

Background: SETMAR is a lysine methyltransferase (KMT) that contributes to DNA repair, but its biochemical function is not well understood.
Results: A novel proteomic strategy identifies splicing factor snRNP70 as a SETMAR substrate.
Conclusion: SETMAR is the first KMT identified to target splicing factors.
Significance: Proteomics can be harnessed to discover methyltransferase substrates. Lysine methylation may be a new mode of regulation for mRNA splicing.
Keywords: Histone Methylation, Mass Spectrometry (MS), Protein Methylation, Proteomics, Substrate Specificity
Abstract
The lysine methyltransferase (KMT) SETMAR is implicated in the response to and repair of DNA damage, but its molecular function is not clear. SETMAR has been associated with dimethylation of histone H3 lysine 36 (H3K36) at sites of DNA damage. However, SETMAR does not methylate H3K36 in vitro. This and the observation that SETMAR is not active on nucleosomes suggest that H3K36 methylation is not a physiologically relevant activity. To identify potential non-histone substrates, we utilized a strategy on the basis of quantitative proteomic analysis of methylated lysine. Our approach identified lysine 130 of the mRNA splicing factor snRNP70 as a SETMAR substrate in vitro, and we show that the enzyme primarily generates monomethylation at this position. Furthermore, we show that SETMAR methylates snRNP70 Lys-130 in cells. Because snRNP70 is a key early regulator of 5′ splice site selection, our results suggest a model in which methylation of snRNP70 by SETMAR regulates constitutive and/or alternative splicing. In addition, the proteomic strategy described here is broadly applicable and is a promising route for large-scale mapping of KMT substrates.
Introduction
SETMAR (also called Metnase) arose in the anthropoid lineage by fusion of a lysine methyltransferase (KMT)6 SET domain to a copy of the Mariner family transposase Hsmar1 (1). SETMAR is implicated in the response to or repair of DNA double strand breaks and in non-homologous end joining through the combined activities of the SET and transposase domains (2–5) and through interactions with factors including topoisomerase IIα (Topo IIα) and PRPF19 (5–7). SETMAR knockout has been reported to delay recovery from DNA damage in a variety of cell lines and increases sensitivity to inhibitors of Topo IIα (8, 9). However, the molecular mechanism by which SETMAR impacts DNA damage responses is not well understood, and, in particular, the enzymatic function of the SET domain remains enigmatic. Specifically, the SET domain of SETMAR has been suggested to dimethylate histone H3 lysine 36 (H3K36), but direct evidence for this activity has not been demonstrated (4). Here we perform a detailed analysis of SETMAR activity on histones and show that although SETMAR can weakly methylate histones H2B and H3 in vitro, this activity does not occur on nucleosomes, the predominant physiologic context of histones. Moreover, SETMAR-catalyzed methylation of histones does not occur on any known physiological sites of histone methylation. This suggests that there may be other substrates, likely non-histone proteins, that contribute to the biological function of SETMAR.
Recent analyses of the methyl-lysine proteome have shown that hundreds or thousands of proteins are modified by lysine methylation (10–13). Only a few of these sites have been associated with specific KMTs, so a large number of enzyme/substrate relationships remain to be discovered (14). Techniques for linking KMTs to non-histone substrates are limited, and this remains a major challenge in understanding how lysine methylation participates in wider signaling processes (15). We recently reported a proteomic strategy to identify and measure lysine methylation by using the three malignant brain tumor domain repeats (3xMBT) of L3MBTL1 as a universal affinity reagent to capture modified proteins (10, 16). Here we extend that strategy into a generalized approach for identifying KMT substrates, and we use it to identify substrates of SETMAR. The approach uses in vitro methylation by an enzyme of interest on cell lysates prepared with stable isotope labeling by amino acids in cell culture (SILAC) (17, 18). SILAC labeling allows lysine methylation to be compared between lysates treated with active or inactive enzyme. Proteins showing increased methylation can be tested as potential substrates through direct molecular approaches. Our proteomic approach identified two candidate SETMAR substrates, and we found that SETMAR methylates the splicing factor snRNP70 (also called U1–70K) at lysine 130 both in vitro and in its cellular context.
EXPERIMENTAL PROCEDURES
Protein Sequences, Plasmids, and Antibodies
Recombinant protein sequences were SETMAR (accession no. NP_006506.3), snRNP70 (accession no. NP_003080.2), and the NSD2 SET domain (19). pGEX-6p-1 (GE Healthcare) was used to express proteins as N-terminal fusions with GST (20). For mammalian cell expression, SETMAR was cloned into pCAG-FLAG and the 3xFLAG snRNP70 expression vector was a gift from Robin Reed (Harvard Medical School). lentiCRISPR v2 (AddGene plasmid 52961) (21) was used for SETMAR knockout. Antibodies used were as follows: SETMAR (Abcam, catalog no. ab129455), FLAG M2 (Sigma, catalog no. F1804), and anti-FLAG M2 magnetic beads (Sigma, catalog no. M8823). Rabbit monoclonal H3K36 methyl-specific antibodies were from Cell Signaling Technology (antibodies 14111S and 2901S). Antibodies against methylated H3K36 were validated by probing a custom peptide array as described previously (22).
Cell Culture, SETMAR Knockout, and SETMAR Reconstitution
HEK 293T, HT-1080, and DLD-1 cells were maintained in high-glucose DMEM supplemented with 10% fetal bovine serum (Gibco), penicillin-streptomycin (Life Technologies), and l-glutamine (Life Technologies). CRISPR/Cas9 knockout used stably expressed lentiCRISPR v2 expressing the indicated guide RNAs. Virus particles were produced by cotransfection of HEK293T cells with pCMV-VSVG (AddGene, plasmid 8454) and pCMV-dR8.2 (AddGene, plasmid 8455). DLD-1 cells were transduced with virus and 4 μg/ml Polybrene (Millipore) followed by selection starting at 48 h with 4 μg/μl puromycin (Sigma). Guide sequences were as follows: 5′-CGCCGGCGCCCTTCCAGGTA-3′ (SETMAR knockout) and 5′-CGGCATACTCACTGCGAGTG-3′ (non-targeting control). Knockdown was verified by quantitative PCR and Western blotting. For quantitative proteomics, HT-1080 cells were prepared using SILAC with light amino acids or heavy amino acids (13C615N2-l-lysine/13C615N4-l-arginine) as described previously (23).
Recombinant Protein Preparation
GST fusion proteins were expressed in BL21 Escherichia coli by overnight culture at 20 °C in LB (10 g/L tryptone, 5 g/L yeast extract, 10 g/L NaCl) supplemented with 50 μm ZnCl2 and 0.1 mm isopropyl 1-thio-β-d-galactopyranoside (Sigma) and purified using glutathione-Sepharose 4B (GE Healthcare) as described previously (16). Where indicated, GST was cleaved by incubation with PreScission protease (GE Healthcare) according to the instructions of the manufacturer. Protein concentrations were measured using the Coomassie Plus assay (Pierce).
In Vitro Methylation and Radiolabeling Reactions
Enzymes and substrates were combined in 25 μl of methyltransferase buffer (50 mm Tris (pH 8.0), 20 mm KCl, 5 mm MgCl2, and 10% glycerol) supplemented with 1 mm S-adenosylmethionine (AdoMet, New England Biolabs) or deuterated AdoMet (C/D/N Isotopes) or 2 μCi of tritiated AdoMet (American Radiolabeled Chemicals Inc.). Reactions were incubated overnight at 37 °C and stopped by addition of Laemmli buffer. Radiolabeling reactions were visualized by SDS-PAGE, followed by transfer to a PVDF membrane, treatment with EN3HANCE spray (PerkinElmer Life Sciences), and exposure to film for at least 24 h at −80 °C.
Histone Peptide Arrays
Histone peptides bearing specific methyl modifications were printed in triplicate on a peptide microarray. Primary antibodies were applied to arrays at 1:500 dilution in PBS + 1% Tween 20 + 10% newborn calf serum and incubated overnight at 4 °C. Chicken anti-rabbit Alexa Fluor 647 secondary antibody (Life Technologies) was applied at 1:500 dilution, also in PBS + 1% Tween 20 + 10% newborn calf serum, for 1 h at room temperature. Arrays were washed five times in PBS + 1% Tween 20 and rinsed in PBS and water to before imaging. Arrays were imaged using the GenePix 4000 axon scanner (Molecular Devices).
FLAG-tagged Protein Transfection and Immunoprecipitation
5 μg of pCAG-FLAG SETMAR or Tyr-262 mutant along with 5 μg of 3xFLAG snRNP70 vector were transfected into HEK293T cells using TransIT-293 transfection reagent (Mirus) according to the instructions of the manufacturer. Two days after transfection, cells were lysed in radioimmune precipitation assay buffer (50 mm Tris (pH 7.5), 1% Nonidet P-40, 0.1% SDS, and 150 mm NaCl) with protease inhibitor mixture (Roche). FLAG-tagged proteins were isolated using Anti-FLAG M2 magnetic beads according to the instructions of the manufacturer.
LC-MS/MS of Recombinant And Immunoprecipitated Protein Methylation
Recombinant histone H3 was treated to propionylate lysine prior to tryptic digestion (24), whereas recombinant SETMAR and snRNP70 were processed by in-solution digestion with trypsin (Promega) or Glu-C (New England Biolabs) according to the instructions of the manufacturer. Immunoprecipitated proteins were separated by SDS-PAGE, stained using the SilverQuest silver staining kit (Invitrogen) according to the instructions of the manufacturer, and processed by in-gel digestion with trypsin or Glu-C (16). Peptides were desalted using C18 stage tips (Thermo Scientific). Peptides were separated by HPLC using an Ekspert nanoLC 420 (AB Sciex) and analyzed with an Orbitrap Elite mass spectrometer (Thermo Scientific). Acquisition used data-dependent selection of the top 20 ions with dynamic exclusion, followed by collision-induced dissociation or higher-energy collisional dissociation and analysis of fragment ions in the Orbitrap. Data were analyzed using MaxQuant version 1.3.0.5 (25) with a 1% false discovery rate for proteins and peptides and allowing as variable modifications methionine oxidation; acetylation of protein N termini; and mono-, di-, and trimethylation of lysine. Candidate methylation sites were verified by manual inspection.
Proteome-wide Labeling and Analysis of SETMAR Substrates
SILAC labeled HT-1080 nuclear extracts were prepared by hypotonic lysis and high salt extraction of nuclei (10). Lysates were dialyzed into KMT reaction buffer (50 mm Tris (pH 8.0), 20 mm KCl, 5 mm MgCl2, and 10% glycerol) using Slide-A-Lyzer MINI dialysis devices with 3.5K molecular weight cutoff (Pierce) and cleared by centrifugation at 15,000 × g for 5 min at 4 °C (we now use 250 mm KCl in the reaction buffer to reduce protein precipitation). 20 μg of recombinant SETMAR or Y262A mutant with N-terminal GST fusion was added to 375 μg of light and heavy lysate supplemented with 100 μm AdoMet. Identical reactions were prepared in parallel, with light and heavy labels reversed. The reactions were incubated overnight at 37 °C and stopped by placing them on ice and adding 10 mm EDTA and 100 μm S-adenosyl homocysteine. Each pair of light and heavy lysates was combined, supplemented with 0.1% Triton-X, and incubated overnight on 30 μl of glutathione-Sepharose saturated with 3xMBT (16). Bound proteins were recovered and analyzed by LC-MS/MS as described above, except that only the top 10 ions were selected for fragmentation, and the ion trap was used for MS/MS to decrease cycle time. Data were processed using MaxQuant as described above. Proteins enriched in SETMAR relative to Tyr-262 control from both labeling directions were selected for validation as novel methylation substrates.
RESULTS
SETMAR Participates in Cellular Resistance to Topoisomerase II Inhibition
SETMAR has been shown to make a small contribution to cell growth in several cell lines, and it contributes to resistance to growth inhibition and cell death when cells are treated with inhibitors of Topo II (8, 9). Previous studies characterized the cellular role of SETMAR in DNA damage using SETMAR overexpression or RNAi-mediated depletion (2, 4). Here we selected DLD-1 colon cancer cells because they strongly express SETMAR by Western blot, and we generated SETMAR KO cells using the CRISPR/Cas9 system with a guide RNA targeting the first exon/intron junction (Fig. 1A, see “Experimental Procedures”) (26). SETMAR KO cells showed a small but significant growth defect relative to control cells during normal growth (Fig. 1B). SETMAR KO cells are also more sensitive to Topo II inhibition by etoposide, with greater reduction in growth than control cells treated with low dose etoposide (Fig. 1, B and C). These results are consistent with results from other cell types published previously and suggest a subtle but consistent role for SETMAR in responding to DNA double strand breaks.
FIGURE 1.
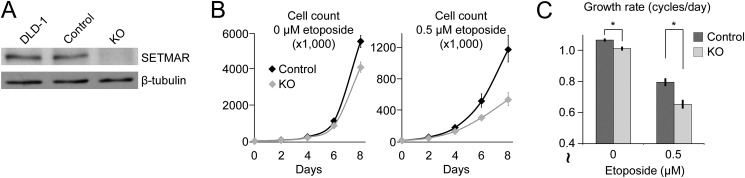
SETMAR contributes to cell growth and resistance to topoisomerase II inhibition. A, SETMAR knockdown using CRISPR/Cas9 in DLD-1 colon cancer cells was verified by Western blotting. B, growth of DLD-1 cells expressing Cas9 and either non-targeting or SETMAR KO guide RNA. Cells were maintained in etoposide at the indicated concentration for 8 days. Each curve represents the average of four independent replicates. Error bars show mean ± S.E. C, growth rates in cycles per day from the growth curves in B. Error bars show mean ± S.E. (n = 4). p values were calculated using two-tailed Student's t test assuming homoscedastic variance. *, p < 0.05.
SETMAR Methylates Histone H3 in Vitro but Not at Lysine 36
To understand how SETMAR enzymatic activity relates to its role in DNA damage responses, we first analyzed potential in vitro histone methylation by recombinant SETMAR. SETMAR methylates the recombinant histones H3 and H2B in radiolabeling experiments with free histones and histone octamers (Fig. 2, A and B). However, SETMAR does not methylate histones in the context of recombinant nucleosomes, a more physiologically relevant model for chromatin (Fig. 2B). Promiscuous activity on free histones but lack of activity on nucleosomes argues that histones may not be physiologically important targets for SETMAR methyltransferase activity.
FIGURE 2.

Histones H2B and H3 are methylated by SETMAR in vitro, but H3K36 is not the target site, and nucleosomes are not appreciably methylated. A, radiolabeling of recombinant histones by GST-SETMAR using tritiated AdoMet and including GST as a negative control. B, radiolabeling of recombinant octamers or nucleosomes by GST-SETMAR with GST as a negative control. Octamer methylation occurs as a doublet for histones H3 and H2B, with a weak signal from histone H4. C, mutation of conserved tyrosines reduces GST-SETMAR activity on histone H2B. D, LC-MS/MS was used to measure lysine methylation of histone H3 after in vitro methylation by GST-SETMAR using deuterated AdoMet (17.0345-Da mass shift). To facilitate the analysis, histones were treated to propionylate non-methyl and monomethyl lysine. HPLC elution profiles show a 10-ppm mass window around peptide masses for the peptide containing H3K36 (peptide sequence KPrSAPATGGVKPrKPrPHR, Lys-36 is underlined, and Pr indicates propionylation; m/z are 801.460, 809.977, and 790.481). E, peptide arrays containing histone peptides with various methyl states were probed to verify antibodies for H3K36 methylation. Each peptide is represented as a set of three adjacent spots. The legend indicates which histone peptide appears at each position. F, histone H3 methylated by GST-SETMAR or the Y262A catalytically inactive mutant and probed with monoclonal antibodies specific for mono- or dimethyl H3K36. Recombinant nucleosomes methylated by NSD2 are shown as a positive control.
Conserved tyrosine residues are often required for SET domain KMT activity. Mutation of the conserved residues Y168A, Y192A, and Y262A greatly reduce SETMAR activity measured in radiolabeling assays with histone H2B (Fig. 2C). We use the Y262A mutant as a catalytically dead control in future experiments because it has the least residual activity, and Y262 aligns with an inactivating mutation in the H3K36 dimethyltransferase NSD2 (19).
Previous data implicating SETMAR in methylation of H3K36 were obtained on the basis of methyl-specific antibodies (2), which often have significant off-target interactions that can lead to misidentification of methylation activity (22, 27). To test whether H3K36 is a direct SETMAR target, we used SETMAR to label recombinant H3 with deuterated AdoMet and analyzed the resulting methylation by tryptic digestion and LC-MS/MS (24). Enzymatic methylation with deuterated AdoMet gives a 17-Da mass shift that is distinct from methylation arising from contaminating chemical processes that could generate methylated residues. Following in vitro methylation, we detected the non-methylated peptide containing Lys-36 with a signal several hundred times the lower limit of detection. No signal was detected for any methylated peptide containing Lys-36 (Fig. 2D). Non-methylated peptides were detected with similar signal strength for every lysine in H3 except Lys-115. We identified monomethylation of Lys-18 by MS/MS, but only at extremely low abundance (<0.1%). To test with an independent method whether SETMAR can methylate H3K36, we first verified the specificity of rabbit monoclonal antibodies for mono- and dimethyl H3K36 (H3K36me1 and H3K36me2) by probing them on an array of histone peptides (Fig. 2E) (22). Western blot analysis with these H3K36me1 and H3K36me2 antibodies showed no detectable signal after overnight reaction with SETMAR. In contrast, a strong signal was observed for H3K36 mono- and dimethylation of recombinant nucleosomes incubated with the NSD2 SET domain, a well characterized H3K36 methyltransferase (Fig. 2F) (19). On the basis of these data we conclude that SETMAR does not methylate H3 at lysine 36.
Automethylation Occurs Primarily at Lysine 335
SETMAR has been reported to methylate itself at Lys-498, and it has been proposed that automethylation has an inhibitory function (5) (note that the NCBI reference sequence NP_006506 has been updated so that SETMAR Lys-498 corresponds to Lys-485 in Ref. 5). To characterize SETMAR automethylation activity, we used LC-MS/MS to examine methylated lysine following in vitro methylation reactions with deuterated SAM. We used separate digestions with trypsin and GluC proteases to map methyl sites. Methylation stoichiometry was estimated from GluC digestion because lysine methylation can affect the efficiency of digestion by trypsin. Trace monomethylation was identified at Lys-498 by MS/MS following tryptic digestion, but the amount of methylation was too low (<0.1%) to be quantified precisely after digestion with GluC (Fig. 3A).
FIGURE 3.

SETMAR automethylation occurs primarily at lysine 335. A, HPLC chromatograms from LC-MS/MS analysis of SETMAR lysine 498 automethylation stoichiometry after overnight in vitro reaction using deuterated AdoMet (17.0345-Da mass shift) and digestion with GluC. The chromatograms show 10 ppm around the expected peptide masses (peptide sequence KWILYDNRRRSAQWLDQE, Lys-498 is underlined; m/z are 595.060, 599.318, and 603.577). B, fragmentation spectrum identifying monomethylation of SETMAR lysine 335 following in vitro automethylation using deuterated AdoMet and digestion with trypsin. The Orbitrap mass analyzer was used to obtain the MS/MS spectrum with high mass accuracy. Kme, monomethyl lysine; asterisk, carbamidomethylation. C, HPLC chromatograms from LC-MS/MS analysis of SETMAR automethylation at lysine 335 following in vitro automethylation using deuterated AdoMet and digestion with GluC (peptide sequence KEPSMC*GSAPSVFPSC*KRLTLE, Lys-335 is underlined; m/z are 621.054, 625.312, and 629.571). D, Lys-to-Arg mutations were made at selected lysine residues in GST-SETMAR, and each enzyme was tested for automethylation in a radiolabeling assay. Radiolabeling of histone H2B is shown to verify that the mutations do not affect enzyme activity. E, FLAG-tagged SETMAR was expressed in HEK 293T cells and analyzed by immunoprecipitation and LC-MS/MS with GluC digestion to determine Lys-335 methylation stoichiometry in cells (the same peptide as in C with a longer HPLC gradient).
We also observed methylation of Lys-335 in tryptic digestion of SETMAR after in vitro automethylation (Fig. 3B). GluC digestion and analysis by LC-MS/MS found about 24% monomethylation and 5% dimethylation relative to unmodified samples at Lys-335 and no trimethylation (Fig. 3C, compare A and C). Consistent with this result, K335R mutation, but not K498R mutation, virtually abrogated SETMAR automethylation in a radiolabeling assay without affecting intrinsic SETMAR catalytic activity on H2B (Fig. 3D). The catalytic mutant Y262A does not automethylate or methylate H2B (Fig. 3D).
To determine whether Lys-335 automethylation occurs in cells, we used LC-MS/MS to analyze the methylation status of ectopically expressed SETMAR isolated from HEK293T cells using an N-terminal FLAG tag. Mono- and dimethylation of Lys-335 occur at about 26% and 2% respectively (Fig. 3E). We also observed monomethylation at Lys-319 and Lys-395 in 293T cells. Because these sites are not observed following in vitro automethylation, they are not likely to be mediated by SETMAR but, rather, a still to be identified KMT.
Methyl-lysine Proteomics Identify Candidate SETMAR Substrates
Because SETMAR does not have a physiological activity on histones, we hypothesized that it may have additional protein substrates beyond automethylation. We recently reported a strategy for quantitative proteome-wide analysis of mono- and dimethylation of lysine (10, 16). The strategy uses the 3xMBT domain of the protein L3MBTL1 as a universal reagent to enrich proteins modified with mono- or dimethylated lysine. Because SETMAR automethylation generates mono- and dimethylation, we reasoned that this proteomic strategy could be used to identify new SETMAR substrates. To identify candidate substrates, we prepared nuclear protein extract from HT-1080 cells grown in medium containing lysine and arginine with normal isotopes or with entirely 13C and 15N SILAC medium. Lysates were treated with SETMAR or Y262A mutant in in vitro methylation reactions and treated. Control lysates were combined, methylated proteins were captured using immobilized 3xMBT, and the relative abundance of proteins between treated and control lysates was determined using LC-MS/MS (Fig. 4A and supplemental Table S1). The experiment was repeated with light and heavy isotopic labels reversed to remove any effects because of differences between the lysates. Methyl-dependent pulldown of snRNP70, a splicing factor, and SPTBN2 increased substantially after treatment with catalytically active SETMAR (Fig. 4B and supplemental Table S1). We confirmed the increase in binding to snRNP70 by detailed manual inspection of the relevant mass spectra (Fig. 4C). We focused validation experiments on snRNP70 because we had identified it previously as a potentially lysine-methylated protein (10).
FIGURE 4.

Proteome-wide analysis of proteins methylated by SETMAR in cell lysate identifies snRNP70 as a candidate substrate. A, schematic of the procedure for in vitro labeling of cell lysates prepared with stable isotopic labeling, enrichment of methylated proteins by the immobilized 3xMBT domain, and quantitative analysis by LC-MS/MS. Candidate substrates are identified by the their enzyme-dependent association with 3xMBT. B, relative enrichment of proteins bound by 3xMBT from lysate methylated by GST-SETMAR compared with the Y262A mutant as a negative control. The axes show log base-2 of relative enrichment, and each axis represents an independent experiment with isotopic labels reversed. Positive values are defined so that candidate substrates are in the top right quadrant. C, an example mass spectrum for a peptide from snRNP70 showing the amount bound by 3xMBT from lysates treated with SETMAR or inactive enzyme. The blue circle indicates peptide from light SILAC lysate (treated with the Y262A mutant), and purple indicates heavy SILAC lysate (treated with active SETMAR).
SETMAR Methylates snRNP70 at Lysine 130
In in vitro radiolabeling reactions, SETMAR directly methylated snRNP70 residues 1–204 expressed in E. coli, suggesting that snRNP70 is a bona fide substrate of SETMAR (Fig. 5A). LC-MS/MS analysis of snRNP70 methylated by SETMAR identified monomethylated Lys-130 along with trace dimethylation (Fig. 5, B and C). Mutating snRNP70 Lys-130 to arginine completely eliminated SETMAR activity toward the substrate, whereas the Y262A SETMAR mutant had no activity on wild-type snRNP70 or on the K130R mutant (Fig. 5D). To determine whether methylation of snRNP70 occurs in cells, we coexpressed 3xFLAG-tagged snRNP70 along with SETMAR or Y262A in HEK 293T cells because these cells do not express SETMAR at an appreciable level (28). FLAG immunoprecipitation followed by in-gel tryptic digestion and LC-MS/MS identified snRNP70 Lys-130 methylated peptide only when coexpressed with wild-type SETMAR. The signal intensity for monomethylated Lys-130 peptide was substantially higher when snRNP70 was coexpressed with active SETMAR compared with the Y262A mutant (0.35% versus 0.05%) (Fig. 5E).
FIGURE 5.

SETMAR methylates snRNP70 at Lys-130. A, radiolabeling assay with GST-SETMAR or the Y262A mutant and snRNP70 (residues 1–204). B, fragmentation spectrum showing in vitro monomethylation of snRNP70 Lys-130 by SETMAR in vitro using deuterated SAM and digested with trypsin. The Orbitrap mass analyzer was used to obtain high-resolution mass accuracy. C, HPLC elution profiles for non-methyl, monomethyl, and dimethyl snRNP70 Lys-130 from tryptic digestion after in vitro methylation by GST-SETMAR using deuterated AdoMet. HPLC elution profiles show a 10-ppm mass window around expected peptide masses (peptide sequence EFEVYGPIKR, Lys-130 is underlined; m/z are 619.332, 628.850, and 636.348). D, radiolabeling of wild-type or K130R mutant snRNP70 (1–204) by GST-SETMAR or the Y262A catalytically inactive mutant. E, snRNP70 was expressed in HEK293T cells with N-terminal 3xFLAG and either active SETMAR or the Y262A mutant. snRNP70 was enriched using FLAG M2 antibody, and methylation at Lys-130 was analyzed by in-gel tryptic digestion and LC-MS/MS. HPLC chromatograms show a 10 ppm mass window around expected peptide masses (the same peptide as in C, methyl mass 14.0157 Da; m/z are 619.332 and 626.340).
DISCUSSION
It has been difficult to identify non-histone substrates of KMTs because techniques were not available for quantitative analysis of the methyl-lysine proteome. We achieved this goal by using 3xMBT as a universal affinity reagent for mono- and dimethylated lysine and have previously demonstrated the proof of principle that 3xMBT can be used for quantitative proteomics (10, 16). Others have recently developed antibodies with a broad specificity for methylated lysine to perform similar proteomic analyses (11, 12) and developed methyl-lysine reader domains as tools to probe lysine methylation (29, 30). Here we applied our approach to show that the mRNA splicing factor snRNP70 is a new non-histone substrate for SETMAR, an orphan KMT.
The strategy described here uses in vitro lysine methylation of cell lysates that are identical except for their isotopic labels. This ensures that protein levels and endogenous methylation are identical between the two conditions. Using in vitro methylation allows substrate methylation to be driven beyond physiological levels, which provides a maximum signal relative to any endogenous methylation. Testing KMT activity in cell lysate also makes it likely that any required cofactors or protein complexes will be present. It is important to note that even relatively small changes in overall methylation, such as the ∼50% increase seen with snRNP70, can point to bona fide KMT substrates. This is because many proteins carry endogenous methylation at multiple lysine residues. Here we mitigate this effect by using cells that express SETMAR at only a low level. In other contexts, background methylation could be minimized by using lysate from KMT knockout cells as the substrate for in vitro labeling reactions. Our approach requires KMTs that are active in cell lysate and cannot be used in cases where methylation only occurs under specific physiological conditions or requires a specific activating signal.
Earlier work linked SETMAR methyltransferase activity to H3K36 dimethylation near DNA double strand breaks (4). Our analysis by LC-MS/MS and rigorously characterized monoclonal antibodies indicates that SETMAR weakly methylates histone H3 at a non-canonical site in vitro but that H3K36 is not methylated by SETMAR. In addition, the inability of the enzyme to methylate nucleosomes suggests that histones may not be physiological substrates. These results argue that if H3K36 methylation is somehow regulated by SETMAR, then the mechanism would be indirect. Our proteome-wide screen identified the mRNA splicing factor snRNP70 as a new SETMAR substrate. We have seen previously that snRNP70 is strongly enriched by binding to 3xMBT relative to a control pulldown, suggesting that it is probably heavily methylated on lysine residues (10). Methylation by SETMAR provides a direct link between lysine methylation of splicing factors and an upstream signaling pathway. Because splicing factors are a major target for lysine methylation, we expect that many more connections remain to be discovered (10). Arginine methylation is already known to regulate splicing (31), and lysine methylation may have a similar role, perhaps forming part of a larger posttranslational “splicing code” analogous to the role of PTMs on histones to regulate chromatin function.
Monomethylation is the primary state created by SETMAR methylation at Lys-335 and at snRNP70 Lys-130, although a small amount of dimethylated product is also observed. A preference for monomethylation as opposed to higher methyl states is consistent with tyrosine at the potential “F/Y switch” residue Tyr-260 (32, 33). Lys-335 methylation does not affect SETMAR catalytic activity, suggesting that it may, instead, regulate protein interactions. The distribution of mono- and dimethylated SETMAR Lys-335 is very similar when the enzyme is overexpressed in HEK 293T cells. We conclude that SETMAR is primarily a monomethyltransferase both in vitro and in its cellular context. We observed an increase in snRNP70 Lys-130 methylation when cells are transfected with SETMAR, although still at a low level. Because SETMAR has been reported to act specifically at sites of DNA damage, it may be that methylation only occurs on a small fraction of the total substrate pool, perhaps to shut down splicing at sites of DNA damage (4). It could also be that SETMAR activity depends on upstream activities, such as phosphorylation by Chk1 (34).
Both snRNP70 Lys-130 and the automethylation site at Lys-335 are followed immediately by arginine and then isoleucine or leucine. This consistent sequence motif suggests that SETMAR recognizes a consensus sequence C-terminal to the target lysine. Our analysis by LC-MS/MS ruled out every site on histone H3 as a major SETMAR target except Lys-115 because this residue is difficult to map by mass spectrometry. Lys-115 is the only lysine in histone H3 followed by Arg, and the valine at position 117 is similar to Ile and Leu. The combination of LC-MS/MS data and sequence motif suggest that Lys-115 is likely to be the residue in histone H3 methylated by SETMAR in vitro, although there is no evidence that methylation at this position occurs in vivo.
The technique described here provides a new proteomic strategy to identify non-histone substrates of KMTs. We expect that this approach and related techniques will be important for identifying KMTs responsible for the many hundreds of proteins modified by lysine methylation. They will be especially useful for uncharacterized or orphan KMT enzymes and for new or emerging KMT families, such as the rapidly growing list of seven-β-strand methyltransferases that target lysine (35).
Supplementary Material
Note Added in Proof
Nicolas Reynoird's contributions to this article fulfill the JBC authorship criteria, but his authorship was inadvertently omitted from the version of the article that was published on March 20, 2015 as a Paper in Press.
This work was supported, in whole or in part, by National Institute of Health Grants R01 GM079641 (to O. G.).
Mass spectrometry data files in the native .raw format and complete search results for proteomic identification of SETMAR substrates have been submitted to the PRIDE proteomics database under accession number PXD001635.

This article contains supplemental Table S1.
- KMT
- lysine methyltransferase
- H3K36
- histone H3 lysine 36
- H3K36me1
- mono-methylated H3K36
- H3K36me2
- di-methylated H3K36
- 3xMBT
- three malignant brain tumor domain repeats
- SILAC
- stable isotope labeling by amino acids in cell culture
- AdoMet
- S-adenosylmethionine.
REFERENCES
- 1. Cordaux R., Udit S., Batzer M. A., Feschotte C. (2006) Birth of a chimeric primate gene by capture of the transposase gene from a mobile element. Proc. Natl. Acad. Sci. U.S.A. 103, 8101–8106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lee S. H., Oshige M., Durant S. T., Rasila K. K., Williamson E. A., Ramsey H., Kwan L., Nickoloff J. A., Hromas R. (2005) The SET domain protein Metnase mediates foreign DNA integration and links integration to nonhomologous end-joining repair. Proc. Natl. Acad. Sci. U.S.A. 102, 18075–18080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beck B. D., Lee S. S., Williamson E., Hromas R. A., Lee S. H. (2011) Biochemical characterization of metnase's endonuclease activity and its role in NHEJ repair. Biochemistry 50, 4360–4370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fnu S., Williamson E. A., De Haro L. P., Brenneman M., Wray J., Shaheen M., Radhakrishnan K., Lee S. H., Nickoloff J. A., Hromas R. (2011) Methylation of histone H3 lysine 36 enhances DNA repair by nonhomologous end-joining. Proc. Natl. Acad. Sci. U.S.A. 108, 540–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Williamson E. A., Rasila K. K., Corwin L. K., Wray J., Beck B. D., Severns V., Mobarak C., Lee S. H., Nickoloff J. A., Hromas R. (2008) The SET and transposase domain protein Metnase enhances chromosome decatenation: regulation by automethylation. Nucleic Acids Res. 36, 5822–5831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beck B. D., Park S. J., Lee Y. J., Roman Y., Hromas R. A., Lee S. H. (2008) Human Pso4 is a metnase (SETMAR)-binding partner that regulates metnase function in DNA repair. J. Biol. Chem. 283, 9023–9030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hromas R., Wray J., Lee S. H., Martinez L., Farrington J., Corwin L. K., Ramsey H., Nickoloff J. A., Williamson E. A. (2008) The human set and transposase domain protein Metnase interacts with DNA ligase IV and enhances the efficiency and accuracy of non-homologous end-joining. DNA Repair 7, 1927–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wray J., Williamson E. A., Sheema S., Lee S. H., Libby E., Willman C. L., Nickoloff J. A., Hromas R. (2009) Metnase mediates chromosome decatenation in acute leukemia cells. Blood 114, 1852–1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wray J., Williamson E. A., Royce M., Shaheen M., Beck B. D., Lee S. H., Nickoloff J. A., Hromas R. (2009) Metnase mediates resistance to topoisomerase II inhibitors in breast cancer cells. PLoS ONE 4, e5323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Moore K. E., Carlson S. M., Camp N. D., Cheung P., James R. G., Chua K. F., Wolf-Yadlin A., Gozani O. (2013) A general molecular affinity strategy for global detection and proteomic analysis of lysine methylation. Mol. Cell 50, 444–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guo A., Gu H., Zhou J., Mulhern D., Wang Y., Lee K. A., Yang V., Aguiar M., Kornhauser J., Jia X., Ren J., Beausoleil S. A., Silva J. C., Vemulapalli V., Bedford M. T., Comb M. J. (2014) Immunoaffinity enrichment and mass spectrometry analysis of protein methylation. Mol. Cell Proteomics 13, 372–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cao X. J., Arnaudo A. M., Garcia B. A. (2013) Large-scale global identification of protein lysine methylation in vivo. Epigenetics 8, 477–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wu Z., Cheng Z., Sun M., Wan X., Liu P., He T., Tan M., Zhao Y. (2015) A chemical proteomics approach for global analysis of lysine mono-methylation. Mol. Cell Proteomics 14, 329–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang X., Wen H., Shi X. (2012) Lysine methylation: beyond histones. Acta Biochim. Biophys. Sin. 44, 14–27 [DOI] [PubMed] [Google Scholar]
- 15. Carlson S. M., Gozani O. (2014) Emerging technologies to map the protein methylome. J. Mol. Biol. 426, 3350–3362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Carlson S. M., Moore K. E., Green E. M., Martín G. M., Gozani O. (2014) Proteome-wide enrichment of proteins modified by lysine methylation. Nat. Protoc. 9, 37–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ong S. E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 18. Ong S. E., Mann M. (2007) Stable isotope labeling by amino acids in cell culture for quantitative proteomics. Methods Mol. Biol. 359, 37–52 [DOI] [PubMed] [Google Scholar]
- 19. Kuo A. J., Cheung P., Chen K., Zee B. M., Kioi M., Lauring J., Xi Y., Park B. H., Shi X., Garcia B. A., Li W., Gozani O. (2011) NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol. Cell 44, 609–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. West L. E., Roy S., Lachmi-Weiner K., Hayashi R., Shi X., Appella E., Kutateladze T. G., Gozani O. (2010) The MBT repeats of L3MBTL1 link SET8-mediated p53 methylation at lysine 382 to target gene repression. J. Biol. Chem. 285, 37725–37732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sanjana N. E., Shalem O., Zhang F. (2014) Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bua D. J., Kuo A. J., Cheung P., Liu C. L., Migliori V., Espejo A., Casadio F., Bassi C., Amati B., Bedford M. T., Guccione E., Gozani O. (2009) Epigenome microarray platform for proteome-wide dissection of chromatin-signaling networks. PLoS ONE 4, e6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ong S. E., Mann M. (2006) A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC). Nat. Protoc. 1, 2650–2660 [DOI] [PubMed] [Google Scholar]
- 24. Garcia B. A., Mollah S., Ueberheide B. M., Busby S. A., Muratore T. L., Shabanowitz J., Hunt D. F. (2007) Chemical derivatization of histones for facilitated analysis by mass spectrometry. Nat. Protoc. 2, 933–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 26. Cong L., Ran F. A., Cox D., Lin S., Barretto R., Habib N., Hsu P. D., Wu X., Jiang W., Marraffini L. A., Zhang F. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fuchs S. M., Krajewski K., Baker R. W., Miller V. L., Strahl B. D. (2011) Influence of combinatorial histone modifications on antibody and effector protein recognition. Curr. Biol. 21, 53–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. De Haro L. P., Wray J., Williamson E. A., Durant S. T., Corwin L., Gentry A. C., Osheroff N., Lee S. H., Hromas R., Nickoloff J. A. (2010) Metnase promotes restart and repair of stalled and collapsed replication forks. Nucleic Acids Res. 38, 5681–5691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Su Z., Boersma M. D., Lee J. H., Oliver S. S., Liu S., Garcia B. A., Denu J. M. (2014) ChIP-less analysis of chromatin states. Epigenetics Chromatin 7, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu H., Galka M., Mori E., Liu X., Lin Y. F., Wei R., Pittock P., Voss C., Dhami G., Li X., Miyaji M., Lajoie G., Chen B., Li S. S. (2013) A method for systematic mapping of protein lysine methylation identifies functions for HP1β in DNA damage response. Mol. Cell 50, 723–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bedford M. T., Clarke S. G. (2009) Protein arginine methylation in mammals: who, what, and why. Mol. Cell 33, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Collins R. E., Tachibana M., Tamaru H., Smith K. M., Jia D., Zhang X., Selker E. U., Shinkai Y., Cheng X. (2005) In vitro and in vivo analyses of a Phe/Tyr switch controlling product specificity of histone lysine methyltransferases. J. Biol. Chem. 280, 5563–5570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dillon S. C., Zhang X., Trievel R. C., Cheng X. (2005) The SET-domain protein superfamily: protein lysine methyltransferases. Genome Biol. 6, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hromas R., Williamson E. A., Fnu S., Lee Y. J., Park S. J., Beck B. D., You J. S., Leitao A., Laitao A., Nickoloff J. A., Lee S. H. (2012) Chk1 phosphorylation of Metnase enhances DNA repair but inhibits replication fork restart. Oncogene 31, 4245–4254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kernstock S., Davydova E., Jakobsson M., Moen A., Pettersen S., Mælandsmo G. M., Egge-Jacobsen W., Falnes P. (2012) Lysine methylation of VCP by a member of a novel human protein methyltransferase family. Nat. Commun. 3, 1038. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.