

Abstract
The role of molecular methods in the diagnosis of head and neck cancer is rapidly evolving and holds great potential for improving outcomes for all patients who suffer from this diverse group of malignancies . However, there is considerable debate as to the best clinical approaches, particularly for Next Generation Sequencing (NGS). The choices of NGS methods such as whole exome, whole genome, whole transcriptomes (RNA-Seq) or multiple gene resequencing panels, each have strengths and weakness based on data quality, the size of the data, the turnaround time for data analysis, and clinical actionability. There have also been a variety of gene expression signatures established from microarray studies that correlate with relapse and response to treatment, but none of these methods have been implemented as standard of care for oropharyngeal squamous cell carcinoma (OPSCC). Because many genomic methodologies are still far from the capabilities of most clinical laboratories, we chose to explore the use of a combination of off the shelf targeted mutation analysis and gene expression analysis methods to complement standard anatomical pathology methods. Specifically, we have used the Ion Torrent AmpliSeq cancer panel in combination with the NanoString nCounter Human Cancer Reference Kit on 8 formalin-fixed paraffin embedded (FFPE) OPSCC tumor specimens, (4) HPV-positive and (4) HPV-negative. Differential expression analysis between HPV-positive and negative groups showed that expression of several genes was highly likely to correlate with HPV status. For example, WNT1, PDGFA and OGG1 were all over-expressed in the positive group. Our results show the utility of these methods with routine FFPE clinical specimens to identify potential therapeutic targets which could be readily applied in a clinical trial setting for clinical laboratories lacking the instrumentation or bioinformatics infrastructure to support comprehensive genomics workflows. To the best of our knowledge, these preliminary experiments are among the earliest to combine both mutational and gene expression profiles using Ion Torrent and NanoString technologies. This reports serves as a proof of principle methodology in OPSCC.
Electronic supplementary material
The online version of this article (doi:10.1007/s12105-014-0566-0) contains supplementary material, which is available to authorized users.
Keywords: Carcinoma of the oropharynx, Cancer mutation profiling, HPV related oropharynx cancer, Head and neck cancer molecular profiling, Next generation sequensing in head and neck cancer, RNA seq in head and neck cancer
Introduction
Over the last decade there has been a noted increased incidence of oropharyngeal carcinoma OPSCC, a subtype of squamous cell carcinoma of the head and neck (SCCHN). Part of the reasons behind this phenomenon is the increased incidence of high risk human papillomavirus (HPV) related OPSCC [1–3]. Interestingly, multiple retrospective studies have shown that patients with HPV-positive OPSCC have a better prognosis compared to patients with HPV-negative tumors [4, 5]. OPSCC accounts for the majority of patients enrolled in therapeutic trials for SCCHN and a growing subgroup of these patients have HPV-related carcinoma. This subgroup is defined by the presence of high-risk types of HPV in tumor cells, predominantly HPV type 16 (HPV-16). The HPV-negative OPSCCs on the other hand are most closely linked to smoking and are reported to have a significantly worse prognosis compared to HPV-positive OPSCC [7].
Expression of viral E6 and E7 encoded by the HPV genome respectively inactivate the tumor-suppressors TP53 and RB, and are necessary for malignant behavior of HPV-positive tumors [6].
Epidemiologic studies suggest little interaction between the two sets of risk factors, suggesting that HPV-positive cancer and HPV-negative cancer may each have a distinct pathogenesis. However, recent data suggest that smoking has an adverse effect on prognosis in both diseases [7].
It is well known that for patients with OPSCC, the overall survival is substantially better among patients with HPV-positive cancer compared to HPV-negative disease [5, 7–10]. In one highly cited clinical trial [7], high risk HPV infection status was demonstrated as an independent prognostic variable. Patients with HPV-positive OPSCC had a 3-year overall survival rate of 82.4 %, compared to 57.1 % among patients with HPV-negative tumors (P < 0.001 by the log-rank test) [7]. After adjustment for age, race, tumor and nodal stage and tobacco exposure, there was a 58 % reduction in the risk of death. It is of note however, that the risk of death significantly increased for the HPV positive patients with each additional pack-year of tobacco smoking, and that tobacco use was an independent prognostic factor for survival. These findings indicate that both HPV status and tobacco use exert significant effects on the outcome of OPSCC. Our goal when performing mutation and transcriptional profiling is to define mutations in patients with HPV-positive or negative OPSCC that will allow development of effective therapeutic interventions.
The type of data generated by performing these mutational and transcriptional profiles may also have implications on better diagnosing or confirming the HPV status in these malignancies. Recent reports have shown that HPV testing by PCR alone is not sufficient for diagnosis of OPSCC [11, 12]. Numerous gene expression studies have been previously used to categorize HPV-positive versus negative OPSCC, and despite numerous reports of the potential clinical benefit of such testing, none of these signatures have been adopted as a routine diagnostic for OPSCC [13].
Recent evidence suggests that targeted next-generation sequencing can offer very useful information that could influence the care of patients with cancer [14]. Molecular alterations are starting to directly affect the approach to patient care and are currently the subject of investigation in numerous clinical trials [15, 16]. We wanted to explore the potential efficacy of implementing higher throughput strategies that combine targeted differential gene expression mutation profiling to help improve stratification of SCCHN by risk categories for both HPV-positive and negative disease.
Materials and Methods
p16 ink4a Immunohistochemical Staining
p16 ink4a (p16) immunohistochemical staining was performed in a standard manner per supplier’s instructions (CINtec 9517, MTM Laboratories, Westborough, MA) on formalin-fixed paraffin embedded (FFPE) tissue sections using the automated, open system immunostainer (Dako Autostainer Link 48). The slides were processed using the 3,3′-diaminobenzidine reagent to visualize the antibody–antigen complex, then counterstained with hematoxylin and subsequently washed and cover-slipped. Both positive and negative control slides were prepared. The proportion of tumor cells demonstrating nuclear and cytoplasmic p16 staining were categorized dichotomously as either p16 positive (>70 % tumor cells exhibiting strong and diffuse nuclear and cytoplasmic staining) or p16 negative (<70 % tumor cells exhibiting strong and diffuse nuclear and cytoplasmic staining) [17].
HPV-16/18 In Situ Hybridization
In situ hybridization (ISH) was performed on five micron sections of FFPE tissue sections and demonstrated using a biotin-labeled HPV-16/18 DNA probe (ENZO Life Sciences, Farmingdale, NY) and GenPoint Tyramide Signal Amplification System and developing the final signal using DAB, and counterstaining with hematoxylin, all in accordance with supplier’s instructions. All steps were performed on the Leica Bond III automated system. Any punctate, dot-like intranuclear staining was interpreted as evidence of DNA integration. A negative control and a positive DNA control were run with each case.
Tissue Acquisition and Processing
A total of 8 FFPE OPSCC tumor specimens, (4) HPV-positive and (4) HPV-negative, were available for this proof of concept study. As part of the routine pathology diagnosis of OPSCC the tumors were evaluated for p16 immunoreactivity and if positive then assessed for high risk HPV by in situ hybridization using the protocols outlined above. The FFPE tumor blocks were de-identified according to IRB approved protocol. 5 μM serial sections were obtained from each block for DNA and RNA isolation.
DNA and RNA Extraction and QC
Genomic DNA and total RNA were isolated from (2) 5 μM sections for each tumor specimen using Omega BioTek chemistries according to the manufacturer’s protocol. DNA was quantitated using NanoDrop and Qubit, and RNA was quantitated using NanoDrop and Agilent BioAnalyzer.
Ion Torrent AmpliSeq Cancer Panel
The Ion Torrent AmpliSeq Cancer Panel (v1) was used to assess mutation frequency and to identify potentially actionable mutations for this study. The AmpliSeq Cancer Panel queries 739 known mutations in 46 well characterized cancer genes listed in the COSMIC database [18]. 200 ng of genomic DNA from each specimen was used to generate uniquely barcoded sequencing libraries. Briefly, libraries were prepared using the Ion DNA Barcoding and Ion Xpress Template kits (Life Technologies, Grand Island, NY, USA) according to the manufacturer’s protocols. Fragment sizes were assessed on an Agilent Bioanalyzer 2100 using the High Sensitivity kit (Agilent, Santa Clara, CA, USA). All eight uniquely barcoded specimen libraries were run on a single 318 chip and sequenced using an Ion PGM 200 sequencing kit.
On-board Torrent Suite v2.0 and Variant Caller v2.0 software were used for data analysis. Barcoded fastq files were aligned to the human genome reference (hg19) followed by coverage analysis and read quality filtering as described elsewhere [19]. Variant calls were performed using a minimum read depth of 500 and variant frequency above 5 %.
NanoString Gene Expression Cancer Panel
Total RNA from two 5 μM sections from each patient sample were isolated and quality assessed as described above. NanoString nCounter data generation was performed by the Oncogenomics Core Facility at the University of Miami. Briefly, two 50 ng aliquots of total RNA from each FFPE OPSCC specimen were run in technical duplicate using the 235-plex Human Cancer Reference kit. Hybridization, sample processing and data acquisition were all performed using vendor protocols. Raw data was delivered as RCC files that were normalized with NanoString controls using nSolver analysis software (v1.1). Data was outputted as text file for PartekExpress software for data analysis. Partek’s pattern discovery tool was used to perform principal component analysis and hierarchical clustering (Fig. 4). One sample (HPV-3) of the eight was removed for further study because six reference control genes were not expressed in both technical replicates. Pearson correlations was calculated for all pairs, and followed by one way ANOVA with no batch affect correction. An unadjusted P value cut-off of 0.05 with fold change cut-off of ≥10 was used to generate a HPV-negative gene list with subpopulation clustering. Due to the highly quantitative and independent nature of the data, false positives were not a concern, and therefore no multiple test correction was applied.
Fig. 4.

Unsupervised clustering of a 230 gene expression panel distinguishes HPV-positive from HPV-negative OPSCC. Total RNA from formalin-fixed paraffin embedded tumor specimens were run in duplicate using the NanoString Human Cancer Reference kit. One HPV-negative (HPV−_3) sample was removed from the clustering due to poor performance of housekeeping genes. Clustering clearly shows the reproducibility of technical replicates as well individual expression pattern of each tumor. Distinct subsets of differentially expressed genes are labeled C1-6 on the y-axis. A full list of these genes and normalized are available in the Supplemental Table (S1)
cBioPortal
Bioinformatics queries of mutational frequencies and differential gene expression were conducted using Head and Neck Squamous Cell Carcinoma (TCGA, in revision July 2014) dataset. This dataset contained sequencing, copy number and RNA-Seq data from 279 Head and Neck Squamous Cell Carcinoma (HNSCC) tumors with known HPV status and clinical outcomes. The cBioPortal can be accessed via: http://www.cbioportal.org/.
Results
We selected 4 HPV-positive and 4 HPV-negative OPSCC specimens in which to assess a combined targeted mutation detection and differential gene expression approach to detect clinically-relevant changes from standard FFPE clinical tumor specimens. These tumors had previously undergone routine pathological diagnosis involving H&E staining, immunohistochemistry for p16 and in situ hybridization for high-risk status (Fig. 1).
Fig. 1.
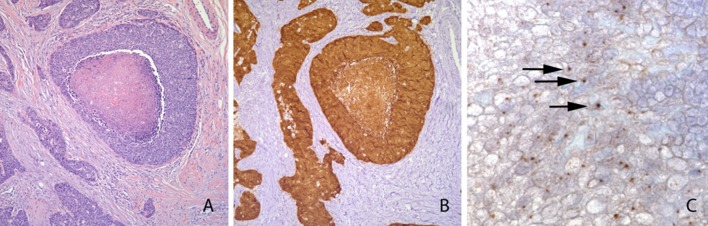
Representative tissue section of an OPSCC specimen used for this analysis. a H&E stain of HPV-positive tumor. b IHC staining using and anti-p16 antibody. c HPV by ISH in an OPSCC
Mutation Profiling
Using The Ion Torrent AmpliSeq Cancer Panel (v1) kit, we queried 739 genomic loci within 46 genes known to harbor hotspot mutations across various cancer types. As shown in Fig. 2, the mutation burden of HPV-negative OPSCC tumor specimens, is much higher than HPV-positive specimens as evidenced by the number of single nucleotide variants (SNVs). Although C to A transversions are considered the hallmark of smoking related cancers [20], 3 out of the 4 HPV-positive tumors show a strong bias of C to T transitions (Fig. 2b). A portion of these variants may be due to sequencing artifacts of FFPE tumor specimens [21], but it is unlikely that a high percentage of these variants would be attributed to formalin fixation exclusively in the HPV-negative tumors.
Fig. 2.

Mutation burden of HPV-positive and negative OPSCC tumor specimens. a Prevalence of SNVs in HPV-positive (blue bars) and negative (red bars) versus total (green bars) number of SNVs queried. b Mutation spectra showing a high prevalence of C to T transitions (purple bars) in the HPV-negative specimens. c Gene specific SNVs in HPV-negative OPSCC. d Gene specific SNVs in HPV-positive OPSCC. All SNVs had a variant frequency greater than 5 % and depth of coverage greater than 500 reads
All SNVs were assessed for likelihood of clinical relevance based on published literature and public databases including dbSNP, 1000 genome, TCGA and COSMIC [22–24]. There were a combined 170 variants (162 HPV−, 7 HPV+) called using the initial criteria of 500 × coverage and 5 % or greater variant frequency. These calls were refined to a list of high confidence variants with greater than 1,000 × coverage and 10 % variant frequency that had published evidence for being a somatic mutation. As shown in Table 1, 4 out of 7 SNVs called by the lower confidence threshold in the HPV-positive samples met the higher confidence threshold, whereas less than 3 % of the HPV-negative tumors met the higher confidence threshold. These mutations were then queried in a provisional TCGA HNSCC dataset with sequencing information from 273 tumors using cBioPortal [25, 26].
Table 1.
Clinically informative mutations in HPV-positive and negative disease
| Sample | Position | Mutation | Ref | Var | (%) Var | # Reads | COSMIC ID | TCGA_HNSCC dataa | Provisional |
|---|---|---|---|---|---|---|---|---|---|
| HPV−_1 | chr3:178936091 | PIK3CA c.1633G>A, p.E545K | G | A | 18.99 | 4,255 | COSM763 | (21) specimens | |
| HPV−_1 | chr7:116411990 | MET c.3029C>T, p.T1010I | C | T | 43.33 | 4,133 | COSM707 | (0) likely SNP | |
| HPV−_3 | chr17:7,577,106 | TP53 c.832C>A, p.P278T | G | T | 40.66 | 2,688 | COSM368635 | (0), but (3) p.T278S | |
| HPV+_1 | chr4:1803568 | FGFR3 c.746C>G, p.S249C | C | G | 17.98 | 1,424 | COSM715 | (1) TCGA-CR-6481 (OPSCC, HPV+) | |
| HPV+_3 | chr4:1803564 | FGFR3 c.742C>T, p.R248C | C | T | 16.98 | 2,091 | COSM714 | (0) specimens | |
| HPV+_3 | chr13:49027168 | RB1 c.1735C>T, p.R579a | C | T | 18.78 | 1,347 | COSM892 | (0), but (8) inactivating mutations | |
| HPV+_4 | chr3:178916876 | PIK3CA c.263G>A, p.R88Q | G | A | 44.92 | 2,560 | COSM746 | (1) TCGA-CR-6471 | |
Using the AmpliSeq Cancer Panel (v1), we identified clinically informative mutations in 50 % (2/4) of the HPV-negative tumors and (3/4) 75 % of the HPV-positive tumors. Specific mutations were compared to the HNSCC TCGA dataset queried using cBioPortal. The numbers of specific mutations which were also found in the TCGA dataset are shown in brackets, and the TCGA case number is shown for tumors where only one tumor shared the same mutation
aProvisional TCGA HNSCC dataset (516 tumors) viewed using cBioPortal July 2014
Potentially clinically informative mutations were identified in five genes (PIK3CA, TP53, FGFR3, RB1 and MET). These genes account for mutations in 84 % (234 tumors) of the total 279 samples in the TCGA HNSCC dataset, with PIK3CA and TP53 accounting for mutations in 21 and 73 % of the tumors, respectively (data not shown). Mutations in the PIK3CA gene were identified in one HPV-negative (HPV-1) and one HPV-positive (HPV+_4) sample. The p.E545K mutation identified in HPV−_1 is one of the most common mutations in HNSCC and together with the p.E542K mutation accounted for 92 % (12/13) of HPV-positive (n = 36) and 44 % (20/45) of the HPV-negative (n = 270) PIK3CA mutations in TCGA HNSCC tumors. Interestingly, the p.R88Q mutation identified in one of our HPV-positive samples (HPV+_4) was found in one tumor in the TCGA dataset, a HPV-positive sample (ID# TCGA-CR-6471) that also had a p.M1043V mutation. The p.R88Q (COSM746) and p.M1043V (COSM12591) have been reported in other cancers [27, 28], but to the best of our knowledge, not as co-occurring in a HNSCC tumor. There was no evidence of the p.M1043V mutation in our HPV+_4 specimen, and therefore the p.R88Q may be recurrent in a minor population of HPV-positive tumors.
Our dataset also appeared to be enriched for FGFR3 mutations in the HPV-positive samples which may also be true for the TCGA dataset (11 % HPV-positive vs. 1 % HPV-negative). The p.S249C (rs121913483, COSM715) variant, which was also observed in an HPV-positive OPSCC tumor (TCGA-CR-6481) in the TCGA dataset, and the p.R248C (rs121913482, COSM714) variant occur in the C-terminus of the FGFR3 protein, outside of the protein tyrosine kinase domain, making their significance uncertain. Likewise a missense change, p.T1010I (rs56391007, COSM707), in the MET gene was identified in one of our HPV-negative tumors. Although this change has been reported as a somatic mutation previously in other cancers [29, 30], it has not been reported in the TCGA HNSCC dataset and is suspected to be a constitutional polymorphism in the individual from which this tumor was derived based on the variant frequency being within the range of 40–60 %. We were unable to acquire additional material (i.e. adjacent normal or blood) from our de-identified cohort to confirm constitutional changes.
Amplicons covering portions of exons 1,4,7,9,13 and 20 of the PIK3CA gene with recurrent mutations in various cancer types are shown in Fig. 3. Only exon 7 performed very poorly (<200 × coverage) across all samples, and potential mutations (e.g. p.C420R, COSM267862) in this region would not have been found using this assay. The PIK3CA p.M1043V mutation occurs in exon 20 and had a minimal depth of coverage of 1,000× across all samples. As for TP53, exons 2–8 and 10 were covered at a minimum depth of 1,000× for all eight samples. At least 11 mutations were identified in exon 9 of TP53 in the TCGA HNSCC dataset, and absence of this exon in this assay may account for one TP53 mutation being identified in our 4 HPV-negative samples.
Fig. 3.

Coverage of the PIK3CA gene sequencing using the AmpliSeqCancer Panel (v1) kit for 8 FFPE OPSCC tumor specimens. a Aligned (hg19) reads of 4 HPV(+) and 4 HPV(−) tumor specimens are shown in relation to the PIK3CA locus and AmpliSeq Cancer (versions 1 and 2) and TruSight tumor library designs (below reads). b Coverage of exon 9 of the PIK3CA gene using the AmpliSeq (v1) kit shows the c.1633G>A (p.E545K) mutation in the HPV−_1 sample
Gene Expression
We obtained total RNA and genomic DNA from serial sections from our 4 HPV-positive and 4 HPV-negative FFPE tumor samples. The genomic DNA was used for mutation profiling described above and the total RNA was analyzed for differential gene expression using the NanoString Cancer Reference kit. NanoString gene expression assays have been reported to be reproducible and informative, particularly in FFPE tumor samples with compromised nucleic acid quality [31, 32]. The Cancer Reference kit was chosen for this study based on its broad range of 230 cancer related genes and cancer relevant processes (i.e. cell cycle, proliferation, apoptosis and immune response), as well as being a low cost alternative to RNA-Seq or gene expression arrays.
Normalized expression values for 230 cancer-relevant genes and five housekeeping genes on the cancer panel were established for our eight samples run in technical duplicate. Unsupervised hierarchical clustering (Fig. 4) and Pierson correlation (Table 2) showed that our replicates for each sample were highly similar and that the HPV-negative tumors were generally more similar to one another than they were to HPV-positive tumors. Clustering also allowed us to stratify samples by gene as well as by HPV status and six groups (C1-6) of differentially expressed genes were identified using this dataset. Clearly HPV+_1 appears to be an outlier compared to the other 3 HPV-positive tumors, and low levels of gene expression in the C3–C5 groups may account for this tumor clustering more closely to the HPV-negative tumors (Fig. 4). It should also be noted that technical replicates of gene expression clearly show that each tumor has a distinct and reproducible pattern of gene expression, which underlies the biology of that tumor and has the potential to be exploited for treatment. A full table of these data is provided in Table 1.
Table 2.
Pierson correlation of FFPE gene expression data demonstrating robust reproducibility of technical replicates
Concordance between gene expression of 230 cancer related genes was greater than 98 % for all technical replicates. One HPV-negative (HPV−_3) sample was removed from the clustering analysis due to poor performance of housekeeping genes
Differential expression analysis between HPV-positive and negative groups showed that expression of several genes was highly likely to correlate with HPV status (Table 3). With this in mind, we chose to see if we could use these genes to identify HPV-positive and HPV-negative subtypes. Our NanoString gene list was sorted by P value and then queried against the TCGA HNSCC RNA-Seq data set using cBioportal [25, 26]. Using the RNA-Seq data from the full HNSCC dataset (n = 279) we were able to filter the list of HPV classifier genes from 30 down to 11. Excluded from the list of 30 genes were: BCL2A1, CSF3, CSF3R, IL1A, NRAS, PLG, PTGS2, TFE3 and TNF, which were not expressed in the total 36 HPV-positive tumors in the 279 sample TGCA dataset; and WNT1, IL4, E2F3, GATA1, RB1, BLM, IRF1, CEBPA, FGR and CSF1R, which appeared to have very similar frequencies of mRNA over expression between the HPV-positive and HPV-negative tumors.
Table 3.
Differentially expressed genes predicting for HPV-positive versus HPV-negative samples
| Gene | This study NanoString P value | TCGA HPV+ (n = 36) % Diff expa | TCGA HPV− (n = 243) % diff expa |
|---|---|---|---|
| OGG1 | 3.89E-05 | 11.1 | 1.4 |
| CSF3R | 0.0002 | 0 | 3.2 |
| WNT10B | 0.0006 | 19.4 | 5.0 |
| WNT1 | 0.0009 | 2.8 | 0.7 |
| PDGFA | 0.0020 | 2.8 | 9.0 |
| IL4 | 0.0035 | 5.6 | 4.3 |
| E2F3 | 0.0057 | 11.1 | 9.3 |
| SIAH1 | 0.0072 | 2.8 | 7.2 |
| GATA1 | 0.0086 | 5.6 | 4.7 |
| RB1 | 0.0091 | 13.9 | 8.6 |
| PTGS2 | 0.0097 | 0 | 2.5 |
| BLM | 0.0112 | 11.1 | 6.5 |
| CDKN2A | 0.0113 | 5.6 | 1.1 |
| IRF1 | 0.0147 | 5.6 | 3.2 |
| TFE3 | 0.0155 | 0 | 6.8 |
| BCL2A1 | 0.0221 | 0 | 1.8 |
| TNF | 0.0223 | 0 | 1.8 |
| REL | 0.0225 | 13.9 | 6.1 |
| CEBPA | 0.0241 | 8.3 | 5.7 |
| FGR | 0.0270 | 5.6 | 4.7 |
| SYK | 0.0275 | 33.3 | 6.5 |
| IL1A | 0.0278 | 0 | 5.7 |
| PLG | 0.0357 | 0 | 2.9 |
| CDKN2C | 0.0360 | 47.2 | 6.5 |
| NRAS | 0.0366 | 0 | 3.6 |
| MAP3K8 | 0.0409 | 16.7 | 5.0 |
| CSF3 | 0.0418 | 0 | 1.8 |
| CSF1R | 0.0443 | 2.8 | 1.8 |
| MST1R | 0.0469 | 11.1 | 3.9 |
| XPC | 0.0493 | 8.3 | 3.2 |
One way ANOVA with no batch affect correction was performed on the NanoString gene expression dataset for the eight samples used in this study as described in the Methods section. An unadjusted P value cut-off of 0.05 with fold change cut-off of ≥10 was used to generate this HPV classifier gene list. Frequency of differential expression by RNA-Seq in the TCGA HNSCC dataset (in revision July 2014) was assessed as separate HPV-positive and HPV-negative cohorts using cBioPortal. This gene list was parsed based on NanoString P values and frequency in HPV-positive and HPV-negative TCGA cohorts to arrive at the 11 gene classifier
aHNSCC RNA-Seq (TCGA, in revision, July 2014)
Combined Gene Expression with Mutation Profiling
The 11 genes chosen (CDKN2C, SYK, WNT10B, CEBPA, MAP3K8, FGR, GATA1, IL4, CDKN2A, E2F211 and PDGFA) were then queried against the RNA-Seq data from the 33 sample oropharyngeal (OPSCC) subset of the full TCGA HNSCC dataset (n = 279). Using a HPV classifier that required over expression of one or more of ten genes (CDKN2C, SYK, WNT10B, CEBPA, MAP3K8, FGR, GATA1, IL4, CDKN2A and E2F211) for HPV-positive status and over expression of PDGFA or no evidence of over expression of the 10 HPV-positive genes, we correctly identified HPV status in 97 % (32/33) of the OPSCC samples. One HPV-positive specimen, TCGA-CR-6472, was misclassified as HPV-negative did not appear to over express any of the 11 genes in the classifier by RNA-Seq (Fig. 5).
Fig. 5.

Eleven gene HPV classifier correctly identifies 97 % (32/33) OPSCC samples in the TCGA HNSCC dataset. OncoPrint output of the TCGA OPSCC subset using our 11 gene expression HPV classifier and clinically relevant mutated gene list (PIK3CA, TP53, FGFR3, RB1 and MET). Over expressed genes used to predict HPV status in our training set shown in Table 3 were parsed to 11 genes and applied to the 33 sample OPSCC subset of the full 279 specimen TCGA HNSCC dataset using cBioPortal. The 11 gene classifier required overexpression of one or more of ten genes (CDKN2C, SYK, WNT10B, CEBPA, MAP3K8, FGR, GATA1, IL4, CDKN2A and E2F2) for HPV-positive status and PDGFA overexpression or absence of overexpression of the 10 HPV-positive genes for HPV-negative status. Only one specimen, TCGA-CR-6472, which was misclassified as HPV-negative, did not over express any of the 11 genes in the classifier by RNA-Seq. Homozygous deletion of CDKN2A and mutations of TP53 are present, as expected, exclusively in the HPV-negative specimens. Mutations, amplifications and over expression of PIK3CA were observed in both HPV-positive and HPV-negative OPSCC tumors
Combining our HPV classifier gene expression list with our clinically relevant mutated gene list (PIK3CA, TP53, FGFR3, RB1 and MET) we then queried the OPSCC TCGA dataset for the full spectrum of mutations, copy number and differential gene expression by RNA-Seq. As expected, homozygous deletion of CDKN2A and mutations of TP53 are present exclusively in the HPV-negative tumors. Mutations, amplifications and over expression of PIK3CA were observed in both HPV-positive and HPV-negative OPSCC tumors and may be more frequent in this histological subtype than for HNSCC tumors grouped by all histological sites. MET over expression was seen in 2 HPV-negative specimens and mutually exclusive of PDGFA over expression or amplification. Likewise CDKN2C and SYK gene expression appeared to be informative in stratifying major subtypes of HPV-positive OPSCC tumors.
Discussion
As evidenced by lung adenocarcinoma, genomics technologies have the potential to inform treatment decision making and improve patient outcomes [33, 34]. However, these technologies have had little impact on standard methods for diagnosis and treatment of HNSCC and many other types of cancer with one or more known mutational or copy number drivers. Despite extensive literature of differential gene expression profiles, mutations and copy number abnormalities in head and neck tumors that could potentially impact future clinical applications, little other than HPV status is done routinely upon diagnosis [7, 10, 11, 13, 15, 24, 32, 35–47]. A main factor contributing to this lack of progress is that no clear directives or actionable guidelines have been adopted for molecularly profiling HNSCCs.
With several highly cited genomic studies in head and neck tumors and a soon to be released TCGA study, it is clear that there is an opportunity to identify mutation, copy number and differential gene expression in these tumors to better inform treatment and prognosis than by HPV status alone [27–29, 48–51]. The mutational spectrum shown in the HPV-negative tumors of this small study demonstrated a high mutational burden in these tumors and a source of potential difficulty in interpreting and reporting using exome or many (>100) gene diagnostic panels which could yield hundreds or thousands of variant calls. Because identifying sequencing artifacts and low level mutations can be more challenging in HPV-negative FFPE specimens, this could be a rate limiting step for implementation by clinical laboratories and oncologists with modest bioinformatics resources. Despite these challenges, we are confident that our data show that routine head and neck tumor clinical samples are amenable to characterization using benchtop Next Generation Sequencing (NGS) technology with simple interpretative software. We should stress that despite finding several mutations, we do think current commercially available somatic mutation library preps are appropriate for HNSCC specimens. We use these data to argue that standardization of minimal criteria for testing eligibility, gene content and reporting of HNSCC mutation assays are required, particularly for patients who fail standard therapy and are appropriate for clinical trials. Several studies have already reported on the results of exome sequencing in SCCHN OPSCC [48]. These studies have identified several genes that have not been previously identified in SCCHN and have started to better identify the genetic landscape of SCCHN [45, 52]. Some of these mutations appear to be more commonly observed in HPV-positive OPSCC [53]. In addition, some of these biomarkers have been shown to be possible predictors of certain therapeutic options in SCCHN [42]. Despite the multiple gene expression signatures noted in OPSCC, namely the NOTCH1 gene [48] HRAS and PTEN [52] as well as PIK3CA which are commonly observed in HPV-positive OPSCC [42], the clinical applications of these have been rather limited. Even though actionable mutations (where clinical intervention is possible) are currently rare in SCCHN, clinical trials that are targeting such mutations, for example PI3K pathway and cMET, have started to accrue patients (NCT01816984; NCT01737450; NCT01696955).
In addition to mutation data, however, we acknowledge that somatic copy number change is a crucial disease mechanism in HNSCC [28, 43]. In our experience, commercially available library kits and vendor supplied data analysis software for benchtop NGS instruments have been inadequate in providing content or analyses that adequately addresses copy number and structural abnormalities that may predict therapeutic response or clinical outcomes. Copy number abnormalities should also be taken into account in standardizing minimal criteria for HNSCC testing.
The goal of molecular medicine is to provide accurate and reproducible information that informs treatment decision making and improves outcomes. This requires an approach to diagnosis and treatment that acknowledges that every tumor has an underlying biology that defines how it will respond or resist standard treatment. This is especially critical for patients with locally advanced disease or distant metastasis. With this in mind, it is clear that gene expression data adds crucial information about biological pathways in individual tumors. In this study, we show that using a multiple gene expression panel provides reproducible gene expression data from total RNA derived from FFPE samples and that information can be used to characterize and to inform distinct biological processes in HPV-positive and HPV-negative OPSCC.
The TCGA OPSCC is limited and requires a much larger sample size with equal numbers of HPV-positive and HPV-negative tumors, but our preliminary findings are provocative in suggesting that the oropharyngeal site may have a much higher frequency of PIK3CA mutations compared to all histological HNSCC sites (52 vs. 21 %). Moreover, the ten exclusive HPV-positive over expressed genes (CDKN2C, SYK, WNT10B, CEBPA, MAP3K8, FGR, GATA1, IL4, PDGFA, CDKN2A, E2F2 and REL) may help to characterize HPV-positive biology in OPSCC. Of particular interest are CDKN2C and SYK co-occurring and mutually exclusive HPV-positive OPSCC and PIK3CA and MET in HPV-negative OPSCC.
CDKN2C is the cyclin-dependent kinase inhibitor 2C, also known as p18, and a member of the INK4 family of cyclin-dependent kinase inhibitors. The CDKN2C protein regulates G1 progression into the cell cycle by binding CDK4 or CDK6, and is upregulated by HPV E6 via TP53 pathway [54]. Overexpression of this gene has been associated with resistance to cisplatin [34] and may be a druggable target in HNSCC using metformin [55]. The SYK (spleen tyrosine kinase) gene is a member of the family of non-receptor type tyrosine protein kinases and high SYK expression is believed to play a role in cell migration and invasion and is significantly correlated with worse survival in HNSCC [56, 57]. This target may be selectively targeted using GS-9973 [58]. PDGFA is a member of the platelet-derived growth factor family and is a mitogenic factor for cells of mesenchymal origin. Overexpression of PDGFA may be targeted via MEK inhibition [59]. Finally, coding mutations are rare in HNSCC, but the OPSCC TCGA dataset suggests that there may be a subset of HPV-negative MET over expressed OPSCC that are mutually exclusive of PIK3CA mutations and PDGFA over expression. MET is a recognized drug target in head and neck cancer [60].
Combining mutation profiling with gene expression is clearly informative in OPSCC tumors, and we are encouraged to test if our HPV-positive classifier will hold up with a larger OPSCC dataset. Furthermore, we have demonstrated the benchtop NGS combined with NanoString gene expression analysis is reproducible and comparable to more comprehensive genomic approaches like whole exome sequencing and RNA-Seq. We are confident that this combined approach has utility in a routine OPSCC diagnostics and can readily be performed in molecular pathology laboratories. We are confident that this information will be particularly meaningful in the context of risk assessment of the individual OPSCC patient and may help us to define treatment modalities within the context of treatment refractory or advanced local disease.
Electronic supplementary material
References
- 1.Simard EP, Torre LA, Jemal A. International trends in head and neck cancer incidence rates: differences by country, sex and anatomic site. Oral Oncol. 2014; Feb 12th. [DOI] [PubMed]
- 2.D’Souza G, Kreimer AR, Viscidi R, et al. Case-control study of human papillomavirus and oropharyngeal cancer. N Engl J Med. 2007;356:1944–56. [DOI] [PubMed]
- 3.Saba NF, Goodman M, Ward K, et al. Gender and ethnic disparities in incidence and survival of squamous cell carcinoma of the oral tongue, base of tongue, and tonsils: a surveillance, epidemiology and end results program-based analysis. Oncology. 2011;81:12–20. [DOI] [PMC free article] [PubMed]
- 4.Chaturvedi AK, Engels EA, Anderson WF, Gillison ML. Incidence trends for human papillomavirus-related and -unrelated oral squamous cell carcinomas in the United States. J Clin Oncol. 2008;26:612–9. [DOI] [PubMed]
- 5.Ragin CC, Taioli E. Survival of squamous cell carcinoma of the head and neck in relation to human papillomavirus infection: review and meta-analysis. Int J Cancer. 2007;121:1813–20. [DOI] [PubMed]
- 6.Rampias T, Sasaki C, Weinberger P, Psyrri A. E6 and E7 gene silencing and transformed phenotype of human papillomavirus 16-positive oropharyngeal cancer cells. J Natl Cancer Inst. 2009;101:412–23. [DOI] [PubMed]
- 7.Ang KK, Harris J, Wheeler R, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med. 2010;363:24–35. [DOI] [PMC free article] [PubMed]
- 8.Fakhry C, Westra WH, Li S, et al. Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. J Natl Cancer Inst. 2008;100:261–269. doi: 10.1093/jnci/djn011. [DOI] [PubMed] [Google Scholar]
- 9.Lau HY, Brar S, Klimowicz AC, et al. Prognostic significance of p16 in locally advanced squamous cell carcinoma of the head and neck treated with concurrent cisplatin and radiotherapy. Head Neck. 2011;33:251–6. [DOI] [PubMed]
- 10.Nichols AC, Faquin WC, Westra WH, et al. HPV-16 infection predicts treatment outcome in oropharyngeal squamous cell carcinoma. Otolaryngol Head Neck Surg. 2009;140:228–234. doi: 10.1016/j.otohns.2008.11.025. [DOI] [PubMed] [Google Scholar]
- 11.Liang C, Marsit CJ, McClean MD, et al. Biomarkers of HPV in head and neck squamous cell carcinoma. Cancer Res. 2012;72:5004–5013. doi: 10.1158/0008-5472.CAN-11-3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holzinger D, Schmitt M, Dyckhoff G, Benner A, Pawlita M, Bosch FX. Viral RNA patterns and high viral load reliably define oropharynx carcinomas with active HPV16 involvement. Cancer Res. 2012;72:4993–5003. doi: 10.1158/0008-5472.CAN-11-3934. [DOI] [PubMed] [Google Scholar]
- 13.Schlecht NF, Burk RD, Adrien L, et al. Gene expression profiles in HPV-infected head and neck cancer. J Pathol. 2007;213:283–293. doi: 10.1002/path.2227. [DOI] [PubMed] [Google Scholar]
- 14.Sehn JK, Hagemann IS, Pfeifer JD, Cottrell CE, LockWood CM. Diagnostic utility of targeted next-generation sequencing in problematic cases. Am J Surg Pathol. 2014;38:534–541. doi: 10.1097/PAS.0000000000000161. [DOI] [PubMed] [Google Scholar]
- 15.Hunt JL, Barnes L, Lewis JS, et al. Molecular diagnostic alterations in squamous cell carcinoma of the head and neck and potential diagnostic applications. Eur Arch Otorhinolaryngol. 2014;271:211–223. doi: 10.1007/s00405-013-2400-9. [DOI] [PubMed] [Google Scholar]
- 16.Brunotto M, Zarate AM, Bono A, Barra JL, Berra S. Risk genes in head and neck cancer: a systematic review and meta-analysis of last 5 years. Oral Oncol. 2014;50:178–188. doi: 10.1016/j.oraloncology.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 17.Lewis JS Jr. p16 Immunohistochemistry as a standalone test for risk stratification in oropharyngeal squamous cell carcinoma. Head Neck Pathol. 2012;6. [DOI] [PMC free article] [PubMed]
- 18.Singh RR, Patel KP, Routbort MJ, et al. Clinical validation of a next-generation sequencing screen for mutational hotspots in 46 cancer-related genes. J Mol Diagn. 2013;15:607–622. doi: 10.1016/j.jmoldx.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 19.Tsongalis GJ, Peterson JD, de Abreu FB, et al. Routine use of the ion torrent AmpliSeq cancer hotspot panel for identification of clinically actionable somatic mutations. Clin Chem Lab Med. 2014;52:707–714. doi: 10.1515/cclm-2013-0883. [DOI] [PubMed] [Google Scholar]
- 20.Jia P, Pao W, Zhao Z. Patterns and processes of somatic mutations in nine major cancers. BMC Med Genomics. 2014;7:11. doi: 10.1186/1755-8794-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Do H, Dobrovic A. Dramatic reduction of sequence artefacts from DNA isolated from formalin-fixed cancer biopsies by treatment with uracil- DNA glycosylase. Oncotarget. 2012;3:546–558. doi: 10.18632/oncotarget.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Forbes SA, Bindal N, Bamford S, et al. COSMIC: mining complete cancer genomes in the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2011;39:D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Genomes Project C. Abecasis GR, Altshuler D, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Y, McGee J, Chen X, et al. Identification of druggable cancer driver genes amplified across TCGA datasets. PLoS One. 2014;9:e98293. doi: 10.1371/journal.pone.0098293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lui VW, Hedberg ML, Li H, et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013;3:761–769. doi: 10.1158/2159-8290.CD-13-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pickering CR, Zhang J, Yoo SY, et al. Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. Cancer Discov. 2013;3:770–781. doi: 10.1158/2159-8290.CD-12-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pickering CR, Zhang J, Neskey DM, et al. Squamous cell carcinoma of the oral tongue in young non-smokers is genomically similar to tumors in older smokers. Clin Cancer Res. 2014;20:3842–3848. doi: 10.1158/1078-0432.CCR-14-0565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pickering CR, Shah K, Ahmed S, et al. CT imaging correlates of genomic expression for oral cavity squamous cell carcinoma. AJNR Am J Neuroradiol. 2013;34:1818–1822. doi: 10.3174/ajnr.A3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beard RE, Abate-Daga D, Rosati SF, et al. Gene expression profiling using nanostring digital RNA counting to identify potential target antigens for melanoma immunotherapy. Clin Cancer Res. 2013;19:4941–4950. doi: 10.1158/1078-0432.CCR-13-1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laborde RR, Wang VW, Smith TM, et al. Transcriptional profiling by sequencing of oropharyngeal cancer. Mayo Clin Proc. 2012;87:226–232. doi: 10.1016/j.mayocp.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nichols AC, Chan-Seng-Yue M, Yoo J, et al. A pilot study comparing HPV-positive and HPV-negative head and neck squamous cell carcinomas by whole exome sequencing. ISRN Oncol. 2012;2012:809370. doi: 10.5402/2012/809370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y, Yuan L, Fu L, Liu C, Liu D, Mei C. Overexpression of p18INK(4)C in LLC-PK1 cells increases resistance to cisplatin-induced apoptosis. Pediatr Nephrol. 2011;26:1291–1301. doi: 10.1007/s00467-011-1877-y. [DOI] [PubMed] [Google Scholar]
- 35.Clark ES, Brown B, Whigham AS, Kochaishvili A, Yarbrough WG, Weaver AM. Aggressiveness of HNSCC tumors depends on expression levels of cortactin, a gene in the 11q13 amplicon. Oncogene. 2009;28:431–444. doi: 10.1038/onc.2008.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bragado P, Estrada Y, Sosa MS, et al. Analysis of marker-defined HNSCC subpopulations reveals a dynamic regulation of tumor initiating properties. PLoS One. 2012;7:e29974. doi: 10.1371/journal.pone.0029974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singh J, Jayaraj R, Baxi S, Mileva M, Curtin J, Thomas M. An Australian retrospective study to evaluate the prognostic role of p53 and eIF4E cancer markers in patients with head and neck squamous cell carcinoma (HNSCC): study protocol. Asian Pac J Cancer Prev. 2013;14:4717–4721. doi: 10.7314/APJCP.2013.14.8.4717. [DOI] [PubMed] [Google Scholar]
- 38.Mahjabeen I, Chen Z, Zhou X, Kayani MA. Decreased mRNA expression levels of base excision repair (BER) pathway genes is associated with enhanced Ki-67 expression in HNSCC. Med Oncol. 2012;29:3620–3625. doi: 10.1007/s12032-012-0300-2. [DOI] [PubMed] [Google Scholar]
- 39.Lemaire F, Millon R, Young J, et al. Differential expression profiling of head and neck squamous cell carcinoma (HNSCC) Br J Cancer. 2003;89:1940–1949. doi: 10.1038/sj.bjc.6601373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Laytragoon-Lewin N, Rutqvist LE, Lewin F. DNA methylation in tumour and normal mucosal tissue of head and neck squamous cell carcinoma (HNSCC) patients: new diagnostic approaches and treatment. Med Oncol. 2013;30:654. doi: 10.1007/s12032-013-0654-0. [DOI] [PubMed] [Google Scholar]
- 41.Hass HG, Schmidt A, Nehls O, Kaiser S. DNA ploidy, proliferative capacity and intratumoral heterogeneity in primary and recurrent head and neck squamous cell carcinomas (HNSCC)–potential implications for clinical management and treatment decisions. Oral Oncol. 2008;44:78–85. doi: 10.1016/j.oraloncology.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 42.Lui VW, Hedberg ML, Li H, et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013;3(7):761–769. doi: 10.1158/2159-8290.CD-13-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walter V, Yin X, Wilkerson MD, et al. Molecular subtypes in head and neck cancer exhibit distinct patterns of chromosomal gain and loss of canonical cancer genes. PLoS One. 2013;8:e56823. doi: 10.1371/journal.pone.0056823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McBride SM, Rothenberg SM, Faquin WC, et al. Mutation frequency in 15 common cancer genes in high-risk head and neck squamous cell carcinoma (HNSCC). Head Neck. 2013. [DOI] [PMC free article] [PubMed]
- 45.Stransky N, Egloff AM, Tward AD, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333(6046):1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jung AC, Job S, Ledrappier S, et al. A poor prognosis subtype of HNSCC is consistently observed across methylome, transcriptome, and miRNome analysis. Clinical Cancer Res. 2013;19:4174–4184. doi: 10.1158/1078-0432.CCR-12-3690. [DOI] [PubMed] [Google Scholar]
- 47.Georgolios A, Batistatou A, Manolopoulos L, Charalabopoulos K. Role and expression patterns of E-cadherin in head and neck squamous cell carcinoma (HNSCC) J Exp Clin Cancer Res. 2006;25:5–14. [PubMed] [Google Scholar]
- 48.Agrawal N, Frederick MJ, Pickering CR, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;6046. [DOI] [PMC free article] [PubMed]
- 49.Agrawal N, Frederick MJ, Pickering CR, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333:1154–1157. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gaykalova DA, Mambo E, Choudhary A, et al. Novel insight into mutational landscape of head and neck squamous cell carcinoma. PLoS One. 2014;9:e93102. doi: 10.1371/journal.pone.0093102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stransky N, Egloff AM, Tward AD, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chiosea SI, Grandis JR, Lui VW, et al. PIK3CA, HRAS and PTEN in human papillomavirus positive oropharyngeal squamous cell carcinoma. BMC Cancer. 2013;13:602. doi: 10.1186/1471-2407-13-602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nichols AC, Palma DA, Chow W, et al. High frequency of activating PIK3CA mutations in human papillomavirus-positive oropharyngeal cancer. JAMA Otolaryngol Head Neck Surg. 2013;139(6):617–622. doi: 10.1001/jamaoto.2013.3210. [DOI] [PubMed] [Google Scholar]
- 54.Wang X, Meyers C, Guo M, Zheng ZM. Upregulation of p18Ink4c expression by oncogenic HPV E6 via p53-miR-34a pathway. Int J Cancer. 2011;129:1362–1372. doi: 10.1002/ijc.25800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sikka A, Kaur M, Agarwal C, Deep G, Agarwal R. Metformin suppresses growth of human head and neck squamous cell carcinoma via global inhibition of protein translation. Cell Cycle. 2012;11:1374–1382. doi: 10.4161/cc.19798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chuang JY, Huang YL, Yen WL, Chiang IP, Tsai MH, Tang CH. Syk/JNK/AP-1 signaling pathway mediates interleukin-6-promoted cell migration in oral squamous cell carcinoma. Int J Mol Sci. 2014;15:545–559. doi: 10.3390/ijms15010545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Luangdilok S, Box C, Patterson L, et al. Syk tyrosine kinase is linked to cell motility and progression in squamous cell carcinomas of the head and neck. Cancer Res. 2007;67:7907–7916. doi: 10.1158/0008-5472.CAN-07-0331. [DOI] [PubMed] [Google Scholar]
- 58.Currie KS, Kropf JE, Lee T, et al. Discovery of GS-9973, a selective and orally efficacious inhibitor of spleen tyrosine kinase. J Med Chem. 2014;57:3856–3873. doi: 10.1021/jm500228a. [DOI] [PubMed] [Google Scholar]
- 59.Worden B, Yang XP, Lee TL, et al. Hepatocyte growth factor/scatter factor differentially regulates expression of proangiogenic factors through Egr-1 in head and neck squamous cell carcinoma. Cancer Res. 2005;65:7071–7080. doi: 10.1158/0008-5472.CAN-04-0989. [DOI] [PubMed] [Google Scholar]
- 60.Seiwert TY, Jagadeeswaran R, Faoro L, et al. The MET receptor tyrosine kinase is a potential novel therapeutic target for head and neck squamous cell carcinoma. Cancer Res. 2009;1:3021–31. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.