

Abstract
Background
The X-linked recessive disease Duchenne muscular dystrophy (DMD) is caused by mutations in the gene encoding the protein dystrophin. Despite its large size, dystrophin is a highly stable protein, demonstrating cooperative unfolding during thermal denaturation as monitored by circular dichroism spectroscopy. In contrast, internal sequence deletions have been associated with a loss of the cooperative unfolding and cause in vitro protein aggregation. Several emerging therapy options for DMD utilize internally deleted micro-dystrophins and multi-exon-skipped dystrophins that produce partially functional proteins, but the stability of such internally truncated proteins has not been investigated.
Methods
In this study, we analyzed the in vitro stability of human dystrophin constructs skipped around exon 45 or exon 51, several dystrophin gene therapy constructs, as well as human full-length and micro-utrophin. Constructs were expressed in insect cells using the baculovirus system, purified by affinity chromatography, and analyzed by high-speed sedimentation, circular dichroism spectroscopy, and differential scanning fluorimetry.
Results
Our results reveal that not all gene therapy constructs display stabilities consistent with full-length human dystrophin. However, all dystrophins skipped in-frame around exon 45 or exon 51 show stability profiles congruent with intact human dystrophin. Similar to previous studies of mouse proteins, full-length human utrophin also displays stability similar to human dystrophin and does not appear to be affected by a large internal deletion.
Conclusions
Our results suggest that the in vitro stability of human dystrophin is less sensitive to smaller deletions at natural exon boundaries than larger, more complex deletions present in some gene therapy constructs.
Keywords: Duchenne muscular dystrophy, Becker muscular dystrophy, Dystrophin, Utrophin, Exon skipping, Gene therapy
Background
The X-linked disease Duchenne muscular dystrophy (DMD) is caused by mutations in the gene encoding the protein dystrophin [1]. Mutations causing this disease are variable with 65% of DMD patients harboring deletions which span exons, 5% to 15% having duplications, and the remaining populations having either point mutations or deep intronic deletions [2]. Becker muscular dystrophy (BMD) is a milder allelic form of dystrophy typically caused by in-frame gene deletions that maintain reading frame but presumably cause disease through diminished abundance or functionality [3].
The dystrophin protein is a critical molecular component of the dystrophin-glycoprotein complex (DGC) that functions to maintain skeletal muscle integrity during contraction [4,5]. Dystrophin provides a structural link between the sub-sarcolemmal cytoskeleton and the extracellular matrix through interactions with actin [6,7], intermediate filaments [8-10], microtubules [11,12], and the membrane-associated dystroglycan complex [13,14]. The observation that milder BMD patients harbor deletions in the central rod domain suggests that dystrophin can tolerate such deletions to some degree and that the central rod domain is less critical to the function of dystrophin.
Two avenues of therapeutic research have focused on producing internally truncated, Becker-like dystrophins in DMD patients. Exon-skipping approaches aim to restore the reading frame of mutated DMD transcripts using antisense oligonucleotides (ASOs) or phosphorodiamidate morpholino oligomers (PMOs), producing an internally truncated but partially functional protein [15-17]. Alternatively, adeno-associated viral (AAV) gene therapy is under active investigation to express miniaturized dystrophin constructs in DMD patients due to the large size of the dystrophin gene and the limited capacity of AAV vectors [18-20].
The stability of the corresponding proteins produced from dystrophin exon skipping or AAV-mediated delivery of micro-dystrophins is unknown and may be an important factor to maximize therapeutic efficacy. Previous in vitro work has demonstrated that the stability of mouse dystrophin was sensitive to disease-causing missense mutations and internal deletion [21,22], raising the question of whether the stabilities of micro-dystrophins or exon-skipped dystrophins relevant to DMD therapies might also be compromised. In contrast, the stability of mouse utrophin, a fetal homologue of dystrophin, was insensitive to both terminal and internal deletion [22].
Here, we expressed and purified five dystrophins skipped around exon 45 or 51 with an exon-43-skipped control, five recombinant dystrophin gene therapy constructs, and two utrophin constructs. The selected constructs represent the leading therapy approaches that have been shown to ameliorate the dystrophic phenotype in mdx mice with transition to clinical trials underway [19,20,23-28]. In the current study, all constructs were expressed from human sequences, as opposed to the mouse constructs used previously [21,22]. Our biophysical analysis revealed that the dystrophin gene therapy constructs exhibited more variable stabilities in vitro while exon-skipped dystrophin constructs showed stabilities not different from full-length dystrophin. Consistent with previous mouse studies, utrophin maintained stability despite internal deletion.
Methods
Cloning
A cDNA corresponding to the sequence of full-length human dystrophin was obtained from the DNASU vector repository in the pE223 Gateway entry vector. Human utrophin and micro-utrophin were cloned from HEK293 cells into the pENTR/D-TOPO vector (Invitrogen™, Waltham, MA, USA) and sequence verified. An eight-amino-acid FLAG-tag (DYKDDDDK) was added to the N-terminus of both human dystrophin and utrophin constructs for use in purification. All human dystrophin deletion constructs were PCR amplified using primers designed around adjacent exons, repeats, or domains for the desired deletion based on reported repeat and domain boundaries [29]. The PCR products were circularized using T4 polynucleotide kinase and T4 DNA ligase (New England BioLabs, Ipswich, MA, USA) and sequence verified. Using the Gateway Recombination system (Life Technologies, Carlsbad, CA, USA), the deletion constructs were recombined into the pDEST8 destination vector and subsequently transformed into DH10Bac-competent Escherichia coli and purified according to the manufacturer’s protocol.
Protein expression and purification
Sf9 insect cells were maintained at 1 × 106 cells/mL in Sf-900™ II SFM (Life Technologies, Carlsbad, CA, USA). Purified baculovirus was transfected using Cellfectin® II (Life Technologies, Carlsbad, CA, USA), and high-titer viral stocks were generated through successive infections of Sf9 cells in 3.5-cm plates (P0), 10-cm plates (P1), and 250 mL of 1 × 106 cells/mL suspended cells (P2). Ten milliliters of P2 virus was used to infect 250 mL of 1 × 106 cells/mL and cultured for 72 h post-infection to maximize protein expression. Infected cells were centrifuged at 1,000 × g for 3 min and re-suspended in phosphate-buffered saline (PBS) containing a cocktail of protease inhibitors (100 nM Aprotinin, 10 mg/mL E-64, 10 μM Leupeptin, 1 mM PMSF, 1 μg/mL Pepstatin). Cells were lysed by sonication, five bursts of 30 s using a BioLogics Ultrasonic Homogenizer set at 30% power. The lysate was centrifuged at 14,000 × g for 10 min at 4°C and the supernatant applied to an anti-FLAG M2 agarose column (Sigma Aldrich, St. Louis, MO, USA). The column was washed with >10 column volumes of PBS and bound protein eluted with PBS-containing protease inhibitors and 100 μg/mL FLAG peptide. After dialysis overnight in 2 L of PBS at pH 7.5, the purified protein was concentrated using the Amicon Centrifugal Filter unit (UFC801024), and protein concentration was determined using A280 and extinction coefficients calculated from the amino acid compositions for each construct. Concentrated proteins were run on a 3% to 12% sodium dodecyl sulfate (SDS) polyacrylamide gradient gel and run at 150 V for 1 h. Gels were stained with Coomassie blue stain and visualized using Licor’s Odyssey® Infrared Imaging System.
Tandem purification of full-length human dystrophin
To optimize the purification of full-length human dystrophin, a Twin-Strep-tag® (IBA Life Sciences, Göttingen, Germany), with amino acid sequence SA-WSHPQFEK(GGGS)2GGSAWSHPQFEK, was cloned onto the C-terminus of pE223 dystrophin in addition to the N-terminal FLAG-tag. The dual-tagged dystrophin was then recombined into the pDEST8 expression vector and expressed in the Sf9 baculovirus system as described above. The cell lysate was applied to a Strep-Tactin ® Superflow ® high-capacity resin (IBA Life Sciences, Göttingen, Germany), eluted with 100 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 2 mM desthiobiotin, pH 8, and the eluent immediately applied to an anti-FLAG M2 agarose column as described above.
Western blotting
Purification fractions from the tandem purification were run on a 3% to 12% SDS polyacrylamide gradient gel at 150 V for 1 h. The gel was transferred onto a polyvinylidene difluoride (PVDF) membrane at 100 V for 1 h. The PVDF membrane was blocked using 5% milk in 1X PBS and 0.1% Tween and blotted using mouse monoclonal anti-FLAG M2 (F1804, Sigma Aldrich, St. Louis, MO, USA) and rabbit polyclonal anti-Strep-tag II (ab76949, Abcam, Cambridge, UK) antibodies at 1:1,000 dilution. The blot was visualized using anti-mouse DyLight™ 800 (green channel) and anti-rabbit DyLight™ 680 (red channel) conjugated antibodies in Licor’s Odyssey® Infrared Imaging System.
High-speed sedimentation
Each purified protein was diluted to 0.3 mg/mL (for exon-51-skipped dystrophins) or 0.5 mg/mL (for all other proteins) with PBS in a final volume of 120 μL, and 60 μL was immediately aliquoted into 12 μL 6X Laemmli sample buffer (LSB) to prepare a ‘total’ fraction. The remaining 60 μL was centrifuged at 100,000 × g for 30 min at 4°C. The supernatant was transferred into 12 μL 6X LSB and the pellet re-suspended in 72 μL of 1X LSB. Triplicate fractions were run on a 3% to 12% gradient polyacrylamide gel at 150 V for 1 h and stained with Coomassie blue stain. Gels were scanned using Licor’s Odyssey® Infrared Imaging System and band density calculated with Odyssey Software v2.1. Full-length human dystrophin was used as a control at both 0.3- and 0.5-mg/mL concentrations and did not show any significant difference in percent aggregation between the different concentrations (17.6% ± 7.39 and 17.4% ± 7.30, respectively).
Circular dichroism
Each purified protein was centrifuged at 14,000 × g for 10 min at 4°C and the supernatant diluted to 0.5 mg/mL (for gene therapy and utrophin proteins) or 0.3 mg/mL (for exon-skipped and full-length dystrophins) using PBS. Absorption spectra were acquired with a Jasco J-815 spectropolarimeter, initially at 20°C as controlled by a Peltier device, from 200- to 260-nm wavelength. Spectra were then acquired at 1°C temperature intervals from 20°C to 90°C and the characteristic ellipticity at alpha-helical wavelength (θ222) recorded. Molar ellipticity, [θ], was calculated using the following equation: [θ] = θ / (10 × c × l) where c is the molar concentration of the sample (mole/L) and l is the path length in cm. Molar ellipticity (with units of degrees, centimeter squared per decimole) was plotted against wavelength for the circular dichroism (CD) spectra. Ellipticity at 222 nm (θ222) was normalized, plotted against temperature, and fit by regression analysis in Sigma Plot (Systat Software, Inc., San Jose, CA, USA) using equations for two-state or three-state unfolding [30].
Differential scanning fluorimetry
Our method closely followed that described by Niesen et al. [31]. Briefly, the fluorescent dye SYPRO Orange (#S6650, Life Technologies™, Carlsbad, CA, USA) was incubated at a ratio of 1:1,000 (w/w) with 0.5 mg/mL (or 0.3 mg/mL for exon-skipped proteins) of purified protein in PBS. The dye/protein solution was aliquoted into 50-μL technical triplicates, and an emission of 610 nm was measured in a real-time PCR instrument (iCycler, Bio-Rad, Hercules, CA, USA) as the temperature was increased from 20°C to 90°C at 1°C temperature intervals. The fluorescence was normalized from 0 to 1, plotted against temperature, and fit by regression analysis in Sigma Plot (Systat Software, Inc., San Jose, CA, USA) using an equation for a two-state unfolding model [30].
Statistical analysis
Data for percentage aggregation and melting temperatures of single-transition melt curves from CD and differential scanning fluorimetry (DSF) were analyzed using a one-way analysis of variance (ANOVA) with Tukey’s post hoc test in Prism software (GraphPad, San Diego, CA, USA), all compared to full-length human dystrophin.
Results
Basis for choice of recombinant proteins and gel analysis
The choice of constructs to analyze was based on current models for exon-skipping and gene therapy in pre-clinical testing and clinical studies (Figure 1). The exon-45-skipped and exon-51-skipped dystrophins were analyzed because they could potentially treat 8% and 13% of DMD patients, respectively [16], and ASO and morpholino drugs targeting these exons are currently in clinical trials [32,33]. Therefore, we expressed a subset of exon-45- and exon-51-skipped human dystrophins: Δex44-45, Δex45-46, Δex45-51, Δex51-52, and Δex51-63, as well as Δex43-44, a control deletion that has been previously speculated to cause decreased stability [34]. While we initially attempted to generate a larger array of exon-51-skipped constructs, we analyzed the three (Δex45-51, Δex51-52, and Δex51-63) that yielded products in the first stages of cloning.
Figure 1.
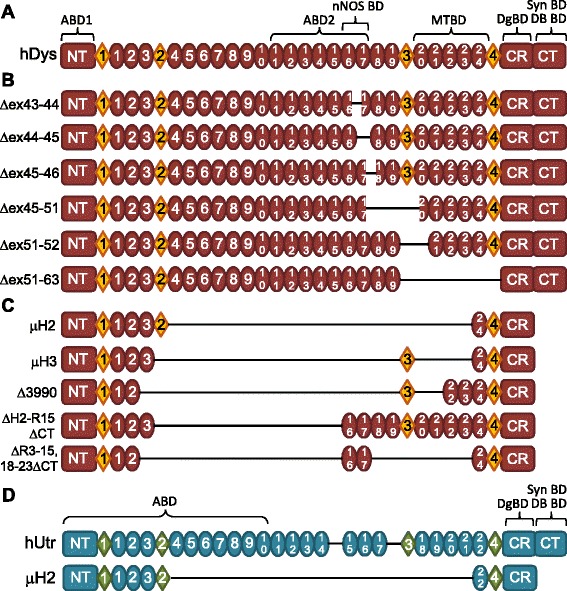
Dystrophin and utrophin constructs analyzed. (A) Diagram of full-length human dystrophin (hDys); NT - N-terminus, CR - cysteine-rich domain, CT - C-terminus, circles - spectrin-like repeats, diamonds - unstructured ‘hinge’ regions, ABD1/2 - actin binding domains, nNOS BD - neuronal nitric oxide synthase binding domain, MTBD - microtubule binding domain, DgBD - dystroglycan binding domain, Syn BD - syntrophin binding domain, DB BD - dystrobrevin binding domain. (B) Diagrams of exon-skipped human dystrophin constructs analyzed. (C) Diagrams of gene therapy human dystrophins analyzed. (D) Diagrams of full-length human utrophin (hUtr) and a micro-utrophin (μH2 hUtr) analyzed.
The gene therapy constructs μH2 human dystrophin (hDys) and μH3 hDys contain spectrin-like repeats (SLRs) 1 to 3 and 24 with hinge 2 or hinge 3, respectively. These constructs have been shown to ameliorate the dystrophic phenotype in mdx mice [27], and μH2 also showed significant expression with muscle improvement in the GRMD dog model of DMD [35,36]. The Δ3990 hDys construct corresponds to the AAV-delivered micro-dystrophin used in a clinical trial that reported minimal recombinant dystrophin expression associated with a strong immune response to dystrophin [20,37]. Constructs ΔH2-R15/ΔCT hDys and ΔR3-15/18-23/ΔCT hDys are miniaturized dystrophins that retain SLRs 16 and 17 necessary for sarcolemmal localization of neuronal nitric oxide synthase (nNOS) [38,39]. Full-length human utrophin (hUtr) and a micro-utrophin (μH2 hUtr), homologous to μH2 hDys, correspond to constructs that are under investigation for gene-, cell-, and protein-based therapies [25,26,28,40].
While gel analysis of the FLAG-affinity-purified recombinant proteins revealed a predominant band of the expected molecular weight for each purified dystrophin gene therapy and utrophin constructs, full-length and exon-skipped human dystrophins exhibited a near-stoichiometric contaminating fragment at approximately 230 kDa (Figure 2A, left panel) that was not previously observed in preparations of full-length mouse dystrophin [21] and was not present in gene therapy or utrophin preparations (Figure 2A, middle and right panels). To identify the contaminating fragment, we generated and expressed a dual-tagged, full-length human dystrophin containing a C-terminal Twin-Strep-tag® (IBA Life Sciences, Göttingen, Germany) in addition to the N-terminal FLAG-tag. Western blot analysis after tandem affinity purification of the dually tagged dystrophin revealed that the near-stoichiometric, approximately 230-kDa contaminating fragment present in the FLAG-affinity-purified samples was an N-terminal fragment (green band, Figure 2B) and was mostly likely caused by a cleavage from a protease in our expression system. The cleavage event was further confirmed by the presence of the corresponding C-terminal fragment in the load and elution fractions of the Strep-tag purification (red band, Figure 2B). The absence of the contaminating N-terminal fragment in constructs with deletions preceding repeat 16 combined with its presence in constructs with deletions after repeat 16 suggests that the proteolytic cleavage site resides within repeat 14 or 15, which would yield the predicted N-terminal fragment of approximately 230 kDa. While the tandem purification was successful in identifying the contaminating fragments and a more purified full-length dystrophin was recovered (yellow band, Figure 2B), the resulting yield was not sufficient to support the planned biochemical or biophysical analyses. Therefore, single FLAG-affinity-purified proteins were used in all subsequent analyses.
Figure 2.
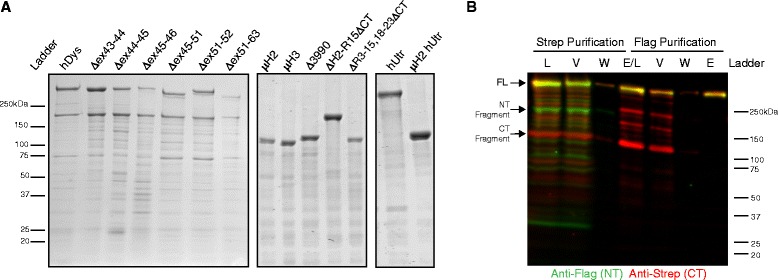
Gel analysis of purified recombinant proteins. (A) Representative Coomassie-stained gels with 5 μg of exon-skipped dystrophins, gene therapy dystrophins, and utrophins loaded for comparison. (B) Western blot of purification fractions from tandem purification of dual-tagged full-length human dystrophin with N-terminal (NT) FLAG-tag (green channel) and C-terminal (CT) Strep-tag (red channel); fractions from Strep affinity purification and FLAG affinity purifications: load (L), void (V), wash (W), and elute (E). hDys - human dystrophin, hUtr - human utrophin.
Protein aggregation
High-speed sedimentation is a facile in vitro technique to quantify aggregation of purified proteins. Full-length human dystrophin exhibited 17.6% aggregation (Figure 3, Table 1), which is similar to the 14% aggregation previously reported for full-length mouse dystrophin [21]. Exon-skipped dystrophin proteins did not show significant increases in aggregation relative to full-length human dystrophin; however, Δex43-44 showed a significant decrease in aggregation (gray bars, Figure 3). Four of the five gene therapy proteins showed a significant increase, with percent aggregation ranging from 31.7% to 44.4% (white bars, Figure 3, Table 1). Interestingly, the Δ3990 protein was the only gene therapy construct that exhibited protein aggregation congruent with full-length dystrophin. Similar to the exon-skipped dystrophins, utrophin and micro-utrophin did not vary significantly from full-length dystrophin aggregation (lined bars, Figure 3).
Figure 3.
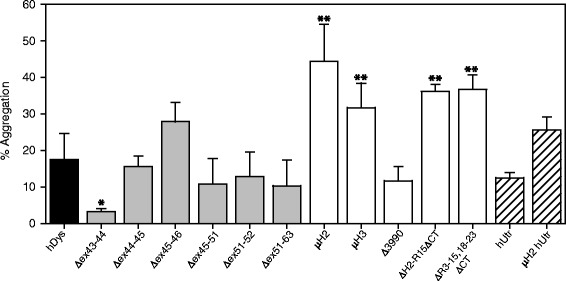
Analysis of protein aggregation by high-speed sedimentation. Quantification of high-speed sedimentation assay of supernatant (S) and pellet (P) fractions where % aggregation = S/(S + P); full-length human dystrophin (hDys) in black bar, exon-skipped dystrophins in gray bars, gene therapy dystrophins in white bars, and utrophins in lined bars; *P < 0.05, **P < 0.0001 using ANOVA statistical analysis compared to full-length human dystrophin. hUtr - human utrophin.
Table 1.
Biophysical properties of human dystrophin and utrophin constructs
| Construct | Molecular weight (kDa) | CD Tm1 (°C) | CD Tm2 (°C) | DSF Tm (°C) | % Aggregation |
|---|---|---|---|---|---|
| hDys | 427 | 48.1 ± 1.17 | - | 45.7 ± 2.62 | 17.6 ± 7.11 |
| Δex43-44 | 414 | 46.7 ± 0.70 | - | 44.3 ± 0.35 | 3.4 ± 0.73* |
| Δex44-45 | 413 | 51.7 ± 1.51 | - | 48.1 ± 1.54 | 15.7 ± 2.83 |
| Δex45-46 | 414 | 48.4 ± 2.38 | - | 46.0 ± 2.60 | 27.9 ± 5.28 |
| Δex45-51 | 385 | 47.9 ± 4.07 | - | 44.6 ± 1.63 | 10.9 ± 7.00 |
| Δex51-52 | 414 | 47.2 ± 0.87 | - | 43.8 ± 0.31 | 12.9 ± 6.70 |
| Δex51-63 | 351 | 48.6 ± 3.08 | - | 43.7 ± 1.15 | 10.4 ± 7.11 |
| μH2 | 139 | 47.3 ± 1.25 | 85.1 ± 4.14 | 46.0 ± 1.28 | 44.4 ± 10.10** |
| μH3 | 137 | 49.5 ± 1.78 | 79.0 ± 2.82 | 45.8 ± 0.86 | 31.7 ± 6.72** |
| Δ3990 | 154 | 56.1 ± 1.97** | - | 50.4 ± 1.91* | 11.7 ± 3.97 |
| ΔH2-R15ΔCT | 242 | 53.7 ± 1.51 | 72.9 ± 4.11 | 49.8 ± 0.54* | 36.2 ± 1.95** |
| ΔR3-15,18-23 ΔCT | 146 | 45.8 ± 2.96 | - | 47.3 ± 0.07 | 36.7 ± 3.96** |
| hUtr | 399 | 46.2 ± 1.42 | - | 43.4 ± 0.36 | 12.5 ± 1.40 |
| μH2 hUtr | 138 | 47.0 ± 1.16 | - | 42.9 ± 0.25 | 25.6 ± 3.58 |
All values are mean values of at least three experiments with standard deviations. CD Tm1 and Tm2: circular dichroism melting temperatures; DSF Tm: differential scanning fluorimetry melting temperatures. *P < 0.05; **P < 0.0001 using ANOVA statistical analysis compared to full-length human dystrophin (hDys). hUtr – human utrophin.
Assessment of secondary structure unfolding
To assess secondary structure and protein unfolding, we analyzed the purified proteins by CD spectroscopy. All of the constructs exhibited CD spectra characteristic of proteins with high alpha-helical content and minima at 208 and 222 nm (Figure 4A,B,C). As the temperature was increased, loss of secondary structure (or unfolding) was monitored at 222 nm to generate melt curves (Figure 4D,E,F) with a calculated melting temperature or temperatures (Table 1). Full-length human dystrophin exhibited a single-transition melt curve with a melting temperature of 48.1°C (Figure 4, Table 1), which is in contrast to the 59.6°C melting temperature reported for full-length mouse dystrophin [21]. Upon closer inspection of the melt curves from the previous report, it is apparent that full-length mouse dystrophin exhibited an additional melting transition similar to the dystrophin isoform Dp260 [22], a property that is absent in CD melt curves of human dystrophin. Exon-skipped dystrophins exhibited single transitions all with comparable melting temperatures to full-length dystrophin (Figure 4D, Table 1). Two of the gene therapy constructs, Δ3990 and ΔR3-15/18-23/ΔCT, also displayed a single transition, but Δ3990 had a significantly higher melting temperature of 56.1°C. However, the other gene therapy constructs displayed a second transition with two calculated melting temperatures ranging from 47°C to 85°C (Figure 4E, Table 1). This indicates that the protein is either not unfolding cooperatively or is composed of two populations of folded and unfolded states. Full-length human utrophin and μH2 hUtr displayed single-transition melt curves with melting temperatures of 46.2°C and 47°C, respectively (Figure 4F, Table 1). These values are not significantly different from full-length dystrophin and are consistent with previously reported melting temperatures for mouse full-length and micro-mouse utrophins [22].
Figure 4.
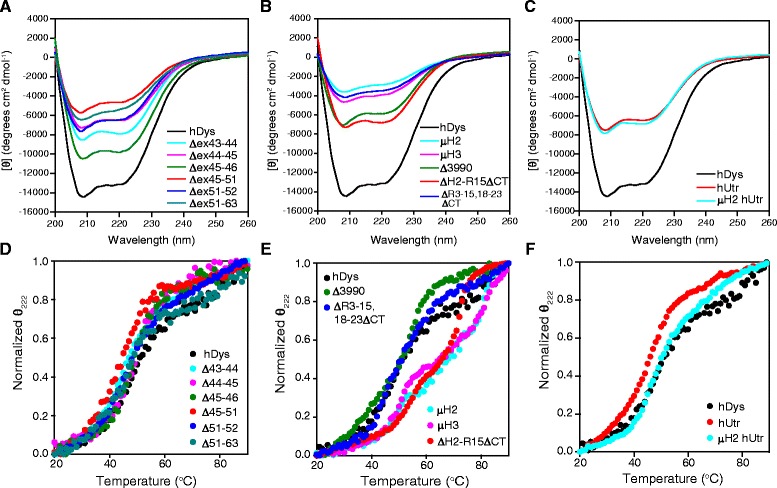
Spectra and melt curves obtained by circular dichroism spectroscopy. (A-C) Circular dichroism absorption spectra from 200 to 260 nm for exon-skipped dystrophins (A), gene therapy constructs (B), and utrophins (C). Molar ellipticity [θ], with units of degrees centimeter squared per decimole, was calculated as θ / (10 × c × l) where c is the molar concentration of the sample (mole/L) and l is the path length in cm. (D-F) CD absorption spectra monitored at 222 nm from 20°C to 90°C for exon-skipped dystrophins (D), gene therapy dystrophins (E), and utrophins (F). Melt curves were normalized to θ222 from 0 to 1 fraction unfolded and a representative curve plotted. See Table 1 for melting temperatures (CD Tm1 and Tm2). hDys - human dystrophin, hUtr - human utrophin.
Assessment of tertiary structure unfolding
DSF utilizes a fluorescent dye that increases its fluorescence emission upon binding to hydrophobic moieties in proteins, which become more exposed and accessible as a protein unfolds during thermal denaturation [31]. Like CD, DSF can be used to obtain protein melt curves, but unlike CD, the signal measured reflects changes in tertiary structure rather than secondary structure. By DSF analysis, full-length human dystrophin displayed a melt curve with a single transition at 45.7°C (Figure 5, Table 1). This temperature was lower than that seen in CD, consistent with the concept that tertiary structure will be lost before secondary structure. Exon-skipped dystrophins displayed single transitions with similar melting temperatures to full-length dystrophin ranging from 43.7°C to 48.1°C (Figure 5A, Table 1). In contrast to the CD data, all of the gene therapy constructs exhibited single transitions with melting temperatures ranging from 45.8°C to 50.4°C (Figure 5B, Table 1). Both Δ3990 and ΔH2-R15/ΔCT hDys had significantly right-shifted melt curves from full-length dystrophin with melting temperatures of 50.4°C and 49.8°C, respectively. Full-length and micro-utrophin both showed a left-shifted melt curve with melting temperatures of 43.4°C and 42.9°C, respectively, but like exon-skipped dystrophins, these values are not significantly different from full-length dystrophin (Figure 5C, Table 1).
Figure 5.
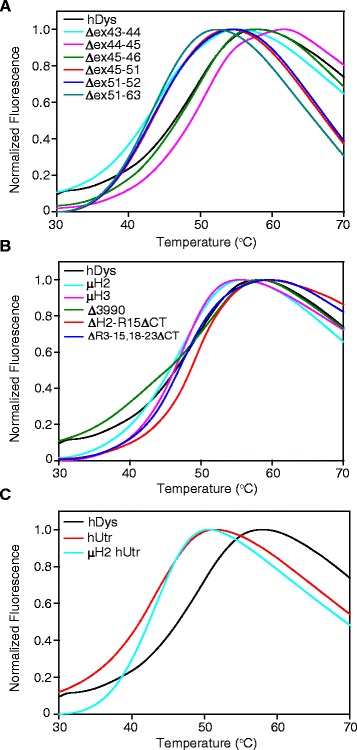
Melt curves obtained by differential scanning fluorimetry. Differential scanning fluorimetry (DSF) melt curves for exon-skipped dystrophins (A), gene therapy dystrophins (B), and utrophins (C). Fluorescence was monitored at 610 nm from 20°C to 90°C, and normalized from 0 to 1 fraction unfolded and a representative curve plotted. See Table 1 for melting temperatures (DSF Tm). hDys - human dystrophin, hUtr - human utrophin.
Discussion
In this study, we have analyzed the biophysical properties of several therapeutically relevant, internally truncated dystrophins and utrophins. Therapies that produce internally deleted dystrophins are based on observations that patients with the milder BMD can harbor large deletions in the central rod domain. In addition to conferring elasticity or flexibility to dystrophin [5,41,42], it is known that the central rod domain encodes a second actin binding domain [6,43,44], as well as domains for localizing nNOS to the sarcolemma [38,39], for in vitro binding to phospholipids [45,46], intermediate filaments [9], and microtubules [11,12].
The biophysical properties of individual and tandem repeats of the rod domain have been extensively investigated, and these findings demonstrate a wide range of stabilities [45-48], whereas full-length dystrophin has remarkable cooperative stability [21]. Additionally, there is evidence that certain internal deletions of the central rod domain are associated with increased protein aggregation and instability [22]. Together, these studies suggest that the stability of individual or tandem repeat fragments does not necessarily reflect that of larger fragments or full-length dystrophin proteins and that protein stability of individual regions within dystrophin is context dependent.
Several groups have investigated the biophysical consequences of exon-skipping on dystrophin fragments within the central rod domain, particularly those spanning exons 43 to 51 [34,49-51]. For constructs skipping exon 51, they concluded that some protein fragments are more stable than others. Our results, however, suggest that for dystrophins skipped around exon 51, there is little measureable change in stability in vitro between exon-skipped proteins and full-length human dystrophin. This conclusion is consistent with the conclusions of a recent report that assessed the structural differences and stability profiles of human dystrophin fragments with deletions around exon 51 [52]. They found that while there were marked structural differences between the different deletion fragments, the stability was not significantly affected.
For the common, out-of-frame deletion of exon 45 (Δ45) in patients, exon-skipping therapies are being designed to either delete exon 44 (Δex44-45) or exon 46 (Δex45-46) to correct the reading frame. Based on another recent report, it was speculated that the Δex45-46 dystrophin protein might be highly unstable because this in-frame deletion is associated with the more severe DMD phenotype in patients [53], and therefore, a Δex44-45-skipping strategy would be more beneficial. However, based on our in vitro data, there does not appear to be any significant difference in stability between Δex44-45 and Δex45-46 and full-length dystrophin proteins. These different conclusions from the clinical and in vitro studies indicate that the source of pathogenesis from the exon 45 to 46 deletion may not depend on the stability of the resulting protein but, perhaps, is caused by a regulatory or functional perturbation.
Interestingly, all of the exon-skipped dystrophins evaluated in our study displayed in vitro stabilities congruent with full-length dystrophin, including the Δex43-44 protein that exhibited decreased stability when previously evaluated in the context of a smaller recombinant fragment encompassing SLRs 16 to 18 [34]. Because dystrophy-causing missense mutations also cause less dramatic instability in full-length dystrophin compared to small fragments [21,54], it seems possible that long-range and cooperative intra-protein communication may serve to buffer dystrophin against the destabilizing effects of sequence changes and deletions.
We also expressed internally deleted human micro-dystrophins that are currently under investigation for gene therapy. We showed that several of these constructs have significantly different stability compared to the full-length human dystrophin protein. Both micro-dystrophins μH2 hDys and μH3 hDys displayed increased aggregation and additional melting transitions upon secondary structure unfolding. One of the sarcolemmal nNOS-localizing constructs, ΔH2-R15/ΔCT hDys, displayed similar behavior. Interestingly, the more truncated nNOS-localizing construct, ΔR3-15/18-23/ΔCT, exhibited a single melting transition similar to full-length dystrophin but also increased aggregation. These data suggest that the in vitro stability of dystrophin gene therapy constructs may be dependent on the stability of the non-native junction created by the internal deletion. The Δ3990 hDys construct was the only gene therapy construct that exhibited wild-type aggregation, and it displayed significantly increased melting temperatures for both CD and DSF, suggesting that the Δ3990 protein is more stable than full-length dystrophin. However, a small clinical trial for AAV-mediated delivery of the Δ3990 was not successful [37].
Utrophin-replacement therapies are also currently under investigation; therefore, we analyzed the stability of full-length human utrophin and a micro-utrophin. Our previous study demonstrated that mouse utrophin is a highly stable protein that does not appear to be sensitive to terminal truncation or internal deletion [22]. Consistent with these results, our data showed that both full-length and micro-utrophin have similar protein aggregation to dystrophin and maintain melting temperatures that are not significantly different from full-length human dystrophin. Investigation of utrophin as a dystrophin substitute therapy for DMD [25,26,28,40] is based on its ability to rescue most phenotypes of the mdx mouse when transgenically overexpressed [55] and also the positive correlation between increased utrophin expression with improved prognosis in a small cohort of DMD patients [56]. Utrophin maintains some functional properties of dystrophin, such as forming an analogous utrophin-glycoprotein complex [57] and binding actin filaments [58], but lacks the ability to localize nNOS to the sarcolemma [39,59] and organize the sub-sarcolemmal microtubule lattice [12]. While utrophin cannot completely substitute for dystrophin in terms of protein-protein interactions, our results suggest that utrophin and micro-utrophin proteins are appealing as therapeutic targets in terms of protein stability, especially when compared to dystrophin gene therapy proteins.
While the recombinant dystrophin and utrophin proteins used in this study were expressed in a eukaryotic cellular environment, their purification and characterization in more simple buffers leaves open the possibility that they exhibit unfolding and aggregation properties in vitro that are different from how they behave in the complex environment of a mammalian muscle cell. It will therefore be important to develop both cell- and tissue-based model systems to better understand how deletions in dystrophins and utrophins affect stability in vivo.
Conclusions
Our in vitro analysis of the biophysical consequences of internal deletion on dystrophin and utrophin suggests that dystrophin stability is context-dependent: relatively unaffected by small deletions at natural exon boundaries but sensitive to larger and more complex rearrangements from deletions present in gene therapy constructs. In contrast, utrophin maintains uniform stability despite large internal deletion. Moreover, our results also highlight the need to better understand how differences in protein stability in vitro translate to therapeutic efficacy in vivo.
Acknowledgements
Supported by a gift from Ryan’s Quest, Muscular Dystrophy Association Grant MDA218545, and National Institutes of Arthritis, Musculoskeletal and Skin Diseases grant RO1 AR042423 to JME. JJB was supported by the NIH Training Program in Muscle Research (AR007612) and a University of Minnesota Doctoral Dissertation Fellowship. JLM was supported by a Ron Edstrom Scholar Award through the Achievement Rewards for College Scientists (ARCS) foundation of Minnesota. DMT was supported by American Heart Association predoctoral fellowship 12PRE12040402.
Abbreviations
- AAV
adeno-associated virus
- ASO
antisense oligonucleotide
- BMD
Becker muscular dystrophy
- CD
circular dichroism
- CT
C-terminal
- DGC
dystrophin-glycoprotein complex
- DMD
Duchenne muscular dystrophy
- DSF
differential scanning fluorimetry
- GRMD
golden retriever muscular dystrophy
- hDys
human dystrophin
- hUtr
human utrophin
- nNOS
neuronal nitric oxide synthase
- NT
N-terminal
- PMO
phosphorodiamidate morpholino oligomer
- SLRs
spectrin-like repeats
- Tm
melting temperature
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JM carried out molecular cloning of exon 51 constructs, performed protein expression/purification and biophysical assays, carried out the data and statistical analysis, and drafted the manuscript. KR carried out protein expression/purification and biophysical assays of gene therapy and utrophin proteins. MJ participated in the molecular cloning of utrophin and dystrophins skipped around exon 45, and protein expression/purification, and biophysical assays for dystrophins skipped around exon 45. JB carried out the molecular cloning for gene therapy and utrophin constructs and edited the manuscript. DT established the DSF instrument and protocol and edited the manuscript. JE participated in the design and coordination of the study and edited the manuscript. All authors have read and approved the final manuscript.
Contributor Information
Jackie L McCourt, Email: mccou013@umn.edu.
Katrina K Rhett, Email: rhett.kr@gmail.com.
Michele A Jaeger, Email: soulistayoga@gmail.com.
Joseph J Belanto, Email: bela0090@umn.edu.
Dana M Talsness, Email: stran386@umn.edu.
James M Ervasti, Email: jervasti@umn.edu.
References
- 1.Hoffman EP, Brown RH, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–28. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 2.Muntoni F, Torelli S, Ferlini A. Review dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. 2003;44:731–40. doi: 10.1016/S1474-4422(03)00585-4. [DOI] [PubMed] [Google Scholar]
- 3.Koenig M, Beggs AH, Moyer M, Scherpf S, Heindrich K, Bettecken T, et al. The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. Am J Hum Genet. 1989;45:498–506. [PMC free article] [PubMed] [Google Scholar]
- 4.Rybakova IN, Ervasti JM. Dystrophin-glycoprotein complex is monomeric and stabilizes actin filaments in vitro through a lateral association. J Biol Chem. 1997;272:28771–8. doi: 10.1074/jbc.272.45.28771. [DOI] [PubMed] [Google Scholar]
- 5.Ervasti JM. Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim Biophys Acta. 2007;1772:108–17 [DOI] [PubMed]
- 6.Rybakova IN, Amann KJ, Ervasti JM. A new model for the interaction of dystrophin with F-actin. J Cell Biol. 1996;135:661–72. doi: 10.1083/jcb.135.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rybakova IN, Patel JR, Ervasti JM. The dystrophin complex forms a mechanically strong link between the sarcolemma and costameric actin. J Cell Biol. 2000;150:1209–14. doi: 10.1083/jcb.150.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stone MR, Neill AO, Catino D, Bloch RJ. Specific interaction of the actin-binding domain of dystrophin with intermediate filaments containing keratin 19. Mol Biol Cell. 2005;16(September):4280–93. doi: 10.1091/mbc.E05-02-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhosle RC, Michele DE, Campbell KP, Li Z, Robson RM. Interactions of intermediate filament protein synemin with dystrophin and utrophin. Biochem Biophys Res Commun. 2006;346:768–77. doi: 10.1016/j.bbrc.2006.05.192. [DOI] [PubMed] [Google Scholar]
- 10.Rezniczek GA, Konieczny P, Nikolic B, Reipert S, Schneller D, Abrahamsberg C, et al. Plectin 1f scaffolding at the sarcolemma of dystrophic (mdx) muscle fibers through multiple interactions with beta-dystroglycan. J Cell Biol. 2007;176:965–77. doi: 10.1083/jcb.200604179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prins KW, Humston JL, Mehta A, Tate V, Ralston E, Ervasti JM. Dystrophin is a microtubule-associated protein. J Cell Biol. 2009;186:363–9. doi: 10.1083/jcb.200905048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belanto JJ, Mader TL, Eckhoff MD, Strandjord DM, Banks GB, Gardner MK, et al. Microtubule binding distinguishes dystrophin from utrophin. Proc Natl Acad Sci U S A. 2014;111:5723–8. doi: 10.1073/pnas.1323842111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jung D, Yang B, Meyer J, Chamberlain JS, Campbell KP. Identification and characterization of the dystrophin anchoring site on beta-dystroglycan. J Biol Chem. 1995;270:27305–10. doi: 10.1074/jbc.270.45.27305. [DOI] [PubMed] [Google Scholar]
- 14.Ishikawa-Sakurai M, Yoshida M, Imamura M, Davies KE, Ozawa E. ZZ domain is essentially required for the physiological binding of dystrophin and utrophin to beta-dystroglycan. Hum Mol Genet. 2004;13:693–702. doi: 10.1093/hmg/ddh087. [DOI] [PubMed] [Google Scholar]
- 15.Van Deutekom JC, Janson AA, Ginjaar IB, Frankhuizen WS, Aartsma-Rus A, Bremmer-Bout M, et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N Engl J Med. 2007;57:2677–86. doi: 10.1056/NEJMoa073108. [DOI] [PubMed] [Google Scholar]
- 16.Aartsma-Rus A, Fokkema I, Verschuuren J, Ginjaar I, van Deutekom J, van Ommen G-J, et al. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum Mutat. 2009;30:293–9. doi: 10.1002/humu.20918. [DOI] [PubMed] [Google Scholar]
- 17.Kinali M, Arechavala-Gomeza V, Feng L, Cirak S, Hunt D, Adkin C, et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009;8:918–28. doi: 10.1016/S1474-4422(09)70211-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harper SQ, Hauser MA, DelloRusso C, Duan D, Crawford RW, Phelps SF, et al. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat Med. 2002;8:253–61. doi: 10.1038/nm0302-253. [DOI] [PubMed] [Google Scholar]
- 19.Sakamoto M, Yuasa K, Yoshimura M, Yokota T, Ikemoto T, Suzuki M, et al. Micro-dystrophin cDNA ameliorates dystrophic phenotypes when introduced into mdx mice as a transgene. Biochem Biophys Res Commun. 2002;293:1265–72. doi: 10.1016/S0006-291X(02)00362-5. [DOI] [PubMed] [Google Scholar]
- 20.Wang B, Li J, Xiao X. Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc Natl Acad Sci U S A. 2000;97:13714–9. doi: 10.1073/pnas.240335297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Henderson DM, Lee A, Ervasti JM. Disease-causing missense mutations in actin binding domain 1 of dystrophin induce thermodynamic instability and protein aggregation. Proc Natl Acad Sci U S A. 2010;107:9632–7. doi: 10.1073/pnas.1001517107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Henderson DM, Belanto JJ, Li B, Heun-Johnson H, Ervasti JM. Internal deletion compromises the stability of dystrophin. Hum Mol Genet. 2011;20:2955–63. doi: 10.1093/hmg/ddr199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu QL, Rabinowitz A, Chen YC, Yokota T, Yin H, Alter J, et al. Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in body-wide skeletal muscles. Proc Natl Acad Sci U S A. 2005;102:198–203. doi: 10.1073/pnas.0406700102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alter J, Lou F, Rabinowitz A, Yin H, Rosenfeld J, Wilton SD, et al. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat Med. 2006;12:175–7. doi: 10.1038/nm1345. [DOI] [PubMed] [Google Scholar]
- 25.Odom GL, Gregorevic P, Allen JM, Finn E, Chamberlain JS. Microutrophin delivery through rAAV6 increases lifespan and improves muscle function in dystrophic dystrophin/utrophin-deficient mice. Mol Ther. 2008;16:1539–45. doi: 10.1038/mt.2008.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sonnemann KJ, Heun-Johnson H, Turner AJ, Baltgalvis KA, Lowe DA, Ervasti JM. Functional substitution by TAT-utrophin in dystrophin-deficient mice. PLoS Med. 2009;6 doi: 10.1371/journal.pmed.1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Banks GB, Judge LM, Allen JM, Chamberlain JS. The polyproline site in hinge 2 influences the functional capacity of truncated dystrophins. PLoS Genet. 2010;6 doi: 10.1371/journal.pgen.1000958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Call JA, Ervasti JM, Lowe DA. TAT-μUtrophin mitigates the pathophysiology of dystrophin and utrophin double-knockout mice. J Appl Physiol. 2011;111:200–5. doi: 10.1152/japplphysiol.00248.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Winder SJ, Gibson TJ, Kendrick-Jones J. Dystrophin and utrophin: the missing links! FEBS Lett. 1995;369:27–33. doi: 10.1016/0014-5793(95)00398-S. [DOI] [PubMed] [Google Scholar]
- 30.Legardinier S, Legrand B, Raguénès-Nicol C, Bondon A, Hardy S, Tascon C, et al. A two-amino acid mutation encountered in Duchenne muscular dystrophy decreases stability of the rod domain 23 (R23) spectrin-like repeat of dystrophin. J Biol Chem. 2009;284:8822–32. doi: 10.1074/jbc.M805846200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc. 2007;2:2212–21. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- 32.Phase IIb Study of PRO045 in Subjects With Duchenne Muscular Dystrophy http://clinicaltrials.gov/show/NCT01826474 Accessed 2 January 2015
- 33.A study of the safety, tolerability & efficacy of long-term administration of drisapersen in US & Canadian subjects https://clinicaltrials.gov/ct2/show/NCT01803412?term=drisapersen&rank=2 Accessed 26 January 2015.
- 34.Ruszczak C, Mirza A, Menhart N. Differential stabilities of alternative exon-skipped rod motifs of dystrophin. Biochim Biophys Acta. 2009;1794:921–8 [DOI] [PMC free article] [PubMed]
- 35.Wang Z, Kuhr CS, Allen JM, Blankinship M, Gregorevic P, Chamberlain JS, et al. Sustained AAV-mediated dystrophin expression in a canine model of Duchenne muscular dystrophy with a brief course of immunosuppression. Mol Ther. 2007;15:1160–6. doi: 10.1038/sj.mt.6300161. [DOI] [PubMed] [Google Scholar]
- 36.Wang Z, Storb R, Halbert CL, Banks GB, Butts TM, Finn EE, et al. Successful regional delivery and long-term expression of a dystrophin gene in canine muscular dystrophy: a preclinical model for human therapies. Mol Ther. 2012;20:1501–7. doi: 10.1038/mt.2012.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mendell J. Dystrophin immunity in Duchenne’s muscular dystrophy. N Engl J Med. 2010;363:1429–37. doi: 10.1056/NEJMoa1000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lai Y, Thomas G, Yue Y, Yang H. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J Clin Investig. 2009;119:624–35. doi: 10.1172/JCI36612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lai Y, Zhao J, Yue Y, Duan D. α2 and α3 helices of dystrophin R16 and R17 frame a microdomain in the α1 helix of dystrophin R17 for neuronal NOS binding. Proc Natl Acad Sci U S A. 2013;110:525–30. doi: 10.1073/pnas.1211431109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Filareto A, Parker S, Darabi R, Borges L, Iacovino M, Schaaf T, et al. An ex vivo gene therapy approach to treat muscular dystrophy using inducible pluripotent stem cells. Nat Commun. 2013;4:1549. doi: 10.1038/ncomms2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koenig M, Kunkel LM. Detailed analysis of the repeat domain of dystrophin reveals four potential hinge segments that may confer flexibility. J Biol Chem. 1990;265:4560–6. [PubMed] [Google Scholar]
- 42.Saadat L, Pittman L, Menhart N. Structural cooperativity in spectrin type repeats motifs of dystrophin. Biochim Biophys Acta. 2006;1764:943–54 [DOI] [PubMed]
- 43.Amann KJ, Renley BA, Ervasti JM. A cluster of basic repeats in the dystrophin rod domain binds F-actin through an electrostatic interaction. J Biol Chem. 1998;273:28419–23. doi: 10.1074/jbc.273.43.28419. [DOI] [PubMed] [Google Scholar]
- 44.Warner LE, DelloRusso C, Crawford RW, Rybakova IN, Patel JR, Ervasti JM, et al. Expression of Dp260 in muscle tethers the actin cytoskeleton to the dystrophin-glycoprotein complex and partially prevents dystrophy. Hum Mol Genet. 2002;11:1095–105. doi: 10.1093/hmg/11.9.1095. [DOI] [PubMed] [Google Scholar]
- 45.Legardinier S, Hubert J-F, Le Bihan O, Tascon C, Rocher C, Raguénès-Nicol C, et al. Sub-domains of the dystrophin rod domain display contrasting lipid-binding and stability properties. Biochim Biophys Acta. 2008;1784:672–82 [DOI] [PubMed]
- 46.Legardinier S, Raguénès-Nicol C, Tascon C, Rocher C, Hardy S, Hubert J-F, et al. Mapping of the lipid-binding and stability properties of the central rod domain of human dystrophin. J Mol Biol. 2009;389:546–58. doi: 10.1016/j.jmb.2009.04.025. [DOI] [PubMed] [Google Scholar]
- 47.Mirza A, Sagathevan M, Sahni N, Choi L, Menhart N. A biophysical map of the dystrophin rod. Biochim Biophys Acta. 2010;1804:1796–809. doi: 10.1016/j.bbapap.2010.03.009. [DOI] [PubMed] [Google Scholar]
- 48.Bhasin N, Law R, Liao G, Safer D, Ellmer J, Discher BM, et al. Molecular extensibility of mini-dystrophins and a dystrophin rod construct. J Mol Biol. 2005;352:795–806. doi: 10.1016/j.jmb.2005.07.064. [DOI] [PubMed] [Google Scholar]
- 49.Krieger CC, Bhasin N, Tewari M, Brown AEX, Safer D, Sweeney HL, et al. Exon-skipped dystrophins for treatment of Duchenne muscular dystrophy: mass spectrometry mapping of most exons and cooperative domain designs based on single molecule mechanics. Cytoskeleton (Hoboken) 2010;67:796–807. doi: 10.1002/cm.20489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sahni N, Mangat K, Le Rumeur E, Menhart N. Exon edited dystrophin rods in the hinge 3 region. Biochim Biophys Acta. 2012;1824:1080–9. doi: 10.1016/j.bbapap.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 51.Nicolas A, Lucchetti-Miganeh C, Ben YR, Kaplan J-C, Chelly J, Leturcq F, et al. Assessment of the structural and functional impact of in-frame mutations of the DMD gene, using the tools included in the eDystrophin online database. Orphanet J Rare Dis. 2012;7:45. doi: 10.1186/1750-1172-7-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nicolas A, Raguenes-Nicol C, Ben Yaou R, Ameziane-Le Hir S, Cheron A, Vie V, et al. Becker muscular dystrophy severity is linked to the structure of dystrophin. Hum Mol Genet. 2015;24:1267–79. doi: 10.1093/hmg/ddu537. [DOI] [PubMed] [Google Scholar]
- 53.Findlay AR, Wein N, Kaminoh Y, Taylor LE, Dunn DM, Mendell JR, et al. Clinical phenotypes as predictors of the outcome of skipping around DMD exon 45. Ann Neurol. 2015;77:668–74. doi: 10.1002/ana.24365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Singh SM, Kongari N, Cabello-Villegas J, Mallela KMG. Missense mutations in dystrophin that trigger muscular dystrophy decrease protein stability and lead to cross-beta aggregates. Proc Natl Acad Sci U S A. 2010;107:15069–74. doi: 10.1073/pnas.1008818107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tinsley J, Deconinck N, Fisher R, Kahn D, Phelps S, Gillis JM, et al. Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat Med. 1998;4:1441–4. doi: 10.1038/4033. [DOI] [PubMed] [Google Scholar]
- 56.Kleopa KA, Drousiotou A, Mavrikiou E, Ormiston A, Kyriakides T. Naturally occurring utrophin correlates with disease severity in Duchenne muscular dystrophy. Hum Mol Genet. 2006;5:1623–8. doi: 10.1093/hmg/ddl083. [DOI] [PubMed] [Google Scholar]
- 57.Matsumura K, Ervasti JM, Ohlendieck K, Kahl SD, Campbell KP. Assocation of dystrophin-related protein with dystrophin-associated proteins in mdx mouse muscle. Nature. 1992;360:588–91. doi: 10.1038/360588a0. [DOI] [PubMed] [Google Scholar]
- 58.Rybakova IN, Humston JL, Sonnemann KJ, Ervasti JM. Dystrophin and utrophin bind actin through distinct modes of contact. J Biol Chem. 2006;281:9996–10001. doi: 10.1074/jbc.M513121200. [DOI] [PubMed] [Google Scholar]
- 59.Li D, Bareja A, Judge L, Yue Y, Lai Y, Fairclough R, et al. Sarcolemmal nNOS anchoring reveals a qualitative difference between dystrophin and utrophin. J Cell Sci. 2010;123:2008–13. doi: 10.1242/jcs.064808. [DOI] [PMC free article] [PubMed] [Google Scholar]