

Abstract
Paragangliomas (PGs) of the head and neck region are typically benign, slow-growing neuroendocrine tumours. At times, they may exhibit unusual histological features, such as prominent stromal sclerosis (sclerosing PG), which may raise concerns of malignancy. We describe a case of sclerosing PG of the carotid body, emphasizing the value of immunohistochemical stains for differential diagnosis. A 43-year-old woman presented with a painless lump on the neck. A magnetic resonance imaging scan demonstrated a hypervascular lesion of the carotid body, which was surgically excised. Grossly, the lesion measured 1.8 cm at maximum diameter. On microscopic examination, irregular nests and tiny bundles of neoplastic cells were found between thick bands of fibrous tissue. Focal nuclear cytomegaly and marked pleomorphism were noted. Neoplastic cells proved to be immunoreactive for chromogranin, synaptophysin and neuron specific enolase, but negative for cytokeratins, smooth muscle actin and CD34. Ultrastructurally, numerous mitochondria, rough endoplasmic reticulum structures and endocrine granules were seen in the cytoplasm of the tumour cells. On consideration of the above-mentioned clinico-pathological and ultrastructural findings a diagnosis of sclerosing PG was established. Sclerosing PG is a rare entity which may mimic a malignant neoplasm. The recognition of this unusual morphological variant of PG, together with appropriate immunostains, leads to the correct diagnosis.
Keywords: Paraganglioma, Sclerosing paraganglioma, Head and neck paraganglioma, Carotid body tumour
Introduction
Paragangliomas (PGs) are neuroendocrine tumours derived from extra-adrenal paraganglionic cells of the autonomic nervous system [1, 2]. When a PG arises in the adrenal gland, it is termed phaeochromocytoma. About 85 % of PGs originate in the abdomen, 12 % develop in the chest and only 3 % in the head and neck region [1]. Although mostly solitary tumours, bilateral and multiple PGs can occur, predominantly in hereditary syndromes [3]. PGs are usually asymptomatic or appear as a painless mass [4]. Electron microscopy has revealed the presence of endocrine granules within the PG cell cytoplams, but only 1–3 % of these tumours are associated with hormonal disorders [5]. In these cases, clinical features may resemble those of phaeochromocytomas. In the majority of cases PGs pursue a benign course and surgery is the gold standard curative therapy [6].
Histologically, PGs display the characteristic “zellballen” pattern, i.e. nests of uniform cells separated by fibrovascular stroma surrounded by sustentacular cells [1]. PGs may also exhibit unusual morphological features, including a blend of large and small cells, clear or spindle cell changes, and a diffuse pattern of growth [1, 7]. Tumour cells are immunoreactive for chromogranin, synaptophysin, neuron specific enolase (NSE), serotonin and neurofilament, whilst negative for S-100 protein. The elongated peripheral sustentacular cells express S-100 protein [4]. Up to 30 % of PGs are associated with inherited conditions, including multiple endocrine neoplasm type 2 (MEN2) syndrome, Von Hippel–Lindau (VHL) disease, neurofibromatosis type 1 (NF1) and familial PG-phaeochromocytoma syndromes (PGL1, PGL2, PGL3 and PGL4) [8]. The latter is the most common hereditary condition, where the PGs are brought about by germline mutations of the SDH genes. In recent years, SDHB immunohistochemistry has been proposed as a reliable diagnostic tool to identify SDH-related tumours [9].
The most common site of PG in the head and neck are carotid body tumours, (CBTs) accounting for approximately 60 % of cases. Patients typically present with a painless swelling in the lateral neck, below the corner of the jaw [4]. Nerve deficits or dysphonia may result from tumour enlargement [10]. The treatment of choice for CBT is surgical resection. Vessel and nerve injury can potentially cause severe morbidity and mortality, the risk increasing with tumour size [4]. Patients unfit for surgery may receive radiotherapy, which has been proposed as an effective alternative [11]. Clinical differential diagnosis includes neural tumours and both benign and malignant lymphadenopathies.
We describe a rare case of sclerosing PG of the carotid body, emphasizing the role of immunohistochemical staining to exclude other entities, namely metastatic carcinomas.
Case Report
A 43-year-old HIV/HCV seropositive woman complained of a lump in the jugulodigastric region. Magnetic resonance imaging (MRI) disclosed a hypervascular carotid body mass. Surgical excision was carried out with complete resection of the tumour.
Gross evaluation of the surgical specimen showed an ill-defined, whitish nodule, measuring 1.8 cm in the greatest dimension. Histologically, the most striking feature was the presence of an abundant sclerotic stroma separating irregular nests and tiny bundles of variably sized epithelioid to spindle cells. Conspicuous nuclear pleomorphism and enlargement was a scattered but significant finding (Fig. 1a, b). Neoplastic cells were immunoreactive for chromogranin, synaptophysin, and NSE. However, no staining occurred with cytokeratins, smooth-muscle-actin or CD34 (Fig. 2a–c).
Fig. 1.
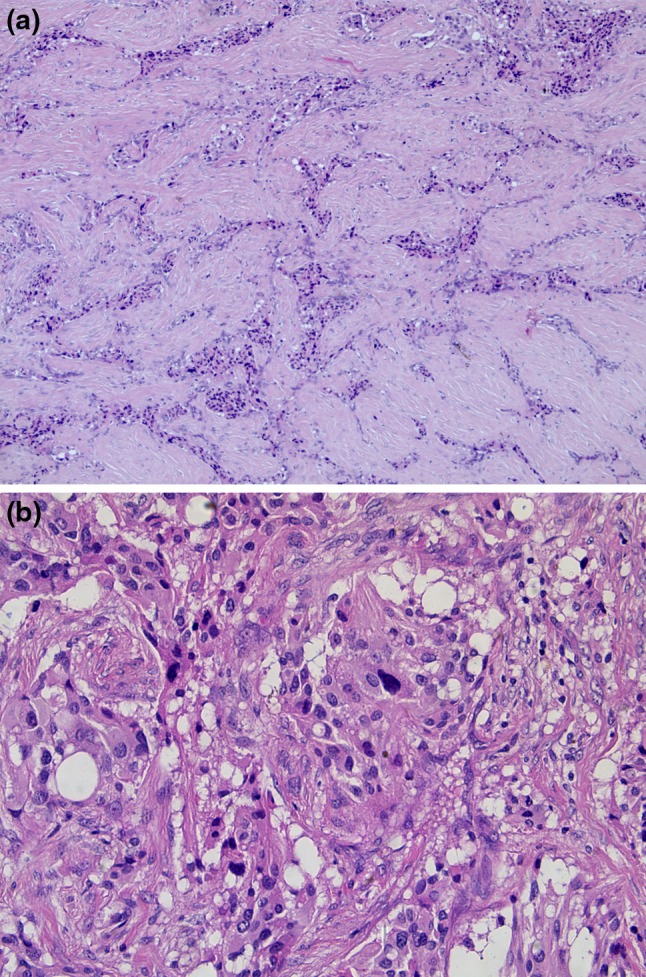
Sclerosing PG showing irregular nests and tiny bundles of epithelioid to spindle cells embedded in a large amount of sclerotic stroma (a) marked nuclear pleomorphism and enlargement was occasionally seen (b)
Fig. 2.
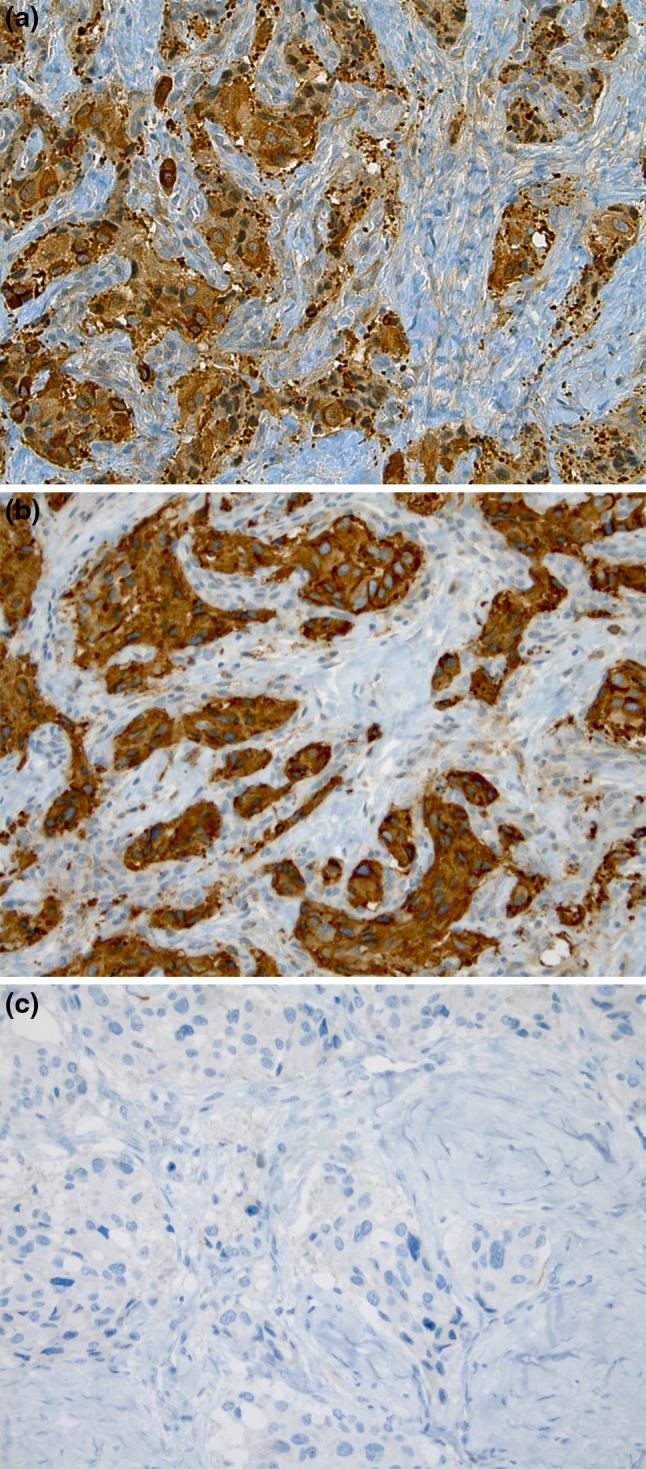
Neoplastic cells were immunoreactive for chromogranin (a) and synaptophysin (b) and negative for cytokeratins (c)
Ultrastructural examination showed that the neoplastic cells were embedded in a large amount of collagen fibre-rich extracellular matrix, either as single elements or in small clusters. Tumour cells were round to oval, with a large cytoplasm containing numerous mitochondria (Fig. 3), small rough endoplasmic reticulum cisternae and compact intracytoplasmic dense core (neuroendocrine) granules measuring 100–150 nm (Fig. 3, inset).
Fig. 3.
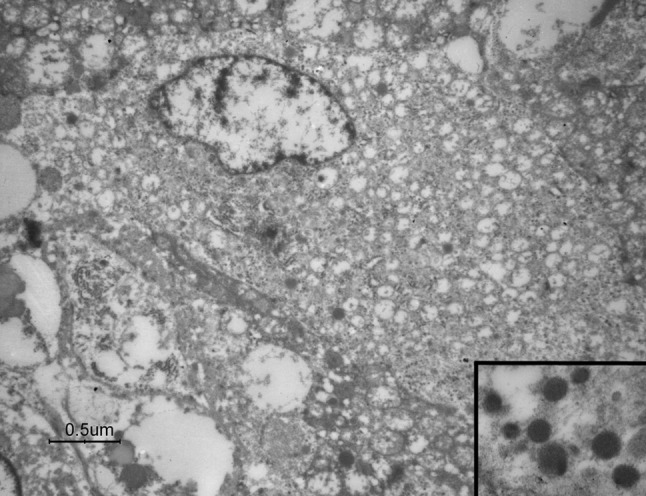
Electron microscopy revealing numerous mitochondria as well as dense core (neuroendocrine) granules (inset) in the cytoplasm of sclerosing PG cells
Based on tumour location and morphological, immunohistochemical and ultrastructural findings, a final diagnosis of sclerosing PG was established. SDHA and SDHB immunohistochemical analysis revealed that the tumour was a non-SDH-related PG (Fig. 4a, b). The patient is well at 12-month follow-up.
Fig. 4.
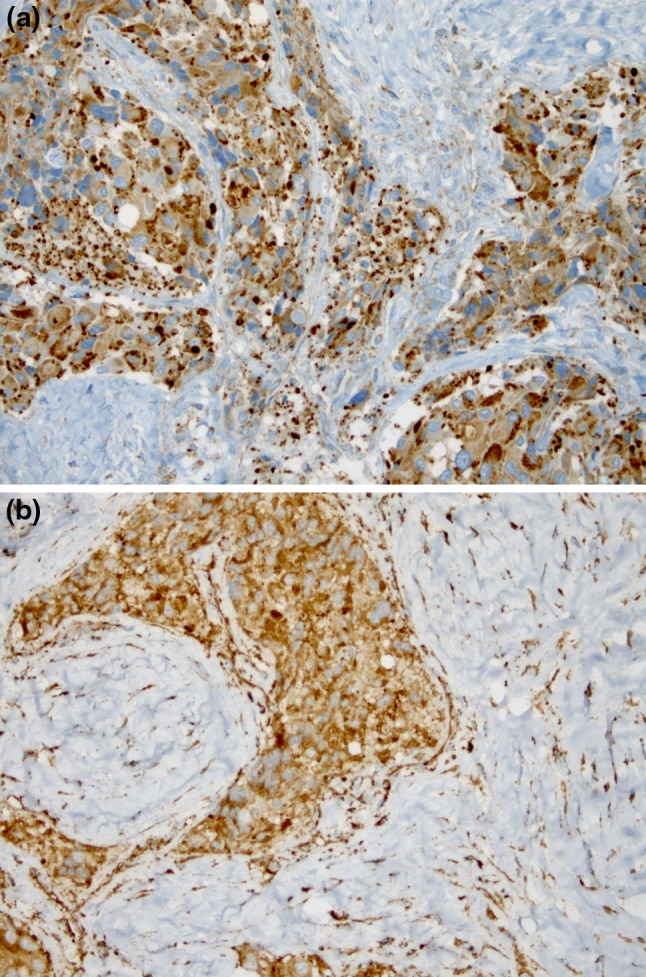
Tumour cells displaying cytoplasmic and distinctly granular reactivity for SDHA (a) and SDHB (b)
Discussion
PGs exhibiting prominent sclerosis are rare entities. The series of 19 sclerosing PGs of the head and neck, published by Plaza and co-workers in 2006, represents the largest series reported to date [12]. Subsequently, an additional case of sclerosing PG in the paratracheal region causing airway-occlusion was described [13]. More recently, Evankovich and co-workers reported a case of sclerosing PG of the thyroid gland [14].
As in the case we describe, the site of the carotid body and onset in middle-aged females seem the typical clinical presentation of sclerosing PG. Indeed, Plaza and co-workers found a remarkable predilection for the carotid body (11/19 cases) in middle-aged women (mean age 50.5 years; 16/19 female patients) [12]. The PG we illustrate developed in an HIV-infected patient with no history of prior malignancy. To the best of our knowledge, only one case of sinonasal PG in an HIV-positive patient has been described, in a 39-year-old female [15]. Although it is widely accepted that HIV infection plays a role in human carcinogenesis as a consequence of the impaired host immune system [16], the literature does not confirm any relationship between PG and HIV, and the connection between the two in the case reported may merely be coincidental [15].
The differential diagnosis of sclerosing PG in the head and neck region can be broad. The stroma embedding the tumour cells, which seem to infiltrate the lesion, together with the presence of cytological atypia, may lead pathologists to consider malignancy. Whilst prominent desmoplastic stroma can also occur in metastatic carcinoma of the lung, breast, larynx and other organs, sclerosing PGs can be distinguished from such tumours by the absence of significant mitotic activity and necrosis. In equivocal cases, immunostains may be employed to reveal diffuse positivity to epithelial markers (e.g. cytokeratins and EMA), in metastatic carcinomas, whereas the majority of PGs are cytokeratin negative [12].
Metastatic neuroendocrine carcinomas in the head and neck region are uncommon, but histologically they can resemble sclerosing PGs [12]. They both share the typical nesting pattern with a trabecular or ribbon-like arrangement. However, PGs generally display a more prominent “zellballen” architecture than carcinomas. Furthermore, although immunoreactive for neuroendocrine markers, the majority of neuroendocrine carcinomas stain with low-molecular weight cytokeratins [17].
Differential diagnosis of sclerosing PG also includes non-epithelial neoplasms. Solitary fibrous tumours may occur in the head and neck region. These tumours consist of a proliferation of spindle cells with various growth patterns: storiform, herringbone, neural with wavy nuclei, and hemangiopericytic with areas of sclerosis [18]. Positive staining for CD34 and bcl-2 can be demonstrated in solitary fibrous tumour cells [12].
Relatively few ultrastructural studies have been conducted on the human carotid body and other head and neck paraganglia [1]. In the present case electron microscopy, besides confirming the neuroendocrine nature of the tumour, showed that sclerosing PG may reveal oncocytic changes. In a previous case report, Robertson and co-workers described a malignant carotid body PG in which so many cells exhibited an abundance of mitochondria that they resembled oncocytes [19].
In our case, both SDHA and SDHB were found to be positive, thus excluding SDH gene mutations. In a familial setting, SDHB-associated PGs are known to often behave in a malignant fashion [20]. Recently, the absence of SDHB expression has been demonstrated to be an excellent indicator of an SDH germline mutation in PGs [9].
In summary, although rare, a sclerosing variant of PG can be a diagnostic pitfall and potentially lead to over-diagnose malignancy. Awareness of this unusual growth pattern of PG and appropriate immunostains allow the pathologist to reach the correct diagnosis.
References
- 1.Lack EE. Atlas of tumor pathology, 4rd Series, fascicle 8. Washington DC: Armed Forces Institute of Pathology; 2007. [Google Scholar]
- 2.DeLellis RA, Lloyd RV, Heitz PU, et al., editors. World Health Organization classification of tumours. Pathology and genetics of tumours of endocrine organs. Lyon: IARC Press; 2004. [Google Scholar]
- 3.Bikhazi PH, Roeder E, Attaie A, Lalwani AK. Familial paragangliomas: the emerging impact of molecular genetics on evaluation and management. Am J Otol. 1999;20:639–643. [PubMed] [Google Scholar]
- 4.Pellitteri PK, Rinaldo A, Myssiorek D, et al. Paragangliomas of the head and neck. Oral Oncol. 2004;40:563–575. doi: 10.1016/j.oraloncology.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 5.Manolidis S, Shohet JA, Jackson CG, Glasscock ME., 3rd Malignant glomus tumors. Laryngoscope. 1999;109:30–34. doi: 10.1097/00005537-199901000-00007. [DOI] [PubMed] [Google Scholar]
- 6.Lim JY, Kim J, Kim SH, et al. Surgical treatment of carotid body paragangliomas: outcomes and complications according to the shamblin classification. Clin Exp Otorhinolaryngol. 2010;3:91–95. doi: 10.3342/ceo.2010.3.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tischler AS. Pheochromocytoma and extra-adrenal paraganglioma: updates. Arch Pathol Lab Med. 2008;132:1272–1284. doi: 10.5858/2008-132-1272-PAEPU. [DOI] [PubMed] [Google Scholar]
- 8.Gill AJ, Benn DE, Chou A, et al. Immunohistochemistry for SDHB triages genetic testing of SDHB, SDHC, and SDHD in paraganglioma-pheochromocytoma syndromes. Hum Pathol. 2010;41:805–814. doi: 10.1016/j.humpath.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 9.van Nederveen FH, Gaal J, Favier J, et al. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis. Lancet Oncol. 2009;10:764–771. doi: 10.1016/S1470-2045(09)70164-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fruhmann J, Geigl JB, Konstantiniuk P, Cohnert TU. Paraganglioma of the carotid body: treatment strategy and SDH-gene mutations. Eur J Vasc Endovasc Surg. 2013;45:431–436. doi: 10.1016/j.ejvs.2013.01.018. [DOI] [PubMed] [Google Scholar]
- 11.Hinerman RW, Mendenhall WM, Amdur RJ, et al. Definitive radiotherapy in the management of chemodectomas arising in the temporal bone, carotid body, and glomus vagale. Head Neck. 2001;23:363–371. doi: 10.1002/hed.1045. [DOI] [PubMed] [Google Scholar]
- 12.Plaza JA, Wakely PE, Jr, Moran C, et al. Report of 19 cases of an unusual variant of neuroendocrine tumor that may be mistaken for an aggressive malignant neoplasm. Am J Surg Pathol. 2006;30:7–12. doi: 10.1097/01.pas.0000174012.37439.c7. [DOI] [PubMed] [Google Scholar]
- 13.Mukhopadhyay S, Naqvi AH, Su W, Landas SK. Airway-occlusive sclerosing paraganglioma presenting with dyspnea. Am J Surg Pathol. 2006;30:1206–1207. doi: 10.1097/01.pas.0000209844.67293.88. [DOI] [PubMed] [Google Scholar]
- 14.Evankovich J, Dedhia RC, Bastaki JM, et al. Primary sclerosing paraganglioma of the thyroid gland: a case report. Ann Otol Rhinol Laryngol. 2012;121:510–515. doi: 10.1177/000348941212100803. [DOI] [PubMed] [Google Scholar]
- 15.Fasunla AJ, Ibekwe TS, Afolabi OA, et al. Sinonasal paraganglioma: a case report. Oral Maxillofac Surg. 2008;12:93–96. doi: 10.1007/s10006-008-0104-x. [DOI] [PubMed] [Google Scholar]
- 16.Chen CJ, Hsu WL, Yang HI, et al. Epidemiology of virus infection and human cancer. Recent Results Cancer Res. 2014;193:11–32. doi: 10.1007/978-3-642-38965-8_2. [DOI] [PubMed] [Google Scholar]
- 17.Milroy CM, Rode J, Moss E. Laryngeal paragangliomas and neuroendocrine carcinomas. Histopathology. 1991;18:201–209. doi: 10.1111/j.1365-2559.1991.tb00827.x. [DOI] [PubMed] [Google Scholar]
- 18.Fletcher CDM, Bridge JA, Hogendoorn PCW, Martens F, editors. World Health Organization classification of tumours of soft tissue and bone. Lyon: IARC Press; 2013. [Google Scholar]
- 19.Robertson DI, Cooney TP. Malignant carotid body paraganglioma: light and electron microscopic study of the tumor and its metastases. Cancer. 1980;46:2623–2633. doi: 10.1002/1097-0142(19801215)46:12<2623::AID-CNCR2820461215>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 20.Timmers HJ, Kozupa A, Eisenhofer G, et al. Clinical presentations, biochemical phenotypes, and genotype-phenotype correlations in patients with succinate dehydrogenase subunit B-associated pheochromocytomas and paragangliomas. J Clin Endocrinol Metab. 2007;92:779–786. doi: 10.1210/jc.2006-2315. [DOI] [PubMed] [Google Scholar]