

Abstract
Purpose
Inhibition of focal adhesion kinase-vascular endothelial growth factor receptor 3 complex by C4 was previously shown to reduce tumor growth alone and synergistically with other chemotherapeutic agents in animal tumor models. Single and multiple dose IV and oral dosing studies were performed in dogs to determine C4 pharmacokinetics.
Methods
C4 was administered to 4 dogs at 1.25 or 2.50 mg/kg IV, or 7.50 mg/kg oral gavage. Single-(IV and oral) and multiple- (IV) dose pharmacokinetic samples were collected on days 1 and 3 at predose and 0.5, 1, 2, 4, 8, 24, 120, 144, and 168 hrs post-dose. C4 concentrations were determined using liquid chromatography with tandem mass spectral detection with a limit of quantitation of 2.50 pg/mL. Pharmacokinetics of C4 was characterized by a 3-compartment model with linear distributional and elimination clearances using Phoenix 64 WinNonlin 6.3.
Results
Mean C4 plasma concentration-time profiles revealed a triexponential decline following either IV or oral administration, independent of dose with no accumulation. For the 2.5 mg/kg dose, the median half-life was approximately 21 hrs. Median Cmax and area-under-the-curve (AUC0-24) was similar for Days 1 and 3. Oral bioavailability for formulations of PBS, TPGS, Maalox®, and Pepcid® was greatest with TPGS (45%), followed by Maalox® (42%), Pepcid® (37%), and PBS (30%)
Conclusions
The pharmacokinetic study revealed C4 has linear pharmacokinetics and does not accumulate following multiple dose administration. Characterization of C4 pharmacokinetics provides a better understanding of the novel targeted agent, which will help facilitate further development of C4.
Keywords: FAK-VEGFR-3, chloropyramine, LC-MS/MS, repurposing, anti-tumor, protein-protein interaction inhibitor
Introduction
Focal adhesion kinase (FAK) protein is a tyrosine kinase that serves as a key signaling orchestrator involved in normal cellular adhesion, motility and survival. Overexpression of FAK in cancer cells allows cells to develop the ability to survive apoptotic stimuli and progress as invasive, metastatic cancer. Multiple studies have confirmed its involvement in the drug resistance of cancer cells, in the support of neovascularization, and the maintenance of cancer stem cells (Zhao and Guan 2011; Frisch et al. 2013). It is well established that in addition to kinase activity, FAK serves as a scaffold for multiple protein signaling complexes, and its scaffolding function is very important in tumor progression (Cance et al. 2013). Targeting such protein complexes allows the possibility to affect, modulate, or simultaneously shut down these multiple signaling pathways.
Several FAK kinase inhibitors have been developed (Schultze and Fiedler 2010; Ward et al. 2013; Kang et al. 2013; Walsh et al. 2013) and some of these inhibitors (PF-00562271, VS-6063, VS-4718) have shown promising results in early clinical trials (Infante et al. 2012; Schultze and Fiedler 2011). We have previously shown that FAK interacts with vascular endothelial growth factor receptor 3 (VEGFR-3) and this complex is critical to tumor progression and host-tumor interactions (Garces et al. 2006). As a result, this interaction serves as a significant platform for the development of targeted drug therapy. In silico screening of the National Cancer Institute's small molecule library for compounds that target the FAK-VEGFR-3 binding site led to the selection of our lead compound, C4 (Kurenova et al. 2009). In vitro and in vivo studies have shown that C4 (N-[(4-chlorophenyl)-methyl]-N-[2-(dimethylamino)-ethyl]-pyridin-2-amine), also commonly known as chloropyramine, is able to disrupt the FAK-VEGFR-3 complex and inhibit tumor growth. This activity has been demonstrated, both alone and synergistically with other chemotherapeutic agents (e.g., doxorubicin and gemcitabine), in multiple mouse tumor models (Kurenova et al. 2009; Stewart et al. 2013; Kurenova et al. 2013). In addition to its anti-tumor activity, C4 is a first generation competitive reversible antagonist, or inverse agonist of histamine receptor H1, synthesized in 1949 (Vaughan et al. 1949) and commonly used in Eastern European countries under the brand name Suprastin for allergic conjunctivitis, allergic rhinitis, bronchial asthma, and other atopic conditions.
Most first-generation H1-antihistamines were introduced before regulatory agencies required extensive clinical pharmacological studies for new pharmaceutical approvals. Although the pharmacokinetics and pharmacodynamics have been determined and published for some of these compounds, it is limited for others. For C4, there is very little published information about the analytical methods to quantitate C4 in biological matrices, or its pharmacokinetic and pharmacodynamic properties. In 1983, Kaverina developed an ion-pair HPLC method with UV detection to determine the pharmacokinetics of C4 (referred to as Suprastin) in rats after a single intragastric 50 mg/kg dose using a 2% starch suspension. The assay had a stated linear range of 0-6.0 μg/mL, but the assay limit of quantitation was not described. Although a chromatogram was reproduced in the manuscript, the peak attributed to Suprastin eluted very early and quite close to the solvent front, which could compromise the selectivity of the methodology. In addition, these earlier analytical methods are generally not as selective and sensitive as newer tandem mass spectrometric technologies.
Although C4 is not approved in the United States, pyrilamine (mepyramine), a structurally related antihistamine, has widespread use in the United States in equine medicine for symptomatic relief of allergic reactions (Dirikolu et al. 2009) and is a component of over-the-counter products such as Midol® for relief of premenstrual/menstrual pain and discomfort, and Deconsal DM®, an antihistamine, nasal decongestant, antitussive combination product. The only structural difference between C4 and pyrilamine is that pyrilamine has a methoxy group in place of the chlorine atom at carbon 4 on the aromatic ring in C4. Dirikolu (2009) described the quantitation and pharmacokinetics of pyrilamine and its major urinary metabolite, O-desmethylpyrilamine (O-DMP), in 4 horses following IV and oral administration (300 mg/horse). The study analyzed both serum and urine. Urinary glucuronides were treated with β-glucuronidase prior to solid phase extraction and derivatization. The resulting sample was analyzed by gas chromatography with mass spectral detection in single ion monitoring mode (GC/MS-SIM) for quantitation. The assay had lower limits of quantitation of 20.0 ng/mL for pyrilamine and 1.0 ng/mL for O-DMP. Pyrilamine had a Cmax of 280 ng/mL at 5 minutes with a half-life (t½) of 1.67 hrs following IV administration, and a Cmax of 36.6 ng/mL with Tmax 0.44 hrs following oral administration, but was not detected in horse serum at 24 hrs by either route of administration. Bioavailability was found to be low (18%). Despite using large sample volumes (1 mL serum and 5 ml urine), the assay's sensitivity and selectivity were limited by using SIM detection rather than multiple reaction monitoring (MRM). The metabolism of pyrilamine in man was examined by Chung (1994) following the administration of a single oral 50 mg dose to 3 males. Urine samples (24 hr collections) were extracted before and after glucuronide hydrolysis, and analyzed by GC/MS following derivatization. Pyrilamine was found to be extensively metabolized since no parent compound was observed in the urine. In addition, little N-demethylation was observed, which is a major metabolic route in rats. Instead, pyrilamine was extensively hydroxylated on the aromatic ring and/or O-demethylated prior to glucuronidation.
Another structurally related antihistamine is tripelennamine (pyrilbenzamine), which has a hydrogen substituted for the chlorine in C4. Similar to pyrilamine, tripelennamine is extensively metabolized and pharmacokinetic data has been reported (Sharma and Hamelin 2003; Chaudhuri et al. 1976, Yeh et al. 1986, Yeh 1990). In the rat, tripelennamine is highly metabolized with less than 2-3% of the dose being recovered unchanged in urine after 24 hrs (Yeh 1990). In humans, following 50 and 100 mg intramuscular doses, Cmax was 105 and 194 ng/mL, respectively, at 0.5 hrs post-dose with t½ of 2.9±0.25 and 4.4±1.3 hrs, respectively (Yeh et al. 1986). Tripelennamine is approved in the United States and available in products such as Pyribenzamine® and Pbz-SR® for the treatment of rhinitis, uticaria and asthma, and is also available in veterinary applications.
Overall, the information available on the pharmacokinetics and metabolism of the classical histamine H1 receptor antagonists is limited. Despite C4's common use as an antihistamine in Eastern European countries, there is an overall lack of published information on the compound. Therefore, the close structural similarity and properties of pyrilamine and tripelennamine to C4, provides important background and insight into the pharmacokinetics, metabolism, and bioavailability of C4. The profiles of these antihistamines and other related compounds are more fully described in a review by Sharma and Hamelin (2003). Since the intent is to develop C4 as an oncolytic agent in the United States, the corresponding preclinical studies need to be performed to define its pharmacokinetics and toxicology. The pharmacokinetics and tissue penetration of C4 in mice following intraperitoneal (IP) administration has been previously reported (Thudium et al. 2011). The goals of this study included the development and validation of a highly sensitive and selective HPLC method with tandem mass spectral detection (LC-MS/MS), application of the method to quantitate C4 concentrations in dog plasma samples following IV and oral administration, and to characterize C4 pharmacokinetics.
Materials and Methods
Materials
Acetonitrile, methanol, methyl-tert-butyl ether (MTBE) were HPLC grade. Other reagents were ACS grade or better. C4 (chloropyramine hydrochloride, C16H20ClN3·HCl, MW 326.3) was obtained from Fluka Analytical (Sigma Aldrich), St. Louis, MO, catalog number C1915-1G (salt conversion factor: 289.81/326.3 = 0.8882 mg of free base/mg of HCl salt) was used as the analytical standard and the preparation of some of the dosing solutions. Chloropyramine hydrochloride (Suprastin, solution for injection, 20 mg/mL, Egis Pharmaceuticals, Budapest, Hungary) was used for preparation of IV dosing solutions. N-Desmethylpyrilamine (N-DMP; C16H21N3O, MW 271.36, free base form) from Toronto Research Chemicals Inc., North York, ON, catalog number M261220, was used as the internal standard (IS). Lithium heparinized beagle plasma was obtained from Bioreclamation, LLC, Hicksville, NY.
Dogs
The source of the animals was Marshall BioResources, North Rose, NY. All dogs used in the study were purpose-bred and naïve beagles. The animals consisted of two males and two females at least 4 months of age. The dogs were single housed in stainless steel cages and acclimated prior to dosing. The protocol was approved by the Roswell Park Cancer Institute IACUC prior to conducting the study.
C4 administration
C4 hydrochloride was dissolved in water and given as an intravenous (IV) infusion through the cephalic vein at 1.25 or 2.50 mg/kg free base over 5 minutes up to three consecutive days. Single dose pharmacokinetic samples were collected on Day 1; multiple dose pharmacokinetic samples were collected on Day 3 after three days of once daily dosing. The single dose pharmacokinetic samples were collected after the 1.25 mg/kg dose on Day 1 of one week, and multiple dose pharmacokinetic samples were collected on Day 3 of the following week after 3 days of consecutive drug administration. Pharmacokinetic samples for the 2.50 mg/kg dosing were collected on Days 1 and 3 of the same week. Following the IV studies, C4 was administered at 7.50 mg/kg free base as a single dose with a 1 week washout between each of the 4 vehicles. Dosing for C4 hydrochloride included: (1) phosphate-buffered saline (PBS), (2) TPGS (d-α-tocopheryl polyethylene glycol succinate), (3) a 10 minute pre-treatment with Maalox® prior to a single dose of C4 in PBS, and (4) a 60 minute pre-treatment with Pepcid® prior to a single dose of C4 in PBS.
Pharmacokinetic sampling and blood collection procedure
Plasma samples for C4 pharmacokinetic analysis were collected at pre-dose, and 0.5, 1, 2, 4, 8, 24, 120, 144, and 168 hrs post-dose after single or multiple (3 days) dosing. The test animals were not fasted before blood collection. One mL of blood was collected into lithium heparin plasma separator tubes. Blood collection tubes were stored on wet ice until centrifuged at 3,000 RPM and 4 °C for 15 minutes. Plasma was transferred to labeled microcentrifuge tubes, capped, frozen in liquid nitrogen, and stored frozen at -80 °C until analyzed.
Calibration standards and quality control (QC) samples
Plasma calibration and quality control (QC) samples were prepared by adding the appropriate spiking solution to blank lithium heparinized dog plasma such that the original matrix was not diluted by more than 5%. After preparing the bulk calibrators and QCs, aliquots were transferred to microcentrifuge tubes and stored frozen at -80 °C until analyzed. The calibrator concentrations ranged from 2.50 - 2,500 pg/mL. Plasma QC samples (7.50, 120 and 1,800 pg/mL) were prepared at concentrations different from the calibrator concentrations and stored in a similar manner.
Plasma sample extractions
A 150 μl aliquot of a matrix calibrator, QC, matrix blank, or study sample was added to a 13 × glass screw-top tube followed by 100 μL of IS solution (1.00 ng/mL N-DMP in 10.0 mM ammonium acetate at pH 5.0), 2.0 mL of 100 mM ammonium hydroxide, and 6.0 mL of MTBE. The tubes were capped using PTFE-lined caps, rotated for 15 minutes, and centrifuged at 3,000 rpm and 4 °C for 20 minutes. The lower aqueous phase was frozen in a dry ice/acetone bath and the MTBE poured into a glass conical tube. The MTBE solvent was evaporated under nitrogen at 37 °C and the residue reconstituted with 70.0 μL of 40:60 methanol:20 mM ammonium acetate (pH 5.0). The reconstituted samples were transferred to 96-well plates, heat sealed, centrifuged for 15 seconds at 1,000 rpm, and injected (20 μL). Samples that had concentrations greater than the upper limit of quantitation were reanalyzed after diluting the sample into the range of the calibration curve with blank dog plasma prior to extraction.
Liquid chromatography/tandem mass spectrometry conditions
LC-MS/MS analysis was performed using a Prominence UFLC System (Shimadzu Scientific Instruments) interfaced with a QTRAP® 5500 mass spectrometer (AB SCIEX). Extracted samples were maintained at 4 °C in the autosampler. Chromatographic separation was achieved using a reverse phase Luna C18(2) column (3 μm, 2.0 mm × 150 mm, part number 00F-4251-B0, Phenomenex, Torrance, CA) preceded by a Phenomenex C18 SecurityGuard guard cartridge. Sample elution was carried out at 40 °C and flow rate of 220 μL/min using a biphasic gradient. Mobile phase A consisted of 30% acetonitrile in 10 mM ammonium acetate (pH 5.0), and mobile phase B was 100% acetonitrile. The linear elution gradient profile was: (1) 0 to 100% Mobile Phase B from 0.0 to 5.7 minutes, (2) 100% Mobile Phase B from 5.7 to 7.0 minutes, (3) 100 to 0% Mobile Phase B from 7.0 to 7.5 minutes, and (4) re-equilibration at 0% Mobile Phase B over 4.5 minutes (total run time = 12 minutes).
The QTRAP® 5500 mass spectrometer was utilized with an electrospray ionization source in positive ion mode using multiple reaction monitoring (MRM) to detect C4 and the IS. AB SCIEX Analyst® software, version 1.5.1, was used for system control, peak area measurement, and quantitation. Mass spectrometer source conditions were ion spray voltage 5300 volts, temperature 725 °C for the turbo gas (gas 2), and settings of 43, 72.5, 23, and medium for nebulizer gas (gas 1), turbo gas (gas 2), curtain gas, and collision-activated dissociation (CAD) gas flows, respectively. Unit mass resolution was used for both Q1 and Q3 mass resolving quadrupoles. Nitrogen gas was employed for the nebulizer, turbo, curtain and collision gases. A divert valve was used to direct flow to the mass spectrometer from 4.5 to 7.5 min; prior to and after this time period, flow was directed to waste. Voltages for maximum parent/fragment ion pair intensities for C4 [290.1 → 245.2 m/z (loss of 45 m/z); DP = 85 V, CE = 23 V and CXP = 13 V] and the IS [272.1 → 241.2 m/z (loss of 31 m/z); DP = 66 V, CE = 15 V and CXP = 16 V] were optimized using individual solutions with direct infusion and flow injection analysis (FIA). The dwell time for data acquisition was 125 msec for both compounds. Three additional transitions were used to monitor potential metabolites based on the fragmentation pattern of C4 and known metabolic routes: 1) hydroxy-C4 at 306.0 → 261.0 m/z (loss of 45 m/z), 2) desmethyl-C4 at 276.0 → 245.0 m/z (loss of 31 m/z), and 3) didesmethyl-C4 at 262.0 → 245.0 m/z (loss of 17 m/z). The declustering potential, collision energy, and exit potential voltages for the proposed metabolites were the same as used for C4.
Qualification of analytical method
The analytical method performance was qualified in lithium heparinized dog plasma over 2 analytical runs to assess linearity, accuracy, precision, sensitivity, selectivity, and reinjection stability. Each of the qualification runs included duplicate calibration curves, two plasma blanks, a reagent blank, and QCs (replicates of 3 in the first run and replicates of 6 in the second). In addition, the QCs in the second run were re-injected to demonstrate sample reinjection stability and system performance. Calibration curves were generated using the analyte/internal standard area response ratios vs. nominal concentrations (ng/mL) with weighted linear regressions using a weighting factor of 1/concentration2. Back-calculated concentrations were generated using the formula x = (y - b)/m where x is the back-calculated concentration, y is the analyte/internal standard ratio, b is the y-intercept, and m is the slope. Calibrator and QC acceptance criteria required all acceptable concentrations to have accuracy deviations of 15% or less from the nominal concentration with overall relative standard deviations (% RSD) of 15% or less, except at the lower limit of quantitation (LLOQ), which was allowed 20% for both parameters. Back-calculated concentrations of the three proposed metabolites were generated using the slope and y-intercept from the C4 calibration curve.
C4 Pharmacokinetic model development
Pharmacokinetic (PK) modeling was performed on individual dog plasma C4 concentration-time data using Phoenix 64 WinNonlin 6.3 (Pharsight Corp., Mountain View, CA). Various compartmental models were tested to characterize the pharmacokinetics of C4. A three compartmental PK model best described C4 disposition in plasma following both single and multiple dose IV and single dose oral administration. The PK model consisted of three compartments, one central plasma compartment where drug was measured and two peripheral tissue compartments, accounting for highly and less perfused organs. Both the IV and oral PK models included linear distributional and elimination clearance terms. Oral absorption was assumed to be linear. The choice of the final three compartment model was based upon evaluation of goodness of fits, the precision of PK parameter estimates, and the Akaike Information Criterion (AIC).
Parameters estimated by the three compartment model were: volume of distribution in the plasma compartment (V1), volume of distribution in the peripheral compartments (V2 and V3), plasma clearance (CL), and distributional clearances (CL2 and CL3). Secondary parameters, including area under the curve (AUC), the half-life of elimination (T1/2γ), and steady-state volume of distribution (Vss), were derived from the primary parameters or calculated using non-compartmental methods.
The following equations were used to describe C4 PK following IV and oral administration:
for oral administration, the above equation will have CGI *(CLpo) as an input into the plasma compartment,
oral administration only, where CGI is the amount of C4 in the gastrointestinal tract.
The primary and secondary PK parameters are defined as follows:
-
AUC0-∞, the area under the plasma concentration-time curve from time zero to infinity, was calculated using the linear trapezoidal rule.
where: ci = the concentration of the ith plasma sample; ti = the time of the ith plasma sample; n = the number of non-missing samples between zero and t hrs, c* = the last measurable concentration, and γ = terminal elimination rate constant.
AUC0-t, calculated from time zero to last measured time point using the linear trapezoidal rule.
Cmax, maximum plasma concentration
T½, the apparent terminal plasma elimination half-life, calculated as ln(2)/γ
V1, the volume of distribution in the plasma compartment
V2, V3, the volumes of distribution in the peripheral compartments
Vss, the volume of distribution at steady state
Vss/F, the apparent volume of distribution at steady state
CL, plasma clearance
-
CL2, CL3, distributional clearances for the tissue compartments
For all volumes of distribution and clearance terms following oral administration, the terms are normalized to F (e.g., CL/F)
Percent (%) fraction metabolized: % of C4 that was converted to a specific metabolite, calculated by AUC0-24 metabolite/AUC0-24 C4 × 100
Bioavailability, F = AUCpo × DoseIV/AUCIV × Dosepo
Results
Method development
The structure of C4 and the IS are shown in Figure 1. A stable-labeled C4 compound was not commercially available; therefore, N-DMP was chosen as the internal standard due to its close structural similarity and chemical behavior. We previously developed a protein precipitation LC-MS/MS assay to measure C4 in mouse plasma with linearity over the calibration range of 0.250 to 100 ng/mL using an AB Sciex API 3000 mass spectrometer (Thudium, 2011). Based on the proposed doses of C4 being administered to dogs, and the observed mouse pharmacokinetic data, a new assay was required with an approximate 40-fold increase in sensitivity (proposed LLOQ ∼6.0 pg/mL) in an attempt to quantify the 24 hr samples since none were detectable using the prior methodology for the mouse samples. Trials applying the original mouse plasma assay procedure to dog plasma resulted in three problems: 1) the sensitivity was insufficient, 2) matrix components specific to dog plasma appeared to cause ionization suppression during the course of an analytical run, and 3) evaporation of the supernatant in 1.0 mL 96-well plates following protein precipitation resulted in sample-to-sample cross-contamination. All issues were resolved using a liquid/liquid MTBE extraction procedure under basic conditions, which provided a cleaner and more concentrated sample for analysis. In addition, the change in the methodology resulted in an overall 100-fold increase in sensitivity when using an AB Sciex 5500 QTrap® mass spectrometer. A comparison of the parameters between the two methods is illustrated in Table 1 with a chromatogram of the LLOQ calibrator [2.50 pg/mL plasma; signal-to-noise (S/N) ratio of 55] using the new methodology shown in Figure 2.
Fig. 1. Structures of C4, its proposed metabolites, and the internal standard N-desmethylpyrilamine.
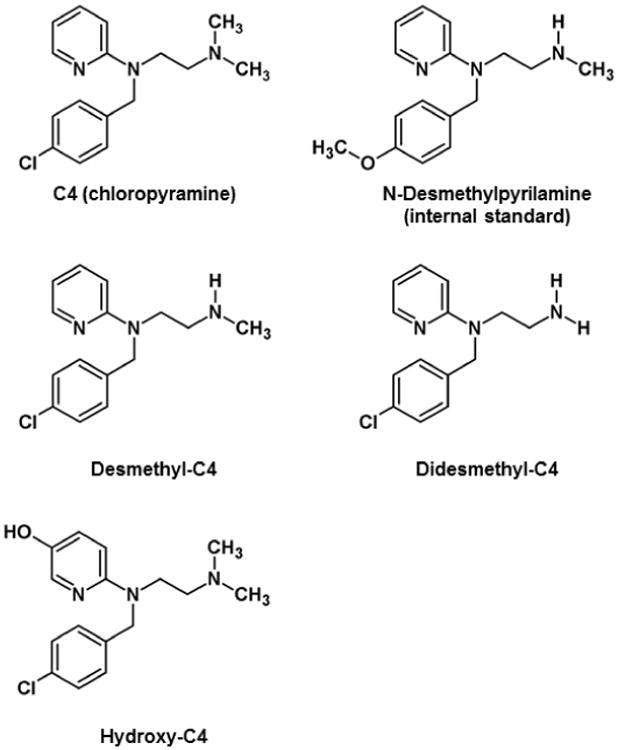
Table 1. A comparison of the LC-MS/MS analytical methods for mouse and dog plasma.
| Parameter | Method for Mouse Plasma | Method for Dog Plasma |
|---|---|---|
| Sample Volume | 100 μL | 150 μL |
| Sample Preparation | Methanol protein precipitation | MTBE liquid/liquid extraction under basic conditions |
| MS Instrumentation | AB Sciex API 3000 | AB Sciex 5500 QTrap® |
| Injection Volume | 20 μL | 20 μL |
| Calibration Range | 0.250 – 100 ng/mL | 2.50 – 2,500 pg/mL |
| Lower Limit of Quantitation | 250 pg/mL | 2.50 pg/mL |
| Run Time | 9.0 min | 12 min |
Fig. 2. LC-MS/MS chromatogram of a dog plasma calibration sample at the lower limit of quantitation (LLOQ = 2.50 pg/mL; signal:noise ratio = 55; on-column mass = 107 fg).
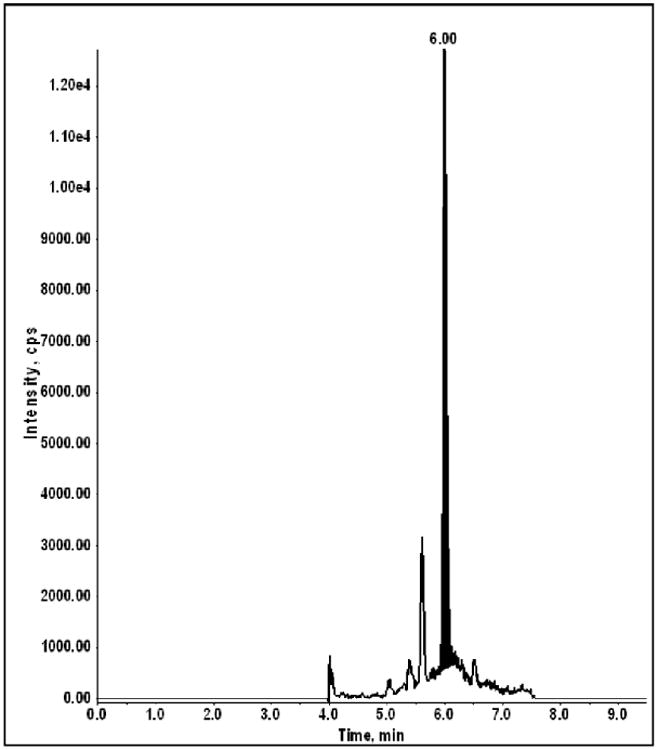
To qualify the analytical method, duplicate plasma calibration curves and replicate QCs were analyzed in each of 2 qualification runs. Calibrator and QC accuracy and precision passed the intended acceptance criteria with mean precision (% RSD) ranging from 2.03 to 8.15% for the calibration samples and from 2.67 to 8.06% for the QCs. Analytical recovery ranged from 92.9 to 107% and 94.0 to 102% for the calibration and QC samples, respectively. Correlation coefficients for C4 were >0.997. Assay specificity was assessed using a reagent blank and pooled plasma blank sample during each qualification run, as well as 3 different lots of blank dog plasma. The reinjection stability study showed no degradation of extracted samples over the reinjection period with samples being stable for up to 18.5 hrs (88 injections).
The samples from the IV and oral phases of the study were analyzed over 4 analytical runs. The calibration standards for the assay ranged from 2.50 to 2,500 pg/mL. The calibration range could not be extended beyond a thousand-fold range to accommodate all expected sample concentrations due to ionization suppression. Therefore, sample concentrations greater than the highest calibrator were reanalyzed after diluting the sample into the range of the calibration curve with blank dog plasma prior to extraction. As a result, the reported QC statistics reflect the precision of the observed concentrations from in-range study samples and out-of-range samples that were diluted in-range. The mean precision (range; as % RSD) for the calibrators was 5.03% (2.62-10.2%), and 5.06% (2.63-7.25%) for the QCs. The percent accuracy (range) was 100% (93.1 to 105%) and 95.5% (93.9 to 96.7%) for the calibrator and QC samples, respectively. The correlation coefficients were >0.9960 and the LLOQ (n=8 values) at 2.50 pg/mL had accuracy and precision of 100% and 10.2%, respectively.
Pharmacokinetic modeling of C4
C4 was given as an IV infusion at 1.25 or 2.50 mg/kg over 5 minutes on 3 consecutive days to determine single and multiple-dose pharmacokinetics, or orally at 7.50 mg/kg using 4 vehicles to determine bioavailability and single oral dose pharmacokinetics. Bioavailability was previously determined as 15.5% in mice comparing oral and IP routes of administration. In this study, C4 was orally administered using four different vehicles in an attempt to increase bioavailability. PBS served as the control. TPGS is a water-soluble derivative of vitamin E that has potential to improve drug delivery and enhance bioavailability of drugs. Maalox® and Pepcid® were used to increase gastrointestinal pH, which has been shown to increase bioavailability of various drug products (Neuvonen and Kivistö, 1994; Akhali and Alavudeen, 2014).
As a result of the increased sensitivity of the assay, C4 concentrations were unexpectedly detected in the pre-dose samples collected in subsequent treatment weeks (either 120, 144, or 168 hrs post-dose, depending on the time of the prior administration). The PK results presented were generated using concentration-time profiles that included the pre-dose sample of the following weeks' treatment as the last time point in the profile for the prior dose administered.
The mean C4 plasma concentration-time profiles revealed a triexponential decline of drug following IV infusion or oral administration (Figures 3-5). A three compartmental PK model described C4 pharmacokinetics following both IV and oral administration well, demonstrating rapid tissue distribution followed by a slow elimination (Figure 6). The estimated median (range) T1/2 was 21.8 hr (17.7 – 27.3) and 21.8 hr (19.5 – 24.0) following 1.25 and 2.50 mg/kg IV, respectively. Similarly, T1/2 was 22.1 hr (18.9 – 25.2), 19.8 hr (19.2 – 20.3), 20.1 hr (18.0 – 22.8), and 13.3 hr (5.34 – 28.6) following 7.50 mg/kg oral administration in PBS, TPGS, Maalox®, and Pepcid®, respectively (Table 2). These results confirmed the long half-life (T1/2) of C4 that has been reported in previous studies (Kurenova et al. 2013).
Fig. 3. Mean plasma pharmacokinetic profile of IV 1.25 mg/kg C4 on Days 1 and 3 (plasma collected at pre-dose and 0.5, 1, 2, 4, 8, and 24 hrs post-dose; each time point represents mean of n=4 dogs; error bars = standard deviation).
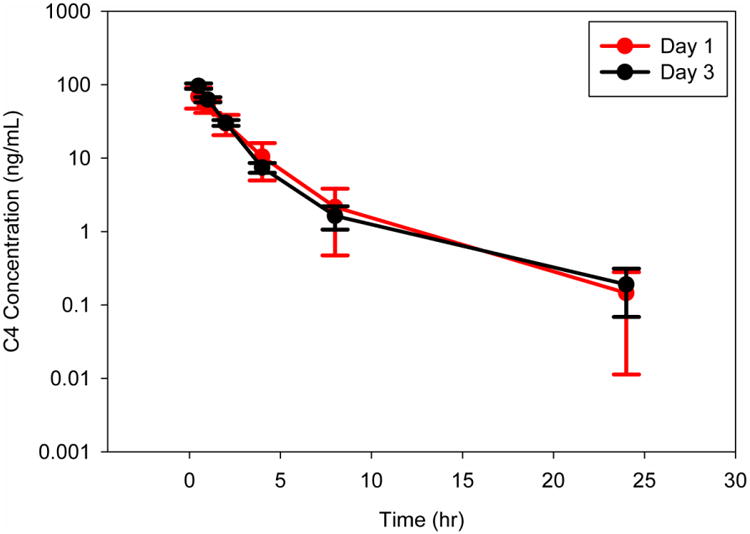
Fig. 5. Comparison of plasma pharmacokinetic profiles of 7.50 mg/kg C4 oral formulations to 24 hrs (top) and 144 hrs (bottom).

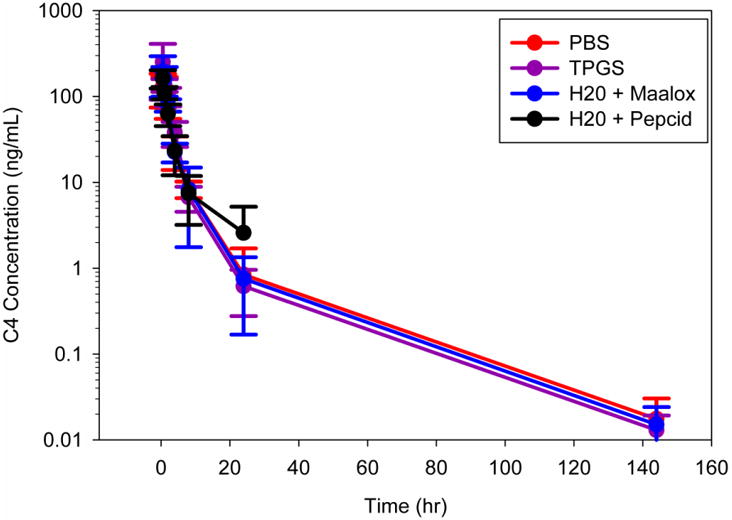
Fig. 6.
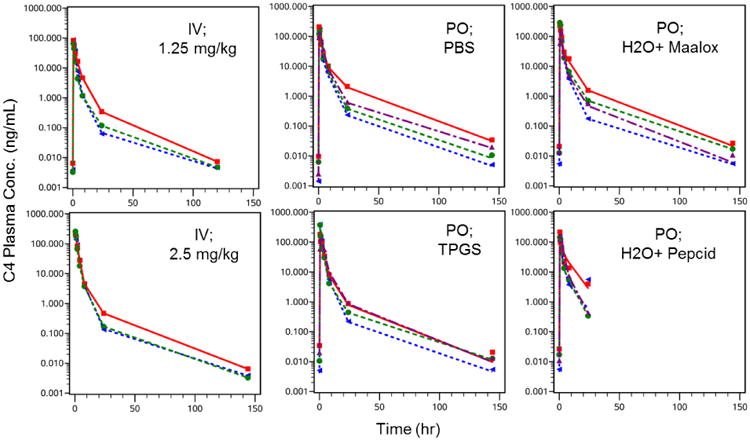
Observed and Model Predicted C4 Concentration-Time Profiles Following Single Dose IV or PO Administration. Individual concentration-time profiles from 0, 0.5, 1, 2, 4, 8, 24, 120 or 144 hr post dose were modeled. C4 was administered either as a single oral dose or a 5 minute IV infusion. The IV dose level (1.25 and 2.50 mg/kg) and oral vehicle (7.50 mg/kg in PBS, TPGS, Maalox®, or Pepcid®) treatments were performed with a week washout in between. C4 concentrations were detectable in pre-dose samples of the subsequent treatment and were included in the previous treatment profile. PBS = phosphate-buffered saline; TPGS = d-α-tocopheryl polyethylene glycol succinate; Maalox® given 10 minute before C4 in PBS; Pepcid® given 60 minutes before C4 in PBS.
Table 2. Model Estimated Secondary PK Parameters [Median (Range)] Following Single Dose IV or PO Administration of C4.
| Single Dose (mg/kg) | Admin. Route | Vehicle | Vss or Vss/F (L/kg) | Cmax (ng/ml) | T1/2 (hr) | AUC0-24hr (hr*ng/ml) | F* (%) |
|---|---|---|---|---|---|---|---|
| 1.25 | IV | Water | 24.2 (15.3 – 30.8) | 122 (103 – 131) | 21.8 (17.7 – 27.3) | 185 (117 – 230 | n/a |
| 2.50 | IV | Water | 13.6 (10.8 – 15.3) | 316 (242 – 402) | 21.8 (19.5 – 24.0) | 461 (455 – 505) | n/a |
| 7.50 | PO | PBS | 88.4 (61.4 – 149) | 151 (122 – 203) | 22.1 (18.9 – 25.2) | 432 (364 – 527) | 30.0 (26.0 – 35.0) |
| 7.50 | PO | TPGS | 120 (14.3 – 162) | 195 (134 – 371) | 19.8 (19.2 – 20.3) | 588 (533 – 772) | 45.0 (35.0 – 56.0) |
| 7.50 | PO | Maalox | 76.6 (71.3 – 99.5) | 234 (124 – 296) | 20.1 (18.0 – 22.8) | 508 (382 – 695) | 42.0 (32.0 – 46.0) |
| 7.50 | PO | Pepcid | 61.8 (54.4 – 124) | 185 (155 – 228) | 13.3 (5.34 – 28.6) | 508 (343 – 582) | 37.0 (25.0 – 38.0) |
AUCs (0-24hr) from the 2.5 mg/kg single dose IV infusion were used as 100% reference point. The four single oral (PO) doses were given at 7 day intervals; PBS = phosphate-buffered saline; TPGS = d-α-tocopheryl polyethylene glycol succinate; Maalox® given 10 minutes before C4 in PBS; Pepcid® given 60 minutes before C4 in PBS.
The median (range) steady state volume of distribution (Vss) of C4 is 24.2 L/kg (15.3 – 30.8) and 13.6 L/kg (10.8 – 15.3) following 1.25 and 2.50 mg/kg IV, respectively. The steady state volume of distribution (Vss/F) of C4 following 7.50 mg/kg PO in PBS,TPGS, Maalox®, and Pepcid® are 88.4 L/kg (61.4 – 149), 120 L/kg (14.3 – 162), 76.6 L/kg (71.3 – 99.5), and 61.8 L/kg (54.4 – 124), respectively (Table 2). The average weights of the dogs ranged from 5.0 - 8.5 kg, making the steady-state volume of distribution approximately ranging from 70 to 750 L. Since the central volume of a dog is around 0.5 L, this large Vss indicates that C4 distributes extensively into various tissues. In addition, C4 does not appear to accumulate following multiple doses, based on the mean C4 plasma concentration-time profiles between Days 1 and 3 and the similarity between the pre-dose sample concentrations. The median (range) plasma clearance (CL) of C4 is similar between the 1.25 and 2.50 mg/kg IV dose levels; 6.42 L/hr/kg (5.30 – 9.63) and 5.04 L/hr/kg (4.70 – 5.36), respectively. In contrast, apparent oral clearance is increased compared to IV; PBS 59.5 L/hr/kg (27.9 – 60.0), TPGS 54.0 L/hr/kg (23.5 – 60.0), Maalox® 76.6 L/hr/kg (32.8 – 60.0), and Pepcid® 53.0 L/hr/kg (25.5 – 60.0), suggesting a high first pass effect by the liver (Table 3). The pharmacokinetics of C4 following multiple dose administration were similar to those following single dose administration (data not shown).
Table 3. Model Estimated Primary PK Parameters [Median (Range)] Following Single Dose IV or PO Administration of C4.
| Single Dose (mg/kg) | Admin Route | Vehicle | CL or CL/F (L/kg/hr) | CL2 or CL2/F (L/kg/hr) | CL3 or CL3/F (L//kg/hr) | V1 or V1/F (L/kg) | V2 or V2/F (L/kg) | V3 or V3/F (L/kg) |
|---|---|---|---|---|---|---|---|---|
| 1.25 | IV | Water | 6.42 (5.30 – 9.63) | 1.61 (0.28 – 4.70) | 0.41 (0.11 – 0.47) | 9.17 (8.81 – 11.59) | 5.16 (1.19 – 5.53) | 9.52 (4.26 – 14.1) |
| 2.50 | IV | Water | 5.04 (4.7 – 5.36) | 0.57 (0.14 – 0.80) | 0.09 (0.09 – 0.25) | 13.6 (6.49 – 16.5) | 0.98 (0.49 – 1.97) | 6.69 (3.02 -2.9) |
| 7.50 | PO | PBS | 59.45 (27.9 – 60.0) | 7.00 (5.29 – 7.00) | 1.43 (0.60 – 1.80) | 40.6 (5.45 – 69.6) | 20.0 (9.92 – 20.0) | 41.2 (19.2 – 60.0) |
| 7.50 | PO | TPGS | 54.0 (23.5 – 60.0) | 5.36 (1.51 – 7.00) | 1.80 (0.13 – 1.80) | 52.2 (3.59 – 92.2) | 20.0 (6.83 – 20.0) | 48.1 (3.90 – 49.8) |
| 7.50 | PO | Maalox | 52.3 (32.8 – 60.0) | 7.00 (5.50 – 7.00) | 1.55 (0.67 – 1.80) | 27.3 (15.75 – 40.3) | 14.2 (11.9 – 20.0) | 42.9 (21.6 – 45.6) |
| 7.50 | PO | Pepcid | 53.0 (25.5 – 60.0) | 7.00 (7.00 – 7.00) | 1.80 (1.49 – 1.80) | 21.8 (5.12 – 52.8) | 15.7 (10.7 – 20.0) | 32.6 (13.4 – 60.0) |
Vehicles: PBS = phosphate-buffered saline; TPGS = d-α-tocopheryl polyethylene glycol succinate; Maalox® given 10 minutes before C4 in PBS; Pepcid® given 60 minutes before C4 in PBS.
The median (range) maximum plasma concentration of C4 (Cmax) following 1.25 and 2.50 mg/kg are 122 ng/ml (103 – 131) and 316 ng/ml (242 – 402), respectively. Following both IV dose levels and the 7.50 mg/kg oral dose level, the Cmax values achieved were similar or greater than the C4 to FAK dissociation constant (Kd), 165 ng/mL, which has been determined previously through binding studies (Kurenova et al. 2013). The median (range) plasma C4 exposure (AUC0-24) values following 1.25 and 2.5 mg/kg IV, are 185 hr*ng/mL (117 – 230) and 461 hr*ng/mL (455 – 505), respectively (Table 2). The relatively proportional increase in Cmax and AUC with the increase in dose from 1.25 to 2.5 mg/kg IV, as well as the dose-normalized concentration-time profiles of C4 (Figure 7) show the pharmacokinetics of C4 to appear linear and dose proportional. Median (range) Cmax and AUC values are comparable among all vehicles (PBS, TPGS, Maalox®, and Pepcid®) with 7.50 mg/kg PO: Cmax values of 151 ng/ml (122 – 203), 195 ng/ml (134 – 371), 234 ng/ml (124 – 296), and 185 ng/ml (155 – 228), respectively; and AUC0-24 values of 432 hr*ng/mL (364 – 527), 588 hr*ng/mL (533 – 772), 508 hr*ng/mL (382 – 695), and 508 hr*ng/mL (343 – 582), respectively (Table 2). The extent (AUC) of exposure of C4 following oral administration was the greatest with the TPGS formulation.
Fig. 7.
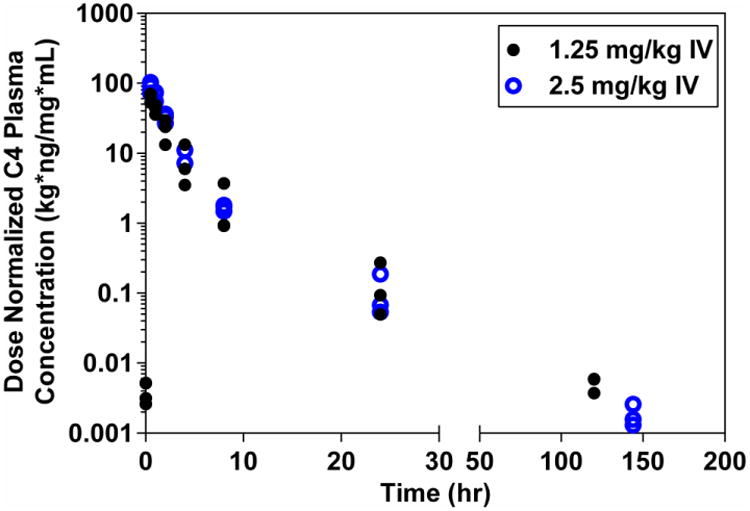
Dose Normalized Observed Plasma C4 Concentration-Time Profiles Following a 1.25 or 2.50 mg/kg Single Dose IV. C4 was administered as a 5 minute IV infusion at two dose levels (1.25 and 2.50 mg/kg) with a week washout in between doses. For all treatments, PK samples were collected at 0.5, 1, 2, 4, 8, 24, 120, and 144 hr post-dose. C4 concentrations were detectable in pre-dose samples of the subsequent treatment and were therefore included in the previous treatment profile. Each point in the figure represents the concentration at a specific time for one dog.
To determine oral bioavailability using each of the four vehicles, the AUC0-24 following a single oral administration of 7.50 mg/kg in the four different vehicles were compared against the AUC0-24 following a single IV administration of 2.5 mg/kg. Oral bioavailability for the formulations of PBS, TPGS, Maalox®, and Pepcid® was the greatest with TPGS (45%), followed by Maalox® (42%), Pepcid® (37%), and PBS (30%) (Table 2).
Three C4 metabolites were also analyzed using ‘virtual’ mass spectral transitions. ‘Virtual’ in this sense is defined as a transition based on theoretical Q1/Q3 masses without the availability or use of authentic compounds. The three metabolites, desmethyl-C4, didesmethyl-C4, and hydroxy-C4 (Figure 1) were based on Phase 1 dealkylation and hydroxylation reactions, and the previously published metabolites for pyrilamine and tripelennamine (Chaudhuri et al. 1976; Sharma and Hamelin 2003). Support for the compounds was obtained from 3-dimensional mass spectral Q1 scans (chromatographic time vs. mass vs. intensity) of liver extracts from an untreated mouse and a 2 hr C4-treated mouse. All three compounds displayed the unique 35Cl/37Cl isotopic pattern and had relative chromatographic retention times (RRT) consistent with the proposed structures and changes in polarity (RRT desmethyl-C4 = 0.94, RRT didesmethyl-C4 = 0.88, and RRT hydroxy-C4 = 0.74 with C4 = 1.0). The concentrations of the metabolites (desmethyl-, didesmethyl-, and hydroxy-C4) in the dog plasma samples were calculated based on the calibration curve of C4. Although these concentrations are not accurate since pure authentic standards weren't available to validate retention times and create individualized calibration curves, the concentrations represent the estimated pharmacokinetic profiles of three potential major metabolites (Figure 8). For the hydroxylated compound, no studies were performed to identify the exact location of the oxygenated site.
Fig. 8. Concentrations of C4 and its metabolites following a single oral 7.50 mg/kg dose in phosphate-buffered saline.
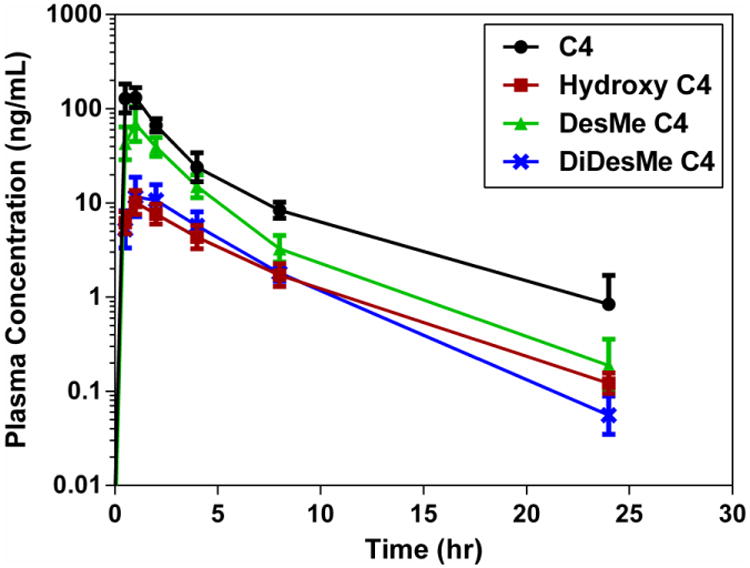
The metabolites were plotted along with the parent C4, analyzed by non-compartmental analysis, and the percent metabolized from parent calculated (Table 4). The percent metabolized for each metabolite was similar for the different C4 IV administrations, but differed from each other. Based on the observed concentrations and the AUC0-24, the hydroxyl- and didesmethyl-C4 metabolites represented less than 5.5% metabolism whereas the desmethyl-C4 represented approximately 15%. C4 was converted to its metabolites to a much greater extent following oral administration, primarily due to first pass metabolism (Table 4). For all oral formulations, hydroxyl- and didesmethyl-C4 represented 15.1% or less of total drug, while desmethyl-C4 was 50%, 48%, 30% and 31% of total drug for the PBS, TPGS, Maalox® and Pepcid® formulations, respectively. These percentages represent the approximate percent metabolism since the metabolite concentrations were back-calculated from the C4 calibration curve and not based on pure authentic standards.
Table 4. C4 metabolite AUC and percent metabolism summary.
| Treatmenta | Day | ID | AUC0-24 (hr*ng/mL) | % Metabolized | |||||
|---|---|---|---|---|---|---|---|---|---|
| C4 | Hydroxy | Des-methyl | Dides-methyl | Hydroxy | Des-methyl | Dides-methyl | |||
| 1.25 mg/kg IV | 1 | n | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| Mean | 171 | 9.59 | 24.1 | 6.05 | 5.50 | 12.7 | 3.29 | ||
| SD | 47.1 | 3.50 | 19.6 | 3.65 | 1.23 | 6.86 | 1.25 | ||
| 3 | n | 4 | 4 | 4 | 4 | 4 | 4 | 4 | |
| Mean | 180 | 8.18 | 25.4 | 6.77 | 4.57 | 14.3 | 3.81 | ||
| SD | 10.2 | 2.07 | 18.8 | 2.72 | 1.26 | 10.9 | 1.62 | ||
| 2.50 mg/kg IV | 1 | n | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| Mean | 456 | 14.5 | 51.6 | 13.7 | 3.18 | 11.0 | 2.96 | ||
| SD | 27.4 | 2.43 | 37.9 | 6.27 | 0.463 | 7.31 | 1.22 | ||
| 3 | n | 4 | 4 | 4 | 4 | 4 | 4 | 4 | |
| Mean | 420 | 15.7 | 64.3 | 16.7 | 3.76 | 15.0 | 3.95 | ||
| SD | 27.2 | 1.14 | 38.9 | 6.33 | 0.356 | 7.97 | 1.38 | ||
| 7.50 mg/kg oral (PBS) | 1 | n | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| Mean | 425 | 53.2 | 211 | 63.0 | 12.8 | 50.2 | 15.1 | ||
| SD | 42.6 | 14.2 | 45.6 | 20.4 | 3.96 | 13.2 | 5.68 | ||
| 7.50 mg/kg oral (TPGS) | 1 | n | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| Mean | 565 | 59.2 | 266 | 75.0 | 10.4 | 47.5 | 13.1 | ||
| SD | 53.7 | 11.3 | 89.5 | 31.1 | 1.21 | 17.0 | 4.64 | ||
| 7.50 mg/kg oral (Maalox®) | 1 | n | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| Mean | 493 | 31.8 | 148 | 52.0 | 6.95 | 30.3 | 11.3 | ||
| SD | 148 | 6.33 | 64.2 | 16.4 | 2.40 | 9.26 | 4.88 | ||
| 7.50 mg/kg oral (Pepcid®) | 1 | n | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| Mean | 424 | 35.8 | 130 | 53.7 | 8.72 | 30.5 | 12.8 | ||
| SD | 89.0 | 16.6 | 58.2 | 23.7 | 3.84 | 10.7 | 4.62 | ||
The four single oral (PO) doses were given at 7 day intervals; PBS = phosphate-buffered saline; TPGS = d-α-tocopheryl polyethylene glycol succinate; Maalox® given 10 minutes before C4 in PBS; Pepcid® given 60 minutes before C4 in PBS.
Discussion
FAK is emerging as a promising cancer target because of its involvement in many aspects of tumor growth, invasion, and metastasis, and is highly expressed in cancer. Inhibition of the FAK catalytic function has led to the development of FAK kinase inhibitors that are currently in clinical trials with promising results. Alternatively, targeting the FAK scaffold is another feasible and promising approach to developing highly specific therapeutics to disrupt the FAK signaling pathways in cancer. We have selected and characterized, preclinically, the small molecule C4 as a potential candidate, which belongs to the first generation of H1-antihistamines. It specifically disrupts the FAK-VEGFR-3 protein complex and inhibits tumor growth in multiple mouse tumor models. C4 is under development for the treatment of pancreatic cancer and was granted Orphan Drug Designation by the U.S. FDA in 2011, a designation assigned to drugs and biologics being developed to treat rare medical conditions that affect less than 200,000 individuals per year in the U.S.
To further develop C4 as an anti-neoplastic agent, characterization of its pharmacokinetics and pharmacodynamics are necessary. An LC-MS/MS method was developed for detection of C4 in mouse plasma (0.250–100 ng/mL) and re-qualified over the concentration range of 0.00250–2.50 ng/mL (100× more sensitive) for analysis of dog plasma samples. In addition, semi-quantitative data was obtained for three C4 metabolites based on ‘virtual’ mass spectral transitions using the C4 calibration curve.
Parameter estimates were similar between dose levels and following single- and multiple-dose IV administration, including the estimated T½ based on a terminal phase out to 120 or 144 hr. The concentrations of C4 remaining in the plasma at these latter time points were low (<35.0 pg/mL), and likely the result of slow C4 release after extensive tissue distribution as suggested by the large Vss. In the absence of tissue data from the current study, results from the IP administration of C4 to mice (Thudium 2011) support this premise. C4 concentrations in mouse plasma and tissues versus time profiles were obtained following 44.6 mg/kg IP administration of C4 free base to CD1 mice (n=3 per time point). C4 reached peak plasma concentrations at 0.67 hrs with mean Cmax of 820 ng/mL and achieved wide tissue distribution with Cmax being reached within 1.0 hr for all tissues. The highest tissue concentration of C4 was achieved in the lungs with mean Cmax (std dev) of 41,600 (20.09) ng/mL.
The mean C4 dog plasma concentration-time profiles revealed a triexponential decline of drug following IV infusion and oral administration. Based on the individual pharmacokinetic profiles at two different IV doses in dogs, and the dose-normalized concentration-time profiles of C4, the pharmacokinetics appear to be linear and dose proportional. The pharmacokinetic parameters are independent of dose, and the effective plasma T½ ranged from 13.3 - 22.1 hrs. Oral clearance was higher compared to plasma clearance, suggesting a pronounced first pass effect by the liver. Oral bioavailability was highest with the TPGS formulation (45%). Maalox® administration also demonstrated increased bioavailability (42%) related to lowered gastrointestinal pH compared to PBS (30%). C4 metabolism in dogs was significantly greater after oral administration as a result of first pass metabolism.
Kaverina (1983) examined the pharmacokinetics of C4 in rats after a single intragastric 50 mg/kg dose (salt vs. free base form not specified) given in 0.5 mL of a 2% starch suspension. The Cmax and T½ were determined to be 0.37 μg/mL and 14.9 hrs, respectively. Serum concentrations were detectable to 24 hrs with a mean concentration of 0.16±0.01 μg/mL; however, the assay limit of quantitation was not described. The rat T½ and terminal concentrations reported by Kaverina are inconsistent with the observations reported in mice by Thudium (2011), who observed a T½ of 0.83±0.34 hrs following a 44.6 mg/kg free base IV dose and no observable concentrations at 24 hrs despite having an analytical method with greater sensitivity. Although the animal species and route of administration are different (gavage in rats vs. IV in mice), some of the observations for the two rodent species are unexpected. Differences in Cmax (0.37 μg/ml for rat gavage vs. 0.82 μg/mL for mouse IV) are explainable due to bioavailability and first-pass metabolism, but the T½ (14.9 hrs) and 24 hr concentrations (0.16 μg/mL) reported by Kaverina are considerably greater than those reported by Thudium (0.83±0.34 hrs and 0 μg/mL, respectively). Such differences could be attributed to either poor assay specificity in the analytical method used by Kaverina vs. the enhanced specificity of newer LC-MS/MS technologies used by Thudium, or some form of enhanced/extended bioavailability of the 2% starch suspension used by Kaverina.
The Kd observed in in vitro C4 binding experiments is in the same range as the Kd predicted by PKPD-PB modeling (results to be published). Since the concentrations of C4 at the extended time points are over 1,000 times lower than the Kd in the terminal slope of the Day 3 profile, the Day 1 drug exposure is more representative of the true pharmacokinetics of C4 when proposing an appropriate dosing strategy.
Overall, this study developed a highly sensitive bioanalytical method for C4 quantitation and a successful PK model, both of which are valuable tools that were used to investigate C4 disposition and pharmacokinetics in dogs. Additionally, the characterization of C4 pharmacokinetics in this study provides insight regarding C4 drug exposure following different routes of administration and oral bioavailability.
Fig. 4. Mean plasma pharmacokinetic profile of IV 2.50 mg/kg C4 on Days 1 and 3 (plasma collected at pre-dose and 0.5, 1, 2, 4, 8, and 24 hrs post-dose; each time point represents mean of n=4 dogs; error bars = standard deviation).

Acknowledgments
Financial Support: This work has been supported by the National Cancer Institute (NCI) grant R01-CA65910 (W. Cance) and by Roswell Park Cancer Institute. This work also utilized core resource activities supported by the NCI Cancer Center Support Grant CA016156 (D. Trump).
Footnotes
Institution Where Work Was Performed: Roswell Park Cancer Institute, Buffalo, New York, USA
Disclosure of Potential Conflicts of Interest: W. Cance and E. Kurenova report grants from National Cancer Institute during the conduct of the study, and are stockholders and founders of CureFAKtor Pharmaceuticals, LLC. University of Florida and Roswell Park Cancer Institute have multiple patents pending and awarded that are based on their work developing FAK inhibitors. Drs. Wilton, Pitzonka, Gaudy, Curtin, Sexton and Fetterly have no disclosures to report.
References
- Akhali K, Alavudeen S. Multiple dose of liquid antacid enhance simvastatin bioavailability in Malaysian male volunteers. Asian J Pharm. 2014;8(2):90–94. [Google Scholar]
- Cance WG, Kurenova E, Marlowe T, Golubovskaya V. Disrupting the Scaffold to Improve Focal Adhesion Kinase-Targeted Cancer Therapeutics. Sci Signal. 2013;6(268):pe10. doi: 10.1126/scisignal.2004021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri NK, Servando OA, Manniello MJ, Luders RC, Chao DK, Bartlett MF. Metabolism of tripelennamine in man. Drug Metab Dispos. 1976;4(4):372–378. [PubMed] [Google Scholar]
- Chung BC, Kim DH, Jung BH, Eom K, Slikker W, Jr, Park J. Identification of urinary metabolites of pyrilamine after oral administration to man. Xenobiotica. 1994;24(5):451–459. doi: 10.3109/00498259409043248. [DOI] [PubMed] [Google Scholar]
- Dirikolu L, Lehner AF, Harkins JD, Woods WE, Karpiesiuk W, Gates RS, Fisher M, Tobin T. Pyrilamine in the horse: detection and pharmacokinetics of pyrilamine and its major urinary metabolite O-desmethylpyrilamine. J Vet Pharmacol Ther. 2009;32:66–78. doi: 10.1111/j.1365-2885.2008.01005.x. [DOI] [PubMed] [Google Scholar]
- FDA. Guidance for Industry: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. 2005 http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm078932.pdf.
- Frisch SM, Schaller M, Cieply B. Mechanisms that link the oncogenic epithelial–mesenchymal transition to suppression of anoikis. J Cell Sci. 2013;126:21–29. doi: 10.242/jcs.120907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garces CA, Kurenova EV, Golubovskaya VM, Cance WG. Vascular endothelial growth factor receptor-3 and focal adhesion kinase bind and suppress apoptosis in breast cancer cells. Cancer Res. 2006;66(3):1446–1454. doi: 10.1158/0008-5472.CAN-05-1661. [DOI] [PubMed] [Google Scholar]
- Infante JR, Camidge DR, Mileshkin LR, Chen EX, Hicks RJ, Rischin D, Fingert H, Pierce KJ, Xu H, Roberts WG, Shreeve SM, Burris HA, Siu LL. Safety, pharmacokinetic, and pharmacodynamic phase I dose-escalation trial of PF-00562271, an inhibitor of focal adhesion kinase, in advanced solid tumors. J Clin Oncol. 2012;30:1527–1533. doi: 10.1200/JCO.2011.38.9346. [DOI] [PubMed] [Google Scholar]
- Kang Y, Hu W, Ivan C, Dalton HJ, Miyake T, Pecot CV, Zand B, Liu T, Huang J, Jennings NB, Rupaimoole R, Taylor M, Pradeep S, Wu SY, Lu C, Wen Y, Huang J, Liu J, Sood AK. Role of focal adhesion kinase in regulating YB-1-mediated paclitaxel resistance in ovarian cancer. J Natl Cancer Inst. 2013;105(19):1485–1495. doi: 10.1093/jnci/djt210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaverina LP, Dorokhov VV, Kholodov LE. Study of the pharmacokinetics of suprastin and fencarol in rats by the method of highly effective liquid chromatography. Pharm Chem J. 1983;17:15–17. [Google Scholar]
- Kurenova EV, Hunt DL, He D, Magis AT, Ostrov DA, Cance WG. The small molecule chloropyramine hydrochloride (C4) targets the binding site of focal adhesion kinase and vascular endothelial growth factor receptor 3 and suppresses breast cancer growth in vivo. J Med Chem. 2009;52:4716–4724. doi: 10.1021/jm900159g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurenova E, Liao J, He DH, Hunt D, Yemma M, Bshara W, Seshadri M, Cance WG. The FAK scaffold inhibitor C4 disrupts FAK-VEGFR-3 signaling and inhibits pancreatic cancer growth. Oncotarget. 2013;4(10):1632–1646. doi: 10.18632/oncotarget.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuvonen PJ, Kivistö KT. Enhancement of drug absorption by antacids. An unrecognised drug interaction. Clin Pharmacokinet. 1994;27(2):120–8. doi: 10.2165/00003088-199427020-00004. [DOI] [PubMed] [Google Scholar]
- Schultze A, Fiedler W. Therapeutic potential and limitations of new FAK inhibitors in the treatment of cancer. Expert Opin Investig Drugs. 2010;19:777–788. doi: 10.1517/13543784.2010.489548. [DOI] [PubMed] [Google Scholar]
- Schultze A, Fiedler W. Clinical importance and potential use of small molecule inhibitors of focal adhesion kinase. Anticancer Agents Med Chem. 2011;11:593–599. doi: 10.2174/187152011796817727. [DOI] [PubMed] [Google Scholar]
- Sharma A, Hamelin BA. Classic histamine H1 receptor antagonists: A critical review of their metabolic and pharmacokinetic fate from a bird's eye view. Curr Drug Metab. 2003;4(2):105–129. doi: 10.2174/1389200033489523. [DOI] [PubMed] [Google Scholar]
- Stewart JE, Ma X, Megison M, Nabers H, Cance WG, Kurenova EV, Beierle EA. Inhibition of FAK and VEGFR-3 binding decreases tumorigenicity in neuroblastoma. Mol Carcinog. 2013 Jul 19; doi: 10.1002/mc.22070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thudium KE, Telang U, Wang P, Diegelman P, Buitrago S, Curtin L, Kurenova E, Cance W, Fetterly G. Pharmacokinetics (PK) and tissue penetration of the novel VEGFR-3/FAK inhibitor, chloropyramine; AACR 102nd Annual Meeting, abstract 4257; Orlando, FL. 2011. [DOI] [Google Scholar]
- Vaughan JR, Jr, Anderson GW, Clapp RC, Clark JH, English JP, Howard KL, Marson HW, Sutherland LH, Denton JJ. Antihistamine agents; halogenated N, N-dimethyl-N-benzyl-N-(2-pyridyl)-ethylenediamines. J Org Chem. 1949;14(2):228–234. doi: 10.1021/jo01154a006. [DOI] [PubMed] [Google Scholar]
- Walsh C, Tanjoni I, Uryu S, Tomar A, Nam JO, Luo H, Phillips A, Patel N, Kwok C, McMahon G, Stupack DG, Schlaepfer DD. Oral delivery of PND-1186 FAK inhibitor decreases tumor growth and spontaneous breast to lung metastasis in pre-clinical models. Cancer Biol Ther. 2010;9(10):778–790. doi: 10.4161/cbt.9.10.11433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward KK, Tancioni I, Lawson C, Miller N, Jean C, Chen XL, Uryu S, Kim J, Tarin D, Stupack D, Plaxe SC, Schlaepfer DD. Inhibition of focal adhesion kinase (FAK) activity prevents anchorage-independent ovarian carcinoma cell growth and tumor progression. Clin Exp Metastasis. 2013;30:579–594. doi: 10.1007/s10585-012-9562-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh SY. N-depyridination and N-dedimethylaminoethylation of tripelennamine and pyrilamine in the rat. New metabolic pathways. Drug Metab Dispos. 1990;18(4):453–461. [PubMed] [Google Scholar]
- Yeh SY, Todd GD, Johnson RE, Gorodetzky CW, Lange WR. The pharmacokinetics of pentazocine and tripelennamine. Clin Pharmacol Ther. 1986;39:669–676. doi: 10.1038/clpt.1986.117. [DOI] [PubMed] [Google Scholar]
- Zhao X, Guan JL. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv Drug Deliv Rev. 2011;63(8):610–615. doi: 10.1016/j.addr.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]