

Background: The B subunit of factor XIII (FXIII-B) was previously thought to inhibit fibrin cross-linking by preventing thrombin-mediated activation of the A subunit (FXIII-A).
Results: FXIII-B accelerated FXIII-A activation and subsequent fibrin cross-linking by formation of an FXIII-A, fibrinogen, and thrombin ternary complex.
Conclusion: FXIII-B accelerated fibrin cross-linking.
Significance: FXIII-B deficiency leads to impaired fibrin stabilization.
Keywords: Blood, Coagulation Factor, Fibrin, Protein Cross-linking, Transglutaminase, Bleeding Disease
Abstract
Covalent cross-linking of fibrin chains is required for stable blood clot formation, which is catalyzed by coagulation factor XIII (FXIII), a proenzyme of plasma transglutaminase consisting of catalytic A (FXIII-A) and non-catalytic B subunits (FXIII-B). Herein, we demonstrate that FXIII-B accelerates fibrin cross-linking. Depletion of FXIII-B from normal plasma supplemented with a physiological level of recombinant FXIII-A resulted in delayed fibrin cross-linking, reduced incorporation of FXIII-A into fibrin clots, and impaired activation peptide cleavage by thrombin; the addition of recombinant FXIII-B restored normal fibrin cross-linking, FXIII-A incorporation into fibrin clots, and activation peptide cleavage by thrombin. Immunoprecipitation with an anti-fibrinogen antibody revealed an interaction between the FXIII heterotetramer and fibrinogen mediated by FXIII-B and not FXIII-A. FXIII-B probably binds the γ-chain of fibrinogen with its D-domain, which is near the fibrin polymerization pockets, and dissociates from fibrin during or after cross-linking between γ-chains. Thus, FXIII-B plays important roles in the formation of a ternary complex between proenzyme FXIII, prosubstrate fibrinogen, and activator thrombin. Accordingly, congenital or acquired FXIII-B deficiency may result in increased bleeding tendency through impaired fibrin stabilization due to decreased FXIII-A activation by thrombin and secondary FXIII-A deficiency arising from enhanced circulatory clearance.
Introduction
Fibrinogen (Fbg),2 a major structural clotting factor, is composed of three types of polypeptide chains, α, β, and γ, that are organized as dimers consisting of one central E- and two outer D-domains. Thrombin cleaves fibrinopeptides A and B in the E-domain of Fbg, thereby converting Fbg to fibrin (Fbn). Fbn spontaneously polymerizes by non-covalent binding between polymerization sites/knobs “A” and “B” (the exposed N termini of α- and β-chains in an E-domain, respectively) of one Fbn molecule and polymerization pockets/holes “a” and “b” in the C-terminal region of γ- and β-chains, respectively, in a D-domain of the other Fbn molecule (1–3). Clots formed by self-assembly of Fbn are insufficient for hemostasis because of their mechanical weakness and susceptibility to proteolysis; a stable hemostatic clot is formed by covalent cross-linking between the γ- and α-chains of Fbn and by cross-linking of the α-chain with α2-plasmin inhibitor, which is catalyzed by coagulation factor XIII (FXIII).
FXIII is a proenzyme of plasma transglutaminase consisting of two enzymatic A subunits (FXIII-A) and two non-catalytic B subunits (FXIII-B). FXIII-A is composed of an N-terminal activation peptide (AP), β-sandwich, core (including catalytic residue Cys-314), and barrel 1 and 2 domains (4). FXIII-B consists of 10 tandem repeat structures called “sushi domains” (5), of which the fourth and ninth contribute to homodimer assembly and the first participates in heterotetramer assembly with FXIII-A (6). Activation of FXIII is a multistep process. The initial step is cleavage of AP by thrombin, which converts FXIII-A to a catalytically inactive intermediate, FXIII-A′ (A2′B2 complex) (7). FXIII-B dissociates from the A2′B2 complex in the presence of calcium, and finally the active form of FXIII (FXIIIa) is yielded. Fbn increases the rate of AP cleavage by thrombin (7, 8) and facilitates dissociation of the A2′B2 complex (7, 9).
There are approximately equal amounts of FXIII-B in the “bound” form (as A2B2) and “free” form (5). In contrast, essentially all FXIII-A (99%) exists in a complex with FXIII-B (10). It has been suggested that FXIII-B is necessary for stable circulation of FXIII-A in blood because it has been observed that FXIII-A levels are markedly low in the plasma of patients with FXIII-B deficiency (11–13) or of FXIII-B knock-out mice (14), although a normal level of FXIII-A exists inside cells, such as monocytes/macrophages and megakaryocytes/platelets of FXIII-B-deficient individuals. However, FXIII-B has long been thought not to contribute to catalytic action of FXIII because it dissociates from FXIIIa at the final step of FXIII activation.
In the present study, we found that cross-linking of Fbn in plasma was delayed in the absence of FXIII-B even in the presence of FXIII-A. We also revealed that FXIII-A did not associate with Fbg in FXIII-B-deficient plasma, although it could directly bind to Fbg under plasma-free conditions (i.e. in a purified system). Herein, we demonstrate that FXIII-B accelerates cross-linking of Fbn via direct interaction with FXIII-A and Fbg.
EXPERIMENTAL PROCEDURES
Recombinant FXIII-A (rFXIII-A) was a kind gift from Zymogenetics (Seattle, WA). Recombinant FXIII-B (rFXIII-B) and its truncation mutants were expressed in a baculovirus system and purified as described previously (6). Anti-FXIII-A monoclonal antibody (mAb) was obtained from Prof. Reed (Massachusetts General Hospital, Boston, MA). Anti-FXIII-A polyclonal antibody (pAb) was generated in-house and affinity-purified using rFXIII-A. Anti-FXIII-B antibody was purchased from Nordic Immunological Laboratories (AX Eindhoven, The Netherlands). Immunoglobulin G (IgG) of anti-FXIII-A and anti-FXIII-B antibodies was purified using Protein A-Sepharose (GE Healthcare) and biotinylated using the ECL protein biotinylation module (GE Healthcare) or coupled to CNBr-activated Sepharose 4B (GE Healthcare). Human Fbg was purchased from Sigma-Aldrich, and contaminating FXIII-B in Fbg was removed using anti-FXIII-B-Sepharose. Rabbit anti-human Fbg antibody and Protein A-coated Staphylococcus aureus (PANSORBIN) were purchased from Merck KGaA. Mouse anti-Fbg mAb (1F3) was obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX). Bovine thrombin, human plasmin, and chymotrypsin were purchased from Sigma-Aldrich. Horseradish peroxidase (HRP)-conjugated anti-rabbit IgG and HRP-conjugated streptavidin were obtained from GE Healthcare. A tetramethylbenzidine peroxidase substrate kit was purchased from Bio-Rad. Immobilon Western chemiluminescent HRP substrate and Zip-Tip C18 were obtained from Millipore (Billerica, MA). Iodoacetamide and trypsin were obtained from Wako Pure Chemical Industries (Osaka, Japan). The Ready-To-Glow secreted luciferase reporter system was purchased from Clontech. The mammalian expression vector pcDNA3 was obtained from Invitrogen.
Fbn Cross-linking in Plasma
FXIII-A- or FXIII-B-depleted plasma was prepared by removal of FXIII-A or -B from pooled normal human plasma using anti-FXIII-A- or anti-FXIII-B-Sepharose, respectively. rFXIII-A (5 μg/ml) and/or rFXIII-B (10 μg/ml) was added to the plasma, and a 10-μl aliquot of the plasma was reacted with 10 units/ml bovine thrombin and 10 mm CaCl2 in a 20-μl mixture at room temperature for the appropriate times. The reaction was terminated by the addition of 50 mm EDTA. Fbn clots were separated from the supernatant by centrifugation, washed two times with 1 ml of 20 mm Tris-HCl (pH 7.5) and 150 mm NaCl (TBS), and dissolved in 40 μl of 8 m urea, 1% SDS, and 50 mm Tris-HCl (pH 8.0) (UST buffer). The sample was boiled with 40 μl of 2% SDS, 0.125 m Tris-HCl (pH 6.8), 15% glycerol, 5% 2-mercaptoethanol, and 0.02% bromphenol blue (SDS-reducing buffer) and electrophoresed using a 10% polyacrylamide gel containing 0.1% SDS. The gel was stained with Coomassie Brilliant Blue R-250. Densitometric analysis was performed using a gel documentation system AE-6932GXCF and CS Analyzer version 2.0 (ATTO, Tokyo, Japan).
Cross-linking Reaction of Purified Human Fbg
FXIII-B-free human Fbg (5 mg/ml) was mixed with 5 μg/ml rFXIII-A with or without 10 μg/ml rFXIII-B. The reaction was started by the addition of 10 units/ml bovine thrombin and 10 mm CaCl2 in a total volume of 20 μl and was terminated by the addition of UST buffer. The sample was boiled with an equal volume of SDS-reducing buffer and electrophoresed on a 10% polyacrylamide gel containing 0.1% SDS.
ELISA for FXIII Remaining in the Supernatant after Cross-linking Reaction
Anti-FXIII-A mAb was immobilized to a 96-well plate for the measurement of FXIII-A, and anti-FXIII-B IgG was used for measurement of FXIII-B. The supernatant from the cross-linking reaction was diluted 1:2,000 using TBS containing 2% bovine serum albumin (BSA), and 0.1 ml of the dilution was incubated in an anti-FXIII-A-mAb- or anti-FXIII-B-IgG-coated plate for 2 h at room temperature. The plate was washed five times with TBS containing 0.1% Tween 20 (TBS-T). To determine FXIII-A and A2B2 complex, anti-FXIII-A pAb and anti-FXIII-B antiserum, respectively, were reacted, followed by reaction with HRP-conjugated anti-rabbit IgG. To determine FXIII-B, biotinylated anti-FXIII-B IgG was reacted followed by reaction with HRP-conjugated streptavidin. The plate was finally reacted with tetramethylbenzidine substrate, and the reaction was terminated by the addition of 0.5 m sulfuric acid. The absorbance at 450 nm was measured.
Western Blotting
The supernatant and Fbn clot of cross-linking reaction that dissolved in UST buffer were boiled with an equal volume of SDS-reducing buffer for the detection of FXIII-A or SDS-nonreducing buffer (SDS-reducing buffer without 2-mercaptoethanol) for FXIII-B. Proteins were electrophoresed using an 8% polyacrylamide gel containing 0.1% SDS and transferred to a nitrocellulose membrane. The membrane was reacted with anti-FXIII-A pAb or anti-FXIII-B antiserum followed by reaction with HRP-conjugated anti-rabbit IgG. Detection was achieved using Immobilon Western chemiluminescent HRP substrate.
Binding of FXIII to Fbg-Sepharose
Fbg-Sepharose (1 mg Fbg/ml) was prepared by coupling CNBr-activated Sepharose 4B with FXIII-B-free Fbg. rFXIII-A (1 μg) and/or rFXIII-B (1 μg) were reacted with 10 μl of Fbg-Sepharose at 4 °C for 1 h. The resin was recovered by centrifugation, washed three times with TBS, and boiled with SDS-nonreducing buffer. Western blotting was conducted using biotinylated anti-FXIII-A and anti-FXIII-B antibodies and HRP-conjugated streptavidin.
ELISA for rFXIII-A and/or rFXIII-B Binding to Fbg
One microgram of Fbg was immobilized on a 96-well plate. One microgram of rFXIII-A and/or rFXIII-B was incubated in the plate at 4 °C for 1 h. The plate was washed five times with TBS and reacted with anti-FXIII-A pAb or anti-FXIII-B antiserum followed by reaction with HRP-conjugated anti-rabbit IgG. The plate was finally reacted with tetramethylbenzidine substrate, and the reaction was terminated by the addition of 0.5 m sulfuric acid. The absorbance at 450 nm was measured.
Immunoprecipitation
Normal, FXIII-A-depleted, or rFXIII-A-containing FXIII-B-depleted plasma was pretreated with Protein A-Sepharose to remove immunoglobulin (Ig). Anti-Fbg antibody was added to Ig-depleted plasma and incubated at room temperature for 1 h. Protein A-Sepharose was then added to the plasma and rotated at 4 °C for 1 h. The resin was collected by centrifugation, washed two times with TBS, and boiled with SDS-nonreducing buffer. FXIII-A and FXIII-B in the immunoprecipitate were detected using biotinylated anti-FXIII-A and anti-FXIII-B antibodies, respectively, and HRP-conjugated streptavidin.
ELISA of FXIII Bound to Fbg in Plasma
Anti-Fbg mAb was immobilized on a 96-well plate. Ten microliters of normal, FXIII-A-depleted, or rFXIII-A-containing FXIII-B-depleted plasma was incubated in the plate at room temperature for 1 h. Detection of FXIII-A or FXIII-B bound to the anti-Fbg mAb-coated plate was performed as described above.
Preparation of Metridia Luciferase (MetLuc) Fused with FXIII-B sushi (BS) Domains
cDNA for MetLuc and each sushi domain of FXIII-B was amplified by PCR using the primers listed in Table 1. Sushi domain cDNA was ligated to the 3′-end of MetLuc cDNA with an XhoI endonuclease site, and the fusion cDNA was inserted into the BamHI and XbaI sites of expression vector pcDNA3. The expression vector was transfected into baby hamster kidney cells by the calcium phosphate precipitation method. The medium was changed to serum-free Dulbecco's modified Eagle's medium 24 h after transfection, and cells were cultured for 24 h. Because the expressed fusion protein (MetLuc-BS) was secreted, its cultured medium was harvested and used as a MetLuc-BS preparation.
TABLE 1.
Primers for the construction of secretory luciferase (MetLuc) fused with the Sushi domain of FXIII-B
| Sushi domain | Sense primera | Antisense primerb |
|---|---|---|
| 1st | ggagaactcgaggcagaagagaaaccctgt | gtcatctagagttcattttttgaagcacct |
| 2nd | aggtgcctcgagaaatgcactaagcctgac | aggtctagatcacgtttcatgttctttcct |
| 3rd | aaagaactcgagacgtgtttggctcctgaa | taatctagatcattttggtgtgagagacca |
| 4th | tggtctctcgagccaaaatgtaccaaatta | cagatctagaggaggtcatctgtttcttct |
| 5th | gaactcgagagaaacagatgtcctcctcca | tggtctagatcaggctaccttctcctgtcc |
| 6th | ggactcgagaaggtagcctgtgaggaacca | aggtctagatcaattctcattattttcaac |
| 7th | gaaaatctcgagaattgtaagcatcctcct | cacatctagagttcatggttccaagcaaac |
| 8th | gtttgcctcgagccatgtactgttaatgtg | aggtctagatgttcacattcctttagattc |
| 9th | gaactcgagggaatgtgcacatctcctcct | agttctagataatgttcatggctctaaaca |
| 10th | ttgtgtctcgagccatgcacatta | gccatctagagtcatgttcttaagggttc |
| MetLuc | ggtcggatccatggacatcaaggtgc | gcggcctcgagcctgtcgccggccatgccd |
a The sense primer for each sushi domain of FXIII-B included an XhoI endonuclease site, shown in italic type.
b The antisense primer for each sushi domain of FXIII-B included a translation termination codon (underlined) and an XbaI endonuclease site (italic type).
c The sense primer for MetLuc contained a BamHI endonuclease site shown in italic type.
d The antisense primer for MetLuc was mutated at the translation termination codon and introduced an XhoI endonuclease site.
Binding of MetLuc-BSs to FXIII-A or Fbg
Each MetLuc-BS preparation was diluted with serum-free medium to adjust MetLuc activity among constructs. One milliliter of a diluted MetLuc sample was incubated with or without 1 μg of rFXIII-A or Fbg in TBS containing 2% BSA at room temperature for 30 min. rFXIII-A or Fbg was recovered by immunoprecipitation using anti-FXIII-A or anti-Fbg antibody and PANSORBIN. The precipitate was washed three times with TBS-T. The MetLuc activity of the precipitate was measured using Ready-To-Glow secreted luciferase substrate.
Kinetic Analysis of Fbg Binding of rFXIII-B
Various concentrations of wild-type, FXIII-B*3 polymorphic variant, or truncated rFXIII-B were incubated with Fbg immobilized on a 96-well plate at room temperature for 1 h, and ELISA for Fbg-bound FXIII-B was performed as described above. The kinetics of Fbg binding was analyzed using the software PRISM version 6.0 (GraphPad Software, Inc., La Jolla, CA) with a fitting model for nonlinear regression of one site-total saturation binding.
Plasmin Digestion of Fbg
FXIII-B-free Fbg (50 μg) was digested with 0.5 μg of plasmin in the absence or the presence of 5 μg of rFXIII-B at 37 °C for 1 h. The reaction was terminated by boiling with an equal volume of SDS-non-reducing or -reducing buffer. The sample was electrophoresed using an 8% polyacrylamide gel containing 0.1% SDS followed by staining with Coomassie Brilliant Blue.
Nanoflow Liquid Chromatography-Tandem Mass Spectrometry (NanoLC-MS/MS)
Coomassie Brilliant Blue-stained protein bands of interest were excised, reductively alkylated, and digested with trypsin as described previously (15, 16). The in-gel digestion was also performed with chymotrypsin (10 μg/ml). The resulting peptides were extracted twice with 5% trifluoroacetic acid containing 50% acetonitrile for 30 min, concentrated using a SpeedVac concentrator, and desalted using a Zip-Tip C18 pipette tip. The desalted peptide solution was diluted 10-fold with solvent A (0.1% formic acid) and analyzed by nanoLC-MS/MS. Separation of the peptides was done using a nanoflow system (EASY-nLC 1000, Thermo Scientific, Hudson, NH) at a flow rate of 300 nl/min on a nanocapillary column (NTTC-360/75–3, Nikkyo Technos, Tokyo, Japan) with a gradient from 0 to 60% solvent B (99.9% acetonitrile, 0.1% formic acid) over 30 min and then to 100% solvent B over 5 min. The nanoflow system was connected to a quadrupole Orbitrap mass spectrometer (Q-Exactive, Thermo Scientific) equipped with a nanoelectrospray emitter. The mass spectrometer was operated in a data-dependent mode to automatically switch between MS and MS/MS acquisition. Survey full-scan spectra (m/z 350–1800) were acquired in the Orbitrap. The 10 most intense ions (intensity threshold, 1.6E+06) were sequentially isolated and fragmented by higher energy C-trap dissociation (17) at 28% normalized collision energy. Peptides with charge state +1 were excluded from fragmentation. Fragment spectra were also acquired in the Orbitrap. A lock mass ion from ambient air (protonated ion of polycyclodimethylsiloxane, m/z 445.12003) was used for internal calibration of measurements.
Peptide Identification
Raw files were searched against the Swiss-Prot human database (542,503 sequences) using Proteome Discoverer (version 1.4; Thermo Scientific) with Mascot (version 2.3; Matrix Science). Mascot defined “semi-specific cleavage” as specific cleavage at one terminus, but where the other terminus may be the result of nonspecific cleavage (see the Matrix Science Web site). Substrates for plasmin and trypsin contain Lys or Arg at the P1 site, but those for chymotrypsin do not. In this study, we identified plasmin-digested protein/peptide bands by in-gel digestion using trypsin or chymotrypsin. To identify plasmin-digested peptides prior to tryptic or chymotryptic digestion, we set the enzyme to trypsin and the semi-specific cleavage to chymotrypsin (termed semi-chymotrypsin) with maximum missed cleavage sites of 2 and 3, respectively. We also set a peptide tolerance of 5 ppm; an MS/MS tolerance of 0.02 Da; and variable modifications for carbamidomethyl cysteine, N-terminal pyroglutaminate, and N-terminal pyrocarbamidomethyl cysteine. The false discovery rate for the identity threshold was in all cases 0% as estimated by the Mascot decoy database function. Peptides with an expectation value less than 0.01 are listed in Tables 2–4.
TABLE 2.
Fibrinogen γ chain (P02679; FIBG_HUMAN)-derived peptides identified by in-gel digestion of γ1
Peptides with expectation values (column 11) less than 0.01 are listed. “Start” (column 1) and “End” (column 2) indicate amino acid residue numbers from the N termini of the mature fibrinogen γ-chain. The enzyme used for in-gel digestion and the number of miscleavages in each reaction are shown in columns 3 and 4, respectively. A peptide whose N- and C-terminal cleavage sites are chymotrypsin FLWY and trypsin KR, respectively, is represented in a pale gray box. “Mr (calc)” (column 7) represents the theoretical monoisotopic molecular mass (Da) based on the peptide sequence. The Mascot score value (column 9) exceeding an identity threshold (column 10) is indicated in boldface type. N-term, N-terminal flanking amino acid; C-term, C-terminal flanking amino acid; Carbamidomethyl (C), carbamidomethyl cysteine; Gln>pyro-Glu (N-term Q), N-terminal pyroglutaminate.
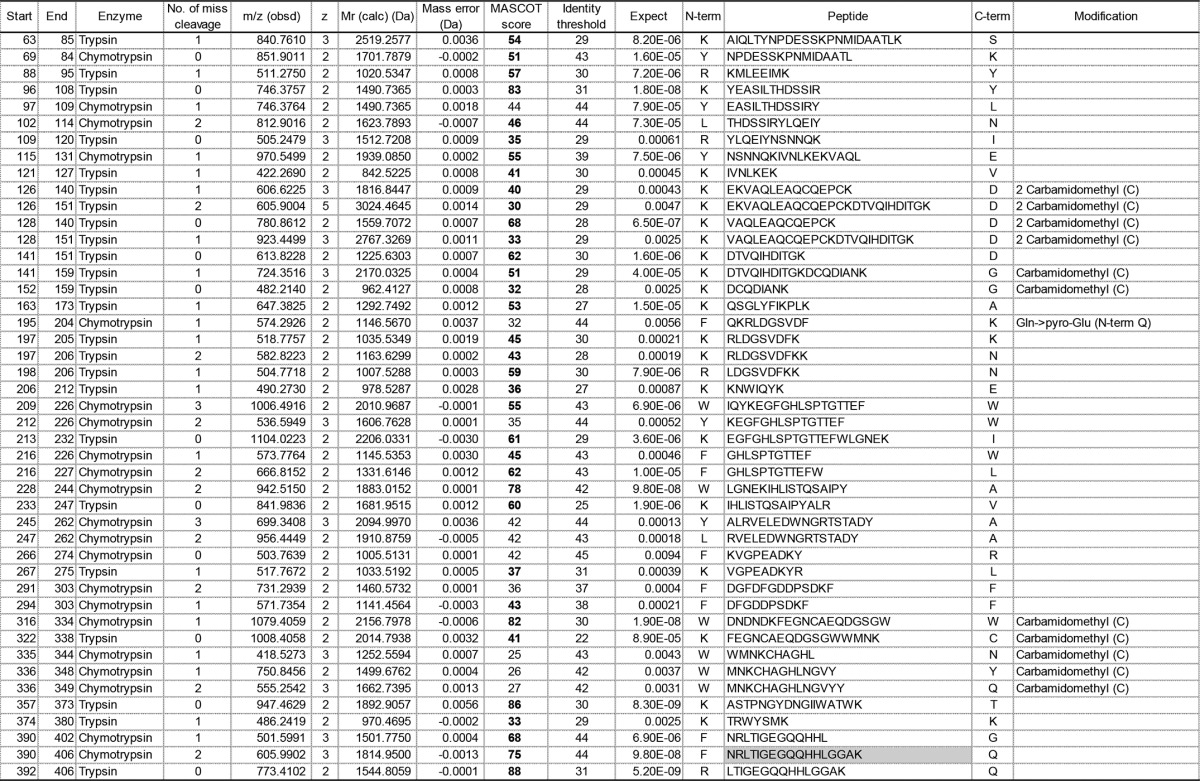
TABLE 3.
Identified fibrinogen γ-chain-derived peptides from in-gel digestion of γ2
Peptides with expectation values (column 11) less than 0.01 are listed. The Mascot score value (column 9) exceeding an identity threshold (column 10) is indicated in boldface type. Carbamidomethyl (C), carbamidomethyl cysteine; Gln>pyro-Glu (N-term Q), N-terminal pyroglutaminate; Pyrocarbamidomethyl (N-term C), N-terminal pyrocarbamidomethyl cysteine.
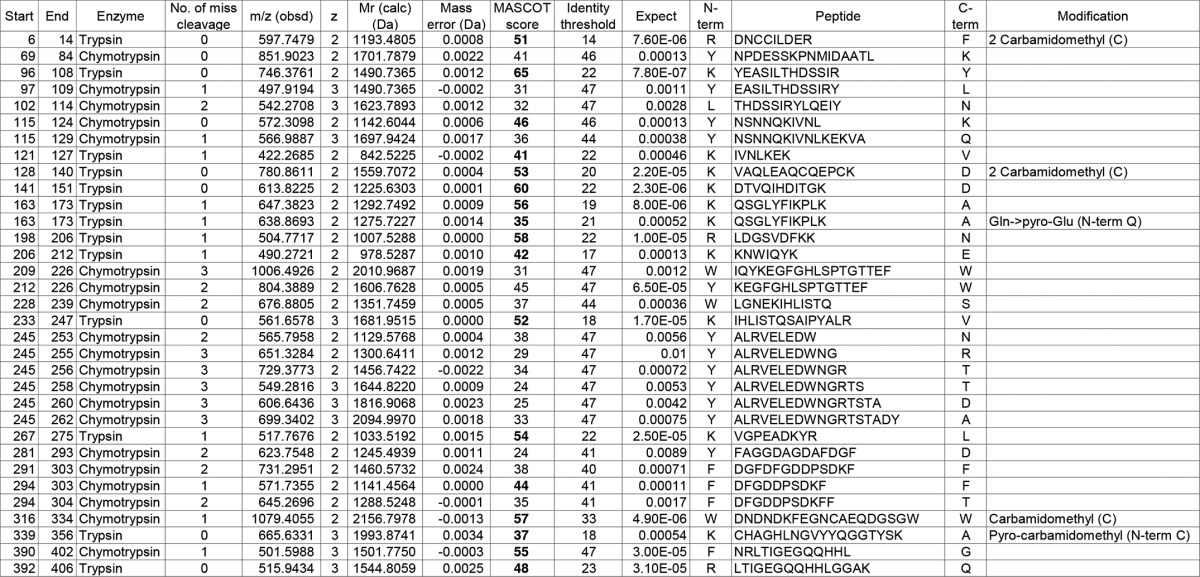
TABLE 4.
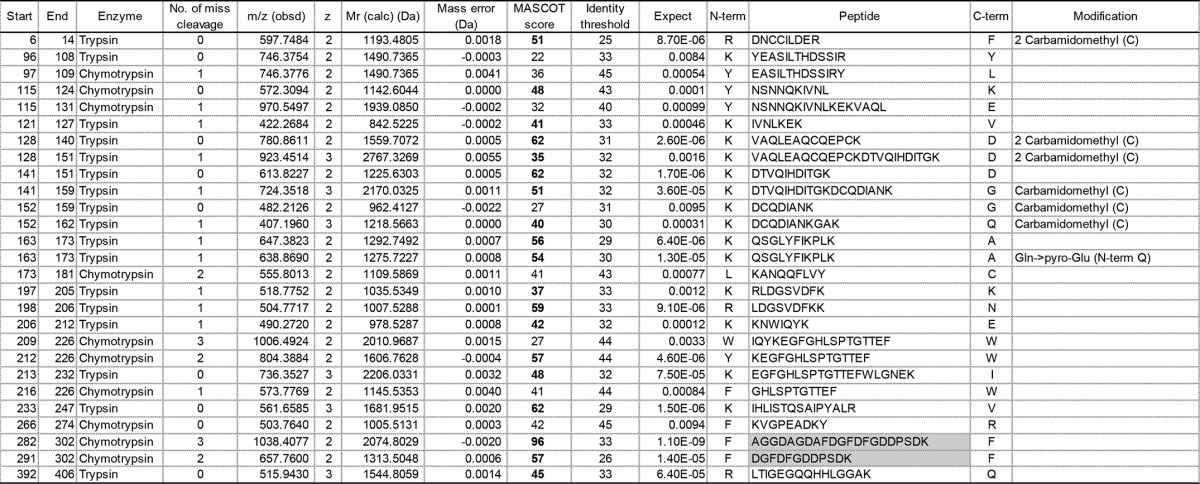
Identification list of fibrinogen γ chain-derived peptides obtained from in-gel digestion of γ3
Peptides with expectation values (column 11) less than 0.01 are listed. Peptides whose N-and C-terminal cleavage sites are chymotrypsin FLWY and trypsin KR, respectively, are represented in pale gray boxes. The Mascot score value (column 9) exceeding an identity threshold (column 10) is indicated in boldface type. Carbamidomethyl (C), carbamidomethyl cysteine; Gln>pyro-Glu (N-term Q), N-terminal pyroglutaminate.
Statistical Analyses
Statistical analyses were performed using JMP software, version 11.0.0 (SAS Institute, Cary, NC) and Prism 6.0e (GraphPad Software Inc.), and results are presented as mean ± S.D. The Mann-Whitney or Kruskal-Wallis test was used to compare groups, and the Spearman coefficient was calculated to measure the correlation between parameters. Differences were deemed statistically significant at a p value <0.05.
RESULTS
FXIII-B Accelerates Cross-linking of Fbn in FXIII-B-deficient Plasma
To examine the contribution of FXIII-B to cross-linking of Fbn in human plasma, either FXIII-A or -B was removed from human normal plasma using antibody-coupled Sepharose beads. Although removal of FXIII-A resulted in undetectable levels of FXIII-A and amine incorporation activity in the plasma, 35% of FXIII-B remained in FXIII-A-depleted plasma. FXIII-A and FXIII-B and amine incorporation activity were not detected in FXIII-B-depleted plasma.
Cross-linking between Fbn γ-chains was almost completed within 1 min in normal plasma, whereas it was hardly observed in both FXIII-A- and -B-depleted plasma (Fig. 1, A–E). Reconstitution with 5 μg/ml rFXIII-A improved γ-γ dimer formation in FXIII-A-depleted plasma to completion within 1 min as in normal plasma, whereas it was insufficient to stimulate γ-γ dimer formation in FXIII-B-depleted plasma. Cross-linking between γ-chains in FXIII-B-depleted plasma was recovered to normal levels by the addition of both rFXIII-A and rFXIII-B. Acceleration of Fbn cross-linking by rFXIII-B was also confirmed in a reaction of purified Fbg with rFXIII-A, thrombin, and calcium (Fig. 1, F and G), indicating that the accelerating effect of FXIII-B on fibrin cross-linking was not mediated by any other plasma components.
FIGURE 1.

Cross-linking of Fbn in the absence of FXIII-B. A–E, FXIII-A or FXIII-B was removed from normal human plasma using anti-FXIII-A or -B antibody-Sepharose. Cross-linking reactions were performed in normal (A), FXIII-A-depleted (B), and FXIII-B-depleted plasma (C) with or without the addition of rFXIII-A and rFXIII-B. Monomer and dimer of γ-chain in FXIII-A-depleted (D) and FXIII-B-depleted plasma (E) were quantified by a densitometer, and γ-γ dimer formation was calculated as dimer/(monomer + dimer). The mean of three independent experiments was plotted. Dotted line, no addition; filled triangle, FXIII-A-depleted plasma with rFXIII-A; open square, FXIII-B-depleted plasma with rFXIII-A; filled square, FXIII-B-depleted plasma with rFXIII-A and rFXIII-B, error bars, S.D. F and G, purified human Fbg was reacted with thrombin in the presence of rFXIII-A without or with rFXIII-B. Densitometric analyses of γ-γ dimer formation in three independent experiments were performed. Open circle, Fbg with rFXIII-A; filled circle, Fbg with rFXIII-A and rFXIII-B. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
FXIII-B Mediates Incorporation of FXIII-A into Fbn Clots and AP Cleavage
FXIII-A is involved in Fbn clot formation and therefore is present at very low levels in serum. Incorporation of FXIII-A into Fbn clots in FXIII-B-depleted plasma supplemented with rFXIII-A (FXIII-B(−)+A plasma) was examined to determine the mechanism of acceleration of Fbn cross-linking by FXIII-B. After the cross-linking reaction, the supernatant was separated from Fbn clots by centrifugation, and the amounts of FXIII-A and -B remaining in the supernatant were quantified by ELISA. More than 60% of FXIII-A was incorporated into Fbn clots in normal plasma within 30 s (Fig. 2A). On the other hand, 65% of supplemented rFXIII-A in FXIII-B(−)+A plasma remained in the supernatant even after 5 min. Western blot analysis confirmed the incorporation of FXIII-A into Fbn clots accompanied by AP cleavage in normal plasma and impaired incorporation and AP cleavage of rFXIII-A in FXIII-B(−)+A plasma (Fig. 2C). When rFXIII-B was added to FXIII-B(−)+A plasma, rFXIII-A incorporation into Fbn and AP cleavage was greatly improved. Enhancement of AP cleavage by rFXIII-B was also confirmed in a reaction with purified Fbg, although rFXIII-B itself did not accelerate AP cleavage in the absence of Fbg (Fig. 2D).
FIGURE 2.

FXIII in Fbn clots from FXIII-B(−)+A plasma. FXIII-A (A) and -B (B) in the supernatant separated from the Fbn clot after the cross-linking reaction were quantified by ELISA, and their amounts are given as a percentage of unreacted plasma. The mean ± S.D. (error bars) of 3–5 independent experiments was plotted. Open circle, normal plasma; open square, FXIII-B(−)+A plasma (FXIII-B-depleted plasma supplemented with rFXIII-A); filled square, FXIII-B(−)+A plasma with rFXIII-B. C, cleavage of the activation peptide of FXIII-A during Fbn cross-linking in FXIII-B(-)+A plasma. FXIII-A in the supernatant and Fbn clot was visualized by Western blotting using an anti-FXIII-A antibody. D, effect of FXIII-B on cleavage of the activation peptide of FXIII-A in a reaction with purified Fbg. Purified human Fbg was reacted with thrombin in the presence of rFXIII-A without or with rFXIII-B, and then samples were analyzed by Western blotting using an anti-FXIII-A antibody. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Cross-linking-dependent Release of FXIII-B from Fbn Clots
Although FXIII-A remained in the Fbn clot, FXIII-B was transiently incorporated into the clot and released within 2 min (Fig. 2B). Consistent with FXIII-B dissociation from activated FXIII-A in the presence of calcium ions, calcium chelator EDTA blocked the release of FXIII-B from Fbn (Fig. 3, A–C). Because EDTA also inhibited the enzymatic activity of activated FXIII-A, the cross-linking reaction might be required for the release of FXIII-B from Fbn clot. When the cross-linking reaction was performed in normal plasma with a transglutaminase inhibitor, iodoacetamide, release of incorporated FXIII-B into the supernatant was markedly repressed (Fig. 3, D–F), indicating that the release of FXIII-B from fibrin was dependent on a transglutaminase reaction.
FIGURE 3.

Release of FXIII-B from Fbn clots. A–C, normal plasma was reacted with 10 units/ml thrombin in the presence of 10 mm CaCl2 (open circle) or 10 mm EDTA (filled triangle) for the indicated time. FXIII-A (A) and FXIII-B (B) remaining in the supernatant were measured by ELISA. Mean ± S.D. (error bars) of three reactions was shown. FXIII-B in the clot was detected by Western blot analysis using an anti-FXIII-B antibody (C). P, non-reacted plasma. D–F, cross-linking-dependent release of FXIII-B from Fbn clot. Normal plasma was reacted with thrombin and CaCl2 in the presence (filled diamond) or the absence (open circle) of 0.5 mm iodoacetamide (IAA) for the indicated time. The Fbn clot was analyzed by SDS-PAGE (D). FXIII-A (E) and FXIII-B (F) remaining in the supernatant were measured by ELISA. The mean ± S.D. of three reactions is shown. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Direct Interaction of FXIII-B with Fbg
Plasma FXIII has been thought to bind to Fbg via FXIII-A because platelet FXIII (FXIII-A) was reported to specifically bind to Fbg (18, 19). FXIII-B incorporated into the Fbn clot was released by calcium-dependent dissociation from FXIII-A and cross-linking of Fbn. This finding implied a possible direct interaction between FXIII-B and Fbg in addition to the heterotetramer assembly with FXIII-A. To explore whether FXIII-B directly bound to Fbg, the interaction of FXIII subunits with Fbg was examined using Fbg-Sepharose. rFXIII-A was collected by Fbg-Sepharose in the absence of rFXIII-B (Fig. 4A). rFXIII-B also bound to Fbg-Sepharose even in the absence of rFXIII-A. Interaction of FXIII with Fbg was quantified by ELISA using a Fbg-immobilized plate. Binding of rFXIII-A to Fbg was reduced in the presence of rFXIII-B (Fig. 4B). Fbg-binding of rFXIII-B tended to decrease in the presence of rFXIII-A, the effect of which was milder than that of rFXIII-B on the rFXIII-A binding to Fbg.
FIGURE 4.

Binding of FXIII to Fbg. A, binding of FXIII to Fbg-Sepharose. rFXIII-A and/or rFXIII-B was reacted with Fbg-Sepharose. The Sepharose of each set of three reactions was analyzed by Western blotting using an anti-FXIII-A antibody (top) or an anti-FXIII-B antibody (bottom). B, ELISA of Fbg-FXIII binding. rFXIII-A and/or rFXIII-B was reacted with Fbg immobilized on a 96-well plate, and ELISA was performed using an anti-FXIII-A antibody (left) or an anti-FXIII-B antibody (right). The mean ± S.D. (error bars) of the amount relative to the reaction with rFXIII-A and rFXIII-B in three reactions is shown. C, co-immunoprecipitation of FXIII with Fbg from plasma. An anti-Fbg antibody (F) or bovine non-immune IgG (N) was added to normal, FXIII-A-depleted, or FXIII-B(−)+A plasma. Immunoprecipitated materials of non-immune IgG and three reactions of anti-Fbg antibody or original plasma (P) were analyzed by Western blotting using an anti-FXIII-A (top) or anti-FXIII-B antibody (bottom). D, ELISA of FXIII bound to Fbg in plasma. Normal (N), FXIII-A-depleted (A(−)), or FXIII-B(−)+rFXIII-A plasma (B(−)+A) was reacted with anti-Fbg antibody immobilized on a 96-well plate, and FXIII-A (left) or FXIII-B (right) bound to the plate was quantitated by ELISA. The mean ± S.D. of the amount relative to normal plasma in three reactions is shown. *, p < 0.05; ***, p < 0.001.
To confirm whether FXIII-B interacted with Fbg in plasma, co-immunoprecipitation of FXIII with Fbg from normal, FXIII-A-depleted, or FXIII-B(−)+A plasma was examined using an anti-Fbg antibody. Both FXIII subunits co-immunoprecipitated with Fbg in normal plasma (Fig. 4C). In FXIII-A-depleted plasma, FXIII-B was recovered with an anti-Fbg antibody, although FXIII-A was absent. However, rFXIII-A in FXIII-B(−)+A plasma was hardly detected in the Fbg immunoprecipitate. ELISA using an anti-Fbg antibody-coated plate revealed an interaction of FXIII-B with Fbg in FXIII-A-depleted plasma and much less binding of rFXIII-A to Fbg in FXIII-B(−)+A plasma (Fig. 4D). Thus, it was confirmed that the FXIII heterotetramer binding to Fbg in plasma is mediated by FXIII-B rather than FXIII-A.
Fbg-binding Domain of FXIII-B
MetLuc-BS fusion proteins were constructed to determine which sushi domain(s) was responsible for the interaction with Fbg. All 10 MetLuc-BSs were confirmed to bind to an anti-FXIII-B antibody, although MetLuc itself did not (Fig. 5A). When each MetLuc-BS was incubated with FXIII-A, only MetLuc carrying the first sushi domain was co-immunoprecipitated with an anti-FXIII-A antibody (Fig. 5B), confirming previous findings that the first sushi domain binds to FXIII-A (6). On the other hand, no MetLuc-BS co-immunoprecipitated with Fbg (Fig. 5C), implying that more than one sushi domain is required for binding between FXIII-B and Fbg.
FIGURE 5.

Determination of the Fbg-binding domain in FXIII-B. A–C, binding of MetLuc fused with each sushi domain of FXIII-B to anti-FXIII-B antibody (A), FXIII-A (B), and Fbg (C). Recovery of MetLuc activity collected with anti-FXIII-B-Sepharose is given as a percentage of starting MetLuc preparation (A), and MetLuc activity collected by anti-FXIII-A antibody (B) or anti-Fbg antibody (C) is given as ratio of co-immunoprecipitation with and without rFXIII-A or Fbg, respectively. (−), wild-type MetLuc (without FXIII-B sushi domain). The mean ± S.D. (error bars) of three independent immunoprecipitations is shown. D, kinetic analysis of Fbg binding to rFXIII-B (filled circle), rFXIII-B*3 (gray circle), rBΔ10th (open triangle), and rBΔ1st (filled triangle). Each set of three reactions was plotted. E, effect of truncated rFXIII-B on Fbn cross-linking in FXIII-B(−)+A plasma. The Fbn cross-linking reaction was performed in FXIII-B(−)+A plasma in the presence of rFXIII-B, rBΔ10th, or rBΔ1st, and the Fbn clot was analyzed by SDS-PAGE stained with Coomassie Brilliant Blue dye. Densitometric analyses of γ-γ dimer formation in three independent experiments were performed. Filled circle, rFXIII-B; open triangle, rBΔ10th; filled triangle, rBΔ1st. F and G, effect of truncated FXIII-B constructs on incorporation of FXIII in Fbn clots in human FXIII-B(−)+A plasma in the absence (open circle) or the presence of 5 μg/ml rFXIII-B (filled circle), rBΔ10th (open triangle), or rBΔ1st (filled triangle); FXIII-A (F) and FXIII-B (G) remaining in the supernatant were measured by ELISA. The mean ± S.D. of three reactions is shown. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
We previously constructed several truncation mutants of FXIII-B and successfully purified rFXIII-Bs missing either the first or the tenth sushi domain (FXIII-B2–10 or FXIII-B1–9; denoted as rBΔ1st or rBΔ10th, respectively) (6). We examined binding of rBΔ1st and rBΔ10th to Fbg. Both truncated rFXIII-B constructs bound to Fbg much less efficiently than wild-type rFXIII-B (Fig. 5D); apparent Kd for wild-type rFXIII-B binding to Fbg was estimated to be 1.54 ± 0.45 × 10−7 m. These results indicated that loss of either the first or the tenth sushi domain resulted in decreased binding ability to Fbg.
There are four isoforms of FXIII-B caused by genetic polymorphisms, one of which (FXIII-B*3) has a distinct C-terminal tail with the same tenth sushi domain resulting from complete allele-specific alternative splicing (20). rFXIII-B*3 showed a Fbg-binding efficiency similar to that of wild-type rFXIII-B (Fig. 5D) because its Kd for the binding to Fbg was estimated to be 3.01 × 10−7 m, excluding the contribution of the C-terminal tail in Fbg binding.
Because truncation of either the first or the tenth sushi domain impaired the interaction to FXIII-A and/or Fbg, the effect of truncated rFXIII-Bs on Fbn cross-linking was examined. When either rBΔ10th or rBΔ1st was added to FXIII-B(−)+A plasma, cross-linking of Fbn γ-chains was nearly unaffected, and incorporation of FXIII-A into Fbn clots was not promoted (Fig. 5, E–G). Only part of rBΔ1st was incorporated and held in the Fbn clot, and rBΔ10th was not incorporated into the clot. In addition, it was previously demonstrated that rBΔ1st could not bind to FXIII-A (6). These results indicate that the interaction of FXIII-B with FXIII-A and Fbg is impaired by deletion of the first or the tenth sushi domain, which delays cross-linking of Fbn.
Possible FXIII-B-binding Region of Fbg
We searched for the region of Fbg that binds FXIII-B. Plasmin digests Fbg to its D- and E-domains (21). When FXIII-B-free Fbg was digested with plasmin, fragments E, D1, D2, and D3 were apparent in SDS-PAGE analysis under the non-reducing condition (Fig. 6A), and γ-chain-derived fragments γ1, γ2, and γ3 were detected under the reducing condition (Fig. 6B). rFXIII-B suppressed the appearance of fragments D2 and D3 (γ2 and γ3) in the digestion of Fbg with plasmin without any effect on the cleavage of either D or E domain, suggesting possible masking of the cleavage site(s) in the γ-chain by the binding of FXIII-B. Therefore, the N and C termini of fragments γ1, γ2, and γ3 were determined by LC-MS/MS analysis. The N termini of fragments γ1, γ2, and γ3 contained a common Tyr-96, and the C termini contained Lys-406, Lys-356, and Lys-302, respectively (Tables 2–4 and Fig. 6, C–E). These results suggest that rFXIII-B protected the γ-chain from cleavage at the carboxyl side of Lys-302 and Lys-356, residues that were located very close to polymerization pocket “a” (22) (Fig. 6F).
FIGURE 6.
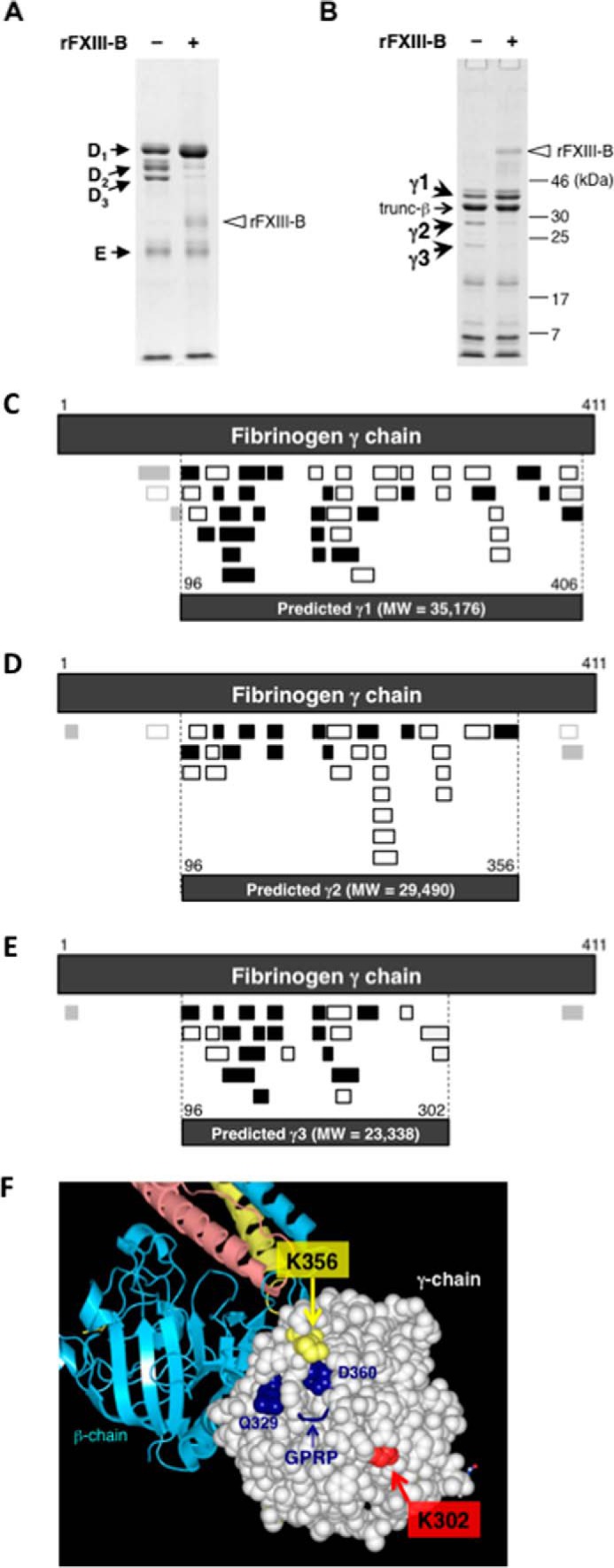
Determination of the FXIII-B-binding region in Fbg. FXIII-B-free Fbg was digested with plasmin in the absence (−) or the presence of rFXIII-B (+) and was analyzed by SDS-PAGE under non-reducing (A) and reducing conditions (B). C–E, prediction of each degradation product (γ1, γ2, and γ3) derived from fibrinogen γ chain using identified peptides from each in-gel digest. LC-MS/MS-identified peptides (Tables 2–4) are represented. Filled boxes, peptide fragments obtained by trypsin digestion; open boxes, fragments obtained by chymotrypsin digestion. Fragments detected with small LC-MS/MS peak areas are shown in light gray boxes. Top dark gray boxes of each panel, mature fibrinogen γ-chain (residues 1–411); bottom dark gray boxes of each panel, predicted plasmin degradation products. C, prediction of γ1. A total of 45 peptides, including 24 tryptic peptides, 20 chymotryptic peptides, and one semi-chymotrypsin peptide, were identified. From these identified peptides, γ1 was predicted to consist of 311 amino acid residues (molecular mass, 35,176 Da) spanning amino acids 96–406 of the mature fibrinogen γ chain. D, prediction of γ2. A total of 33 peptides, including 13 tryptic peptides and 20 chymotryptic peptides, were identified. From these identified peptides, γ2 was predicted to consist of 261 amino acid residues (molecular mass, 29,490 Da) spanning residues 96–356 of the mature protein, whereas at least three distinct peptide regions, two at the N terminus and one at the C terminus, besides the predicted region were identified. E, prediction of γ3. A total of 27 peptides, including 17 tryptic peptides, 8 chymotryptic peptides, and 2 semi-chymotrypsin peptides, were identified. From these identified peptides, γ3 was predicted to consist of 207 amino acid residues (molecular mass, 23,338 Da) spanning amino acids 96–302 of the mature protein, whereas at least two distinct peptide regions, one in each of the N- and C termini, besides the predicted region were identified. F, structural positions of γ-chain Lys-302 and Lys-356 in Fbg (Protein Data Bank code 3GHG). The D-domain is shown as drawn in Waals (Altif Laboratories). The γ-chain in the D-domain is represented with space-filling residues, and the β-chain is represented with ribbons (cyan). Residues Lys-302 and Lys-356 are shown in light green and light yellow, respectively. The structural data do not include the C-terminal region of the γ-chain (residues 395–411).
DISCUSSION
The role of FXIII-B in the Fbn cross-linking reaction has been overlooked for a long time because FXIII-B is not catalytic and dissociates from active FXIII-A at the end of the activation process. Instead, FXIII-B has been thought to slow down the overall conversion of FXIII (A2B2) to FXIIIa (A*2) by masking the active center of FXIII-A (23). In the present study, we demonstrated, for the first time, that FXIII-B accelerated cross-linking of Fbn.
Plasma FXIII is thought to circulate in a complex with Fbg based on observations that FXIII co-precipitated and migrated with plasma Fbg in immunoelectrophoretic or immunodiffusion experiments (24). It has been suggested that binding of plasma FXIII to Fbg was mediated by FXIII-A because platelet FXIII (FXIII-A) specifically bound to Fbg (18, 19). In the present study, however, FXIII-A in FXIII-B-deficient plasma was not recovered by immunoprecipitation using an anti-Fbg antibody. In contrast, FXIII-B co-immunoprecipitated with Fbg from FXIII-A-deficient plasma, indicating that plasma FXIII interacts with Fbg via FXIII-B. It is therefore understandable that loss of FXIII-B delays the access of FXIII-A to the Fbn substrate, causing delay of Fbn cross-linking.
Digestion of Fbg with plasmin in the presence of FXIII-B suggested that FXIII-B bound to the C-terminal part of the γ-chain in the D-domain, which is near the polymerization pocket “a” (Fig. 6F). It is possible that the polymerization pocket “a” of Fbg (and the Fbn monomer) was not masked in the complex with FXIII-B because FXIII-B remained bound to Fbn clots in the presence of the transglutaminase inhibitor iodoacetamide.
Siebenlist et al. (25) reported that plasma FXIII (A2B2) specifically bound to Fbg containing a splicing variant of the γ-chain, γ′ (γA/γ′), and suggested that the interaction between Fbg (γA/γ′) and plasma FXIII was mediated by FXIII-B because placental or platelet FXIII (A2) did not bind to γA/γ′. We constructed a fusion protein of secreted luciferase with γ-chain (residues 391–411) or γ′-chain (residues 391–427) C termini, but direct binding of these fusion proteins to FXIII-B was not observed.3 It is difficult to conclude whether FXIII-B binds directly to the C-terminal tail of γ′-chain; the other C-terminal part of γ (γ′)-chain together with the γ′-tail may be required for the tight binding with FXIII-B. On the other hand, Moaddel et al. (26) demonstrated that plasma FXIII formed complexes with both predominant Fbg (γA/γA) and γA/γ′, although it bound to γA/γ′ ∼20-fold more tightly than to γA/γA. Because the D-domain of almost all Fbgs (mixture of γA/γA and γA/γ′) was protected from digestion with plasmin by FXIII-B, FXIII-B seems to bind to both forms of Fbg.
Both the first and tenth sushi domains of FXIII-B probably contribute to the binding to Fbg, although neither of them alone bound Fbg. Our previous study demonstrated that FXIII-B formed a homodimer through the fourth and ninth sushi domains and assembled a hetrotetramer with FXIII-A through the first sushi domain (6). Herein, we confirmed that the first sushi domain binds FXIII-A. Although the exact structure of FXIII-B is still unknown because of the absence of crystallographic data, we propose a model that the first (or first through third) and tenth sushi domains of FXIII-B bind to a D-domain nodule of Fbg in a clip-like structure and that the FXIII-A connection to Fbg is mediated by the first sushi domain of FXIII-B (Fig. 7, preactivation stage). Moaddel et al. (26) proposed that one FXIII molecule forms a complex with two Fbg molecules. It is possible that a FXIII-B homodimer has two clips at either end and that the C termini of two D-domains are connected by FXIII-B in an FXIII-Fbg complex, which is advantageous for rapid cross-linking between γ-chains immediately after the activation of FXIII.
FIGURE 7.

Schematic illustration of possible FXIII-Fbg complex during cross-linking reaction. The FXIII-B dimer is drawn as a series of squares (the second to ninth sushi domains) with two small rectangles (filled rectangle, first sushi domain; dark dotted rectangle, tenth sushi domain). In plasma, the first and tenth sushi domains of a single FXIII-B molecule clip D-domains of two Fbg molecules; thus, these Fbg molecules are connected by FXIII-B (preactivation stage, top). FXIII-A (gray rectangles, the back of FXIII-B) binds to the first sushi domain of FXIII-B. When thrombin (FIIa) is generated to convert Fbg to Fbn by cleaving off fibrinopeptides (Fps) A and B at the amino termini of α and β chains in the Fbg E-domain, thrombin formerly bound to a central E-domain of one Fbn molecule binds to polymerization pocket “a” in a D-domain of another Fbn molecule, allowing thrombin to cleave AP of FXIII-A (activation stage, middle). When FXIII-A* cross-links between C-terminal tails of two Fbn γ-chains, FXIII-B finally dissociates from Fbn (cross-linking stage, bottom). FPs, fibrinopeptides.
Lack of FXIII-B in plasma also delayed AP cleavage of FXIII-A by thrombin. Fbn (Fbg) is known to accelerate activation of FXIII, including the AP cleavage reaction (7, 8). Greenberg et al. (27) demonstrated that the activation of FXIII was promoted by the formation of a complex of thrombin, Fbn, and plasma FXIII (A2B2). Thus, it is understandable that binding of FXIII-B to Fbg and FXIII-A enhances AP cleavage by thrombin. Greenberg and Shuman (28) also demonstrated acceleration of thrombin-catalyzed FXIII activation by polymerization of Fbn. Thrombin interacts with the central E-domain of Fbg and remains bound to Fbn after fibrinopeptides are removed (29, 30). Therefore, it is hypothesized that thrombin bound with a central E-domain of one Fbn approaches FXIII-A that is bound near the polymerization pocket “a” of another Fbn by FXIII-B, leading to the effective cleavage of AP (Fig. 7, activation stage).
FXIII-B dissociates from AP-cleaved FXIII-A (FXIII-A′) in the presence of calcium ion (converting to FXIII-A*), and Fbg lowers the concentration of calcium required for the dissociation of FXIII-B from FXIII-A* (31). Therefore, FXIII-B remains bound to FXIII-A′ in Fbn clots formed in the presence of EDTA (32). Surprisingly, inhibition of cross-linking by iodoacetamide also blocked the dissociation of FXIII-B from Fbn clots even in the presence of calcium concentrations adequate for dissociation. Because iodoacetamide does not inhibit the calcium-dependent dissociation of FXIII-B from FXIII-A (33), FXIII-B probably maintains binding to non-cross-linked Fbn but not to FXIII-A*. In turn, FXIII-B dissociation from Fbn depends on cross-linking between γ-chains; calcium-dependent dissociation of A*2B2 and cross-linking between two γ-tails may coincide with “unclipping” of FXIII-B from Fbn (Fig. 7, cross-linking stage). FXIII-A* is retained in Fbn clots after dissociation of FXIII-B as a “true” enzyme-substrate complex and continues further cross-linking between α-chains, α-chain and α2-plasmin inhibitor, and other Fbn elements.
In the presence of FXIII-B (normal plasma), γ-chains are cross-linked by FXIII-A* more rapidly than α-chains, possibly due to guidance of FXIII-B as described above. Accordingly, cross-linking of Fbn chains in the absence of FXIII-B may occur in random order. However, at least under the conditions studied herein, cross-linking of α-chains in FXIII-B-depleted plasma was delayed from the formation of γ-γ dimers. Because AP cleavage was almost negligible in the supernatant of FXIII-B-depleted plasma, a D-domain of polymerized Fbn may be the location for AP cleavage of FXIII-B-free FXIII-A by E-domain-bound thrombin.
In the purified system, both the FXIII-A activation and fibrin-cross-linking reaction were delayed in the absence of FXIII-B when compared with those in the presence of FXIII-B, although FXIII-A could bind to Fbg. Mary et al. (19) reported that platelet FXIII (FXIII-A) bound to the Aα and Bβ-chains in the D-domain of Fbg. The position(s) in Fbg to which FXIII-A bound in the absence of FXIII-B might be improper for FXIII-A to contact with thrombin, and thus the FXIII-A activation was not enhanced. On the other hand, both the FXIII-A activation and fibrin-cross-linking reaction were enhanced in the presence of FXIII-B, although FXIII-B reduced the binding of FXIII-A to Fbg. Therefore, FXIII-B may bring FXIII-A to a proper position(s) of Fbg to be activated by thrombin.
However, FXIII-A by itself could not bind to Fbg in plasma, suggesting that this hypothetical potential binding site(s) on Fbg may be occupied other plasma proteins, such as α2-plasmin inhibitor and plasminogen (34), fibronectin, von Willebrand factor, etc. In plasma, because FXIII-B exists in excess of FXIII-A, all FXIII-A is in a complex with FXIII-B. Therefore, FXIII-B may bring FXIII-A to Fbg to be readily activated by thrombin, as demonstrated by the current studies.
Among Asian Indians, FXIII-B*3 is associated with a lower FXIII activity than FXIII-B*1 (35). However, the differences in the C-terminal tail of FXIII-B*1 and FXIII-B*3 did not affect binding to Fbg. There may be other unknown function(s) of FXIII-B in FXIII-mediated catalysis.
In conclusion, FXIII-B essentially accelerates cross-linking of Fbn via direct interactions with Fbg and FXIII-A whereby FXIII-B localizes FXIII-A to the D-domain of Fbg close to the Fbn polymerization pocket and the C-terminal tail of the γ-chain, which is responsible for activation of FXIII-A and cross-linking between γ-chains. In addition to the conservation of FXIII-A in blood, this accelerating effect of FXIII-B on Fbn cross-linking is expected to be responsible for maintaining hemostasis because FXIII-B deficiency results in an increased bleeding tendency despite having a milder phenotype than FXIII-A deficiency (11, 36).
This study was supported by a research grant from the Japanese Ministry of Education, Culture, Sports, Science, and Technology and was presented in part at the 35th Annual Meeting of the Japanese Society on Thrombosis and Hemostasis in Yamagata in May 2013 and at the 24th Congress of the International Society on Thrombosis and Hemostasis in Amsterdam in July 2013.
M. Souri, T. Osaki, and A. Ichinose, unpublished data.
- Fbg
- fibrinogen
- Fbn
- fibrin
- FXIII
- coagulation factor XIII
- FXIII-A
- FXIII A subunit
- FXIII-B
- FXIII B subunit
- AP
- activation peptide of FXIII-A
- FXIIIa
- active form of FXIII
- rFXIII-A
- recombinant FXIII-A
- rFXIII-B
- recombinant FXIII-B
- pAb
- polyclonal antibody
- MetLuc
- Metridia luciferase
- MetLuc-BS
- MetLuc fusion protein with sushi domain of FXIII-B
- nanoLC
- nanoflow liquid chromatography.
REFERENCES
- 1. Blombäck B., Hessel B., Hogg D., Therkildsen L. (1978) A two-step fibrinogen-fibrin transition in blood coagulation. Nature 275, 501–505 [DOI] [PubMed] [Google Scholar]
- 2. Laudano A. P., Doolittle R. F. (1978) Synthetic peptide derivatives that bind to fibrinogen and prevent the polymerization of fibrin monomers. Proc. Natl. Acad. Sci. U.S.A. 75, 3085–3089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Olexa S. A., Budzynski A. Z. (1980) Evidence for four different polymerization sites involved in human fibrin formation. Proc. Natl. Acad. Sci. U.S.A. 77, 1374–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yee V. C., Pedersen L. C., Le Trong I., Bishop P. D., Stenkamp R. E., Teller D. C. (1994) Three-dimensional structure of a transglutaminase: human blood coagulation factor XIII. Proc. Natl. Acad. Sci. U.S.A. 91, 7296–7300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ichinose A., McMullen B. A., Fujikawa K., Davie E. W. (1986) Amino acid sequence of the B subunit of human coagulation factor XIII, a protein composed of ten repetitive segments. Biochemistry 25, 4633–4638 [DOI] [PubMed] [Google Scholar]
- 6. Souri M., Kaetsu H., Ichinose A. (2008) Sushi domains in the B subunit of factor XIII responsible for oligomer assembly. Biochemistry 47, 8656–8664 [DOI] [PubMed] [Google Scholar]
- 7. Janus T. J., Lewis S. D., Lorand L., Shafer J. A. (1983) Promotion of thrombin-catalyzed activation of factor XIII by fibrinogen. Biochemistry 22, 6269–6272 [DOI] [PubMed] [Google Scholar]
- 8. Lewis S. D., Janus T. J., Lorand L., Shafer J. A. (1985) Regulation of the formation of factor XIIIa by its fibrin substrates. Biochemistry 24, 6772–6777 [DOI] [PubMed] [Google Scholar]
- 9. Radek J. T., Jeong J. M., Wilson J., Lorand L. (1993) Association of the A subunits of recombinant placental factor XIII with the native carrier B subunits from human plasma. Biochemistry 32, 3527–3534 [DOI] [PubMed] [Google Scholar]
- 10. Katona E., Pénzes K., Csapó A., Fazakas F., Udvardy M. L., Bagoly Z., Orosz Z. Z., Muszbek L. (2014) Interaction of factor XIII subunits. Blood 123, 1757–1763 [DOI] [PubMed] [Google Scholar]
- 11. Saito M., Asakura H., Yoshida T., Ito K., Okafuji K., Yoshida T., Matsuda T. (1990) A familial factor XIII subunit B deficiency. Br. J. Haematol. 74, 290–294 [DOI] [PubMed] [Google Scholar]
- 12. Izumi T., Hashiguchi T., Castaman G., Tosetto A., Rodeghiero F., Girolami A., Ichinose A. (1996) Type I factor XIII deficiency is caused by a genetic defect of its b subunit: insertion of triplet AAC in exon III leads to premature termination in the second Sushi domain. Blood 87, 2769–2774 [PubMed] [Google Scholar]
- 13. Koseki S., Souri M., Koga S., Yamakawa M., Shichishima T., Maruyama Y., Yanai F., Ichinose A. (2001) Truncated mutant B subunit for factor XIII causes its deficiency due to impaired intracellular transportation. Blood 97, 2667–2672 [DOI] [PubMed] [Google Scholar]
- 14. Souri M., Koseki-Kuno S., Takeda N., Degen J. L., Ichinose A. (2008) Administration of factor XIII B subunit increased plasma factor XIII A subunit levels in factor XIII B subunit knock-out mice. Int. J. Hematol. 87, 60–68 [DOI] [PubMed] [Google Scholar]
- 15. Yuasa Y., Osaki T., Makino H., Iwamoto N., Kishimoto I., Usami M., Minamino N., Harada-Shiba M. (2014) Proteomic analysis of proteins eliminated by low-density lipoprotein apheresis. Ther. Apher. Dial. 18, 93–102 [DOI] [PubMed] [Google Scholar]
- 16. Shevchenko A., Tomas H., Havlis J., Olsen J. V., Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 17. Olsen J. V., Macek B., Lange O., Makarov A., Horning S., Mann M. (2007) Higher-energy C-trap dissociation for peptide modification analysis. Nat. Methods 4, 709–712 [DOI] [PubMed] [Google Scholar]
- 18. Greenberg C. S., Shuman M. A. (1982) The zymogen forms of blood coagulation factor XIII bind specifically to fibrinogen. J. Biol. Chem. 257, 6096–6101 [PubMed] [Google Scholar]
- 19. Mary A., Achyuthan K. E., Greenberg C. S. (1987) Factor XIII binds to the A α- and B β- chains in the D-domain of fibrinogen: an immunoblotting study. Biochem. Biophys. Res. Commun. 147, 608–614 [DOI] [PubMed] [Google Scholar]
- 20. Iwata H., Kitano T., Umetsu K., Yuasa I., Yamazaki K., Kemkes-Matthes B., Ichinose A. (2009) Distinct C-terminus of the B subunit of factor XIII in a population-associated major phenotype: the first case of complete allele-specific alternative splicing products in the coagulation and fibrinolytic systems. J. Thromb. Haemost. 7, 1084–1091 [DOI] [PubMed] [Google Scholar]
- 21. Pizzo S. V., Taylor L. M., Jr., Schwartz M. L., Hill R. L., McKee P. A. (1973) Subunit structure of fragment D from fibrinogen and cross-linked fibrin. J. Biol. Chem. 248, 4584–4590 [PubMed] [Google Scholar]
- 22. Pratt K. P., Côté H. C. F., Chung D. W., Stenkamp R. E., Davie E. W. (1997) The primary fibrin polymerization pocket: three-dimensional structure of a 30-kDa C-terminal γ-chain fragment complexed with the peptide Gly-Pro-Arg-Pro. Proc. Natl. Acad. Sci. U.S.A. 94, 7176–7181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Curtis C. G., Brown K. L., Credo R. B., Domanik R. A., Gray A., Stenberg P., Lorand L. (1974) Calcium-dependent unmasking of active center cysteine during activation of fibrin stabilizing factor. Biochemistry 13, 3774–3780 [DOI] [PubMed] [Google Scholar]
- 24. Loewy A. G., Dahlberg A., Dunathan K., Kriel R., Wolfinger H. L., Jr. (1961) Fibrinase: II. Some physical properties. J. Biol. Chem. 236, 2634–2643 [PubMed] [Google Scholar]
- 25. Siebenlist K. R., Meh D. A., Mosesson M. W. (1996) Plasma factor XIII binds specifically to fibrinogen molecules containing γ′ chains. Biochemistry 35, 10448–10453 [DOI] [PubMed] [Google Scholar]
- 26. Moaddel M., Farrell D. H., Daugherty M. A., Fried M. G. (2000) Interactions of human fibrinogens with factor XIII: roles of calcium and the γ′ peptide. Biochemistry 39, 6698–6705 [DOI] [PubMed] [Google Scholar]
- 27. Greenberg C. S., Achyuthan K. E., Fenton J. W. (1987) Factor XIIIa formation promoted by complexing of α-thrombin, fibrin, and plasma factor XIII. Blood 69, 867–871 [PubMed] [Google Scholar]
- 28. Greenberg C. S., Miraglia C. C. (1985) The effect of fibrin polymers on thrombin-catalyzed plasma factor XIIIa formation. Blood 66, 466–469 [PubMed] [Google Scholar]
- 29. Binnie C. G., Lord S. T. (1993) The fibrinogen sequences that interact with thrombin. Blood 81, 3186–3192 [PubMed] [Google Scholar]
- 30. Pechik I., Madrazo J., Mosesson M. W., Hernandez I., Gilliland G. L., Medved L. (2004) Crystal structure of the complex between thrombin and the central “E” region of fibrin. Proc. Natl. Acad. Sci. U.S.A. 101, 2718–2723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Credo R. B., Curtis C. G., Lorand L. (1978) Ca2+-related regulatory function of fibrinogen. Proc. Natl. Acad. Sci. U.S.A. 75, 4234–4237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Greenberg C. S., Dobson J. V., Miraglia C. C. (1985) Regulation of plasma factor XIII binding to fibrin in vitro. Blood 66, 1028–1034 [PubMed] [Google Scholar]
- 33. Lorand L. (2001) Factor XIII: structure, activation, and interactions with fibrinogen and fibrin. Ann. N.Y. Acad. Sci. 936, 291–311 [DOI] [PubMed] [Google Scholar]
- 34. Tsurupa G., Yakovlev S., McKee P., Medved L. (2010) Non-covalent interaction of α2-antiplasmin with fibrin(ogen): localization of α2-amtiplasmin binding sites. Biochemistry 49, 7643–7651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Saha N., Aston C. E., Low P. S., Kamboh M. I. (2000) Racial and genetic determinants of plasma factor XIII activity. Genet. Epidemiol. 19, 440–455 [DOI] [PubMed] [Google Scholar]
- 36. Ivaskevicius V., Biswas A., Loreth R., Schroeder V., Ohlenforst S., Rott H., Krause M., Kohler H. P., Scharrer I., Oldenburg J. (2010) Mutations affecting disulphide bonds contribute to a fairly common prevalence of F13B gene defects: results of a genetic study in 14 families with factor XIII B deficiency. Haemophilia 16, 675–682 [DOI] [PubMed] [Google Scholar]