

Background: Discrimination of cytokine receptor signaling pathways is poorly understood.
Results: Manipulation of extracellular prolactin receptor interface residues selectively affected activation of the MAPK pathway but not the STAT5 pathway.
Conclusion: MAPK pathway activation correlated with apparent ligand·receptor complex lifetimes suggesting pathway-specific lifetime thresholds for activation.
Significance: Discrimination of prolactin receptor signaling is controlled by the extracellular homodimer receptor interface.
Keywords: Cell Proliferation, Cell Surface Receptor, Cytokine, Extracellular Signal-regulated Kinase (ERK), Nuclear Magnetic Resonance (NMR), STAT Transcription Factor
Abstract
Cytokine receptors elicit several signaling pathways, but it is poorly understood how they select and discriminate between them. We have scrutinized the prolactin receptor as an archetype model of homodimeric cytokine receptors to address the role of the extracellular membrane proximal domain in signal transfer and pathway selection. Structure-guided manipulation of residues involved in the receptor dimerization interface identified one residue (position 170) that in cell-based assays profoundly altered pathway selectivity and species-specific bio-characteristics. Subsequent in vitro spectroscopic and nuclear magnetic resonance analyses revealed that this residue was part of a residue quartet responsible for specific local structural changes underlying these effects. This included alteration of a novel aromatic T-stack within the membrane proximal domain, which promoted selective signaling affecting primarily the MAPK (ERK1/2) pathway. Importantly, activation of the MAPK pathway correlated with in vitro stabilities of ternary ligand·receptor complexes, suggesting a threshold mean lifetime of the complex necessary to achieve maximal activation. No such dependence was observed for STAT5 signaling. Thus, this study establishes a residue quartet in the extracellular membrane proximal domain of homodimeric cytokine receptors as a key regulator of intracellular signaling discrimination.
Introduction
Cytokine receptors activate many different intracellular pathways, including JAK/STAT, MAPK, Src, and PI3K/Akt, resulting in a wide variety of cellular responses (1–3). Besides cytokines, various stimuli have been shown to trigger signaling by these receptors, including antibodies, small molecule agonists, or even activating mutations intrinsic to the receptors (see below). However, several studies have shown that the same cytokine receptor can elicit different cellular responses depending on the nature of these stimuli, which implies signaling discrimination between agonistic signals. Typical examples include the family of type I IFNs, whose numerous members exhibit markedly different antiviral versus antiproliferative properties despite acting through the same heterodimeric receptor. In-depth analysis of structures, energetics, and dynamics of ligand·receptor complexes revealed that this functional diversity was mainly driven by different complex lifetimes and ligand affinities more than by specific conformational differences (4). Recently, a polyclonal anti-idiotypic antibody directed against porcine growth hormone (GH)4 was shown to trigger activation of the GH receptor (GHR) expressed in rat hepatocytes (5). However, only a partial response was observed as just the JAK/STAT, but not the MAPK (i.e. ERK1/2), pathway was activated. The molecular mechanism involved was not elucidated (5). Antibody-mediated activation of the thrombopoietin receptor also resulted in a different signaling profile compared with thrombopoietin stimulation, which translated into the promotion of cell proliferation at the expense of cell differentiation (6). Activation of the erythropoietin (EPO) receptor (EPOR) using an antibody binding to an epitope of the extracellular domain (ECD) distinct from the EPO-binding site stimulated in vitro erythropoiesis, but nevertheless displayed altered STAT signaling (7). In line with this, the constitutively active EPORR129C variant (Arg-129 within the ECD replaced with Cys) was shown to activate mainly STAT5B, whereas EPO-triggered EPORWT activates both A and B isoforms of STAT5 (8). These examples illustrate that in addition to binding circulating ligands, one function of cytokine receptor ECDs may be to participate in selective activation of intracellular signaling pathways. However, the structural and mechanistic features of the ECD regulating this process are poorly understood. Deciphering precise molecular mechanisms of signaling selectivity will no doubt result in substantial advances in our understanding of several diseases and allow the development of a new generation of drugs that exhibit well controlled signaling properties, including pathway selectivity.
The prolactin receptor (PRLR) and the GHR represent archetypes of the so-called homodimeric hematopoietic cytokine receptors (9), i.e. the simplest of type 1 cytokine receptors, made of two identical transmembrane chains exhibiting minimal structural complexity compared with other family members. Their ECDs contain a single cytokine receptor homology module made of two fibronectin type III domains, termed D1 (membrane-distal) and D2 (membrane-proximal) (Fig. 1A). The latter contains the conserved WS motif (Trp-Ser-Xaa-Trp-Ser sequence), a hallmark of type 1 cytokine receptors (10, 11). Under physiological conditions, signaling requires the formation of ternary complexes involving one ligand interacting with both receptor chains via two asymmetrical binding sites, termed sites 1 and 2 (Fig. 1A). Cell-based studies have shown that at least parts of the GHR and PRLR population expressed in eukaryotic cells are pre-dimerized in the absence of ligand, suggesting that signaling activation involves ligand-induced conformational changes within the receptor homodimer rather than alteration of receptor stoichiometry (12–14). For the PRLR, we recently used nuclear magnetic resonance (NMR) spectroscopy to show that in the unbound state the two tryptophan residues of the WS motif adopt a T-stack conformation that stabilizes the receptor off-state, whereas in the ligand-bound state these residues form a Trp-Arg ladder characteristic of the receptor on-state (15). Chemical shift perturbation studies following PRL binding to the D2 domain further point to structural rearrangements involving various residues, including specific sites within the receptor-receptor dimerization interface, also called site 3 (Fig. 1B).
FIGURE 1.

PRLR homodimer interface (site 3). A, representation of the 2:1 rPRLR·hPRL complex structure (PDB accession code 3NPZ) (25). The two receptors (rPRLR1 and rPRLR2) are shown in gray, and their D1 and D2 domains are labeled in light and dark blue, respectively, and hPRL is shown in pale cyan. The three intermolecular interaction sites are indicated in the structure (sites 1 and 2 between hPRL and the receptor and site 3 between the two receptors). B, magnification of site 3 with blue sticks (light blue on rPRLR1 and dark blue on rPRLR2) identifying interacting residues at the interface, which are at focus in the current work. The A to G strands are identified (see Ref. 15, 25 for structural details).
In this work, we discovered that a residue quartet positioned at the receptor dimerization interface tunes the level of MAPK signaling while not affecting STAT5 signaling. This quartet includes position 170 that is either a Leu or a Phe in the human (h) and rat (r) PRLRs, respectively (Fig. 1B). Optical and NMR spectroscopic analyses of soluble receptor domains (ECD and D2, both WT and variants) revealed underlying specific local structural changes. These included alteration of a novel aromatic T-stack between residues from the E- and B-strands of the D2 domain (see Fig. 1B), which promoted selective signaling affecting primarily the MAPK pathway. Furthermore, based on real time analysis of the PRL/PRLR-ECD interactions, in vitro stability of the ternary ligand·receptor complex appeared to correlate with both structural (T-stack) and functional (the level of MAPK activation) properties, suggesting the lifetime of the ternary complex to be deterministic for activation. Thus, using the PRLR as a prototypic homodimeric cytokine receptor, this study establishes the dimerization interface as a key regulator of intracellular signaling discrimination. These findings may have a huge impact on future knowledge-based drug development aimed at providing pathway-selective treatment strategies.
EXPERIMENTAL PROCEDURES
Plasmids
The pQE-70-rPRLR-ECD and pET11a-hPRLR-D2 prokaryotic expression plasmid, pcDNA3(+)-hPRLRWT and -rPRLRWT eukaryotic expression vectors (long PRLR isoforms), and the lactogenic hormone-response element-luciferase (firefly) reporter plasmid were described previously (15–17). Interface substitutions were introduced using the QuikChange II mutagenesis kit (Stratagene), and coding sequences were verified on both strands. Primers were from Eurogentec (Oligold quality) or from TAGCopenhagen (sequences available upon request).
Hormones and Receptors
Recombinant hPRL and rPRL, hPRLR/rPRLR-ECDs (WT and variants), hPRLR-D2 (WT and variants, unlabeled and stable isotope-labeled), and the PRLR antagonist Del1–9-G129R-hPRL were produced in Escherichia coli and purified as described (15, 17, 18). Pituitary bPRL was obtained from Dr. Parlow (NIDDK, National Institutes of Health).
Cell Cultures and Transfections
Routine culture conditions, cell transfections, generation of stable populations, and luciferase and proliferation assays have been extensively described (19). The iterative selection scheme used for Ba/F3 cell populations was recently described (20).
SureFire® pSTAT5 and pERK1/2 Assays
All reagents were supplied in the SureFire® pSTAT5 or pERK1/2 assay kits (PerkinElmer Life Sciences). Various amounts of fresh cell lysates (4–8 μg of protein content, ∼1–2 × 105 cells) were added per well of 384-well plates. Five μl of acceptor mix (activation buffer, reaction buffer, and acceptor beads) were added to cell lysates. The plate was incubated under low light conditions for 2 h at room temperature and then 2 μl of donor mix (dilution buffer and donor beads) were added. After 2 h of incubation, the plate was read on an EnVisionTM device (PerkinElmer Life Sciences).
Signaling Studies
We followed recently described procedures (19). Briefly, cells were starved in 1% FCS medium overnight (HEK293) or for 5 h (Ba/F3) and subsequently stimulated using 1 μg/ml PRL (origin as indicated). Immunoblotting of total cell lysates involved anti-pSTAT5 (C11C5, Cell Signaling, 1:1,000), anti-pERK1/2 (D13.14.4E, Cell Signaling, 1:1,000), anti-ERK1/2 (Millipore, 1:1,000), anti-STAT5 (C-17, Santa Cruz Biotechnology, 1:1,000), and various anti-PRLR antibodies directed against the hPRLR-ECD (clone 1A2B1, Zymed Laboratories Inc., 1:1,000, hPRLR-specific), the rPRLR-ECD (clone U5, Abcam, 1:1,000, rPRLR-specific), or the intracellular domain (ICD) of the hPRLR (polyclonal H300, Santa Cruz Biotechnology, 1;1,000; note that this mAb recognizes both hPRLR and rPRLR). Antigen·antibody complexes were revealed using horseradish peroxidase conjugated to anti-rabbit/-mouse antibodies as appropriate (GE Healthcare, 1:3,000).
Cell Surface PRLR Binding
Recombinant hPRL was fluorescently labeled using the DyLight 650 NHS Ester kit (catalog 62266) from the ThermoScientific Kit (Pierce). We strictly followed the instructions of the manufacturer. Briefly, hPRL suspended in 50 mm sodium borate, pH 8.5 (2 μg/ml), was mixed with fluorescent DyLight 650 dyes (1 vial) for 1 h at room temperature, and then the mix was dialyzed overnight in PBS to eliminate free dyes. After measuring A280 (protein) and A650 (fluorescent dye) using a Nanodrop, the degree of labeling of fluorescent hPRL (hPRLfluo) was calculated as ∼0.75 (mol of dye/mol of protein). For cell surface PRLR binding experiments, stable Ba/F3 populations were starved in 1% FCS medium for ∼2 h, and then 2·106 live cells (per condition) were washed and resuspended in 500 μl of RPMI 1640 medium, 0.5% BSA. Unlabeled hPRL (3 μl from 2 μg/ml stock), hPRLfluo (3 μl from 2 μg/ml stock), or both (3.5 molar excess of hPRL over hPRLfluo) were added to each well. After 210 min of incubation at 4 °C (to avoid receptor internalization) in the dark, cells were pelleted in 1 ml of cold PBS, 2% BSA and added 1 μl of SYTOX® Blue before FACS analysis (LSR Fortessa SORP, BD Biosciences).
Circular Dichroism (CD) Spectroscopy
Far-UV CD spectra were recorded of hPRLR-D2WT (1.6 μm), hPRLR-D2L170F (3.2 μm), hPRLR-D2L170A (2.3 μm), and hPRLR-D2K168A (2.3 μm) from 250 to 200 nm at 20 nm/min, bandwidth 1 nm, 2-s response time at 25 °C using a Jasco-810 spectropolarimeter in a 1-mm quartz cuvette, 10 mm NaH2PO4-NaOH, pH 7.4. The spectra were averaged over 10 scans with corresponding spectra of the buffer subtracted and smoothed using a fast Fourier transform algorithm.
Fluorescence Spectroscopy
Conformational stability was measured from GdmCl-induced changes in intrinsic tryptophan and tyrosine fluorescence with excitation at 280 nm, protein concentrations of 1–3 μm (rECD and variants) or 5 μm (hD2 and variants) in a 0.5-cm quartz cuvette, 10 μm NaH2PO4/Na2HPO4, pH 7.4, 25 °C. The concentration of GdmCl was measured by refractive index. The fluorescence emission intensity at unfolded λmax was plotted as a function of increasing GdmCl concentration, normalized to fraction of unfolded population, and fitted to a two state unfolding as described (21).
NMR Spectroscopy
The NMR samples contained 10% (v/v) D2O, 10 mm NaH2PO4/Na2HPO4, 10 mm tris(2-carboxyethyl)phosphine, 1 mm 2,2-dimethyl-2-silapentane-5-sulfonic acid, and 0.02% (v/v) NaN3 at pH 7.4. The samples were prepared at concentrations (for 15N-labeled) of 0.15 mm hPRLR-D2WT, 0.28 mm hPRLR-D2L170F, 0.28 mm hPRLR-D2K168A, and 0.15 mm hPRLR-D2Y122A, respectively. All experiments were conducted at 25 °C on either an 800 or 750 MHz Varian INOVA spectrometer. 1H,15N HSQC spectra were recorded for all samples. hPRLR-D2K168A and hPRLR-D2L170F were reassigned using 13C-correlated HNCACB and CACB(CO)NH spectra recorded on 0.33 mm 13C,15N-hPRLR-D2L170F and 0.28 mm 13C,15N-hPRLR-D2K168A samples with nonlinear sampling to 20%. NMR spectra were referenced to 2-dimethyl-2-silapentane-5-sulfonic acid at 0.00 ppm (1H) and transformed using weighted Fourier transformation and two-dimensional spectra transformed using NMRPipe (22). The three-dimensional spectra were reconstructed from nonlinear sampling using qMDD with compressed sensing. The spectra were phased on the direct dimension and analyzed in CcpNmr analysis using the assignment of hPRLR-D2 (BMRB accession code 17752 (15)) as a starting point. Changes in chemical shifts obtained from 1H,15N HSQC spectra were analyzed using the formula (Δδ = ((ΔδH)2 + (0.154·ΔδN)2)0.5). The chemical shift changes were analyzed in the context of the structure of hPRLR-D2 (PDB accession code 2LFG).
Surface Plasmon Resonance
rPRLR-ECDWT and rPRLR-ECDL170F were immobilized covalently onto nitrilotriacetic acid-derivatized sensor chips, as described (23–25). Surface densities of 3,000–4,000 resonance units (resonance unit ≈ pg mm−2) were obtained. Binding assays were performed in triplicate on a BIAcore 2000 instrument equilibrated at 25 °C. The PRLR-ECD surfaces were first saturated with hPRL (250 nm), and then rPRLR-ECDs (0.4–50 μm) were injected for 3 min at 20 μl/min onto the preformed PRLR-ECD·PRL complexes, followed by a 5-min dissociation period. The site 2 + 3 association and dissociation profiles were double referenced using the Scrubber 2.0 software (Bio-Logic Software) (i.e. both the signals from the reference surface (ethanolamine-derivatized) and the signals from blank experiments (using running buffer instead of protein) were subtracted). The site 2 + 3 steady-state SPR responses (Req) were plotted against the concentration of rPRLR-ECD (site 2) and fitted using Equation 1,
where Kd is the equilibrium dissociation constant and Rmax is the maximal binding capacity of the surface.
Statistics
Statistical analyses were performed using GraphPad Prism software. As indicated in the figure legends, we used one-way ANOVA followed by Dunnett's post hoc test or two-way ANOVA followed by Bonferroni post hoc test. p values are represented using 1 (p < 0.05), two (p < 0.01), or three (p < 0.001) symbols (*).
RESULTS
PRLR Interface Residues Control Signaling Selectivity
Using oriented SPR to measure ternary complex stabilities (24), we recently identified Lys-168 and Phe-170 as hot spot residues of the rPRLR dimerization site (Fig. 1B) (25). To investigate the role of these inter-molecular contacts for signal transfer, alanine substitutions were generated in the full-length receptor (rPRLRK168A and rPRLRF170A), and their capacity to activate the canonical STAT5 pathway in transiently transfected HEK293 fibroblasts was analyzed (Fig. 2A). No STAT5 phosphorylation was detected in the absence of PRL, ruling out ligand-independent activity of these receptor variants. When stimulated with 1 μg/ml hPRL for 15 min, all variants retained the ability to induce strong STAT5 phosphorylation similarly to that of the rPRLRWT (Fig. 2A). Using the classical LHRE-luciferase reporter gene assay to monitor STAT5 transcriptional activity (16), we obtained overlapping curves in PRL dose-response experiments irrespective of the receptor variant (Fig. 2B). These observations demonstrated that activation of the STAT5 pathway was not altered by these interface substitutions. However, in contrast to the STAT5 pathway, the ERK1/2 pathway was not activated by hPRL stimulation in HEK293 cells (Fig. 2A, bottom), in agreement with our previous observations (19, 20, 26).
FIGURE 2.

Bioactivity of rPRLR interface variants in HEK293 and Ba/F3 cells. A, HEK293 cells transiently transfected with expression vectors encoding rPRLRWT or rPRLR variants were starved and then stimulated with 1 μg/ml hPRL (15 min). Total PRLR (U5 mAb), phosphorylated and total STAT5 (from the same gel), and phosphorylated and total ERK1/2 (from a different gel) were analyzed by immunoblotting. B, HEK293 cells were transiently co-transfected using expression vectors encoding rPRLRWT or rPRLR variants, and the LHRE-luciferase reporter plasmid. For each variant, the luciferase values obtained at varying PRL concentrations were normalized to the unstimulated condition for comparison (means ± S.E., n = 3 in triplicate). C, stable populations of Ba/F3 cells expressing rPRLRWT or rPRLR variants were obtained and selected as described in the text and Table 1. Proliferation of populations 2 in response to a range of hPRL concentrations was measured (WST-1 reagent) in three independent experiments performed in triplicate (mean ± S.E.). D, starved cells were treated using 1 μg/ml hPRL (15 min), then analyzed for phosphorylated and total STAT5 (upper), and ERK1/2 (lower) by immunoblotting (two noncontiguous parts from the same immunoblot are shown and separated by a dividing line). E, densitometric analysis of STAT5 and ERK1/2 activation was averaged from five separate immunoblotting experiments. The phosphorylated/total ratio obtained in Ba/F3-rPRLRWT cells stimulated with hPRL was set to 100% (mean ± S.E.). F, basal and stimulated levels of STAT5 and ERK1/2 activation were determined using semi-quantitative SureFire® homogeneous assays (AlphaScreen® technology, n = 3). E and F, stimulated conditions were compared by one-way ANOVA. p values are represented by asterisks, using *, p < 0.05; **, p < 0.01.
We previously reported that stably transfected Ba/F3 cells express low receptor density and thus constitute a much more sensitive bioassay to discriminate bioactivity of mutated PRLs or PRLRs (27, 28). To generate stable cell populations expressing rPRLRK168A or rPRLRF170A, we followed the three-step selection scheme recently described (20) with survival data summarized in Table 1. All receptor plasmids were successfully stabilized in Ba/F3 cells upon G418 addition, keeping IL-3 as the survival/growth factor (selection 1). When hPRL was substituted for IL-3 (selection 2), all cell populations could be propagated in routine culture conditions, confirming that interface rPRLR variants triggered sufficient levels of intracellular signaling to ensure some degree of cell survival/proliferation. However, hPRL dose-response proliferation assays performed in low serum conditions (Fig. 2C) revealed that cells expressing rPRLRF170A or rPRLRK168A exhibited ∼5- to ∼17-fold higher EC50 values compared with Ba/F3-rPRLRWT cells, respectively (Table 2). When stimulated with 1 μg/ml hPRL (corresponding to the plateau in proliferation assays for all receptor variants, see Fig. 2C), the interface variants induced similar levels of STAT5 phosphorylation compared with rPRLRWT (Fig. 2D), suggesting that PRLRs present at the cell surface were functional. We therefore hypothesized that differential intracellular signaling could explain the observed proliferative differences. In contrast to HEK293 cells, Ba/F3 cells expressing hPRLRWT exhibited strong activation of the ERK1/2 pathway in response to hPRL stimulation (19, 20, 26), although other PRLR pathways (STAT3, Akt, and Src) displayed much more discrete responses (data not shown). Interestingly, Ba/F3 cells expressing rPRLRF170A or rPRLRK168A exhibited markedly lower levels of ERK1/2 phosphorylation compared with rPRLRWT (Fig. 2D). Densitometric quantification of five independent immunoblotting experiments (Fig. 2E) were fully confirmed by semi-quantitative SureFire® homogeneous assays using AlphaScreen® technology (Fig. 2F). These results demonstrated that PRLR interface residues selectively control activation of the MAPK pathway. This was further confirmed by the experiments described below.
TABLE 1.
Survival of Ba/F3 cell populations at each step of selection
Table indicates the survival/growth (+) or death (−) of stable Ba/F3 cell populations expressing the PRLR (WT or variants, as indicated) after each step of selection (#1, G418 addition; #2, substitution of hPRL for IL-3; and #3, hPRL withdrawal).

1 Responsiveness to bPRL present in culture medium (fetal calf serum) is shown.
TABLE 2.
Sensitivity of rPRLR variants to PRL of various origins
Dose-response proliferation assays of stable Ba/F3 cells (population 2) expressing rPRLR (WT or variant, as indicated) in response to PRL of human or bovine origin were performed as described in Fig. 2C. EC50 values are given as PRL concentration (ng/ml) and as ratio versus rPRLRWT (bold). The right column reflects the relative sensitivity of PRLR variants to PRLs of either origin.
| rPRLR | EC50 |
||||
|---|---|---|---|---|---|
| Human PRL |
Bovine PRL |
Ratio of EC50 for bovine vs human PRL | |||
| ng/ml | Mutant vs. WT | ng/ml | Mutant vs. WT | ||
| WT | 0.9 | 1 | 1.8 | 1 | 1.9 |
| K168A | 16.1 | 17.5 | 22.2 | 12.7 | 1.4 |
| F170A | 4.4 | 4.8 | 13.3 | 7.6 | 3.0 |
Residue 170 Directs the PRLR Species-specific Signaling Profile in Response to Ligands of Various Origins
As shown in Table 1, rPRLRWT was able to promote Ba/F3 cell survival across PRL withdrawal (selection 3). Intriguingly, this property was lost in both rPRLR interface variants (Table 1). Similarly, cells expressing hPRLRWT did not survive in the absence of hPRL supply, which emphasized a marked difference between receptor species. We suspected that the receptor-specific capacity to support cell survival might reflect a different sensitivity to bovine (b) PRL present in the culture medium via the addition of serum. To address this point, we compared dose-response proliferation assays performed in low serum conditions (1% FCS) using hPRL versus bPRL (Table 2). First, these assays revealed a higher sensitivity of rPRLRWT to bPRL compared with the rPRLR variants, providing a strong rationale for the survival of Ba/F3-rPRLRWT cells in hPRL-free growth medium (see Table 1). Second, comparison of the EC50 values of proliferation curves involving hPRL versus bPRL revealed that in contrast to rPRLRK168A, the growth-promoting properties of rPRLRF170A were highly dependent on the ligand origin (Table 2, right column). As shown below, investigation of rPRLRF170A signaling properties confirmed that this variant was indeed much less responsive than rPRLRWT to bPRL and, notably, to the homologous rPRL.
Based on these findings, we aimed to further investigate the importance of the residue located at position 170, Leu in the hPRLR versus Phe in the rPRLR. Although the data described above indicated that the F170A substitution conferred human receptor-like properties to the rPRLR, we found that the L170F substitution conferred rat bio-characteristics to the hPRLR. Indeed, Ba/F3-hPRLRL170F cells (i) survived and proliferated in growth medium without hPRL supplementation (Fig. 3A), (ii) exhibited detectable STAT5 signaling in response to serum (Fig. 3B, lanes 6 and 7), and (iii) lost the species-specific activation profiles of STAT5 and ERK1/2 in response to PRLs of various origins (Fig. 3B, lanes 8–10). None of these properties were observed for cells expressing hPRLRWT (Fig. 3, A and B, lanes 1–5). The basal activation of STAT5 signaling observed in Ba/F3-hPRLRL170F cells in the presence of serum (no hPRL added) was abolished by treating them with the pure hPRLR antagonist Del1–9-G129R-hPRL (Fig. 3C), confirming a PRLR-mediated mechanism (18). These data collectively showed that the residue at position 170 directly controls the species-specific properties of the PRLRs.
FIGURE 3.

Residue 170 at the PRLR interface directs signaling cascade selectivity. A, basal proliferation of Ba/F3-hPRLRWT (population 2), Ba/F3-hPRLRL170F (population 3), and parental Ba/F3 cells in growth medium (10% FCS, no PRL added) was monitored using WST-1 reagent. Values (means ± S.E., n = 3 in triplicates) were compared by two-way ANOVA. p values are represented as follows: **, p < 0.01; ***, p < 0.001. B, starved Ba/F3-hPRLRWT and Ba/F3-hPRLRL170F cells were treated with 1 or 10% FCS or 1 μg/ml hPRL, rPRL, or bPRL for 15 min. Phosphorylated versus total STAT5 and ERK1/2 were analyzed by immunoblotting. C, basal phosphorylation of STAT5 in Ba/F3-hPRLRL170F observed in 10% FCS growth medium was inhibited in a time-dependent manner by the addition of the pure PRLR antagonist Del1–9-G129R-hPRL (5 μg/ml). D, densitometric quantification of ERK1/2 activation in Ba/F3-rPRLRWT and Ba/F3-rPRLRL170F in response to PRL of human, rat, or bovine origin is shown. Values were normalized to the phosphorylated/total ratio obtained in Ba/F3-hPRLRWT cells stimulated with hPRL (mean ± S.E.) and compared by two-way ANOVA. NS, not significant.
The most remarkable observation made from these immunoblot experiments was that PRL stimulation (1 μg/ml, i.e. at the plateau) induced much stronger ERK1/2 activation via hPRLRL170F (3.3-fold versus basal) compared with hPRLRWT (2.2-fold versus basal) (Fig. 3B). This was statistically significant irrespective of the ligand origin (Fig. 3D). It was also in line with the much higher intrinsic level of ERK1/2 activation triggered by rPRLRWT (4.6-fold versus basal) (Fig. 2E, left bars) compared with hPRLRWT (2.2-fold versus basal) (Fig. 3B). Furthermore, pathway-specific and dose-response profiling of these receptors confirmed that manipulation of the sole position 170 discriminated the profiles of the PRLR responsiveness to various PRLs (Fig. 4); those receptors harboring a Phe at this position (i.e. rPRLRWT and hPRLRL170F) exhibited strong activation of both ERK1/2 and STAT5 pathways whatever the ligand origin, and those harboring an aliphatic residue (i.e. rPRLRF170A and hPRLRWT) displayed marked species preferences toward the ligand and reduced ERK1/2 activation, although STAT5 activation was unaffected. This underlines the key role of the residue at position 170 in the selective control of PRLR signaling properties.
FIGURE 4.

Position 170 directs species specificity and signaling selectivity. Ba/F3 cells stably expressing rPRLRWT (A), rPRLRF170A (B), hPRLRL170F (C), or hPRLRWT (D) were stimulated for 15 min with various concentrations of PRL of human, rat, or bovine origin, and activation of ERK1/2 and STAT5 pathways was monitored. The qualitative profile of responsiveness of each cascade to the various stimulations is schematized below each panel by black triangles.
Residue 170 Controls PRLR Turnover in Ba/F3 Cells
Because changes in signaling intensity may be linked to receptor turnover, we subsequently investigated whether manipulation of position 170 affected the amount of receptors available at the cell membrane. First, we performed FACS analyses using hPRLfluo (see “Experimental Procedures”) to assess cell surface expression of the PRLR variants. Fluorescent signal specificity was assessed by competing hPRLfluo with 3.5-fold excess unlabeled hPRL (Fig. 5A). Interestingly, much lower cell surface hPRLfluo binding in Ba/F3-hPRLRL170F cells compared with Ba/F3-hPRLRWT cells was observed, although it was the opposite for Ba/F3-rPRLRF170A cells compared with Ba/F3-rPRLRWT cells (Fig. 5B). Because lower cell surface binding was not correlated with lower PRLR affinity (see below), but rather the reverse, cell surface expression and degradation may be intimately linked to receptor activation and thus indirectly to position 170. We therefore investigated the degradation profiles of the different receptor variants using immunoblotting with different antibodies directed against the hPRLR-ECD (1A2B1) or -ICD (H300) or the rPRLR-ECD (U5) (Fig. 5, C and D). In routine culture conditions, the degradation patterns of hPRLRWT and hPRLRL170F appeared to be similar. The smear at ∼60–70 kDa was recognized by the H300 antibody (Fig. 5C) but not by the 1A2B1 mAb (data not shown), indicating that it corresponded to the ICD of the receptor. Of note, the band corresponding to full-length hPRLR (∼90 kDa) was much less intense for hPRLRL170F than for hPRLRWT (Fig. 5C), suggesting that L170F promoted faster receptor degradation. Accordingly, densitometric analysis of two independent experiments confirmed the lower full-length/degraded receptor ratio of hPRLRL170F compared with hPRLRWT (data not shown). In agreement with this, the F170A substitution in the rPRLR had the opposite effect as it clearly prevented degradation of the full-length receptor (Fig. 5C). Finally, PRLR degradation profiles were not altered by PRL stimulation (Fig. 5D) suggesting that they are intrinsically linked to receptor species and specifically to the residue present at position 170.
FIGURE 5.

Substituting residue 170 alters cell surface expression and degradation profile of the PRLR. A, representative FACS profile of Ba/F3-rPRLRWT cells incubated with unlabeled hPRL (background fluorescence, orange) or hPRLfluo (PRLR-binding fluorescence, cyan). Displacement of the curve obtained when hPRLfluo was competed with 3.5 molar excess of unlabeled hPRL assessed the specificity of the hPRLfluo signal (blue). B, specific binding for the four Ba/F3 cell populations was calculated as the difference between FACS values obtained with hPRL and hPRLfluo as described in A. C, degradation profiles of rPRLR and hPRLR (WT and variants, as indicated) stably expressed in Ba/F3 cells cultured in growth medium. Membranes were blotted using antibodies directed against the hPRLR-ICD (H300) or the rPRLR-ECD (U5). Arrowheads show PRLR-related bands (full-length or degraded) and asterisks indicate nonspecific bands that were identified using parental Ba/F3 cells (Par.) lacking endogenous PRLR. D, same Ba/F3 cell populations as above were starved for 5 h and then stimulated with hPRL (1 μg/ml, 15 min) before the degradation profiles of the various PRLRs were analyzed as described in C.
Longer Persistence Time of the Ternary Complex Parallels Efficient ERK1/2 Activation
We have previously shown using our established SPR setup that the F170A substitution was detrimental to ternary hPRL·rPRLR-ECD complex formation due to ∼10-fold reduced affinity at sites 2 + 3 (25). We hypothesized that this feature could be correlated with the reduced ability of rPRLRF170A to activate the ERK1/2 pathway observed in this study. We therefore investigated whether the L170F substitution that increased the ability of the hPRLR to activate ERK1/2 also increased the site 2 + 3 affinity of the ternary complex. Indeed, SPR experiments showed that hPRLR-ECDL170F had an increased affinity for the preformed 1:1 hPRL·rPRLR-ECD complex by >2-fold (n = 3) irrespective of the nature of the immobilized ECD (WT or L170F) (Table 3). This increase in affinity could be correlated to an ∼2-fold increase in the lifetime of the complex (Fig. 6). Although the gain in affinity provided by the L170F substitution was less pronounced than the loss of affinity resulting from the F170A substitution, these results strongly suggested that the longer the lifetime of the ternary complex, the higher the level of ERK1/2 activation. Thus, by modulating complex lifetimes, the chemical structure of the residue at position 170 appears to play a key role in regulating intracellular signaling properties.
TABLE 3.
Equilibrium dissociation constants, Kd (in μm), for the interactions between PRLR-ECD in solution and immobilized PRLR-ECD·hPRL 1:1 complex
The scheme in the upper left corner describes the experimental setting (see Refs. 22–24 for details). The experiment was performed using hPRLR and rPRLR ECDs (WT and variants), as indicated.

FIGURE 6.

Real time SPR characterization of site 2 + 3 interactions involving hPRLR-ECDs. hPRLR-ECDWT (A) or hPRLR-ECDL170F (B) were immobilized on a nickel-nitrilotriacetic acid surface and saturated with PRL (250 nm) as described under “Experimental Procedures.” Representative sensorgrams corresponding to the injection of hPRLR-ECDWT (81 μm; blue) or hPRLR-ECDL170F (63.6 μm; red) onto preformed 1:1 complexes are shown. C and D, PRLR-ECD2 concentration dependence of the steady-state SPR responses, from which the Kd values were determined (see Table 3), is shown for all complexes investigated (n = 2 experiments) as follows: C, hPRLR-ECDWT/hPRLR-ECDWT (blue) and hPRLR-ECDL170F/hPRLR-ECDWT (red); D, hPRLR-ECDWT/hPRLR-ECDL170F (blue) and hPRLR-ECDL170F/hPRLR-ECDL170F (red).
Residue Quartet Modulates Structure and Stability of the ECD
To understand why PRLR signaling was so affected by single substitutions in the D2 domain, we first analyzed the structures and stabilities of the ECD variants in the rPRLR background. Previously, the overall fold of the rPRLR-ECD interface variants (K168A and F170A) were evaluated from their far-UV circular dichroism (CD) spectra (25). Because of the presence of aromatic residues that are spatially close to each other in the ECD, this gives rise to a through-space coupling of their electric dipoles. This results in complex and characteristic CD spectra with a characteristic splitting. Such coupling, known as exciton coupling, occurs in the D2 domain and stems from residues Trp-191 and Trp-194 of the WS motif. Thus, the CD spectrum of rPRLR-ECD has a positive ellipticity at 230 nm, which obscures the signature from the dominant β-strand structure (15). Interestingly, both rPRLR-ECDK168A and rPRLR-ECDF170A gave rise to a smaller contribution from aromatic exciton couplings, suggesting the following: (i) the substitutions interfered with the organization of the WS motif; (ii) a contribution from additional aromatic exciton couplings of an opposite sign at the same wavelength appeared, or (iii) couplings of a similar sign disappeared. To address whether substitutions affected protein stabilities, we measured the conformational stabilities of rPRLR-ECDWT versus rPRLR-ECDK168A and rPRLR-ECDF170A using fluorescence spectroscopy (Fig. 7A and Table 4). The conformational stability of rPRLR-ECDWT was large, ΔGN-DH2O = −32 ± 5 kJ mol−1, suggesting a stable scaffold. The rPRLR-ECD responded remarkably differently to the substitutions. Whereas, the stability of rPRLR-ECDK168A was not significantly altered compared to the WT receptor, rPRLR-ECDF170A was severely destabilized by 20 kJ mol−1 (Table 4).
FIGURE 7.

Conformational stability of rPRLR-ECD and hPRLR-D2 variants. The conformational stability of rPRLR-ECD (A) and hPRLR-D2 WT (B) and variants was measured by intrinsic fluorescence emission and normalized to the fraction of-folded protein. A: circle, rPRLR-ECDWT; triangle, rPRLR-ECDK168A; diamond, rPRLR-ECDF170A. The best fits to two-state unfolding are shown as solid line (rPRLR-ECDWT), dashed line (rPRLR-ECDK168A), or dotted line (rPRLR-ECDF170A). B: circle, hPRLR-D2WT; square, hPRLR-D2L170F; triangle, hPRLR-D2F170A. The best fits to two-state unfolding are shown as a solid line (hPRLR-D2WT), dashed line (hPRLR-D2L170F), or dotted line (hPRLR-D2L170A).
TABLE 4.
Thermodynamic stabilities of rPRLR-ECDs and hPRLR-D2 domains measured by intrinsic fluorescence emission
| Cm | m | ΔG | ΔΔG (mutant-WT) | |
|---|---|---|---|---|
| M | kJ mol−1 m−1 | kJ mol−1 | kJ mol−1 | |
| rPRLR-ECD | ||||
| rPRLR-ECDWT | 2.59 ± 0.04 | 12.2 ± 2.0 | −32 ± 5 | |
| rPRLR-ECDK168A | 2.64 ± 0.05 | 13.4 ± 2.5 | −35 ± 7 | −3 ± 8 |
| rPRLR-ECDF170A | 1.9 ± 0.2 | 5.9 ± 1.2 | −11 ± 3 | 20 ± 7 |
| hPRLR-D2 | ||||
| hPRLR-D2WT | 3.83 ± 0.03 | 12.3 ± 1.2 | −47 ± 5 | |
| hPRLR-D2L170A | 4.35 ± 0.04 | 10.8 ± 1.1 | −47 ± 5 | |
| hPRLR-D2L170F | 3.81 ± 0.04 | 12.1 ± 1.7 | −46 ± 6 | |
To obtain site-specific insight into the possible conformational changes of the variants and to the extent of the involvement of the WS motif, we transferred the substitutions into the hPRLR-D2 scaffold for which NMR assignments are available (which is not the case for rPRLR-D2) (15). To match the alanine and rat/human substitutions characterized in functional assays, we created hPRLR-D2K168A, hPRLR-D2L170A, and hPRLR-D2L170F. These substitutions led to changes in the CD spectrum similarly to the rPRLR-ECD variants (25) with a pronounced effect on the aromatic exciton band at 230 nm (Fig. 8A). However, whereas hPRLR-D2K168A and hPRLR-D2L170A both led to a reduction in ellipticity compared with hPRLR-D2WT, hPRLR-D2L170F led to an increase at 230 nm. Inspection of the structural environment around Lys-168 and Leu-170 in the three-dimensional structures identified two aromatic residues, Tyr-122 and Trp-124 (Fig. 8B), which could contribute to the observed change in the CD spectrum. Substitution of Tyr-122 with alanine led to a significant decrease of the positive ellipticity at 230 nm (Fig. 8A), confirming a contribution from this residue to the aromatic exciton couplings observed in the CD spectrum.
FIGURE 8.
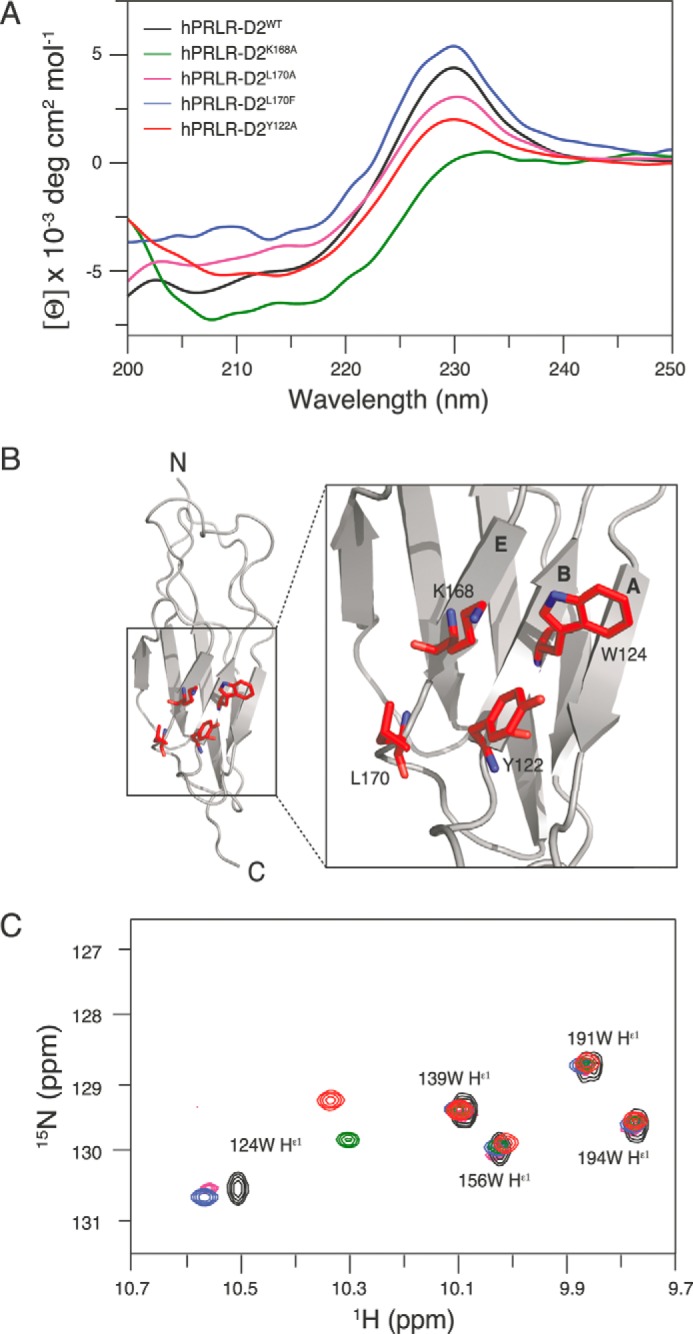
Structural analyses of the human D2 domains of PRLR interface variants. A, far-UV CD spectra of hPRLR-D2WT and variants. A large positive ellipticity was observed at ∼230 nm for hPRLR-D2WT (black), hPRLR-D2L170A (magenta), and hPRLR-D2L170F (blue) originating from the exciton coupling of the WS motif, although it was almost nonexistent for hPRLR-D2K168A (green). The residual exciton coupling from the WS motif was still present in hPRLR-D2Y122A (red). B, positions of Lys-168, Leu-170, Tyr-122, and Trp-124 in the solution structure of unbound hPRLR-D2WT (gray, PDB accession code 2LFG) are shown as red sticks on the zoom-in. Nitrogen atoms are shown in blue. The A, B, and E strands are indicated on the structure. C, overlay of the Trp indole region of the 1H, 15N HSQC spectra of hPRLR-D2WT (black), hPRLR-D2K168A (green), hPRLR-D2Y122A (red), hPRLR-D2L170A (magenta), and hPRLR-D2L170F (blue). Only small changes in chemical shifts were seen for residues Trp-139, Trp-156, Trp-194, and Trp-191 located in the WS motif. In contrast, the chemical shifts of the Trp-124 indole were significantly changed for all variants.
To explore in detail the involvement of these aromatic residues and to locate whether the site-specific changes imposed by the interface substitutions (residues 168 and 170) also involved the WS motif, we measured the changes in NMR chemical shifts of the indoles and of backbone amides of the three hPRLR-D2 variants in comparison with hPRLR-D2WT. Such changes report on both chemical and conformational changes imposed by the substitutions. The indole NH region of the spectra clearly showed that Trp-191 and Trp-194 of the WS motif were unaffected by the substitutions (Fig. 8C). Instead, the chemical shifts of Trp-124 indole changed significantly as suggested from the CD data (Fig. 8A), implicating Trp-124 to be related to the changes observed in the CD spectra. Thus, the CD and NMR analyses confirmed conformational changes of Tyr-122 and Trp-124 in response to the hot spot interface substitutions.
To locate the regions affected by the substitutions, we then compared the backbone chemical shifts of the three variants to those of hPRLR-D2WT (Fig. 9). As expected from the indole analysis, no significant changes were seen in the WS motif (Fig. 9, marked by the red bar) in accordance with the fact that none of the variants exhibited ligand-independent activity, and hence that the off-state of the receptor was in principle preserved (15). From the combined changes in chemical shifts, residues whose chemical shifts were affected more than one S.D. from the trimmed average were highlighted on the structure of hPRLR-D2WT (Fig. 9, right panels). For all three variants, changes were observed at the site of the substituted residue and locally within 5 Å from this. Moreover, all three substitutions resulted in chemical shift changes for residues of the A- and B-strands (in particular of Trp-124 and Tyr-122) and the loop connecting them (Fig. 9, green underlines). Interestingly, the C-terminal membrane proximal residues were also affected in all variants.
FIGURE 9.

Chemical shift analyses of hPRLR-D2 variants. Comparison of the combined amide proton and nitrogen chemical shift differences for hPRLR-D2WT and hPRLR-D2K168A (A), hPRLR-D2L170A (B), and hPRLR-D2L170F (C) are shown. Each substitution site is identified by black arrowheads; prolines and unassigned residues are marked with red dots. The vertical gray bars indicate the A-G strands, the red bar locates the WS motif, and the asterisks identify residues Tyr-122 and Trp-124. The horizontal red line represents the trimmed average chemical shift difference (AVG), and the dotted line the trimmed average plus 1 S.D. (AVG+1σ). Residues with chemical shift differences higher than the trimmed average plus 1 S.D. are marked in red on the structure of unbound hPRLR-D2WT (PDB accession code 2LFG). Residues more than 5 Å of the substituted residue are shown in red sticks. The substituted residues are shown as red space-filling atoms; the green bars indicate regions with chemical shift changes above average plus 1 S.D. and include the A-B strand and loop as well as the C terminus.
Because the chemical shifts from the side chain of Trp-124 were significantly affected in the variants, particularly in hPRLR-D2K168A (Fig. 8C), and because the ellipticity at 230 nm for this variant almost disappeared (Fig. 8A), this suggested the presence of an exciton coupling with the opposite sign. Thus, removal of the lysine side chain in hPRLR-D2K168A would allow room for alterations in the interactions between Tyr-122 and Trp-124, where a rotation of Tyr-122 would lead to the mirror image and hence exciton couplings of the opposite sign. In contrast, in hPRLR-D2L170F the side chain of Trp-124 was mostly unaffected, but an increase in ellipticity at 230 nm was observed. This suggests a stabilization of the interaction between Tyr-122 and Trp-124. By the same reasoning, the L170A substitution would then lead to a destabilization of their interaction and hence a lowering of the 230 nm intensity. This is indeed what was observed and correlates well with the strong destabilization seen in the rPRLR-ECD upon substitution of Phe-170 by alanine (Table 4). We subsequently measured the stabilities by chemical denaturation of hPRLR-D2L170F and hPRLR-D2L170A and compared them with hPRLR-D2WT (Fig. 7B and Table 4) (15). None of these substitutions significantly destabilized the human D2 domain. For hPRLR-D2L170A the slope of the transition (m), which reports on the degree of newly exposed surface area, was smaller than for hPRLR-D2WT, and the midpoint of the transition (Cm) larger. Thus, fewer surfaces were exposed, suggesting that the folded state had some local destabilization around the mutation site, which was compensated for by an increase in entropy, resulting in the same global stability.
In summary, the interface substitutions in both rPRLR and hPRLR strongly affected the local structural geometry in a quartet of highly conserved residues consisting of Tyr-122, Trp-124, Lys-168, and Leu/Phe-170 (Fig. 8B). This quartet connects the E- and B-strands of the receptor ECD. Consistently, structural effects were noticed in the A-strand, B-strand, the AB-loop, and in the C-terminal of the D2 domain, and these latter two parts of the receptor ECD are notably closest to the membrane. Remarkably, the substitution-specific effects on ERK1/2 signaling nicely paralleled the structural alterations of this quartet.
DISCUSSION
Despite the core knowledge obtained over recent years on cytokine receptor activation (3, 29), the molecular mechanisms and the structural details linking ligand binding to intracellular signaling events are still poorly understood. More enigmatic is to comprehend ligand-specific signaling profiles through the exact same receptor. Using the PRLR as a prototypic homodimeric cytokine receptor, we here demonstrate that the receptor interface selectively controls activation of downstream signaling pathways. Specifically, alanine substitution of two interface residues (Lys-168, Leu/Phe-170) left the STAT5 pathway unaffected, although signaling through ERK1/2 was significantly decreased (Fig. 2, D–F). This underlines the critical role of these residues in signaling pathway discrimination. In contrast to position 168, position 170 is occupied by different amino acids in the rat (Phe) versus the human (Leu) PRLR. Remarkably, we showed the following: (i) this position conferred the species with intrinsically different levels of ERK1/2 activation (Figs. 2E versus 3D); (ii) the very same position also conferred hormone species specificity to the receptors (Fig. 4); and (iii) these intrinsic functional properties could be transferred from one receptor species to the other (and vice versa) by changing only the chemistry at this position. Importantly, the effects of substitutions on ERK1/2 activation were inversely correlated to the PRL binding capacity at the cell surface (Fig. 5B) as well as receptor turnover (Fig. 5, C and D), preventing biased interpretation of the results. Thus, together these data identify a key role of position 170 in the control of intrinsic species-specific signaling properties of the PRLR.
A parallel between our findings on the PRLR and those recently reported by Waters and co-workers (30) for the GHR can be made. This group reported that mutations introduced within the F′G′ loop of the GHR-ECD also altered MAPK signaling without affecting the STAT5 pathway (30). Because the regions and residues involved in both receptors are distinct, we aimed to understand the structural mechanisms involved in the PRLR. To that end, NMR and other spectroscopic data were collected on WT and variants of the D2 domains and ECDs from human and rat PRLRs, respectively. The structural data suggested at least two different ways of modulating intracellular signaling. The first involved destabilization of the ECD/D2 scaffold as seen severely for rPRLR-ECDF170A and more locally for hPRLR-D2L170A. The second and most predominant way was through alteration of the local structure around Tyr-122 and Trp-124 as observed for the K168A and L170F substitutions. From solving the NMR structure of hPRLR-D2WT, we observed that Lys-168 and Tyr-122 interacted strongly, e.g. they had a number of mutual strong nuclear Overhauser effects (NOEs) (15). In contrast, we did not observe NOEs from these residues to the ring of Trp-124, indicating that this ring points away from the quartet. Thus, the Lys-168 and Tyr-122 interaction ensures a fixed orientation of the aromatic ring of Tyr-122. When the lysine side chain is removed in hPRLR-D2K168A, the two aromatic rings of Tyr-122 and Trp-124 are free to reorient. This reorientation manifests in diagnostic changes in the CD spectrum revealing exciton couplings with negative ellipticity in hPRLR-D2K168A, canceling the positive one originating from the WS motif. This structure coincides with decreased MAPK signaling (alanine substitutions). When Leu-170 is replaced by a Phe, the interaction between Tyr-122 and Trp-124 is instead stabilized, which is concluded from the increase in the positive exciton coupling. This structure coincides with increased MAPK signaling (human/rat substitutions). Remarkably, alanine substitution at position 170 destabilizes either the entire scaffold (rPRLR) or the local structure (hPRLR), leading to a destabilization of the Tyr-122/Trp-124 interaction. Again, this destabilization coincides with decreased MAPK signaling. The intrinsic structure and interaction network involving this residue quartet (Tyr-122, Trp-124, Lys-168, and Leu/Phe-170) thus seems to tightly regulate the maximal intensity of MAPK signaling triggered by ligand-bound PRLR variants. In the ternary PRL·receptor complex (Fig. 1B), Trp-124 is only involved in the receptor interface from PRLR2 (25); therefore, it is likely that the regulatory mechanism involving the residue quartet only applies to that receptor.
Signaling is tuned by the affinities between the ligand and the receptor complex. These affinities, or more righteously the off-rates (koff), determine the mean lifetime of a given complex (t½ = ln2/koff; mean lifetime = 1/koff), which must be long enough to trigger signaling. The two signaling pathways monitored here differ significantly in the number of steps involved. It is well known that the JAK2 tyrosine kinase is constitutively bound to the PRLR-ICD (31), and that STAT5 recruitment occurs very rapidly once the receptor is tyrosine-phosphorylated (32). In contrast, MAPK signaling requires multiple protein/protein interactions in the upstream cascade (Lyn/Fyn, PLCγ, Grb2, SHC, SOS, Ras, Raf, MEK, and ERK) (30, 33). Accordingly, the time for half-maximal stimulation of STAT5 by the PRLR was recently established as ∼3 times shorter compared with ERK1/2 (∼3 min versus ∼10 min) with a small lag phase for ERK1/2 activation (33). This indicates either that signal relay takes time or that a longer lifetime of the functional PRL·PRLR ternary complex is needed to elicit maximal ERK1/2 activation compared with STAT5 activation, or both. The PRLR interface substitutions selectively altered activation of the ERK1/2 pathway. To demonstrate that this results from different lifetimes of the ternary complexes, precise measurements of the off-rates of PRLR2 interaction (sites 2 + 3) would ideally be needed. However, these dissociation rates are in all cases higher than the limit accessible to SPR measurements (>0.25 s−1) (23–25). If we assume that the difference in Kd values between the variants and the WT receptors (Table 3) originate exclusively from variations in koff (with kon being unaffected by the substitutions), we tentatively set an upper ∼10-fold limit for the differences in the apparent mean lifetime of the ternary complexes involving rPRLRK168A or rPRLRF170A compared with rPRLRWT. In line with this, we would therefore expect that hPRLRL170F, which exhibited a stronger ERK1/2 phosphorylation than hPRLRWT, would have a lower Kd value at sites 2 + 3 and hence result in a longer complex lifetime compared with WT. This was indeed confirmed (Table 3).
The complex lifetime concept was previously proposed as a mechanism for the functional diversity of the multiple type I IFNs (4). For the homodimeric cytokine receptor family, previous studies involving ligands and receptors of the PRL/GH family have supported that a minimal time of homodimer persistence was needed to elicit cellular responses (34–37). As it was subsequently demonstrated that both the GHR (12) and the PRLR (13, 14) are pre-dimerized at the cell membrane, this should translate into minimal lifetime of ternary complex. Although this concept was built on the analysis of global cell responses (e.g. cell proliferation), we suggest a model where the complex lifetime of homodimeric cytokine type 1 receptors discriminates downstream signaling pathways (Fig. 10). The proposed model defines two different ranges of receptor signaling sensitivity. One range, which we term the dynamic range, will not respond to an increase in the complex lifetime as maximum activation has already been reached. In the second range called the regulatory range, any change to the lifetime will impact signaling. With respect to the PRLR (rat or human), the lifetimes of the WT complexes are in the dynamic range for STAT5 activation, although they are within the regulatory range for MAPK activation. This model integrates the intrinsically higher MAPK activation by the rat compared with the human PRLR as well as the modulation (increased for hPRLR or decreased for rPRLR) of MAPK signaling by the interface hot spot variants. Likewise, because STAT5 activation is far within the dynamic range for both species, no effects were observed by the substitutions. This model may also explain why MAPK activation is lost first when unconventional receptor agonists are used (5).
FIGURE 10.

Model of signaling pathway activation as a function of ligand·receptor complex lifetime. For STAT5 activation, the WT lifetimes in this work are within the dynamic range, whereas for MAPK activation, the WT lifetimes are within the regulatory range (see “Discussion” for explanations). This could explain the intrinsically higher MAPK activation by the rat receptor and the modulation observed by the hot spot receptor variants. Likewise, because affinities leading to maximal STAT5 activation are far within the dynamic range, no effects of substitutions on activity were observed. The different PRLR variants examined are indicated in the figure according to the provided legend.
The results presented in this study establish the dimerization interface as a key regulator of signaling pathway discrimination. For the PRLR, we propose a structural mechanism involving a residue quartet among which the residue occupying position 170 appears to play a major regulatory role. The regulation by the four residues seems to originate from the PRLR2 receptor in the ternary complex. Although this residue quartet is not conserved among homodimeric type 1 cytokine receptors (38), it is striking that the residue topologically equivalent to position 170 in the GHR, Tyr-200, is one of the four hot spot residues defining the central core of the GHR dimerization interface (39). This clearly suggests common molecular mechanisms. Whether GHRTyr-200 also specifically tunes MAPK signaling is unknown because the functional importance of this residue was revealed using a c-fos luciferase reporter construct that integrates both STAT5 and ERK1/2 signaling (40). With respect to the PRLR, it is noteworthy that three of the four residues constituting the quartet were fully conserved across evolution, whereas the regulatory position 170 harbors either a Phe (rat, mouse, cow, sheep, red deer, and possum) or a Leu (primates, pig, dog, and rabbit) (41). This raises the question of the potential physiological benefit of having intrinsically higher levels of ERK1/2 activation in some species versus others. Finally, the proposed relationship between complex lifetime, signaling potency, and discrimination may also have pathophysiological relevance. In tumor contexts, the high glycolytic activity and proton extrusion by cancer cells is known to acidify the extracellular milieu (42). Knowing that PRL binding to the PRLR is strongly pH-dependent (43–45), the reduction of complex lifetime due to tumor acidification should reduce signaling to such an extent that it ultimately brings STAT5 activation into the regulatory range (Fig. 10). In good agreement, a recent study showed that acidosis, pH 6.5, disrupted PRLR signaling in various breast cancer cell lines, providing a mechanistic rationale for the loss of STAT5 phosphorylation frequently observed in aggressive breast cancers (42). Whether STAT5 and ERK1/2 pathways exhibit differential sensitivity to progressive acidosis remains unknown, but such results may further corroborate our model (Fig. 10).
Finally, from a decomposition of function, structure, and affinities of PRLRs and interface variants, we propose that within homodimeric type 1 cytokine receptor ECDs, so-called signaling hot spot residues control the extent and duration of the structural propagations that are necessary to disseminate the signal from the ECD to the ICD. One set of these residues is part of the dimerization interface of the ECDs, and they play key roles in determining the off-rates (koff) and hence the complex lifetime. The identification of such residues in different cytokine receptors will have huge potential. Generally, in light of our findings they will significantly affect future structure-directed drug development strategies aimed at providing pathway-selective treatment strategies.
Acknowledgments
We are grateful to Signe A. Sjørup for generation of mutant plasmids, to Corinne Cordier for help with FACS experiments, to Philippe Monget for sequence comparisons, to Lucila Sackmann Sala for help with statistical analyses, and to Johan G. Olsen for discussions.
This work was supported by the Danish Cancer Society grants (to B. B. K.), Danish Research Councils for Health and Disease Grants 09-072179 and 12-125862 (to B. B. K.), the Novo Nordic Foundation (to B. B. K.), Ligue Contre le Cancer Grant RS11/75-26 (to I. B.), University Paris Descartes Grant 990 UMRS 845/2011/02 (to V. G. and I. B.), and the Agence Nationale de la Recherche Grant ANR-07-PCVI-0029 (to V. G.).
- GH
- growth hormone
- GHR
- GH receptor
- PDB
- Protein Data Bank
- EPO
- erythropoietin
- EPOR
- EPO receptor
- ECD
- extracellular domain
- PRL
- prolactin
- PRLR
- PRL receptor
- r
- rat
- h
- human
- b
- bovine
- SPR
- surface plasmon resonance
- ICD
- intracellular domain
- ANOVA
- analysis of variance
- GdmCl
- guanidinium chloride.
REFERENCES
- 1. Karnitz L. M., Abraham R. T. (1995) Cytokine receptor signaling mechanisms. Curr. Opin. Immunol. 7, 320–326 [DOI] [PubMed] [Google Scholar]
- 2. Chang F., Steelman L. S., Lee J. T., Shelton J. G., Navolanic P. M., Blalock W. L., Franklin R. A., McCubrey J. A. (2003) Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: potential targeting for therapeutic intervention. Leukemia 17, 1263–1293 [DOI] [PubMed] [Google Scholar]
- 3. Stark G. R., Darnell J. E., Jr. (2012) The JAK-STAT pathway at twenty. Immunity 36, 503–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Piehler J., Thomas C., Garcia K. C., Schreiber G. (2012) Structural and dynamic determinants of type I interferon receptor assembly and their functional interpretation. Immunol. Rev. 250, 317–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li W., Lan H., Liu H., Fu Z., Yang Y., Han W., Guo F., Liu Y., Zhang H., Liu J., Zheng X. (2013) The activation and differential signalling of the growth hormone receptor induced by pGH or anti-idiotypic monoclonal antibodies in primary rat hepatocytes. Mol. Cell. Endocrinol. 376, 51–59 [DOI] [PubMed] [Google Scholar]
- 6. Millot G. A., Vainchenker W., Duménil D., Svinarchuk F. (2004) Differential signalling of NH2-terminal flag-labelled thrombopoietin receptor activated by TPO or anti-FLAG antibodies. Cell. Signal. 16, 355–363 [DOI] [PubMed] [Google Scholar]
- 7. Liu Z., Stoll V. S., Devries P. J., Jakob C. G., Xie N., Simmer R. L., Lacy S. E., Egan D. A., Harlan J. E., Lesniewski R. R., Reilly E. B. (2007) A potent erythropoietin-mimicking human antibody interacts through a novel binding site. Blood 110, 2408–2413 [DOI] [PubMed] [Google Scholar]
- 8. Moucadel V., Constantinescu S. N. (2005) Differential STAT5 signaling by ligand-dependent and constitutively active cytokine receptors. J. Biol. Chem. 280, 13364–13373 [DOI] [PubMed] [Google Scholar]
- 9. Bazan J. F. (1989) A novel family of growth factor receptors: a common binding domain in the growth hormone, prolactin, erythropoietin and IL-6 receptors, and p75 IL-2 receptor b-chain. Biochem. Biophys. Res. Commun. 164, 788–795 [DOI] [PubMed] [Google Scholar]
- 10. Bazan J. F. (1990) Structural design of molecular evolution of a cytokine receptor superfamily. Proc. Natl. Acad. Sci. U.S.A. 87, 6934–6938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Olsen J. G., Kragelund B. B. (2014) Who climbs the tryptophan ladder? On the structure and function of the WSXWS motif in cytokine receptors and thrombospondin repeats. Cytokine Growth Factor Rev. 25, 337–341 [DOI] [PubMed] [Google Scholar]
- 12. Brown R. J., Adams J. J., Pelekanos R. A., Wan Y., McKinstry W. J., Palethorpe K., Seeber R. M., Monks T. A., Eidne K. A., Parker M. W., Waters M. J. (2005) Model for growth hormone receptor activation based on subunit rotation within a receptor dimer. Nat. Struct. Mol. Biol. 12, 814–821 [DOI] [PubMed] [Google Scholar]
- 13. Tallet E., Fernandez I., Zhang C., Salsac M., Gregor N., Ayoub M. A., Pin J. P., Trinquet E., Goffin V. (2011) Investigation of prolactin receptor activation and blockade using time-resolved fluorescence resonance energy transfer. Front. Endocrinol. 2, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gadd S. L., Clevenger C. V. (2006) Ligand-independent dimerization of the human prolactin receptor isoforms: functional implications. Mol. Endocrinol. 20, 2734–2746 [DOI] [PubMed] [Google Scholar]
- 15. Dagil R., Knudsen M. J., Olsen J. G., O'Shea C., Franzmann M., Goffin V., Teilum K., Breinholt J., Kragelund B. B. (2012) The WSXWS motif in cytokine receptors is a molecular switch involved in receptor activation: insight from structures of the prolactin receptor. Structure 20, 270–282 [DOI] [PubMed] [Google Scholar]
- 16. Goffin V., Kinet S., Ferrag F., Binart N., Martial J. A., Kelly P. A. (1996) Antagonistic properties of human prolactin analogs that show paradoxical agonistic activity in the Nb2 bioassay. J. Biol. Chem. 271, 16573–16579 [DOI] [PubMed] [Google Scholar]
- 17. Teilum K., Hoch J. C., Goffin V., Kinet S., Martial J. A., Kragelund B. B. (2005) Solution structure of human prolactin. J. Mol. Biol. 351, 810–823 [DOI] [PubMed] [Google Scholar]
- 18. Bernichtein S., Kayser C., Dillner K., Moulin S., Kopchick J. J., Martial J. A., Norstedt G., Isaksson O., Kelly P. A., Goffin V. (2003) Development of pure prolactin receptor antagonists. J. Biol. Chem. 278, 35988–35999 [DOI] [PubMed] [Google Scholar]
- 19. Goffin V., Bogorad R. L., Touraine P. (2010) Identification of gain-of-function variants of the human prolactin receptor. Methods Enzymol. 484, 329–355 [DOI] [PubMed] [Google Scholar]
- 20. Zhang C., Cherifi I., Nygaard M., Haxholm G. W., Bogorad R. L., Bernadet M., England P., Broutin I., Kragelund B. B., Guidotti J. E., Goffin V. (2015) Residue 146 regulates prolactin receptor-folding, basal activity and ligand-responsiveness: Potential implications in breast tumorigenesis. Mol. Cell. Endocrinol. 401, 173–188 [DOI] [PubMed] [Google Scholar]
- 21. Maxwell K. L., Wildes D., Zarrine-Afsar A., De Los Rios M. A., Brown A. G., Friel C. T., Hedberg L., Horng J. C., Bona D., Miller E. J., Vallée-Bélisle A., Main E. R., Bemporad F., Qiu L., Teilum K., et al. (2005) Protein-folding: defining a “standard” set of experimental conditions and a preliminary kinetic data set of two-state proteins. Protein Sci. 14, 602–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 23. Broutin I., Jomain J. B., Tallet E., van Agthoven J., Raynal B., Hoos S., Kragelund B. B., Kelly P. A., Ducruix A., England P., Goffin V. (2010) Crystal structure of an affinity-matured prolactin complexed to its dimerized receptor reveals the topology of hormone binding site 2. J. Biol. Chem. 285, 8422–8433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jomain J. B., Tallet E., Broutin I., Hoos S., van Agthoven J., Ducruix A., Kelly P. A., Kragelund B. B., England P., Goffin V. (2007) Structural and thermodynamic bases for the design of pure prolactin receptor antagonists. X-ray structure of Del1–9-G129R-hPRL. J. Biol. Chem. 282, 33118–33131 [DOI] [PubMed] [Google Scholar]
- 25. van Agthoven J., Zhang C., Tallet E., Raynal B., Hoos S., Baron B., England P., Goffin V., Broutin I. (2010) Structural characterization of the stem-stem dimerization interface between prolactin receptor chains complexed with the natural hormone. J. Mol. Biol. 404, 112–126 [DOI] [PubMed] [Google Scholar]
- 26. Courtillot C., Chakhtoura Z., Bogorad R., Genestie C., Bernichtein S., Badachi Y., Janaud G., Akakpo J. P., Bachelot A., Kuttenn F., Goffin V., Touraine P., and Benign Breast Diseases Study Group (2010) Characterization of two constitutively active prolactin receptor variants in a cohort of 95 women with multiple breast fibroadenomas. J. Clin. Endocrinol. Metab. 95, 271–279 [DOI] [PubMed] [Google Scholar]
- 27. Bernichtein S., Jeay S., Vaudry R., Kelly P. A., Goffin V. (2003) New homologous bioassays for human lactogens show that agonism or antagonism of various analogs is a function of assay sensitivity. Endocrine 20, 177–190 [DOI] [PubMed] [Google Scholar]
- 28. Bogorad R. L., Courtillot C., Mestayer C., Bernichtein S., Harutyunyan L., Jomain J. B., Bachelot A., Kuttenn F., Kelly P. A., Goffin V., Touraine P. (2008) Identification of a gain-of-function mutation of the prolactin receptor in women with benign breast tumors. Proc. Natl. Acad. Sci. U.S.A. 105, 14533–14538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Atanasova M., Whitty A. (2012) Understanding cytokine and growth factor receptor activation mechanisms. Crit. Rev. Biochem. Mol. Biol. 47, 502–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rowlinson S. W., Yoshizato H., Barclay J. L., Brooks A. J., Behncken S. N., Kerr L. M., Millard K., Palethorpe K., Nielsen K., Clyde-Smith J., Hancock J. F., Waters M. J. (2008) An agonist-induced conformational change in the growth hormone receptor determines the choice of signalling pathway. Nat. Cell Biol. 10, 740–747 [DOI] [PubMed] [Google Scholar]
- 31. Lebrun J. J., Ali S., Sofer L., Ullrich A., Kelly P. A. (1994) Prolactin-induced proliferation of Nb2 cells involves tyrosine phosphorylation of the prolactin receptor and its associated tyrosine kinase JAK2. J. Biol. Chem. 269, 14021–14026 [PubMed] [Google Scholar]
- 32. Lebrun J. J., Ali S., Goffin V., Ullrich A., Kelly P. A. (1995) A single phosphotyrosine residue of the prolactin receptor is responsible for activation of gene transcription. Proc. Natl. Acad. Sci. U.S.A. 92, 4031–4035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Aksamitiene E., Achanta S., Kolch W., Kholodenko B. N., Hoek J. B., Kiyatkin A. (2011) Prolactin-stimulated activation of ERK1/2 mitogen-activated protein kinases is controlled by PI3-kinase/Rac/PAK signaling pathway in breast cancer cells. Cell. Signal. 23, 1794–1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pearce K. H., Jr., Cunningham B. C., Fuh G., Teeri T., Wells J. A. (1999) Growth hormone binding affinity for its receptor surpasses the requirements for cellular activity. Biochemistry 38, 81–89 [DOI] [PubMed] [Google Scholar]
- 35. Wan Y., McDevitt A., Shen B., Smythe M. L., Waters M. J. (2004) Increased site 1 affinity improves biopotency of porcine growth hormone. Evidence against diffusion dependent receptor dimerization. J. Biol. Chem. 279, 44775–44784 [DOI] [PubMed] [Google Scholar]
- 36. Helman D., Herman A., Paly J., Livnah O., Elkins P. A., de Vos A. M., Djiane J., Gertler A. (2001) Mutations of ovine and bovine placental lactogens change, in different ways, the biological activity mediated through homologous and heterologous lactogenic receptors. J. Endocrinol. 169, 43–54 [DOI] [PubMed] [Google Scholar]
- 37. Gertler A., Djiane J. (2002) Mechanism of ruminant placental lactogen action: molecular and in vivo studies. Mol. Genet. Metab. 75, 189–201 [DOI] [PubMed] [Google Scholar]
- 38. Boulay J. L., O'Shea J. J., Paul W. E. (2003) Molecular phylogeny within type I cytokines and their cognate receptors. Immunity 19, 159–163 [DOI] [PubMed] [Google Scholar]
- 39. Bernat B., Pal G., Sun M., Kossiakoff A. A. (2003) Determination of the energetics governing the regulatory step in growth hormone-induced receptor homodimerization. Proc. Natl. Acad. Sci. U.S.A. 100, 952–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen C., Brinkworth R., Waters M. J. (1997) The role of receptor dimerization domain residues in growth hormone signaling. J. Biol. Chem. 272, 5133–5140 [DOI] [PubMed] [Google Scholar]
- 41. Li Y., Wallis M., Zhang Y. P. (2005) Episodic evolution of prolactin receptor gene in mammals: coevolution with its ligand. J. Mol. Endocrinol. 35, 411–419 [DOI] [PubMed] [Google Scholar]
- 42. Yang N., Liu C., Peck A. R., Girondo M. A., Yanac A. F., Tran T. H., Utama F. E., Tanaka T., Freydin B., Chervoneva I., Hyslop T., Kovatich A. J., Hooke J. A., Shriver C. D., Rui H. (2013) Prolactin-Stat5 signaling in breast cancer is potently disrupted by acidosis within the tumor microenvironment. Breast Cancer Res. 15, R73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kulkarni M. V., Tettamanzi M. C., Murphy J. W., Keeler C., Myszka D. G., Chayen N. E., Lolis E. J., Hodsdon M. E. (2010) Two independent histidines, one in human prolactin and one in its receptor, are critical for pH-dependent receptor recognition and activation. J. Biol. Chem. 285, 38524–38533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hansen M. J., Olsen J. G., Bernichtein S., O'Shea C., Sigurskjold B. W., Goffin V., Kragelund B. B. (2010) Development of prolactin receptor antagonists with reduced pH-dependence of receptor binding. J. Mol. Recognit. 124, 533–547 [DOI] [PubMed] [Google Scholar]
- 45. Keeler C., Jablonski E. M., Albert Y. B., Taylor B. D., Myszka D. G., Clevenger C. V., Hodsdon M. E. (2007) The kinetics of binding human prolactin, but not growth hormone, to the prolactin receptor vary over a physiologic pH range. Biochemistry 46, 2398–2410 [DOI] [PubMed] [Google Scholar]