007; Monto et al., 2010; Zoulim and Locarnini, 2009), and is confirmed by genetic analyses of viral DNA in the liver during therapy that explicitly demonstrate replenishment of the cccDNA even in the absence of clinically detectable viremia (Coffin et al., 2011). This opens an opportunity for improving antiviral therapy by suppressing HBV replication further than is currently possible.
3. RNaseH enzymes
3.1. General features of RNaseH enzymes
RNaseHs (Hostomsky et al., 1993) cleave RNA when the RNA is bound to DNA in a heteroduplex. They help destroy failed transcription products, remove RNA primers during DNA synthesis, and contribute to reverse transcription of viral and retrotransposon genomes. They belong to the nucleotidyl transferase enzyme superfamily whose members share a similar protein fold and similar enzymatic mechanisms (Nowotny, 2009; Yang and Steitz, 1995). This family includes E. coli RNaseH I and II (Katayanagi et al., 1990; Lai et al., 2000), human RNaseH 1 and 2 (Lima et al., 2001), retroviral RNaseH and integrases (Dyda et al., 1994), and many other nucleases and transposases. The canonical RNaseH structure contains about 100 amino acids with four conserved carboxylates (the “DEDD” motif) that coordinate two divalent cations (Nowotny et al., 2005). Integrases have a similar protein fold, but they employ only three carboxylates to coordinate the essential divalent cations (the “DDE” motif) (Nowotny, 2009). The nucleic acid cleavage mechanism is believed to require both divalent cations to promote a hydroxyl-mediated nucleophilic scission reaction (Klumpp et al., 2003; Nowotny and Yang, 2006; Yang and Steitz, 1995).
3.2. The HBV RNaseH
The HBV RNaseH was originally identified by sequence homology to known RNaseH enzymes (Khudyakov and Makhov, 1989; Schodel et al., 1988), and its existence was confirmed by evaluating the effects on viral replication of mutating the predicted RNaseH motifs (Chang et al., 1990; Chen and Marion, 1996; Gerelsaikhan et al., 1996). Ablating HBV RNaseH activity causes accumulation of long RNA:DNA heteroduplexes, truncates most minus-polarity DNA strands, and blocks production of the plus-polarity DNA strand. Virions can still be secreted when the RNaseH activity is blocked, but they contain defective RNA:DNA heteroduplex genomes (Gerelsaikhan et al., 1996; Wei et al., 1996). These genomes are biologically inert due to the severe damage to the viral DNA, especially the suppression of the second and third strand transfer reactions needed to produce mature genomes. Consequently, inhibiting the RNaseH would block both release of infectious virions and replenishment of the nuclear cccDNA (Fig. 1). The RNaseH has not been analyzed biochemically because the full-length polymerase protein is unable to accept exogenous RNaseH substrates (Gong et al., 2001), and because production of active recombinant HBV RNaseH has been a significant challenge.
The HBV and Human Immunodeficiency Virus (HIV) RNaseHs are only distantly related to one another. The genetic distance between the HBV and HIV RNaseHs is so large that the HBV enzyme is genetically about equally related to the HIV RNaseH and integrase, sharing 23% and 19% identity, respectively, in the core catalytic domains (Tavis et al., 2013). Despite this distance, the HBV and HIV RNaseHs perform the equivalent biological roles during viral replication (Freed and Martin, 2007; Tavis and Badtke, 2009).
3.3. HIV RNaseH and integrase as drug targets
The HIV RNaseH has attracted much attention as a drug target, with over 100 compounds based on a variety of chemical scaffolds being reported to have anti-HIV RNaseH activity (Cao et al., 2014; Chung et al., 2011; Esposito and Tramontano, 2014; Klumpp and Mirzadegan, 2006; Le Grice, 2012). Most of these inhibitors act by binding to the RNaseH active site, in part through direct interactions of three adjacent Lewis basic moieties on the inhibitors with the divalent cations in the active site (Billamboz et al., 2011; Chung et al., 2011; Kirschberg et al., 2009; Su et al., 2010). IC50 values for the majority of the inhibitors are in the low micromolar or high nanomolar range. This limited efficacy appears to be primarily due to the shallow, open shape of the HIV RNaseH catalytic groove which provides few contact sites for the inhibitors other than interactions with the two metal ions (Billamboz et al., 2011; Chung et al., 2011; Kirschberg et al., 2009; Su et al., 2010). The inhibitors usually induce modest cytotoxicity, leading to therapeutic index (TI) values that are often <10. Despite these limitations, compounds with efficacy and TI values appropriate for a drug have been reported (Himmel et al., 2006; Williams et al., 2010).
HIV integrase inhibitors are based on multiple chemical scaffolds, especially the diketo- and β-keto acids (Li et al., 2014). Three drugs have been approved: raltegravir, elvitegravir, and dolutegravir (Mesplede et al., 2014). Like the RNaseH inhibitors, the integrase inhibitors usually function by binding to and chelating the divalent cations in the active site (Agrawal et al., 2012; Espeseth et al., 2000; Hare et al., 2010; Hazuda et al., 2009), although binding of the inhibitors is strengthened in many cases by interactions with both the enzyme and the DNA substrate.
As predicted from their common membership in the nucleotidyl transferase superfamily (Nowotny, 2009; Yang and Steitz, 1995) and similar orientation of the two cations in their active sites (Hare et al., 2012), some anti-HIV RNaseH compounds inhibit the integrase, and some anti-HIV integrase compounds inhibit the RNaseH (Billamboz et al., 2008; Klarmann et al., 2002; Williams et al., 2010). This ability of some compounds to cross-inhibit distal members of the nucleotidyl transferase superfamily prompted us to develop the means to screen HIV RNaseH and integrase inhibitors against the HBV RNaseH.
4. Development of a low throughput anti-HBV RNaseH screening pipeline
4.1. Production of active recombinant HBV RNaseH
Although a few reports of active recombinant HBV RNaseH were published prior to our efforts (Choi et al., 2002; Lee et al., 1997; Potenza et al., 2007; Wei and Peterson, 1996), these systems were not developed far enough to be employed for antiviral drug discovery. Consequently, no anti-HBV RNaseH drug discovery efforts have been reported by groups other than our own, although unpublished efforts may have been conducted by the pharmaceutical industry.
We expressed the HBV RNaseH in E. coli as a C-terminally hexahistidine-tagged protein (Tavis et al., 2013). The RNaseH was recovered following nickel-affinity purification at very low levels that were detectable only by western analysis. RNaseH activity was evaluated using an oligonucleotide-directed RNA cleavage assay. In this assay, a DNA oligonucleotide is annealed to a uniformly 32P-labeled RNA to create an RNA:DNA heteroduplex. Cleavage of the RNA in the heteroduplex yields two RNA fragments that are resolved by electrophoresis Fig. 2A. Wild-type HBV RNaseH could cleave the RNA, cleavage required a complementary oligonucleotide, and the size of the RNA products varied as predicted when the oligonucleotide annealing site on the RNA was altered, formally proving RNaseH activity. Most of this activity was due to the HBV enzyme because mutating essential DEDD residues in the active site sharply reduced RNaseH activity. The enzyme had the predicted Mg++ dependence and could tolerate up to 2% DMSO (Tavis et al., 2013). We recently produced Coomassie-stainable amounts of the RNaseH as a maltose-binding protein fusion with a hexahistidine tag at its C-terminus (unpublished). HBV is a genetically diverse virus with eight or nine genotypes (A – I) that differ from each other by ≥8% at the nucleotide level (Schaefer, 2007). The RNaseH sequences differ between genotypes by ~6% at the amino acid level (Hayer et al., 2014). Currently, active HBV RNaseH can be produced from HBV genotypes B, C, D, and H.
Fig. 2. Principles of the assays to study RNaseH inhibitors.

A. Oligonucleotide-directed RNA cleavage assay to measure RNaseH activity. The substrate (S) is 32P-labeled RNA (black line) annealed to a complimentary DNA oligo (grey line). The RNaseH cleaves the RNA within the RNA/DNA heteroduplex and forms two products (P1 and P2). The reaction products are resolved by denaturing PAGE and detected by autoradiography. B. Basis of the strand-preferential quantitative PCR assay used to measure effects of RNaseH inhibitors on viral replication. The minus-polarity DNA strand (grey line) is detected by amplifying the sequence close to its 5′ end. The plus-polarity DNA strand (black line) is detected by amplifying across the gap in the minus-polarity DNA strand. Modified from (Cai et al., 2014).
This recombinant HBV RNaseH has been employed successfully in low throughput drug screening efforts (Cai et al., 2014; Hu et al., 2013; Lu et al., 2015; Tavis et al., 2013), but it remains far from ideal. Although we can produce large amounts of protein at relatively high purity, its specific activity is very low, and its specificty for RNA:DNA heteroduplexes is reduced by oxidizing conditions, divalent cations other than Mg++, and low ionic strength. Furthermore, the yield of RNAseH activity is roughly proportional to the size of the bacterial culture from which the protein is derived, not yield of the RNaseH protein. This implies that an unidentified bacterial component is limiting in formation of active enzyme. High throughput screening against the HBV RNaseH will not be feasible until these challenges are surmouonted. Ongoing efforts to solve these problems include optimization of the expression and purification protocols, truncating the enzyme’s C-terminus which aligns poorly with other RNaseHs and is predicted to be intrinsically disordered, and identifying the limiting component in the bacteria.
4.2. Cell-based HBV RNaseH inhibition assays
Inhibition of HBV replication by interfering with the RNaseH activity is best assessed by measuring preferential inhibition of HBV plus-polarity DNA strand synthesis. This is because the plus-polarity DNA cannot be made if the RNaseH has not removed the viral RNA from the newly synthesized minus-polarity DNA strand. In contrast, production of the 5′ half of the minus-polarity DNA strand is largely unaffected (Cai et al., 2014; Gerelsaikhan et al., 1996; Hu et al., 2013). Therefore, we developed a strand-preferential quantitative PCR assay [Fig. 2B; (Cai et al., 2014)]. Replication inhibition is usually measured in HepDES19 cells, which contain a tetracycline-repressible expression cassette for a replication-competent HBV genotype D genome (Guo et al., 2007). Antiviral activity of the RNaseH inhibitors is tested by treating cells replicating HBV with compounds for three days, followed by quantitative PCR detection of the HBV plus- and minus-polarity DNA strands within viral capsid particles (Cai et al., 2014; Lu et al., 2015).
4.3. Low-throughput screening pipeline for HBV RNaseH inhibitors
These assays have been integrated into a low-throughput drug screening pipeline (Fig. 3). Compounds are initially evaluated in a semi-quantitative biochemical assay against HBV RNaseH from a genotype D isolate using the oligonucleotide-directed cleavage assay. Primary hits are confirmed by screening against enzyme from HBV genotype B or C isolates to eliminate hits specific for only one of HBV’s genotypes. Confirmed hits are counter-screened against recombinant human RNaseH1 to identify compounds with a high probability of having unacceptable toxicity. IC50 values are then measured for compounds active against RNaseHs from two or more genotypes but have minimal activity against the human enzyme. HBV inhibitors with IC50 values <30 μM are selected for evaluation against HBV replication in cell culture. Compounds showing selective suppression of the HBV plus-polarity DNA strand in cells using a semi-quantitative form of the strand-preferential quantitative PCR assay are then evaluated for cytotoxicity, and EC50 values are determined for compounds with low toxicity at the effective concentrations. Compounds with favorable therapeutic indexes then undergo in vitro and in vivo absorption, distribution, metabolism, and excretion studies, plus basic pharmacokinetic and toxicity analyses in mice. Finally, promising compounds undergo efficacy studies in HBV transgenic mice that produce authentic HBV in their livers (Dandri et al., 2006), using the strand-preferential quantitative PCR assay as a readout. The leading anti-RNaseH compounds are just beginning the in vivo analyses.
Fig. 3.

Low-throughput screening pipeline for HBV RNaseH inhibitors.
5. α-Hydroxytropolones as example HBV RNaseH inhibitors
5.1. Discovery of α-hydroxytropolone inhibitors of HBV RNaseH
α-hydroxytropolones (αHT) have a seven-membered aromatic ring with three adjacent hydroxyl and carbonyl moieties oriented so that they can chelate the divalent cations of nucleotidyl transferase superfamily enzymes. Consequently, αHT compounds can suppress replication of HIV, the Herpes Simplex Viruses, many bacteria, and some tumors (Meck et al., 2014; Tavis et al., 2014; Zhao, 2007). Fifty-one αHT compounds or related troponoids were purchased, obtained from the NCI Developmental Therapeutics compound repository, provided by Dr. Stuart Le Grice at the National Cancer Institute, or synthesized by Dr. Ryan Murelli at the City University of New York (Chung et al., 2011; Hirsch et al., 2014; Meck et al., 2014; Meck et al., 2012; Williams et al., 2013). Key αHT compounds are in Fig. 4. Thirteen αHTs inhibited the HBV RNaseH at ≤60 μM, with five inhibiting at ≤20 μM in the primary screening assays (compounds #46, 106, 107, 110, and 113) (Lu et al., 2015). The best IC50 against the genotype D enzyme was 5.9 μM for compound #46 (β-thujaplicinol) (Table 1). Eleven αHTs were tested against HBV replication in cell culture, and seven suppressed HBV replication, six with EC50 values below 10 μM. The best EC50 values for αHTs against viral replication were 0.34 for #110 and 1.0 μM for #46. CC50 values by MTT assays ranged from 25 to 79 μM, leading to TI values from 3.8 up to a high of 94 for compound #110 (Lu et al., 2015).
Fig. 4. Select HBV RNaseH inhibitors.

Derivatization sites on the tropolone ring are numbered in red for compound #46.
Table 1.
Inhibitors of the HBV RNaseH. Data for compound #1 are in (Cai et al. 2014); qualitative data for #12 are in (Tavis et al., 2013); data for #46 – 120 are in (Hu et al. 2013) and (Lu et al. 2015). Unpublished data are the quantitative values for #12, all values for #153, and the cell-based results for #120. Quantitative data are not available for all compounds. −, inactive; +, active at 60 μM; ++, active at 5 μM; t, toxic at 20 μM. gtD, genotype D.
| Compound number | Name | Chemical class | IC50, gtD (μM) | EC50 (μM) | CC50 (μM) | TI |
|---|---|---|---|---|---|---|
| 1 | 2-Hydroxyisoquinoline-1,3(2H,4H)-dione | N-hydroxyisoquinolinedione | 28 | 4.2 | 75 | 18 |
| 12 | CWHM-000618 | Napthyridinone | 5.7 | 3.4 | 7.1 | 2.1 |
| 153 | CWHM-000613 | 4.1 | 4.4 | 6.1 | 1.4 | |
| 46 | beta-thujaplicinol | αHydroxytropolone | 5.9 | 1 | 25 | 25 |
| 56 | Manicol | 9.1 | 35 | 3.8 | ||
| 106 | CM1012-6a | 29.6 | 2.7 | 38 | 14 | |
| 110 | CM1012-6e | 34.6 | 0.34 | 32 | 94 | |
| 112 | CM1012-6i | >100 | 2.5 | 79 | 32 | |
| 113 | RM-YM-1-0613 | >100 | 4.2 | 66 | 16 | |
| 120 | RM-MD-1-0713 | + | ++ | t |
5.2. Preliminary structure-activity relationship αHTs against the HBV RNaseH
Four constraints on the structure-activity relationship for the αHTs are apparent. First, the intact α-tropolone moiety is needed because deleting one of the three oxygens on the tropolone ring ablates inhibition (Lu et al., 2015). This implies that αHTs inhibit the HBV RNaseH by the metal-chelating mechanism employed against the HIV RNaseH (Budihas et al., 2005; Chung et al., 2011; Didierjean et al., 2005). Second, at least one appendage must exist at positions R1, R2, or R3 on the αHT scaffold for activity [unsubstituted α-hydroxytropolone was inactive but 13 substituted derivatives were active (Lu et al., 2015); the positions of the R groups are indicated in Fig. 4]. Third, a wide variety of polar, aliphatic, or aromatic groups are permitted in the appendages. Finally, the appendages at R1, R2, and R3 had to be less than about four atoms in length for substantial activity (Lu et al., 2015). This size limitation implies that the HBV RNaseH active site is probably narrower than the HIV RNaseH active site. The probability of a narrow active site is reinforced by our studies on the N-hydroxyisoquinolinediones, where only the smallest compound tested worked against the HBV enzyme (compound #1) despite many of the larger molecules being active against the HIV RNaseH (Cai et al., 2014). A narrower active site would imply that designing compounds with adequate specificity for the RNaseH will be easier for HBV than HIV. Unfortunately, the breadth of the HBV RNaseH active site cannot be experimentally confirmed because no crystal structure exists.
6. Known anti-HBV RNaseH compounds
Over 190 compounds in the αHT, N-hydroxyisoquinolinedione, napthyridinone, dioxobutanoic acid, hydroxychromenone, Elvitegravir, Raltegravir, aminocyanothiophene, hydroxyxanthenone, cyanopyran, and thienopyrimidinone chemical families have been screened against the HBV RNaseH. Nineteen compounds inhibited the HBV RNaseH at ≤20 μM, with the best hits having low micromolar IC50 values [(Cai et al., 2014; Hu et al., 2013; Lu et al., 2015; Tavis et al., 2013) and unpublished]. Strong hits were observed among the αHTs, N-hydroxyisoquinolinediones, napthyridinones, and hydroxychomenones. In all cases, the compounds had similar activities against HBV genotypes C, D, and/or H. The inhibitors appear to act by chelating the divalent cations in the RNaseH active site because removing one of the three Lewis basic moieties or altering their angles relative to each other ablates inhibition [(Cai et al., 2014; Lu et al., 2015) and unpublished]. Counter-screening against human RNaseH1 revealed that most of these screening hits were also active against the human enzyme, but differences in the sensitivity of the two enzymes were apparent (Lu et al., 2015). This indicates that increasing selectivity for the HBV enzyme must be a major focus during chemical optimization.
Thirty-seven compounds have been tested for ability to suppress HBV replication in culture; 10 inhibited HBV replication at ≤20 μM (Table 1). Preferential suppression of the viral plus-polarity DNA strand confirmed that inhibition was due to blocking the viral RNaseH activity for all compounds except #56. Moderate cytotoxicity was observed for all active compounds, with CC50 values ranging from 6.1 to 79 μM. Therefore, it will be essential to minimize cytotoxicity during development of RNaseH inhibitors. The EC50 values for most of these inhibitors was substantially lower than their IC50 values. The cause for this quantitative discrepancy is unknown, but it could be due to accumulation of the compounds in cells, metabolism of the inhibitors to more active forms, or most likely, that the recombinant RNaseH does not recapitulate all aspects of the enzyme in its natural context as part of the full-length HBV polymerase protein. Nevertheless, strong inhibition of the recombinant enzyme is an excellent predictor of a compound’s potential to inhibit HBV replication in cells.
7. Outlook
The HBV RNaseH is a promising but unexploited target for antiviral drug development. Anti-RNaseH drugs are envisioned to be used in combination with the existing nucleos(t)ide analogs to suppress HBV replication much further than is currently possible. The goal will be to suppress viral replication enough to block replenishment of the cccDNA in the liver, permitting turnover of the cccDNA and elimination of infected cells by the immune system to clear the infection. The long apparent halflife of the cccDNA implies that therapy will be prolonged and may well benefit from co-administration with novel immune-stimulating therapies that are under development (Bertoletti and Gehring, 2013; Gehring et al., 2014). Therefore, minimizing toxicity and optimizing specificity and pharmacokinetic properties of potential RNaseH drugs will be especially important.
HIGHLIGHTS.
The HBV ribonuclease H is a promising but unexploited drug target.
Recent technical advances permit low-throughput screening for ribonuclease H inhibitors.
Some of the known HIV ribonuclease H inhibitors also inhibit HBV.
Combining ribonuclease H inhibitors with existing or novel drugs may significantly improve treatment efficacy for HBV.
Acknowledgments
Writing of this review was supported in part by R01 AI104494 to J.T. and an ancillary study within the NIDDK-sponsored Hepatitis B Virus Research Network (U01 DK082871 to Adrian Di Bisceglie and J.T.). We thank our collaborators Drs. Stuart Le Grice, Ryan Murelli, Fabrice Bailly, Philippe Cotelle, and Marvin Meyers for their many contributions to this RNaseH project. Use of some of these compounds against HBV are covered by U.S. Patent Application 13/072201 (Pending).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Addison WR, Walters KA, Wong WW, Wilson JS, Madej D, Jewell LD, Tyrrell DL. Half-life of the duck hepatitis B virus covalently closed circular DNA pool in vivo following inhibition of viral replication. J Virol. 2002;76:6356–6363. doi: 10.1128/JVI.76.12.6356-6363.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal A, DeSoto J, Fullagar JL, Maddali K, Rostami S, Richman DD, Pommier Y, Cohen SM. Probing chelation motifs in HIV integrase inhibitors. Proc Natl Acad Sci USA. 2012;109:2251–2256. doi: 10.1073/pnas.1112389109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoletti A, Gehring AJ. Immune therapeutic strategies in chronic hepatitis B virus infection: virus or inflammation control? PLoS pathogens. 2013;9:e1003784. doi: 10.1371/journal.ppat.1003784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billamboz M, Bailly F, Barreca ML, De LL, Mouscadet JF, Calmels C, Andreola ML, Witvrouw M, Christ F, Debyser Z, Cotelle P. Design, synthesis, and biological evaluation of a series of 2-hydroxyisoquinoline-1,3(2H,4H)-diones as dual inhibitors of human immunodeficiency virus type 1 integrase and the reverse transcriptase RNase H domain. J Med Chem. 2008;51:7717–7730. doi: 10.1021/jm8007085. [DOI] [PubMed] [Google Scholar]
- Billamboz M, Bailly F, Lion C, Touati N, Vezin H, Calmels C, Andreola ML, Christ F, Debyser Z, Cotelle P. Magnesium chelating 2-hydroxyisoquinoline-1,3(2H,4H)-diones, as inhibitors of HIV-1 integrase and/or the HIV-1 reverse transcriptase ribonuclease H domain: discovery of a novel selective inhibitor of the ribonuclease H function. J Med Chem. 2011;54:1812–1824. doi: 10.1021/jm1014692. [DOI] [PubMed] [Google Scholar]
- Block TM, Gish R, Guo H, Mehta A, Cuconati A, Thomas London W, Guo JT. Chronic hepatitis B: What should be the goal for new therapies? Antiviral Res. 2013;98:27–34. doi: 10.1016/j.antiviral.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budihas SR, Gorshkova I, Gaidamakov S, Wamiru A, Bona MK, Parniak MA, Crouch RJ, McMahon JB, Beutler JA, Le Grice SF. Selective inhibition of HIV-1 reverse transcriptase-associated ribonuclease H activity by hydroxylated tropolones. Nucleic Acids Res. 2005;33:1249–1256. doi: 10.1093/nar/gki268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai CW, Lomonosova E, Moran EA, Cheng X, Patel KB, Bailly F, Cotelle P, Meyers MJ, Tavis JE. Hepatitis B virus replication is blocked by a 2-hydroxyisoquinoline-1,3(2H,4H)-dione (HID) inhibitor of the viral ribonuclease H activity. Antiviral Res. 2014;108:48–55. doi: 10.1016/j.antiviral.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Mills C, Yu W, Yan R, Aldrich CE, Saputelli JR, Mason WS, Xu X, Guo JT, Block TM, Cuconati A, Guo H. Identification of disubstituted sulfonamide compounds as specific inhibitors of hepatitis B virus covalently closed circular DNA formation. Antimicrob Agents Chemother. 2012;56:4277–4288. doi: 10.1128/AAC.00473-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Song W, De Clercq E, Zhan P, Liu X. Recent progress in the research of small molecule HIV-1 RNase H inhibitors. Curr Med Chem. 2014;21:1956–1967. doi: 10.2174/0929867321666140120121158. [DOI] [PubMed] [Google Scholar]
- Chang LJ, Hirsch RC, Ganem D, Varmus HE. Effects of insertional and point mutations on the functions of the duck hepatitis B virus polymerase. J Virol. 1990;64:5553–5558. doi: 10.1128/jvi.64.11.5553-5558.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Marion PL. Amino Acids Essential for RNAse H Activity of Hepadnaviruses Are Also Required for Efficient Elongation of Minus-Strand DNA. J Virol. 1996;70:6151–6156. doi: 10.1128/jvi.70.9.6151-6156.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng PN, Liu WC, Tsai HW, Wu IC, Chang TT, Young KC. Association of intrahepatic cccDNA reduction with the improvement of liver histology in chronic hepatitis B patients receiving oral antiviral agents. J Med Virol. 2011;83:602–607. doi: 10.1002/jmv.22014. [DOI] [PubMed] [Google Scholar]
- Choi J, Kim EE, Park YI, Han YS. Expression of the active human and duck hepatitis B virus polymerases in heterologous system of Pichia methanolica. Antiviral Res. 2002;55:279–290. doi: 10.1016/s0166-3542(02)00023-2. [DOI] [PubMed] [Google Scholar]
- Chung S, Himmel DM, Jiang JK, Wojtak K, Bauman JD, Rausch JW, Wilson JA, Beutler JA, Thomas CJ, Arnold E, Le Grice SF. Synthesis, activity, and structural analysis of novel alpha-hydroxytropolone inhibitors of human immunodeficiency virus reverse transcriptase-associated ribonuclease H. J Med Chem. 2011;54:4462–4473. doi: 10.1021/jm2000757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffin CS, Mulrooney-Cousins PM, Peters MG, van MG, Roberts JP, Michalak TI, Terrault NA. Molecular characterization of intrahepatic and extrahepatic hepatitis B virus (HBV) reservoirs in patients on suppressive antiviral therapy. J Viral Hepat. 2011;18:415–423. doi: 10.1111/j.1365-2893.2010.01321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox N, Tillmann H. Emerging pipeline drugs for hepatitis B infection. Expert Opin Emerg Drugs. 2011;16:713–729. doi: 10.1517/14728214.2011.646260. [DOI] [PubMed] [Google Scholar]
- Dandri M, Lutgehetmann M, Volz T, Petersen J. Small animal model systems for studying hepatitis B virus replication and pathogenesis. Semin Liver Dis. 2006;26:181–191. doi: 10.1055/s-2006-939760. [DOI] [PubMed] [Google Scholar]
- Dandri M, Volz TK, Lutgehetmann M, Petersen J. Animal models for the study of HBV replication and its variants. J Clin Virol. 2005;34(Suppl 1):S54–S62. doi: 10.1016/s1386-6532(05)80011-3. [DOI] [PubMed] [Google Scholar]
- Didierjean J, Isel C, Querre F, Mouscadet JF, Aubertin AM, Valnot JY, Piettre SR, Marquet R. Inhibition of human immunodeficiency virus type 1 reverse transcriptase, RNase H, and integrase activities by hydroxytropolones. Antimicrob Agents Chemother. 2005;49:4884–4894. doi: 10.1128/AAC.49.12.4884-4894.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienstag JL. Benefits and risks of nucleoside analog therapy for hepatitis B. Hepatology. 2009;49:S112–121. doi: 10.1002/hep.22920. [DOI] [PubMed] [Google Scholar]
- Dyda F, Hickman AB, Jenkins TM, Engelman A, Craigie R, Davies DR. Crystal structure of the catalytic domain of HIV-1 integrase: similarity to other polynucleotidyl transferases. Science. 1994;266:1981–1986. doi: 10.1126/science.7801124. [DOI] [PubMed] [Google Scholar]
- Espeseth AS, Felock P, Wolfe A, Witmer M, Grobler J, Anthony N, Egbertson M, Melamed JY, Young S, Hamill T, Cole JL, Hazuda DJ. HIV-1 integrase inhibitors that compete with the target DNA substrate define a unique strand transfer conformation for integrase. Proc Natl Acad Sci USA. 2000;97:11244–11249. doi: 10.1073/pnas.200139397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito F, Tramontano E. Past and future. Current drugs targeting HIV-1 integrase and reverse transcriptase-associated ribonuclease H activity: single and dual active site inhibitors. Antiviral chemistry & chemotherapy. 2014;23:129–144. doi: 10.3851/IMP2690. [DOI] [PubMed] [Google Scholar]
- Freed EO, Martin MA. HIVs and their replication. In: Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE, editors. Fields Virology. Lippincott Williams & Wilkins; Philadelphia: 2007. pp. 2107–2185. [Google Scholar]
- Gehring A, Bertoletti A, Tavis JE. Host factor-targeted hepatitis B virus therapies. Intervirology. 2014;57:158–162. doi: 10.1159/000360938. [DOI] [PubMed] [Google Scholar]
- Gerelsaikhan T, Tavis JE, Bruss V. Hepatitis B Virus Nucleocapsid Envelopment Does Not Occur without Genomic DNA Synthesis. J Virol. 1996;70:4269–4274. doi: 10.1128/jvi.70.7.4269-4274.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghany M, Liang TJ. Drug targets and molecular mechanisms of drug resistance in chronic hepatitis B. Gastroenterology. 2007;132:1574–1585. doi: 10.1053/j.gastro.2007.02.039. [DOI] [PubMed] [Google Scholar]
- Gong Y, Yao E, Tavis JE. Evidence that the RNAseH activity of the duck hepatitis B virus is unable to act on exogenous substrates. BMC Microbiology. 2001;1:12. doi: 10.1186/1471-2180-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Jiang D, Zhou T, Cuconati A, Block TM, Guo JT. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J Virol. 2007;81:12472–12484. doi: 10.1128/JVI.01123-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature. 2010;464:232–236. doi: 10.1038/nature08784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare S, Maertens GN, Cherepanov P. 3′-Processing and strand transfer catalysed by retroviral integrase in crystallo. EMBO J. 2012;31:3020–3028. doi: 10.1038/emboj.2012.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayer J, Rodriguez C, Germanidis G, Deleage G, Zoulim F, Pawlotsky JM, Combet C. Ultradeep pyrosequencing and molecular modeling identify key structural features of hepatitis B virus RNase H, a putative target for antiviral intervention. J Virol. 2014;88:574–582. doi: 10.1128/JVI.03000-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazuda D, Iwamoto M, Wenning L. Emerging pharmacology: inhibitors of human immunodeficiency virus integration. Annu Rev Pharmacol Toxicol. 2009;49:377–394. doi: 10.1146/annurev.pharmtox.011008.145553. [DOI] [PubMed] [Google Scholar]
- Himmel DM, Sarafianos SG, Dharmasena S, Hossain MM, Coy-Simandle K, Ilina T, Clark AD, Jr, Knight JL, Julias JG, Clark PK, Krogh-Jespersen K, Levy RM, Hughes SH, Parniak MA, Arnold E. HIV-1 reverse transcriptase structure with RNase H inhibitor dihydroxy benzoyl naphthyl hydrazone bound at a novel site. ACS Chem Biol. 2006;1:702–712. doi: 10.1021/cb600303y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch DR, Cox G, D’Erasmo MP, Shakya T, Meck C, Mohd N, Wright GD, Murelli RP. Inhibition of the ANT(2″)-Ia resistance enzyme and rescue of aminoglycoside antibiotic activity by synthetic alpha-hydroxytropolones. Bioorganic & medicinal chemistry letters. 2014;24:4943–4947. doi: 10.1016/j.bmcl.2014.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hostomsky Z, Hostomska Z, Matthews DA. Ribonuclease H. In: Linn SM, Lloyd RS, Roberts RJ, editors. Nulceases. Cold Spring Harbor Laboratory Press; Plainview, NY: 1993. pp. 341–376. [Google Scholar]
- Hu Y, Cheng X, Cao F, Huang A, Tavis JE. beta-Thujaplicinol inhibits hepatitis B virus replication by blocking the viral ribonuclease H activity. Antiviral Res. 2013;99:221–229. doi: 10.1016/j.antiviral.2013.06.007. [DOI] [PubMed] [Google Scholar]
- Katayanagi K, Miyagawa M, Matsushima M, Ishikawa M, Kanaya S, Ikehara M, Matsuzaki T, Morikawa K. Three-dimensional structure of ribonuclease H from E. coli. Nature. 1990;347:306–309. doi: 10.1038/347306a0. [DOI] [PubMed] [Google Scholar]
- Khudyakov YE, Makhov AM. Prediction of terminal protein and ribonuclease H domains in the gene P product of hepadnaviruses. FEBS letters. 1989;243:115–118. doi: 10.1016/0014-5793(89)80110-3. [DOI] [PubMed] [Google Scholar]
- Kirschberg TA, Balakrishnan M, Squires NH, Barnes T, Brendza KM, Chen X, Eisenberg EJ, Jin W, Kutty N, Leavitt S, Liclican A, Liu Q, Liu X, Mak J, Perry JK, Wang M, Watkins WJ, Lansdon EB. RNase H active site inhibitors of human immunodeficiency virus type 1 reverse transcriptase: design, biochemical activity, and structural information. J Med Chem. 2009;52:5781–5784. doi: 10.1021/jm900597q. [DOI] [PubMed] [Google Scholar]
- Klarmann GJ, Hawkins ME, Le Grice SF. Uncovering the complexities of retroviral ribonuclease H reveals its potential as a therapeutic target. AIDS Rev. 2002;4:183–194. [PubMed] [Google Scholar]
- Klumpp K, Hang JQ, Rajendran S, Yang Y, Derosier A, Wong KI, Overton H, Parkes KE, Cammack N, Martin JA. Two-metal ion mechanism of RNA cleavage by HIV RNase H and mechanism-based design of selective HIV RNase H inhibitors. Nucleic Acids Res. 2003;31:6852–6859. doi: 10.1093/nar/gkg881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumpp K, Mirzadegan T. Recent progress in the design of small molecule inhibitors of HIV RNase H. Curr Pharm Des. 2006;12:1909–1922. doi: 10.2174/138161206776873653. [DOI] [PubMed] [Google Scholar]
- Kwon H, Lok AS. Hepatitis B therapy. Nat Rev Gastroenterol Hepatol. 2011;8:275–284. doi: 10.1038/nrgastro.2011.33. [DOI] [PubMed] [Google Scholar]
- Lai L, Yokota H, Hung LW, Kim R, Kim SH. Crystal structure of archaeal RNase HII: a homologue of human major RNase H. Structure. 2000;8:897–904. doi: 10.1016/s0969-2126(00)00179-9. [DOI] [PubMed] [Google Scholar]
- Lavanchy D. Worldwide epidemiology of HBV infection, disease burden, and vaccine prevention. Journal of clinical virology: the official publication of the Pan American Society for Clinical Virology. 2005;34(Suppl 1):S1–3. doi: 10.1016/s1386-6532(05)00384-7. [DOI] [PubMed] [Google Scholar]
- Le Grice SF. Human immunodeficiency virus reverse transcriptase: 25 years of research, drug discovery, and promise. The Journal of biological chemistry. 2012;287:40850–40857. doi: 10.1074/jbc.R112.389056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YI, Hong YB, Kim Y, Rho HM, Jung G. RNase H activity of human hepatitis B virus polymerase expressed in Escherichia coli. Biochem Biophys Res Commun. 1997;233:401–407. doi: 10.1006/bbrc.1997.6467. [DOI] [PubMed] [Google Scholar]
- Li Y, Xuan S, Feng Y, Yan A. Targeting HIV-1 integrase with strand transfer inhibitors. Drug discovery today Dec. 2014;6:S1359–6446. doi: 10.1016/j.drudis.2014.12.001. [DOI] [PubMed] [Google Scholar]
- Liaw YF. Impact of therapy on the outcome of chronic hepatitis B. Liver international: official journal of the International Association for the Study of the Liver. 2013;33(Suppl 1):111–115. doi: 10.1111/liv.12057. [DOI] [PubMed] [Google Scholar]
- Lima WF, Wu H, Crooke ST. Human RNases H. Methods Enzymol. 2001;341:430–440. doi: 10.1016/s0076-6879(01)41168-2. [DOI] [PubMed] [Google Scholar]
- Lu G, Lomonosova E, Cheng X, Moran EA, Meyers MJ, Le Grice SF, Thomas CJ, Jiang JK, Meck C, Hirsch DR, D’Erasmo MP, Suyabatmaz DM, Murelli RP, Tavis JE. Hydroxylated Tropolones Inhibit Hepatitis B Virus Replication by Blocking the Viral Ribonuclease H Activity. Antimicrob Agents Chemother. 2015;59:1070–1079. doi: 10.1128/AAC.04617-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui YY, Tsoi KK, Wong VW, Kao JH, Hou JL, Teo EK, Mohamed R, Piratvisuth T, Han KH, Mihm U, Wong GL, Chan HL. Cost-effectiveness analysis of roadmap models in chronic hepatitis B using tenofovir as the rescue therapy. Antivir Ther. 2010;15:145–155. doi: 10.3851/IMP1496. [DOI] [PubMed] [Google Scholar]
- Marcellin P, Asselah T. Viral hepatitis: impressive advances but still a long way to eradication of the disease. Liver international: official journal of the International Association for the Study of the Liver. 2014;34(Suppl 1):1–3. doi: 10.1111/liv.12422. [DOI] [PubMed] [Google Scholar]
- Meck C, D’Erasmo MP, Hirsch DR, Murelli RP. The biology and synthesis of alpha-hydroxytropolones. Med Chem Comm. 2014;5:842–852. doi: 10.1039/C4MD00055B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meck C, Mohd N, Murelli RP. An oxidopyrylium cyclization/ring-opening route to polysubstituted alpha-hydroxytropolones. Organic letters. 2012;14:5988–5991. doi: 10.1021/ol302892g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesplede T, Quashie PK, Zanichelli V, Wainberg MA. Integrase strand transfer inhibitors in the management of HIV-positive individuals. Annals of medicine. 2014;46:123–129. doi: 10.3109/07853890.2014.883169. [DOI] [PubMed] [Google Scholar]
- Monto A, Schooley RT, Lai JC, Sulkowski MS, Chung RT, Pawlotsky JM, McHutchison JG, Jacobson IM. Lessons from HIV therapy applied to viral hepatitis therapy: summary of a workshop. Am J Gastroenterol. 2010;105:989–1004. doi: 10.1038/ajg.2009.726. [DOI] [PubMed] [Google Scholar]
- Moraleda G, Saputelli J, Aldrich CE, Averett D, Condreay L, Mason WS. Lack of effect of antiviral therapy in nondividing hepatocyte cultures on the closed circular DNA of woodchuck hepatitis virus. J Virol. 1997;71:9392–9399. doi: 10.1128/jvi.71.12.9392-9399.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotny M. Retroviral integrase superfamily: the structural perspective. EMBO Rep. 2009;10:144–151. doi: 10.1038/embor.2008.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotny M, Gaidamakov SA, Crouch RJ, Yang W. Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell. 2005;121:1005–1016. doi: 10.1016/j.cell.2005.04.024. [DOI] [PubMed] [Google Scholar]
- Nowotny M, Yang W. Stepwise analyses of metal ions in RNase H catalysis from substrate destabilization to product release. EMBO J. 2006;25:1924–1933. doi: 10.1038/sj.emboj.7601076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potenza N, Salvatore V, Raimondo D, Falanga D, Nobile V, Peterson DL, Russo A. Optimized expression from a synthetic gene of an untagged RNase H domain of human hepatitis B virus polymerase which is enzymatically active. Protein Expr Purif. 2007;55:93–99. doi: 10.1016/j.pep.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Radziwill G, Tucker W, Schaller H. Mutational analysis of the hepatitis B virus P gene product: domain structure and RNase H activity. J Virol. 1990;64:613–620. doi: 10.1128/jvi.64.2.613-620.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer S. Hepatitis B virus taxonomy and hepatitis B virus genotypes. World J Gastroenterol. 2007;13:14–21. doi: 10.3748/wjg.v13.i1.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schodel F, Weimer T, Will H, Sprengel R. Amino acid sequence similarity between retroviral and E. coli RNase H and hepadnaviral gene products. AIDS Research and Human Retroviruses. 1988;4:9–11. doi: 10.1089/aid.1988.4.ix. [DOI] [PubMed] [Google Scholar]
- Seeger C, Zoulim F, Mason WS. Hepadnaviruses. In: Knipe DM, Howley P, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE, editors. Fields Virology. Lippincott Williams & Wilkins; Philadelphia: 2007. pp. 2977–3029. [Google Scholar]
- Seeger C, Zoulim F, Mason WS. Hepadnaviruses. In: Knipe DM, Howley PM, editors. Fields Virology. 6. Lippincott Williams & Wilkins; Philadelphia PA: 2013. pp. 2185–2221. [Google Scholar]
- Sorrell MF, Belongia EA, Costa J, Gareen IF, Grem JL, Inadomi JM, Kern ER, McHugh JA, Petersen GM, Rein MF, Strader DB, Trotter HT. National Institutes of Health Consensus Development Conference Statement: management of hepatitis B. Ann Intern Med. 2009;150:104–110. doi: 10.7326/0003-4819-150-2-200901200-00100. [DOI] [PubMed] [Google Scholar]
- Su HP, Yan Y, Prasad GS, Smith RF, Daniels CL, Abeywickrema PD, Reid JC, Loughran HM, Kornienko M, Sharma S, Grobler JA, Xu B, Sardana V, Allison TJ, Williams PD, Darke PL, Hazuda DJ, Munshi S. Structural basis for the inhibition of RNase H activity of HIV-1 reverse transcriptase by RNase H active site-directed inhibitors. J Virol. 2010;84:7625–7633. doi: 10.1128/JVI.00353-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers J, Mason WS. Replication of the Genome of a Hepatitis B-Like Virus by Reverse Transcription of an RNA Intermediate. Cell. 1982;29:403–415. doi: 10.1016/0092-8674(82)90157-x. [DOI] [PubMed] [Google Scholar]
- Tavis JE, Badtke MP. Hepadnaviral Genomic Replication. In: Cameron CE, G”tte M, Raney KD, editors. Viral Genome Replication. Springer Science+Business Media, LLC; New York: 2009. pp. 129–143. [Google Scholar]
- Tavis JE, Cheng X, Hu Y, Totten M, Cao F, Michailidis E, Aurora R, Meyers MJ, Jacobsen EJ, Parniak MA, Sarafianos SG. The hepatitis B virus ribonuclease h is sensitive to inhibitors of the human immunodeficiency virus ribonuclease h and integrase enzymes. PLoS pathogens. 2013;9:e1003125. doi: 10.1371/journal.ppat.1003125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavis JE, Wang H, Tollefson AE, Ying B, Korom M, Cheng X, Cao F, Davis KL, Wold WS, Morrison LA. Inhibitors of nucleotidyl transferase superfamily enzymes suppress herpes simplex virus replication. Antimicrob Agents Chemother. 2014;58:7451–7461. doi: 10.1128/AAC.03875-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bommel F, De Man RA, Wedemeyer H, Deterding K, Petersen J, Buggisch P, Erhardt A, Huppe D, Stein K, Trojan J, Sarrazin C, Bocher WO, Spengler U, Wasmuth HE, Reinders JG, Moller B, Rhode P, Feucht HH, Wiedenmann B, Berg T. Long-term efficacy of tenofovir monotherapy for hepatitis B virus-monoinfected patients after failure of nucleoside/nucleotide analogues. Hepatology. 2010;51:73–80. doi: 10.1002/hep.23246. [DOI] [PubMed] [Google Scholar]
- Wei X, Peterson DL. Expression, purification, and characterization of an active RNase H domain of the hepatitis B viral polymerase. J Biol Chem. 1996;271:32617–32622. doi: 10.1074/jbc.271.51.32617. [DOI] [PubMed] [Google Scholar]
- Wei Y, Tavis JE, Ganem D. Relationship between Viral DNA Synthesis and Virion Envelopment in Hepatitis B Viruses. J Virol. 1996;70:6455–6458. doi: 10.1128/jvi.70.9.6455-6458.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werle-Lapostolle B, Bowden S, Locarnini S, Wursthorn K, Petersen J, Lau G, Trepo C, Marcellin P, Goodman Z, Delaney WE, Xiong S, Brosgart CL, Chen SS, Gibbs CS, Zoulim F. Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy. Gastroenterology. 2004;126:1750–1758. doi: 10.1053/j.gastro.2004.03.018. [DOI] [PubMed] [Google Scholar]
- Williams PD, Staas DD, Venkatraman S, Loughran HM, Ruzek RD, Booth TM, Lyle TA, Wai JS, Vacca JP, Feuston BP, Ecto LT, Flynn JA, DiStefano DJ, Hazuda DJ, Bahnck CM, Himmelberger AL, Dornadula G, Hrin RC, Stillmock KA, Witmer MV, Miller MD, Grobler JA. Potent and selective HIV-1 ribonuclease H inhibitors based on a 1-hydroxy-1,8-naphthyridin-2(1H)-one scaffold. Bioorg Med Chem Lett. 2010;20:6754–6757. doi: 10.1016/j.bmcl.2010.08.135. [DOI] [PubMed] [Google Scholar]
- Williams YD, Meck C, Mohd N, Murelli RP. Triflic acid-mediated rearrangements of 3-methoxy-8-oxabicyclo[3.2.1]octa-3,6-dien-2-ones: synthesis of methoxytropolones and furans. The Journal of organic chemistry. 2013;78:11707–11713. doi: 10.1021/jo401617r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong DK, Yuen MF, Ngai VW, Fung J, Lai CL. One-year entecavir or lamivudine therapy results in reduction of hepatitis B virus intrahepatic covalently closed circular DNA levels. Antivir Ther. 2006;11:909–916. [PubMed] [Google Scholar]
- Woo G, Tomlinson G, Nishikawa Y, Kowgier M, Sherman M, Wong DK, Pham B, Ungar WJ, Einarson TR, Heathcote EJ, Krahn M. Tenofovir and entecavir are the most effective antiviral agents for chronic hepatitis B: a systematic review and Bayesian meta-analyses. Gastroenterology. 2010;139:1218–1229. doi: 10.1053/j.gastro.2010.06.042. [DOI] [PubMed] [Google Scholar]
- Yang W, Steitz TA. Recombining the structures of HIV integrase, RuvC and RNase H. Structure. 1995;3:131–134. doi: 10.1016/s0969-2126(01)00142-3. [DOI] [PubMed] [Google Scholar]
- Zhao J. Plant troponoids: chemistry, biological activity, and biosynthesis. Curr Med Chem. 2007;14:2597–2621. doi: 10.2174/092986707782023253. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Yamamoto T, Cullen J, Saputelli J, Aldrich CE, Miller DS, Litwin S, Furman PA, Jilbert AR, Mason WS. Kinetics of hepadnavirus loss from the liver during inhibition of viral DNA synthesis. J Virol. 2001;75:311–322. doi: 10.1128/JVI.75.1.311-322.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoulim F. Hepatitis B virus resistance to antiviral drugs: where are we going? Liver international: official journal of the International Association for the Study of the Liver. 2011;31(Suppl 1):111–116. doi: 10.1111/j.1478-3231.2010.02399.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoulim F, Locarnini S. Hepatitis B virus resistance to nucleos(t)ide analogues. Gastroenterology. 2009;137:1593–1608. doi: 10.1053/j.gastro.2009.08.063. [DOI] [PubMed] [Google Scholar]