

Background: Lipoprotein lipase (LPL) function is modified by interactions with its transporter GPIHBP1 and the inhibitor angiopoietin-like 4 (ANGPTL4).
Results: ANGPTL4 inactivated GPIHBP1-bound LPL. Inactivated LPL could not bind GPIHBP1.
Conclusion: ANGPTL4 inactivation of LPL reduces the affinity of LPL for GPIHBP1 causing dissociation.
Significance: Understanding ANGPTL4's interactions with LPL in a physiological context is vital to clarifying ANGPTL4's role in triglyceride metabolism.
Keywords: Endothelial Cell, Lipase, Lipolysis, Lipoprotein Metabolism, Triglyceride
Abstract
The release of fatty acids from plasma triglycerides for tissue uptake is critically dependent on the enzyme lipoprotein lipase (LPL). Hydrolysis of plasma triglycerides by LPL can be disrupted by the protein angiopoietin-like 4 (ANGPTL4), and ANGPTL4 has been shown to inactivate LPL in vitro. However, in vivo LPL is often complexed to glycosylphosphatidylinositol-anchored high density lipoprotein-binding protein 1 (GPIHBP1) on the surface of capillary endothelial cells. GPIHBP1 is responsible for trafficking LPL across capillary endothelial cells and anchors LPL to the capillary wall during lipolysis. How ANGPTL4 interacts with LPL in this context is not known. In this study, we investigated the interactions of ANGPTL4 with LPL-GPIHBP1 complexes on the surface of endothelial cells. We show that ANGPTL4 was capable of binding and inactivating LPL complexed to GPIHBP1 on the surface of endothelial cells. Once inactivated, LPL dissociated from GPIHBP1. We also show that ANGPTL4-inactivated LPL was incapable of binding GPIHBP1. ANGPTL4 was capable of binding, but not inactivating, LPL at 4 °C, suggesting that binding alone was not sufficient for ANGPTL4's inhibitory activity. We observed that although the N-terminal coiled-coil domain of ANGPTL4 by itself and full-length ANGPTL4 both bound with similar affinities to LPL, the N-terminal fragment was more potent in inactivating both free and GPIHBP1-bound LPL. These results led us to conclude that ANGPTL4 can both bind and inactivate LPL complexed to GPIHBP1 and that inactivation of LPL by ANGPTL4 greatly reduces the affinity of LPL for GPIHBP1.
Introduction
Metabolic homeostasis depends on the proper delivery of dietary and liver-synthesized fat to the appropriate tissues. Delivery of dietary fat is crucially dependent on the activity of lipoprotein lipase (LPL),2 which hydrolyzes plasma triglycerides, liberating fatty acids for tissue uptake. LPL deficiency in humans results in severe hypertriglyceridemia and can cause eruptive xanthoma, lipemia retinalis, and pancreatitis (1). Moreover, LPL activity levels are altered in human obese and diabetic patients as well as in rodent models of metabolic disease, suggesting that LPL might play a role in the development of insulin resistance and diabetes (2–5). LPL is regulated at multiple levels, including transcriptionally, translationally, and post-translationally (6–8). Importantly, after its secretion by parenchymal cells (e.g. adipocytes and cardiomyocytes), LPL must be transported across capillary endothelial cells into the vascular lumen to become physiologically functional. This transport is carried out by GPIHBP1, which binds LPL secreted by parenchymal cells and transcytotically transports it to the capillary lumen (9–11). In GPIHBP1-deficient mice, LPL accumulates in the interstitial space and is absent from the capillary lumen, resulting in a dramatic reduction in triglyceride processing and severely elevated plasma triglyceride levels (9, 10, 12–18).
In addition to GPIHBP1, LPL interacts with a number of extracellular factors that modulate its activity and function (6, 7, 19). One of these factors, ANGPTL4 (angiopoietin-like 4), has emerged as an important regulator of triglyceride clearance. ANGPTL4 is a potent inhibitor of LPL in vitro, and ANGPTL4 overexpression leads to increased levels of plasma triglycerides, whereas lower than normal plasma triglycerides are observed in ANGPTL4-deficient mice (20–22). ANGPTL4 is secreted by several tissues, including adipose tissue and the liver, and is also present in the plasma (20, 23). Thus, ANGPTL4 could potentially interact with LPL both in the interstitial space and in the vascular lumen. However, whether ANGPTL4 can actually bind and inhibit LPL at each of these locations remains unclear (24).
One source of this uncertainty is that the direct interactions of ANGPTL4 with LPL have generally been characterized in vitro using free LPL in solution. However, in vivo much of the LPL in peripheral tissues is bound to endothelial cells by GPIHBP1 (9, 25). Not only does GPIHBP1 transport LPL across capillary endothelial cells, but after transport, LPL remains anchored to GPIHBP1 on the capillary wall, allowing LPL to bind and process triglyceride-rich lipoproteins (25). Thus, it seems certain that in vivo ANGPTL4 encounters LPL that is bound to or will soon be bound to endothelial cells by GPIHBP1. Because it is important to understand the ability of ANGPTL4 to interact with LPL in physiological contexts, we investigated how ANGPTL4 interacts with LPL bound to the surface of endothelial cells by GPIHBP1, as well as how these interactions modify LPL-GPIHBP1 complexes.
EXPERIMENTAL PROCEDURES
Cell Lines
Rat heart microvessel endothelial cells (RHMVECs; VEC Technologies) were grown in MCDB-131 base medium (Gene Depot) supplemented with 10 mm l-glutamine, 1% PenStrep antibiotic solution (10,000 units/ml penicillin and 10,000 μg/ml streptomycin, Gibco), 5% fetal bovine serum (Atlanta Biologicals), 1 μg/ml hydrocortisone (Sigma), 10 μg/ml human epidermal growth factor (Gibco and Life Technologies, Inc.), and 12 μg/ml bovine brain extract (Lonza). Because endothelial cells lose expression of GPIHBP1 when cultured (26), lentiviruses encoding S-protein-tagged mouse GPIHBP1 were transduced into endothelial cells and selected for stable transduction with puromycin as described previously (9). We have previously shown that GPIHBP1 expression levels in lentivirus-transduced RHMVECs are similar to those found in endothelial cells in vivo (9).
Production of LPL Conditioned Media
A construct expressing FLAG-tagged human LPL, pSS1, was generated by replacing the V5 tag of a human LPL construct (27) with the FLAG tag using site-directed mutagenesis. FLAG-tagged human LPL was concentrated from the medium of a Chinese hamster ovary cell line (CHO-K1) stably expressing FLAG-tagged human LPL as described previously (28). The presence of LPL in the conditioned media was assessed by Western blotting using a mouse antibody against the FLAG tag (1:5000; Sigma). LPL activity was assessed by a lipase activity assay (see below).
Production of ANGPTL4-conditioned Media
A construct expressing full-length human ANGPTL4 (pXC2) was generated by amplifying full-length ANGPTL4 cDNA (OpenBiosystems) and using it to replace the FLAG-tagged LPL of pSS1 using In-Fusion cloning (Clontech). A V5 tag was appended to the C terminus of the open reading frame using Phusion site-directed mutagenesis (New England Biolabs) to create a V5-tagged version of human ANGPTL4 (pHS2). To generate conditioned media containing V5-tagged ANGPTL4, 293T cells were transfected with pHS2. 24 h post-transfection, the media were switched to serum-free DMEM containing protease inhibitors and grown for 2 days. The media were then collected, and the concentration of ANGPTL4 in the conditioned media was determined using a human ANGPTL4 ELISA kit (Sigma). Conditioned media prepared from untransfected cells were used as a control for all ANGPTL4 experiments.
Generation of Truncated and Cleavage-resistant ANGPTL4
Truncated V5-tagged human ANGPTL4 (amino acids 1–160) was generated by deleting residues 161–406 from pHS2 by site-directed mutagenesis. To generate a full-length cleavage-resistant V5-tagged human ANGPTL4 construct, the highly conserved cleavage motif 161RRKR164 was mutated to 161GSGS164 using site-directed mutagenesis. Production of conditioned media containing truncated and cleavage-resistant ANGPTL4 was performed as described above.
LPL Activity Assay
Lipase activity was assayed using EnzChek lipase fluorescent substrate (Molecular Probes) as described previously (29). Briefly, 50 μl of sample was mixed with 25 μl of 4× assay buffer (0.6 m NaCl, 80 mm Tris-HCl, pH 8.0, and 6% fatty acid-free BSA). 25 μl of substrate solution containing 2.48 μm EnzChek lipase fluorescent substrate, 0.05% 3-(N,N-dimethylmyristylammonio)propanesulfonate Zwittergent detergent (Acros Organics) in 1% methanol was then added to each sample. Samples were then incubated at 37 °C with fluorescence (485 nm excitation/528 nm emission) read every minute for 30 min with a Synergy Neo multimode plate reader (BioTek). Relative lipase activity was calculated by first subtracting background (calculated by reading fluorescence of a sample with no LPL) and then calculating the slope of the curve between the 10- and 15-min reads.
LPL and ANGPTL4 Binding Assays
For LPL binding assays, RHMVECs expressing GPIHBP1 grown on fibronectin-coated plates or coverslips were incubated with control or ANGPTL4-treated LPL at 4 °C for 3.5 h. Following incubation, unbound LPL was removed by washing extensively with PBS. For ANGPTL4 binding assays, GPIHBP1-expressing RHMVECs were likewise incubated with LPL at 4 °C for 3.5 h. After washing off unbound LPL, cells were incubated with ANGPTL4 at 4 °C for 30 min or at 37 °C for 15 min. For all assays, cells were then either lysed with radioimmunoprecipitation assay buffer (1% Nonidet P-40 substitute, 0.5% sodium deoxycholate, and 0.1% SDS in PBS) for Western blotting or fixed with 4% paraformaldehyde for immunofluorescence (see below).
Immunofluorescence
Cells grown on fibronectin-coated glass coverslips were fixed with 4% paraformaldehyde for 15 min. After washing with PBS, cells were blocked with 0.2% BSA in PBS and then incubated with primary antibody diluted in blocking buffer. GPIHBP1 was detected with goat anti-S-protein tag (1:1000; Abcam). LPL was detected with anti-LPL monoclonal 5D2 (1:100; Santa Cruz Biotechnology) or mouse monoclonal anti-FLAG tag (1:1000; Sigma). ANGPTL4 was detected with a rabbit anti-hANGPTL4 antibody (1:1400; Fisher). Following another set of PBS washes, cells were incubated with secondary antibodies diluted in blocking buffer. Secondary antibodies were AlexaFluor-488 donkey anti-mouse IgG (Life Technologies, Inc.), AlexaFluor-555 donkey anti-rabbit IgG (Life Technologies, Inc.), and Dylight-650 donkey anti-goat IgG (Thermo Scientific). Coverslips were then mounted on glass slides with ProLong Gold antifade reagent with DAPI (Molecular Probes). Quantification of immunofluorescence was performed using Leica LAS AF software. ANGPTL4 binding was quantitated by subtracting background fluorescence and then normalizing the ANGPTL4 signal to LPL signal.
Western Blot
Protein samples were size-fractionated on SDS-12% polyacrylamide gels and then transferred to a nitrocellulose membrane. Membranes were blocked with casein buffer (1% casein, Fisher Science Education). Primary antibodies were diluted in casein buffer + 0.2% Tween. Primary antibody dilutions were 1:5000 for a mouse monoclonal antibody against the FLAG tag (Sigma), 1:5000 for a mouse monoclonal antibody against the V5 tag (Invitrogen), 1:10,000 for a goat antibody against the S-protein tag (Abcam), 1:400 for a mouse monoclonal antibody 5D2 against LPL (Santa Cruz Biotechnology), and 1:2500 for a goat antibody against actin (Santa Cruz Biotechnology). After washing with PBS + 0.1% Tween (PBS-T), membranes were incubated with Dylight680- or Dylight800-labeled secondary antibodies (Thermo Scientific) diluted 1:5000 in casein buffer + 0.2% Tween. After washing with PBS-T, antibody binding was detected using an Odyssey Infrared Scanner (LI-COR). Quantification of Western blots was performed using Li-Cor Image Studio software. GPIHBP1 was used as the control for quantification.
Detection of LPL Activity on the Cell Surface
To detect LPL activity on the cell surface, RHMVECs expressing GPIHBP1 were incubated with LPL at 4 °C for 3–4 h. After washing off unbound LPL, cells were treated with ANGPTL4 or control conditioned media. After again washing the cells with PBS, cells were treated with heparin (100 units/ml in H2O) for 10 min at room temperature to release surface-bound LPL. Released LPL was collected and immediately assayed for lipase activity. Samples were also analyzed for LPL protein mass by Western blotting.
In some cases, LPL was released from the cell surface in complex with GPIHBP1 using phosphatidylinositol phospholipase C (PIPLC). In these experiments, after treating the cells with ANGPTL4 and washing with PBS, cells were treated with PIPLC (5 units/ml in H2O; Life Technologies) for 10 min at 37 °C. Released LPL-GPIHBP1 complexes were collected and immediately assayed for lipase activity.
Separation of LPL Monomers and Dimers
To separate LPL monomers from dimers, LPL samples were applied to a HiTrap Heparin HP 1-ml column (GE Healthcare). The column was then equilibrated with 10 ml of 20 mm Tris-HCl, pH 7.4, 0.15 m NaCl, and 20% glycerol at a flow rate of 1 ml/min. LPL was then eluted in two steps as described previously (30). Briefly, 4 ml of equilibration buffer containing 0.6 m NaCl was applied to the column to elute LPL monomers, and then LPL dimers were eluted with 4 ml of 1.6 m NaCl. Elutions were collected in 1-ml fractions. Protein in each fraction was precipitated by adding trichloroacetic acid to a final concentration of 10% (w/v) and incubating on ice for 1 h. After centrifugation at 20,000 × g for 5 min at 4 °C, pellets were washed three times with cold acetone and then dried at room temperature for 30 min. Samples were resuspended in 2× SDS sample buffer and analyzed for LPL protein by Western blotting.
Enzyme-linked Immunosorbent Assays (ELISA)
The concentration of full-length ANGPTL4 in conditioned media was determined using a human ANGPTL4 ELISA kit (Sigma). Molar concentrations of truncated and cleavage-resistant ANGPTL4 conditioned media were measured using a direct ELISA against the V5 tag. V5-tagged full-length ANGPTL4-conditioned media with a known concentration of 6 nm was used to generate a standard curve. The pH of conditioned media was adjusted to 9.45 by adding sodium bicarbonate to a final concentration of 0.1 m. 96-Well flat-bottom microtiter plates (Greiner Bio-One) were coated with 100 μl of pH-adjusted conditioned media overnight at 4 °C with gentle rocking. After washing three times with PBS-T, wells were blocked with 1% BSA in PBS for 2 h at room temperature, washed, and then incubated with a mouse monoclonal antibody against the V5 tag (1:1000; Invitrogen). Wells were then washed four times with PBS-T, incubated with HRP-conjugated goat anti-mouse secondary antibody (1:1000; Thermo Scientific) for 30 min at room temperature, and then washed again four times with PBS-T. A developing solution was made by adding 1 mg/ml o-phenylenediamine dihydrochloride (Thermo Scientific) to a pH 5 buffer containing 50 mm citric acid and 50 mm sodium phosphate. Hydrogen peroxide was added to a final percentage of 0.03% (v/v). 100 μl of this developing solution was added to each well and incubated for 15 min. Absorbance was read at 450 nm, and concentrations of truncated and cleavage-resistant ANGPTL4 were calculated by fitting A450 to the standard curve.
RESULTS
Ability of ANGPLT4 to Inactivate LPL
We produced V5-tagged ANGPTL4 conditioned media and tested its ability to inactivate LPL. As expected, when ANGPTL4 was incubated with LPL in solution at 37 °C, LPL activity was reduced in a dose- and time-dependent manner (Fig. 1A).
FIGURE 1.

Inhibition of LPL by ANGPTL4. FLAG-tagged human LPL was incubated with the indicated concentrations of ANGPTL4 for 0–60 min at either 37 °C (A) or 4 °C (B and C). Following incubation, samples were immediately assayed for lipase activity. Graphs show activity (mean ± S.E.) of triplicate assays relative to control treated LPL (**, p < 0.01; ***, p < 0.001 for time points compared at the same concentration).
The time-dependent nature of LPL inactivation was consistent with a catalytic mechanism (22). To explore this issue further, we asked whether inactivation was also temperature-dependent. Consistent with a previous report (22), when ANGPTL4 was incubated with LPL at 4 °C, ANGPTL4-mediated LPL inactivation was greatly blunted (Fig. 1B). Moreover, in contrast to the situation at 37 °C, no time-dependent component of inhibition was observed when ANGPTL4 and LPL were incubated together at 4 °C (Fig. 1B). In fact, when we repeated our time course experiment and included a 0-min time point (where cold LPL and ANGPTL4 were mixed and immediately assayed for activity), we found that similar inhibition could be observed even when incubation time at 4 °C was reduced to virtually zero (Fig. 1C). Because lipase activity is measured at 37 °C (even after incubation with ANGPTL4 at 4 °C), we think it likely that little or no ANGPTL4-mediated inhibition occurred at 4 °C even at high concentrations of ANGPTL4. Rather, the reduction in LPL activity we measured was likely a result of ANGPTL4-mediated inactivation occurring as the samples warmed during the lipase activity assay.
Binding of Inactive LPL to GPIHBP1
We next asked whether inactivation of LPL by ANGPTL4 changed the ability of LPL to bind its endothelial cell transporter GPIHBP1. Although heat-denaturing LPL has been previously shown to abolish its ability to bind GPIHBP1 (31), the effect of ANGPTL4 inhibition on GPIHBP1 binding and trafficking has not been reported. To test the ability of inactive LPL to bind GPIHBP1 on the surface of endothelial cells, LPL was inactivated either by treatment with ANGPTL4 for 30 min or by heat inactivation (60 min at 50 °C). LPL activity was abolished by both ANGPTL4 treatment and heat inactivation (Fig. 2A), but the levels of full-length LPL protein remained unchanged (Fig. 2B). Both ANGPTL4-treated and heat-inactivated LPL were unable to bind GPIHBP1-expressing endothelial cells, as judged by Western blotting, whereas control-treated LPL bound these cells robustly (Fig. 2C). As expected, endothelial cells lacking GPIHBP1 bound little LPL (Fig. 2C, rightmost three lanes). LPL treated with ANGPTL4 at 4 °C instead of 37 °C retained both its activity (Fig. 2D) and its ability to bind GPIHBP1 on the surface of endothelial cells (Fig. 2E).
FIGURE 2.

Binding of ANGPTL4-inactivated LPL to GPIHBP1. FLAG-tagged human LPL was incubated with 60 ng/ml V5-tagged human ANGPTL4 or control (CTRL) media for 30 min at 37 °C (A–C) or 4 °C (D and E). Samples were then tested for lipase activity (A and D), full-length LPL protein levels (B), and ability to bind GPIHBP1-expressing cells (C and E). LPL that had been inactivated by heating at 50 °C for 60 min (H) was used as a negative control. Lipase activity (A and D) is shown relative to control-treated media (mean ± S.E. of triplicate assays). LPL protein levels (B) were measured by Western blotting with an anti-FLAG tag antibody. To assess the binding of treated LPL to GPIHBP1-expressing endothelial cells (C and E), the cells were incubated with ANGPTL4-treated, control-treated, or heat-inactivated LPL for 3.5 h at 4 °C. After removal of unbound LPL, cell lysates were collected and blotted for LPL (using an antibody against the FLAG tag), GPIHBP1 (using an antibody against the S tag), and actin. Blots show biological triplicates. Blot in C is representative of four independent experiments.
The ability of inactive LPL to bind GPIHBP1-expressing endothelial cells was also tested by immunofluorescence. Again, both heat inactivation at 50 °C and ANGPTL4 treatment at 37 °C abolished lipase activity (Fig. 3A), but ANGPTL4 treatment at 4 °C did not (Fig. 3B). GPIHBP1-expressing cells bound control-treated LPL robustly as judged by immunofluorescence staining of cells (Fig. 3C, top row), but this binding was greatly attenuated when LPL was pretreated with ANGPTL4 at 37 °C or heat-inactivated (Fig. 3C, middle and bottom rows). Again, LPL incubated with ANGPTL4 at 4 °C retained its ability to bind GPIHBP1 (Fig. 3D, middle row).
FIGURE 3.
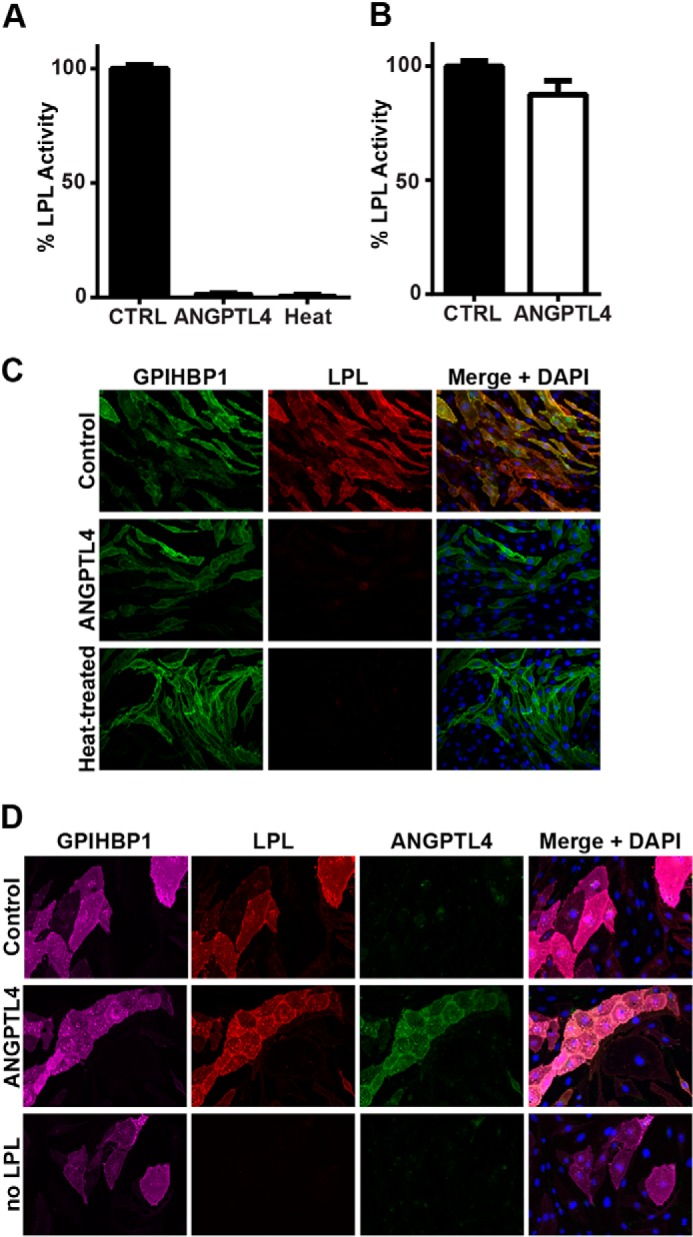
Binding of ANGPTL4-inactivated LPL to GPIHBP1. FLAG-tagged human LPL was incubated with 60 ng/ml V5-tagged human ANGPTL4 or control media for 30 min at 37 °C (A and C) or 4 °C (B and D). Samples were then tested for lipase activity (A and B) and ability to bind GPIHBP1-expressing cells (C and D). LPL that had been inactivated by heating at 50 °C for 60 min (Heat) was used as a negative control. Lipase activity is shown relative to control-treated media (mean ± S.E. of triplicate assays). To assess the binding of treated LPL to GPIHBP1-expressing endothelial cells (C and D), the cells were incubated with ANGPTL4-treated, control-treated, or heat-inactivated LPL for 3.5 h at 4 °C. After removal of unbound LPL, cells were fixed and stained with antibodies against the S tag (GPIHBP1), LPL, or ANGPTL4. Nuclei were visualized by staining with DAPI. Micrographs are representative of multiple fields of view from biological triplicates.
Binding of ANGPTL4 to LPL-GPIHBP1 Complexes
Interestingly, when LPL was incubated with ANGPTL4 at 4 °C and then added to GPIHBP1-expressing cells, not only did LPL bind robustly to cells, but ANGPTL4 binding was detected as well (Fig. 3D). This observation raised the possibility that ANGPTL4 might bind but not inhibit LPL at 4 °C. ANGPTL4 was not detected in the absence of LPL (Fig. 3D, bottom row), suggesting that ANGPTL4 bound cells through LPL.
To further test the ability of ANGPLT4 to bind GPIHBP1-bound LPL, we incubated LPL with GPIHBP1-expressing endothelial cells to form LPL-GPIHBP1 complexes on the cell surface. Unbound LPL was removed by washing, and cells were incubated with ANGPTL4 and then washed again. The binding of ANGPTL4 to these cells was tested both by Western blotting of cell extracts and immunofluorescence. ANGPTL4 bound GPIHBP1-expressing cells that had been incubated with LPL in a concentration-dependent manner and could bind surface-bound LPL both at 4 °C (Fig. 4, A, B, D, and E) and 37 °C (Fig. 4, C and F). However, ANGPTL4 had no capacity to bind GPIHBP1-expressing cells in the absence of LPL (Fig. 4B, bottom row). Because cell-bound ANGPTL4 was not detected in the absence of LPL and because little LPL bound endothelial cells in the absence of GPIHBP1 (see Fig. 2, C and E), we concluded that the ANGPTL4 we observed was bound to LPL complexed with GPIHBP1. Thus, LPL could be bound by both GPIHBP1 and ANGPTL4 simultaneously.
FIGURE 4.

Binding of ANGPTL4 to LPL-GPIHBP1 complexes. RHMVECs expressing GPIHBP1 were incubated with FLAG-tagged human LPL for 3.5 h at 4 °C. After washing off unbound LPL, the indicated concentrations of ANGPTL4 were added to the cells, and cells were incubated for an additional 30 min at 4 °C (A and B) or an additional 15 min at 37 °C (C). The binding of ANGPTL4 to cells was then assessed by Western blotting (A) or immunofluorescence (B and C) using the appropriate antibodies. Blot shows biological triplicates and is representative of two independent experiments. Micrographs are representative of multiple fields of view from five (B) or three (C) biological replicates. Graphs show quantification of ANGPTL4 binding (mean ± S.E.) from two independent Western blots (D), micrographs (n = 4/concentration) from immunofluorescence after 4 °C binding (E), and micrographs (n = 9/concentration) from immunofluorescence after 37 °C binding (F). AU, arbitrary units.
We also attempted to visualize binding of ANGPTL4 to LPL-GPIHBP1 complexes in vivo in mice. However, none of the several commercially available antibodies we tested bound specifically to mouse ANGPTL4 in control experiments (data not shown).
ANGPTL4 Inhibition of GPIHBP1-bound LPL
A previous report found that GPIHBP1-bound LPL is protected from ANGPTL4 inactivation, but those studies were carried out in cell-free solutions with truncated versions of ANGPTL4 and GPIHBP1 (32). To determine whether ANGPTL4 could inactivate LPL bound to GPIHBP1 on the surface of endothelial cells, we again formed LPL-GPIHBP1 complexes by incubating GPIHBP1-expressing endothelial cells with LPL. After washing off unbound LPL, cells were treated with ANGPTL4 for 10 min at 37 °C and then washed. LPL was released from the cell surface using heparin (100 units/ml) and immediately assayed for lipase activity. LPL recovered from the surface of ANGPTL4-treated cells was significantly less active than LPL recovered from control-treated cells (Fig. 5A), even though similar levels of LPL were recovered from the cell surface (Fig. 5B), indicating that ANGPTL4 was capable of inactivating LPL bound by GPIHBP1 in a dose-dependent manner. Although inactivation of GPIHBP1-bound LPL was easily observable, it is important to note that the concentration of ANGPTL4 required to inactivate GPIHBP1-bound LPL appeared to be 3–5-fold greater than the concentration of ANGPTL4 needed to inactivate LPL in solution (compare Figs. 1 and 5).
FIGURE 5.
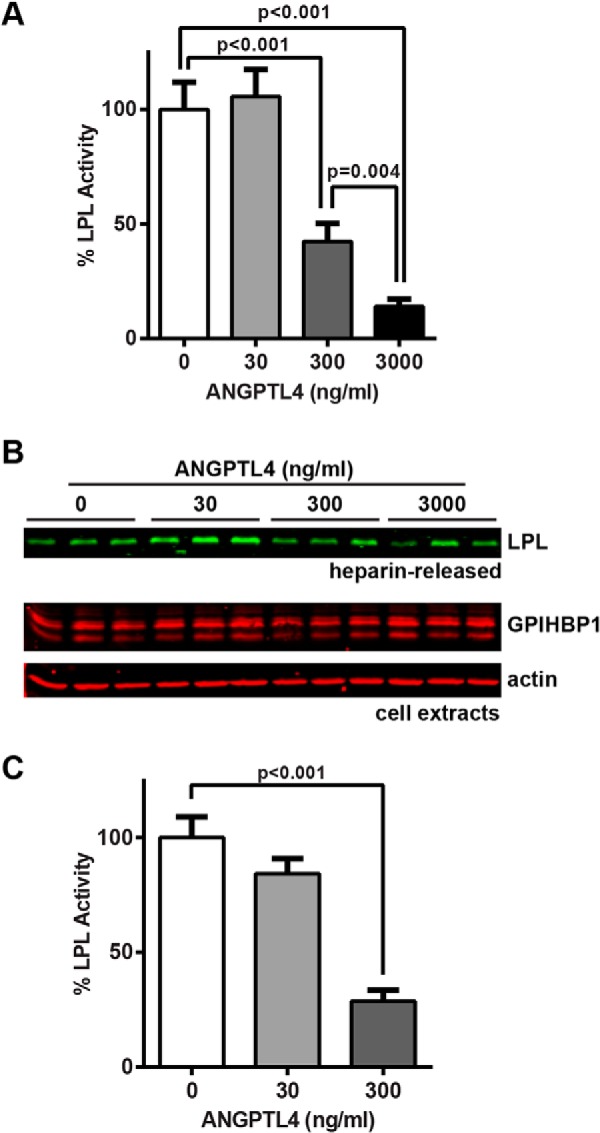
Inhibition of GPIHBP1-bound LPL by ANGPTL4. RHMVECs expressing GPIHBP1 were incubated with FLAG-tagged human LPL for 3.5 h at 4 °C. After washing off unbound LPL, the indicated concentrations of ANGPTL4 were then added to the cells, and cells were incubated for an additional 10 min at 37 °C. Cells were then washed and treated with 100 units/ml heparin to release surface-bound LPL. Released LPL was then immediately assayed for lipase activity (A). Both heparin-released LPL and cell extracts were also subjected to Western blotting with the appropriate antibodies (B). C, RHMVECs expressing GPIHBP1 were likewise incubated with LPL, washed, and incubated with the indicated concentrations of ANGPTL4 for 30 min at 37 °C. Cells were then washed and treated with 5 units/ml PIPLC to release surface-bound LPL-GPIHBP1 complexes. Lipase activity was immediately assayed. For both A and C, graphs show activity (mean ± S.E.) of three independent experiments, each performed in triplicate. Means are plotted relative to control-treated LPL. Blot (B) show biological triplicates and is representative of three independent experiments.
Because LPL is released from GPIHBP1 by heparin prior to assaying lipase activity, it is formally possible that ANGPTL4 acts to inhibit LPL only after LPL has been released from GPIHBP1. To address this possibility, we performed experiments where LPL-GPIHBP1 complexes were released from the surface of cells using PIPLC. PIPLC cleaves the GPI anchor of GPIHBP1, releasing GPIHBP1 from the plasma membrane (10). Thus, under these conditions LPL is released from the cell surface but remains bound to GPIHBP1 throughout the activity assay. In these experiments, we also observed dose-dependent inactivation of LPL by ANGPTL4 (Fig. 5C).
A previous study demonstrated that ANGPTL4 has a greater affinity for active LPL dimers than for inactive monomers and that ANGPTL4 promotes the conversion of active LPL dimers to inactive monomers (22). These findings have been questioned by a more recent study that suggested that ANGPTL4 is a reversible inhibitor of LPL (33). To determine whether ANGPTL4-mediated inhibition of GPIHBP1-bound LPL altered the ratio of LPL dimers to monomers, we used heparin-Sepharose affinity chromatography and stepwise elution by NaCl (30). In this procedure, LPL bound to a heparin-Sepharose column is first eluted with 0.6 m NaCl, releasing LPL monomers, and then with 1.6 m NaCl, which elutes LPL dimers (30). Incubation of LPL with ANGPTL4 in solution resulted in the inactivation of LPL and an increase in the percentage of LPL monomers (Fig. 6, A and C). ANGPTL4 treatment (3000 ng/ml for 10 min at 37 °C) of LPL bound to the surface of endothelial cells by GPIHBP1 also resulted in LPL inactivation and an increase in LPL monomers when compared with control-treated LPL (Fig. 6, B and D). These results indicate that ANGPTL4 inactivation of LPL increases the inactive monomeric form of LPL regardless of whether LPL is in solution or bound to GPIHBP1.
FIGURE 6.

ANGPTL4-mediated conversion of LPL dimers to monomers. A and C, FLAG-tagged human LPL was incubated with either control conditioned media (CTRL) or 60 ng/ml of ANGPTL4 for 30 min at 37 °C. Samples were assayed for lipase activity (A) and dimerization state (C). B and D, RHMVECs expressing GPIHBP1 were incubated with FLAG-tagged human LPL for 3.5 h at 4 °C. After washing off unbound LPL, cells were incubated with 3000 ng/ml ANGPTL4 or control media for 10 min at 37 °C. Cells were then washed and treated with 100 units/ml heparin to release the surface-bound LPL. LPL was then assayed for lipase activity (B) and for dimerization state (D). Lipase activity graphs (A and B) show activity (mean ± S.E.) relative to control-treated LPL. To assess the dimerization state (C and D), samples were applied to a heparin-Sepharose column. LPL monomers and dimers were eluted with 0.6 and 1.6 m NaCl, respectively. Three fractions of each salt concentration were collected and precipitated with TCA and then subjected to Western blotting with an antibody against the FLAG tag. Blots are representative of two (C) or four (D) independent experiments. FT = flow-through; S = starting material.
ANGPTL4 inactivation of GPIHBP1-bound LPL was also time-dependent, as incubation with ANGPTL4 for longer times at 37 °C decreased GPIHBP1-bound LPL activity to a greater extent (Fig. 7A). Incubation of cell-bound LPL-GPIHBP1 complexes with ANGPTL4 at 4 °C resulted in little inhibition of heparin-releasable LPL activity (Fig. 7B).
FIGURE 7.

Time-dependent inhibition of GPIHBP1-bound LPL by ANGPTL4. RHMVECs expressing GPIHBP1 were incubated with FLAG-tagged human LPL for 3.5 h at 4 °C. After washing off unbound LPL, the indicated concentrations of ANGPTL4 were then added to the cells, and cells were incubated for an additional 10–30 min at 37 °C (A and C) or 4 °C (B and D). Cells were then washed and treated with 100 units/ml heparin to release surface-bound LPL. Released LPL was then immediately assayed for lipase activity (A and B). Graphs show activity (mean ± S.E.) of three independent experiments, each done in triplicate. Means are plotted relative to control-treated LPL (*, p < 0.05; ***, p < 0.001 when compared with control-treated LPL at the same time point). Both heparin-released LPL and cell extracts were subjected to Western blotting with the appropriate antibodies (C and D). Blots show biological triplicates and are representative of all three independent experiments.
Dissociation of ANGPTL4-inactivated LPL from GPIHBP1
In addition to testing LPL activity in the above experiments, we also measured the relative levels of LPL protein released from cells by Western blotting. Interestingly, at later time points we observed a marked reduction in total LPL released from the cells treated with ANGPTL4 (Fig. 7C). This reduction appeared to be dose-dependent and was not observed when cells were incubated with ANGPTL4 at 4 °C (Fig. 7D). As this reduction in cell-surface LPL protein lagged behind the reduction in LPL activity, we suspected that once GPIHBP1-bound LPL was inactivated by ANGPTL4, the inactive LPL had reduced affinity for GPIHBP1, leading to dissociation of the LPL-GPIHBP1 complex. To further test this idea, we performed a similar experiment in which GPIHBP1-expressing endothelial cells were incubated with LPL to form LPL-GPIHBP1 complexes. Cells were then treated with ANGPTL4 for 10–30 min at 37 °C. Instead of releasing LPL with heparin, we prepared cell lysates and assayed total cell-associated LPL levels using Western blotting. Again, ANGPTL4 treatment resulted in dissociation of LPL from GPIHBP1 in a time- and dose-dependent manner (Fig. 8).
FIGURE 8.
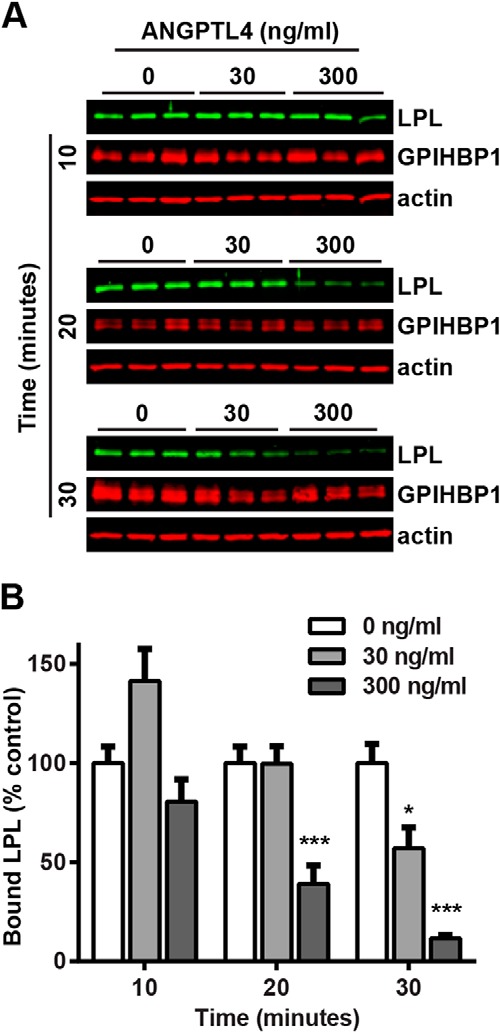
ANGPTL4 dissociates LPL from GPIHBP1. RHMVECs expressing GPIHBP1 were incubated with FLAG-tagged human LPL for 3.5 h at 4 °C. After washing off unbound LPL, the indicated concentrations of ANGPTL4 were then added to the cells, and cells were incubated for an additional 10–30 min at 37 °C. Cells were then washed, and cell lysates were collected and blotted for LPL (using an antibody against the FLAG tag), GPIHBP1 (using an antibody against the S-tag), and actin. Blots (A) are representative of two independent experiments, each performed in triplicate. Graphs (B) show quantification of LPL signal from all experiments (mean ± S.E.) relative to control-treated LPL at the same time point (*, p < 0.05; ***, p < 0.001).
Relative Effects of Full-length ANGPTL4 and Its N-terminal Cleavage Product on LPL Function
Full-length ANGPTL4 consists of both an N-terminal coiled-coil domain and a C-terminal fibrinogen-like domain (23). These two domains can be separated by the action of proprotein proteases that recognize the highly conserved cleavage site 161RRKR164 (34–37). Although the C-terminal fibrinogen-like domain has no ability to inhibit LPL on its own, the N-terminal coiled-coil domain is both necessary and sufficient for LPL inhibition in vitro (22, 38). Western blot analysis showed that the ANGPTL4-conditioned media we generated from a construct coding full-length human ANGPTL4 contained a mixture of full-length and cleaved ANGPTL4 (Fig. 9A; note that because the V5 tag is on the C terminus, only the C-terminal fragment is visible on the blot).
FIGURE 9.
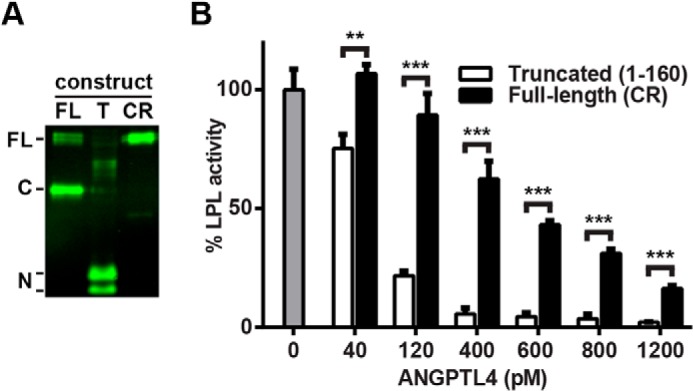
Inhibition of LPL by truncated and cleavage-resistant ANGPTL4. A, conditioned media from 293T cells transfected with V5-tagged full-length (FL), truncated (T), or full-length cleavage-resistant (CR) ANGPTL4 were Western-blotted using antibodies against the V5 tag. Full-length (FL), C-terminal (C), and N-terminal fragments (N) are indicated. B, FLAG-tagged human LPL was incubated with the indicated concentrations of truncated or cleavage-resistant (CR) ANGPTL4 for 10 min at 37 °C. Following incubation, samples were immediately assayed for lipase activity. Graphs show activity (mean ± S.E.) of triplicate assays relative to control-treated LPL (**, p < 0.01; ***, p < 0.001).
To compare the interactions of full-length ANGPTL4 and the N-terminal coiled-coil domain of ANGPTL4 with GPIHBP1-bound LPL, we generated V5-tagged versions of both truncated (amino acids 1–160) and cleavage-resistant (161RRKR164 → 161GSGS164) ANGPTL4. Western blot analysis showed the expected molecular weights (Fig. 9A). The molarity of the two different versions of ANGPTL4 was determined using a direct ELISA against the V5 tag and normalizing against a known concentration of full-length ANGPTL4 (300 ng/ml full-length ANGPTL4 equals ∼6 nm).
We compared the ability of equivalent amounts of truncated and cleavage-resistant full-length ANGPTL4 to inhibit LPL activity in solution. In agreement with what has been reported previously (36, 37), the N-terminal portion of ANGPTL4 was a more potent inactivator of LPL than full-length ANGPTL4 (Fig. 9B). The ability of both the N-terminal fragment and full-length cleavage-resistant ANGPTL4 to inhibit LPL complexed with GPIHBP1 on the surface of endothelial cells was also tested. Again, the N-terminal fragment of ANGPTL4 was more effective at inactivating LPL (Fig. 10, A and B). Over time, inactivation of LPL by either the N-terminal fragment or full-length cleavage-resistant ANGPTL4 led to dissociation of LPL from GPIHBP1, consistent with what we previously observed with our full-length ANGPTL4 construct (Fig. 10, C and D).
FIGURE 10.

Binding and inhibition of GPIHBP1-bound LPL by truncated and cleavage-resistant ANGPTL4. RHMVECs expressing GPIHBP1 were incubated with FLAG-tagged human LPL for 3.5 h at 4 °C. After washing off unbound LPL, the indicated concentrations of truncated (A and C) or cleavage-resistant (B and D) ANGPTL4 were then added to the cells, and cells were incubated for an additional 10–30 min at 37 °C (A–D) or 30 min at 4 °C (E). A–D, following LPL and ANGPTL4 incubations, cells were washed and treated with 100 units/ml heparin to release surface-bound LPL. Released LPL was then immediately assayed for lipase activity (A and B). Graphs show activity (mean ± S.E.) of three independent experiments, each done in triplicate. Means are plotted relative to control-treated LPL at 10 min (***, p < 0.001 when compared with control-treated LPL at the same time point). Both heparin-released LPL and cell extracts were also subjected to Western blotting with the appropriate antibodies (C and D). Blots are representative of three independent experiments, each performed in triplicate. E, to assess ANGPTL4 binding, after the LPL and ANGPTL4 incubations, cells were washed, and cell lysates were collected. The binding of truncated (left) and cleavage-resistant (right) ANGPTL4 to cells was then assessed by Western blotting using the appropriate antibodies. Blots show biological triplicates and are representative of two independent experiments.
We speculated that the increased inhibitory efficiency of the N-terminal cleavage product of ANGPTL4 was due to an enhanced ability to bind LPL. To test this idea, GPIHBP1-expressing endothelial cells were incubated with LPL to form LPL-GPIHBP1 complexes on the cell surface. Cells were then incubated with either truncated or cleavage-resistant full-length ANGPTL4. Binding was assessed by Western blotting of cell extracts. Surprisingly, we observed similar binding of truncated and full-length ANGPTL4 to LPL-GPIHBP1 complexes (Fig. 10E), suggesting that the greater ability of the N-terminal cleavage product to inactivate LPL was not due to an enhanced ability to bind LPL, but rather an increased ability to inactivate LPL once bound.
DISCUSSION
In this study, we investigated the interactions of ANGPTL4, GPIHBP1, and LPL on the surface of endothelial cells. Our major findings were as follows: 1) ANGPTL4 could bind to and inactivate LPL complexed to GPIHBP1 on the surface of endothelial cells; 2) ANGPTL4-inactivated LPL had a greatly reduced capacity to bind GPIHBP1; 3) once GPIHBP1-bound LPL was inactivated by ANGPTL4, it tended to dissociate from GPIHBP1; 4) binding of ANGPTL4 to LPL and the inhibition of LPL by ANGPTL4 were separable activities—binding could occur efficiently at 4 °C but inhibition could not; 5) the N-terminal domain of ANGPTL4 alone was a more potent inhibitor of LPL than full-length ANGPTL4, whether or not LPL was bound to GPIHBP1.
A previous study reported that in solution a soluble version of GPIHBP1 offered substantial protection against ANGPTL4, with 20-fold greater concentrations of ANGPTL4 needed to inactivate LPL when it was bound to GPIHBP1 (32). When we tested LPL bound to GPIHBP1 on the surface of endothelial cells, we also found that GPIHBP1-bound LPL was more resistant to ANGPTL4 inactivation than LPL in solution, but the difference was much less profound (only 3–5-fold different). Importantly, even though there was some level of protection, ANGPTL4 retained a substantial capacity to inactivate GPIHBP1-bound LPL. Moreover, it should be noted that the level of protection offered by GPIHBP1 is more than offset by the gain in ANGPTL4 efficacy that occurs when full-length ANGPTL4 is proteolytically cleaved. The mechanism behind GPIHBP1-mediated protection is not clear. It is possible that GPIHBP1-binding stabilizes LPL in such a way that makes it resistant to ANGPTL4-induced inactivation. Alternatively, the binding of LPL to GPIHBP1 could reduce the ability of ANGPTL4 to bind LPL. We found that binding of LPL to GPIHBP1 did not prevent ANGPTL4 binding, but our results do not exclude the possibility that the affinity of ANGPTL4 for LPL was reduced when LPL was complexed to GPIHBP1.
The precise physiological locations where ANGPTL4 acts on LPL have not been established, however ANGPTL4 could possibly act in both a paracrine and endocrine fashion. Our studies shed some light on what the possible efficacy of ANGPTL4 might be in different physiological contexts. It has been previously suggested that the most prevalent site of ANGPTL4-mediated LPL inactivation may be in the interstitial space (11, 39). Concentrations of ANGPTL4 in the interstitial space of various tissues are not known, but they are likely to be high in tissues such as adipose where gene expression is high (20). Interestingly, adipose tissue appears to secrete mostly full-length ANGPTL4 (40). Although our studies show that full-length ANGPTL4 is substantially less inhibitory than the N-terminal cleavage product, in the interstitial space ANGPTL4 could inactivate LPL before it binds GPIHBP1. Thus, it would not have to overcome the protection GPIHBP1 provides. It should be noted that our data suggest that even full-length ANGPTL4, when present at high concentrations, can inactivate LPL regardless of whether it was bound to GPIHBP1 or not. Although less effective at inactivating LPL, full-length ANGPTL4, by retaining the C-terminal fibrinogen-like domain, can bind the extracellular matrix, keeping it anchored near the place of expression (35, 41). Thus, full-length ANGPTL4 appears to be well suited for local, tissue-specific regulation of LPL (24).
Our data imply that LPL inactivated by ANGPTL4 in the interstitial space is unable to bind GPIHBP1 and thus would not be transported to the capillary lumen. This observation may appear to have little relevance to LPL function as inactivated LPL would be unable to hydrolyze triglycerides even if it were to be transported. However, it is important to note that nonenzymatic functions, such as the bridging of lipoproteins particles to relevant receptors, have been attributed to LPL (42–44). These nonenzymatic functions also require the presence of LPL in the capillary lumen. Thus, ANGPTL4 acting in the interstitial space would not only inactivate LPL enzymatic activity but, by preventing binding to GPIHBP1, would also reduce the ability of LPL to perform its nonenzymatic functions.
The contribution of plasma ANGPTL4 to triglyceride metabolism remains enigmatic. Increasing plasma levels of ANGPTL4, either by overexpression in the liver or by direct injection of ANGPTL4 into the bloodstream, can reduce plasma triglycerides in mice (21, 45), suggesting that ANGPTL4 can act on the LPL-GPIHBP1 complexes in the vasculature. Yet initial studies in human subjects reported no correlation between plasma ANGPTL4 concentrations and plasma triglycerides (46, 47). In these studies, ANGPTL4 concentrations ranged from 2 to 232 ng/ml (46, 47). In the circulation, ANGPTL4 is most likely to encounter LPL bound to GPIHBP1 on the surface of endothelial cells (9, 10, 25). Our data suggest that only at the upper ranges of these concentrations would ANGPTL4 be able to significantly inhibit LPL, and this only in the absence of triglyceride-rich lipoproteins, which have been shown to protect LPL from ANGPTL4 (39). A more recent study reported higher levels of plasma ANGPTL4 across all subjects and a modest correlation between ANGPTL4 concentrations and plasma triglyceride levels (48). However, a major caveat of all these correlation studies is that none report the relative levels of full-length ANGPTL4 and the N-terminal cleavage product. In fact, it has recently come to light that the ELISAs used for measuring ANGPTL4 in human plasma do not detect the N-terminal cleavage product at all (49, 50). This is a significant concern as both full-length ANGPTL4 and the N-terminal cleavage product are present in plasma (40). In fact, the liver, which is believed to be the major endocrine source of ANGPTL4, primarily secretes the cleaved form (40). Our data show that the N-terminal cleavage product is much more effective at inactivating LPL and that this greater potency offsets the protection provided by GPIHBP1. Thus, this unmeasured N-terminal cleavage product likely plays a crucial role in LPL regulation. In the future, it will be imperative to measure both forms of ANGPTL4 in the plasma. Assessing the tissues in which ANGPTL4 binds to LPL-GPIHBP1 complexes in vivo will also be important once an appropriate antibody has been generated. It will also be important to develop methodologies for gauging ANGPTL4 levels in the interstitial space with the ultimate goal of matching changes in ANGPTL4 levels with changes in functional LPL levels.
Both the irreversible conversion of active LPL dimers to inactive monomers (22) and reversible noncompetitive inhibition of LPL (33) have been proposed as mechanisms for ANGPTL4's inhibitory effect. Although this study did not set out to address the mechanism of ANGPTL4 action, several of our findings are directly relevant to this issue. First, we found that ANGPTL4 binding alone was not sufficient to inhibit LPL. When incubated with LPL at 4 °C, ANGPTL4 could bind but not inhibit LPL. We also found that ANGPTL4-mediated inhibition at 37 °C was not just dose-dependent but also time-dependent, with lower concentrations of ANGPTL4 able to inhibit significant fractions of LPL activity given sufficient time. Finally, we found that ANGPTL4 treatment reduced the ratio of active LPL dimers to inactive monomers regardless of whether LPL was in solution or bound to cells. These data are consistent with those reported by Sukonina et al. (22) but appear to conflict with the report of Lafferty et al. (33) who found that all ANGPTL4-mediated inhibition was concentration-dependent but not time-dependent. In the Lafferty et al. (33) study, high concentrations of ANGPTL4 were needed for LPL inhibition, and all inhibition occurred before the first time point (15 min) regardless of ANGPTL4 concentration, suggesting that there was no catalytic component to ANGPTL4-mediated inhibition. These apparent contradictory results may be explained by considering the temperature at which LPL was incubated with ANGPTL4. Lafferty et al. (33) incubated LPL and ANGPTL4 together on ice and then performed lipase activity assays at 25 °C. Our results suggest that at 4 °C ANGPTL4 binds LPL, but little or no inhibition occurs. The apparent inhibition we observed at 4 °C with higher concentrations of ANGPTL4 appeared to occur only as a result of warming the samples during the lipase activity assay. When interpreted using this premise, i.e. binding at 4 °C but no inhibition until the temperature increases at the onset of the activity assay, many of the observations made by Lafferty et al. (33) no longer exclude a model invoking catalytic conversion of active dimers to inactive monomers. Thus, although our study did not initially seek to clarify the mechanism of ANGPTL4 action, our results are consistent with a catalytic model (22) and, at the same time, potentially explain the apparently contradictory results reported by Lafferty et al. (33).
This study clarifies the mechanism by which ANGPTL4 inactivates LPL and reveals the interactions of ANGPTL4 with LPL when LPL is complexed with GPIHBP1 on the surface of endothelial cells. Although these studies provide insight into how ANGPTL4 may function in different physiological contexts, the precise role of ANGPTL4 in regulating triglyceride metabolism remains to be elucidated. In the future, it will be important to combine molecular studies and in vivo models to dissect ANGPTL4 function across various tissues and physiological sites.
This work was supported by a grant from the American Heart Association Scientist Development (to B. S. J. D.).
- LPL
- lipoprotein lipase
- GPIHBP1
- glycosylphosphatidylinositol-anchored HDL-binding protein 1
- ANGPTL4
- angiopoietin-like 4
- RHMVEC
- rat heart microvessel endothelial cells
- PIPLC
- phosphatidylinositol phospholipase C.
REFERENCES
- 1. Reina M., Brunzell J. D., Deeb S. S. (1992) Molecular basis of familial chylomicronemia: mutations in the lipoprotein lipase and apolipoprotein C-II genes. J. Lipid Res. 33, 1823–1832 [PubMed] [Google Scholar]
- 2. Taylor K. G., Galton D. J., Holdsworth G. (1979) Insulin-independent diabetes: a defect in the activity of lipoprotein lipase in adipose tissue. Diabetologia. 16, 313–317 [DOI] [PubMed] [Google Scholar]
- 3. Kroupa O., Vorrsjö E., Stienstra R., Mattijssen F., Nilsson S. K., Sukonina V., Kersten S., Olivecrona G., Olivecrona T. (2012) Linking nutritional regulation of Angptl4, Gpihbp1, and Lmf1 to lipoprotein lipase activity in rodent adipose tissue. BMC Physiol. 12, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mitrou P., Boutati E., Lambadiari V., Maratou E., Komesidou V., Papakonstantinou A., Sidossis L., Tountas N., Katsilambros N., Economopoulos T., Raptis S. A., Dimitriadis G. (2010) Rates of lipid fluxes in adipose tissue in vivo after a mixed meal in morbid obesity. Int. J. Obes. 34, 770–774 [DOI] [PubMed] [Google Scholar]
- 5. Wang Y., Puthanveetil P., Wang F., Kim M. S., Abrahani A., Rodrigues B. (2011) Severity of diabetes governs vascular lipoprotein lipase by affecting enzyme dimerization and disassembly. Diabetes 60, 2041–2050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang H., Eckel R. H. (2009) Lipoprotein lipase: from gene to obesity. Am. J. Physiol. Endocrinol. Metab. 297, E271–E288 [DOI] [PubMed] [Google Scholar]
- 7. Olivecrona T., Olivecrona G. (2009) in Cellular Lipid Metabolism (Ehnholm C., ed) pp. 315–369, Springer, Berlin [Google Scholar]
- 8. Davies B. S., Beigneux A. P., Fong L. G., Young S. G. (2012) New wrinkles in lipoprotein lipase biology. Curr. Opin. Lipidol. 23, 35–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Davies B. S., Beigneux A. P., Barnes R. H., 2nd, Tu Y., Gin P., Weinstein M. M., Nobumori C., Nyrén R., Goldberg I., Olivecrona G., Bensadoun A., Young S. G., Fong L. G. (2010) GPIHBP1 is responsible for the entry of lipoprotein lipase into capillaries. Cell Metab. 12, 42–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Beigneux A. P., Davies B. S., Gin P., Weinstein M. M., Farber E., Qiao X., Peale F., Bunting S., Walzem R. L., Wong J. S., Blaner W. S., Ding Z.-M., Melford K., Wongsiriroj N., Shu X., et al. (2007) Glycosylphosphatidylinositol-anchored high density lipoprotein-binding protein 1 plays a critical role in the lipolytic processing of chylomicrons. Cell Metab. 5, 279–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Davies B. S., Goulbourne C. N., Barnes R. H., 2nd, Turlo K. A., Gin P., Vaughan S., Vaux D. J., Bensadoun A., Beigneux A. P., Fong L. G., Young S. G. (2012) Assessing mechanisms of GPIHBP1 and lipoprotein lipase movement across endothelial cells. J. Lipid Res. 53, 2690–2697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Beigneux A. P., Franssen R., Bensadoun A., Gin P., Melford K., Peter J., Walzem R. L., Weinstein M. M., Davies B. S., Kuivenhoven J. A., Kastelein J. J., Fong L. G., Dallinga-Thie G. M., Young S. G. (2009) Chylomicronemia with a mutant GPIHBP1 (Q115P) that cannot bind lipoprotein lipase. Arterioscler. Thromb. Vasc. Biol. 29, 956–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Olivecrona G., Ehrenborg E., Semb H., Makoveichuk E., Lindberg A., Hayden M. R., Gin P., Davies B. S., Weinstein M. M., Fong L. G., Beigneux A. P., Young S. G., Olivecrona T., Hernell O. (2010) Mutation of conserved cysteines in the Ly6 domain of GPIHBP1 in familial chylomicronemia. J. Lipid Res. 51, 1535–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Franssen R., Young S. G., Peelman F., Hertecant J., Sierts J. A., Schimmel A. W., Bensadoun A., Kastelein J. J., Fong L. G., Dallinga-Thie G. M., Beigneux A. P. (2010) Chylomicronemia with low postheparin lipoprotein lipase levels in the setting of GPIHBP1 defects. Circ. Cardiovasc. Genet. 3, 169–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Coca-Prieto I., Kroupa O., Gonzalez-Santos P., Magne J., Olivecrona G., Ehrenborg E., Valdivielso P. (2011) Childhood-onset chylomicronaemia with reduced plasma lipoprotein lipase activity and mass: identification of a novel GPIHBP1 mutation. J. Intern. Med. 270, 224–228 [DOI] [PubMed] [Google Scholar]
- 16. Charrière S., Peretti N., Bernard S., Di Filippo M., Sassolas A., Merlin M., Delay M., Debard C., Lefai E., Lachaux A., Moulin P., Marçais C. (2011) GPIHBP1 C89F neomutation and hydrophobic C-terminal domain G175R mutation in two pedigrees with severe hyperchylomicronemia. J. Clin. Endocrinol. Metab. 96, E1675–E1679 [DOI] [PubMed] [Google Scholar]
- 17. Rios J. J., Shastry S., Jasso J., Hauser N., Garg A., Bensadoun A., Cohen J. C., Hobbs H. H. (2012) Deletion of GPIHBP1 causing severe chylomicronemia. J. Inherit. Metab. Dis. 35, 531–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weinstein M. M., Tu Y., Beigneux A. P., Davies B. S., Gin P., Voss C., Walzem R. L., Reue K., Tontonoz P., Bensadoun A., Fong L. G., Young S. G. (2010) Cholesterol intake modulates plasma triglyceride levels in glycosylphosphatidylinositol HDL-binding protein 1-deficient mice. Arterioscler. Thromb. Vasc. Biol. 30, 2106–2113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kersten S. (2014) Physiological regulation of lipoprotein lipase. Biochim. Biophys. Acta 1841, 919–933 [DOI] [PubMed] [Google Scholar]
- 20. Kersten S., Mandard S., Tan N. S., Escher P., Metzger D., Chambon P., Gonzalez F. J., Desvergne B., Wahli W. (2000) Characterization of the fasting-induced adipose factor FIAF, a novel peroxisome proliferator-activated receptor target gene. J. Biol. Chem. 275, 28488–28493 [DOI] [PubMed] [Google Scholar]
- 21. Köster A., Chao Y. B., Mosior M., Ford A., Gonzalez-DeWhitt P. A., Hale J. E., Li D., Qiu Y., Fraser C. C., Yang D. D., Heuer J. G., Jaskunas S. R., Eacho P. (2005) Transgenic angiopoietin-like (angptl)4 overexpression and targeted disruption of angptl4 and angptl3: regulation of triglyceride metabolism. Endocrinology 146, 4943–4950 [DOI] [PubMed] [Google Scholar]
- 22. Sukonina V., Lookene A., Olivecrona T., Olivecrona G. (2006) Angiopoietin-like protein 4 converts lipoprotein lipase to inactive monomers and modulates lipase activity in adipose tissue. Proc. Natl. Acad. Sci. U.S.A. 103, 17450–17455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yoon J. C., Chickering T. W., Rosen E. D., Dussault B., Qin Y., Soukas A., Friedman J. M., Holmes W. E., Spiegelman B. M. (2000) Peroxisome proliferator-activated receptor γ target gene encoding a novel angiopoietin-related protein associated with adipose differentiation. Mol. Cell. Biol. 20, 5343–5349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dijk W., Kersten S. (2014) Regulation of lipoprotein lipase by Angptl4. Trends Endocrinol. Metab. 25, 146–155 [DOI] [PubMed] [Google Scholar]
- 25. Goulbourne C. N., Gin P., Tatar A., Nobumori C., Hoenger A., Jiang H., Grovenor C. R., Adeyo O., Esko J. D., Goldberg I. J., Reue K., Tontonoz P., Bensadoun A., Beigneux A. P., Young S. G., Fong L. G. (2014) The GPIHBP1-LPL complex is responsible for the margination of triglyceride-rich lipoproteins in capillaries. Cell Metab. 19, 849–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Davies B. S., Waki H., Beigneux A. P., Farber E., Weinstein M. M., Wilpitz D. C., Tai L.-J., Evans R. M., Fong L. G., Tontonoz P., Young S. G. (2008) The expression of GPIHBP1, an endothelial cell binding site for lipoprotein lipase and chylomicrons, is induced by peroxisome proliferator-activated receptor-γ. Mol. Endocrinol. 22, 2496–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ben-Zeev O., Mao H. Z., Doolittle M. H. (2002) Maturation of lipoprotein lipase in the endoplasmic reticulum concurrent formation of functional dimers and inactive aggregates. J. Biol. Chem. 277, 10727–10738 [DOI] [PubMed] [Google Scholar]
- 28. Beigneux A. P., Gin P., Davies B. S., Weinstein M. M., Bensadoun A., Fong L. G., Young S. G. (2009) Highly conserved cysteines within the Ly6 domain of GPIHBP1 are crucial for the binding of lipoprotein lipase. J. Biol. Chem. 284, 30240–30247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Basu D., Manjur J., Jin W. (2011) Determination of lipoprotein lipase activity using a novel fluorescent lipase assay. J. Lipid Res. 52, 826–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bergö M., Olivecrona G., Olivecrona T. (1996) Forms of lipoprotein lipase in rat tissues: in adipose tissue the proportion of inactive lipase increases on fasting. Biochem. J. 313, 893–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gin P., Beigneux A. P., Voss C., Davies B. S., Beckstead J. A., Ryan R. O., Bensadoun A., Fong L. G., Young S. G. (2011) Binding preferences for GPIHBP1, a glycosylphosphatidylinositol-anchored protein of capillary endothelial cells. Arterioscler. Thromb. Vasc. Biol. 31, 176–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sonnenburg W. K., Yu D., Lee E.-C., Xiong W., Gololobov G., Key B., Gay J., Wilganowski N., Hu Y., Zhao S., Schneider M., Ding Z.-M., Zambrowicz B. P., Landes G., Powell D. R., Desai U. (2009) GPIHBP1 stabilizes lipoprotein lipase and prevents its inhibition by angiopoietin-like 3 and angiopoietin-like 4. J. Lipid Res. 50, 2421–2429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lafferty M. J., Bradford K. C., Erie D. A., Neher S. B. (2013) Angiopoietin-like protein 4 inhibition of lipoprotein lipase: evidence for reversible complex formation. J. Biol. Chem. 288, 28524–28534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ge H., Yang G., Huang L., Motola D. L., Pourbahrami T., Li C. (2004) Oligomerization and regulated proteolytic processing of angiopoietin-like protein 4. J. Biol. Chem. 279, 2038–2045 [DOI] [PubMed] [Google Scholar]
- 35. Chomel C., Cazes A., Faye C., Bignon M., Gomez E., Ardidie-Robouant C., Barret A., Ricard-Blum S., Muller L., Germain S., Monnot C. (2009) Interaction of the coiled-coil domain with glycosaminoglycans protects angiopoietin-like 4 from proteolysis and regulates its antiangiogenic activity. FASEB J. 23, 940–949 [DOI] [PubMed] [Google Scholar]
- 36. Yin W., Romeo S., Chang S., Grishin N. V., Hobbs H. H., Cohen J. C. (2009) Genetic variation in ANGPTL4 provides insights into protein processing and function. J. Biol. Chem. 284, 13213–13222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lei X., Shi F., Basu D., Huq A., Routhier S., Day R., Jin W. (2011) Proteolytic processing of angiopoietin-like protein 4 by proprotein convertases modulates its inhibitory effects on lipoprotein lipase activity. J. Biol. Chem. 286, 15747–15756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ge H., Cha J.-Y., Gopal H., Harp C., Yu X., Repa J. J., Li C. (2005) Differential regulation and properties of angiopoietin-like proteins 3 and 4. J. Lipid Res. 46, 1484–1490 [DOI] [PubMed] [Google Scholar]
- 39. Nilsson S. K., Anderson F., Ericsson M., Larsson M., Makoveichuk E., Lookene A., Heeren J., Olivecrona G. (2012) Triacylglycerol-rich lipoproteins protect lipoprotein lipase from inactivation by ANGPTL3 and ANGPTL4. Biochim. Biophys. Acta 1821, 1370–1378 [DOI] [PubMed] [Google Scholar]
- 40. Mandard S., Zandbergen F., Tan N. S., Escher P., Patsouris D., Koenig W., Kleemann R., Bakker A., Veenman F., Wahli W., Müller M., Kersten S. (2004) The direct peroxisome proliferator-activated receptor target fasting-induced adipose factor (FIAF/PGAR/ANGPTL4) is present in blood plasma as a truncated protein that is increased by fenofibrate treatment. J. Biol. Chem. 279, 34411–34420 [DOI] [PubMed] [Google Scholar]
- 41. Cazes A., Galaup A., Chomel C., Bignon M., Bréchot N., Le Jan S., Weber H., Corvol P., Muller L., Germain S., Monnot C. (2006) Extracellular matrix-bound angiopoietin-like 4 inhibits endothelial cell adhesion, migration, and sprouting and alters actin cytoskeleton. Circ. Res. 99, 1207–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Merkel M., Kako Y., Radner H., Cho I. S., Ramasamy R., Brunzell J. D., Goldberg I. J., Breslow J. L. (1998) Catalytically inactive lipoprotein lipase expression in muscle of transgenic mice increases very low density lipoprotein uptake: direct evidence that lipoprotein lipase bridging occurs in vivo. Proc. Natl. Acad. Sci. U.S.A. 95, 13841–13846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Merkel M., Heeren J., Dudeck W., Rinninger F., Radner H., Breslow J. L., Goldberg I. J., Zechner R., Greten H. (2002) Inactive lipoprotein lipase (LPL) alone increases selective cholesterol ester uptake in vivo, whereas in the presence of active LPL it also increases triglyceride hydrolysis and whole particle lipoprotein uptake. J. Biol. Chem. 277, 7405–7411 [DOI] [PubMed] [Google Scholar]
- 44. Olivecrona G., Olivecrona T. (1995) Triglyceride lipases and atherosclerosis. Curr. Opin. Lipidol. 6, 291–305 [DOI] [PubMed] [Google Scholar]
- 45. Yoshida K., Shimizugawa T., Ono M., Furukawa H. (2002) Angiopoietin-like protein 4 is a potent hyperlipidemia-inducing factor in mice and inhibitor of lipoprotein lipase. J. Lipid Res. 43, 1770–1772 [DOI] [PubMed] [Google Scholar]
- 46. Smart-Halajko M. C., Robciuc M. R., Cooper J. A., Jauhiainen M., Kumari M., Kivimaki M., Khaw K.-T., Boekholdt S. M., Wareham N. J., Gaunt T. R., Day I. N., Braund P. S., Nelson C. P., Hall A. S., Samani N. J., et al. (2010) The relationship between plasma angiopoietin-like protein 4 levels, angiopoietin-like protein 4 genotype, and coronary heart disease risk. Arterioscler. Thromb. Vasc. Biol. 30, 2277–2282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Robciuc M. R., Tahvanainen E., Jauhiainen M., Ehnholm C. (2010) Quantitation of serum angiopoietin-like proteins 3 and 4 in a Finnish population sample. J. Lipid Res. 51, 824–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mehta N., Qamar A., Qu L., Qasim A. N., Mehta N. N., Reilly M. P., Rader D. J. (2014) Differential association of plasma angiopoietin-like proteins 3 and 4 with lipid and metabolic traits. Arterioscler. Thromb. Vasc. Biol. 34, 1057–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Adhikary T., Brandt D. T., Kaddatz K., Stockert J., Naruhn S., Meissner W., Finkernagel F., Obert J., Lieber S., Scharfe M., Jarek M., Toth P. M., Scheer F., Diederich W. E., Reinartz S., et al. (2013) Inverse PPARβ/δ agonists suppress oncogenic signaling to the ANGPTL4 gene and inhibit cancer cell invasion. Oncogene 32, 5241–5252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jonker J. T., Smit J. W., Hammer S., Snel M., van der Meer R. W., Lamb H. J., Mattijssen F., Mudde K., Jazet I. M., Dekkers O. M., de Roos A., Romijn J. A., Kersten S., Rensen P. C. (2013) Dietary modulation of plasma angiopoietin-like protein 4 concentrations in healthy volunteers and in patients with type 2 diabetes. Am. J. Clin. Nutr. 97, 255–260 [DOI] [PubMed] [Google Scholar]