

Background: Proprotein convertases (PCs) control the maturation of several A Disintegrin and Metalloprotease (ADAM) proteases.
Results: Mutating a newly identified upstream PC site interferes with activation of ADAMs 9, 10, and 17.
Conclusion: Processing at the upstream PC site is important for the activation of these ADAMs.
Significance: The upstream PC site in several ADAMs suggests a novel general mechanism for their maturation.
Keywords: ADAM, Epidermal Growth Factor Receptor (EGFR), Furin, Metalloprotease, Shedding, Proenzyme Processing, Proprotein Convertase
Abstract
Proenzyme maturation is a general mechanism to control the activation of enzymes. Catalytically active members of the A Disintegrin And Metalloprotease (ADAM) family of membrane-anchored metalloproteases are synthesized as proenzymes, in which the latency is maintained by their autoinhibitory pro-domains. A proteolytic processing then transforms the proenzyme into a catalytically active form. The removal of the pro-domain of ADAMs is currently thought to depend on processing at a canonical consensus site for the proprotein convertase Furin (RXXR) between the pro- and the catalytic domain. Here, we demonstrate that this previously described canonical site is a secondary cleavage site to a prerequisite cleavage in a newly characterized upstream PC site embedded within the pro-domain sequence. The novel upstream regulatory site is important for the maturation of several ADAM proenzymes. Mutations in the upstream regulatory site of ADAM17, ADAM10, and ADAM9 do not prevent pro-domain processing between the pro- and metalloprotease domain, but nevertheless, cause significantly reduced catalytic activity. Thus, our results have uncovered a novel functionally relevant PC processing site in the N-terminal part of the pro-domain that is important for the activation of these ADAMs. These results suggest that the novel PC site is part of a general mechanism underlying proenzyme maturation of ADAMs that is independent of processing at the previously identified canonical Furin cleavage site.
Introduction
The term proenzyme refers to an inactive precursor of an enzyme, in which the N-terminal portion of the protein acts as an autoinhibitor to keep the protein in its latent form. The secretion of inactive enzymes helps to prevent unwanted consequences such as pre-mature or inappropriate activity of the enzyme. A Disintegrin And Metalloprotease (ADAM)2 family members are typically produced as proenzymes and require limited proteolysis at selected sites to generate the active form. ADAMs are multidomain type I transmembrane proteases responsible for ectodomain processing of membrane-tethered proteins. These proteins play important roles in many biological processes such as development (1–3), inflammatory responses (4, 5), pathogenesis of cancer (4, 6), and Alzheimer disease (7–10). Despite their importance as major proteases responsible for the secretion and maturation of vital membrane proteins in vivo, the activation mechanism of ADAMs by processing of their pro-domains is not fully understood.
Catalytically active ADAMs are generated as inactive proenzymes, in which the N-terminal pro-domain is responsible for inhibiting the activity via the “cysteine switch” mechanism (11), where a cysteine residue in the pro-domain coordinates the zinc ion in the catalytic site, preventing proteolytic activity. One of the most important post-translational modifications of ADAMs is thought to be their pro-domain removal in the trans-Golgi network, a process that results in the mature form of the enzyme. The role of the pro-domain in the biosynthesis of the enzyme has been demonstrated to be more than keeping the enzyme in its latent form. In fact, the presence of the pro-domain was also shown to be required for proper intracellular trafficking of ADAM10 and -17 (12, 13), presumably because it also acts as an intramolecular chaperone to ensure correct protein folding and to protect against degradation (14, 15).
Most members of the ADAM family of metalloproteases contain a proprotein convertase (PC) consensus cleavage site, characterized by a dibasic motif (16, 17) RXXR, at the boundary between the pro-domain and the catalytic domain. The involvement of PC or Furin, a member of the PC family, in the activation of ADAMs has been demonstrated in several studies. For example, mutations in the putative Furin site located between the pro-domain and the catalytic domain of ADAM10, ADAM12, and ADAM19 prevent the release of the pro-domain, thus keeping the enzyme latent (12, 18, 19). In addition, inhibitors of the early secretory pathway, brefeldin A and monensin, have been shown to block the processing of ADAM9 and ADAM15 in the Golgi apparatus (14, 20). This intracellular location where the maturation of ADAMs takes place is consistent with the localization of Furin and other PCs (17). However, autocatalytic removal of the pro-domain has been shown for maturation of ADAM8 and ADAM28 (21, 22).
Members of the PC family are serine proteases that include PACE (Furin), PACE4, PC5/PC6, and PC7/PC8. These enzymes recognize dibasic motifs and cleave the peptide bond on the carboxyl side (16, 17). As a major PC, Furin is concentrated in the trans-Golgi network and cycles between this compartment and the cell surface through the endocytic pathway. Numerous studies have shown that Furin activates a large number of proproteins in multiple compartments (23). The minimal recognition sequence for Furin requires basic residues at P1 and P4, but the consensus sequence for efficient cleavage by Furin is RXXR, although an amino acid with a hydrophobic aliphatic side chain is not suitable at the P1′ position (17).
In this report we show that the removal of the ADAM pro-domain occurs via cleavage of two different sites, in which, the boundary canonical sites are secondary to upstream and unstructured regions, bearing regulatory cleavage sites embedded within the enzyme pro-domain. Processing at the upstream regulatory site in the pro-domain is mandatory for proenzyme maturation and full enzyme activation. Mutations in the unstructured upstream regulatory site did not prevent pro-domain processing at the canonical site, but nevertheless, gave rise to ADAMs in a pseudo mature form on the cell surface with significantly reduced activity compared with controls. Thus, our results present a novel and potentially general mechanism underlying the maturation of pro-ADAMs that deviates from the current canonical model of activation by processing at the boundary between the pro- and metalloprotease domains.
EXPERIMENTAL PROCEDURES
Sequence Alignment and Prediction of Disordered Sequences and Proprotein Convertase Cleavage Sites in ADAM Pro-domains
Sequence alignment of all the pro-domains of members of the ADAM family of membrane-anchored metalloproteases (including the signal peptide) were computed by using the ClustalW2 server. The alignment was cross-referenced with the propeptide cleavage potential predicted by the ProP server (24), and the ADAM members, which have been identified to have an upstream PC site around the same region, were picked for further analysis. The following ADAM members were found to possess an upstream PC site: ADAM8-RVRR44/A45, ADAM9-RERR56/E57 ADAM10-RAKR51/E52, ADAM11-RLVR55/E56, and ADAM17-RKR58/D59. The pro-domains of these ADAMs were analyzed for their potential to be disordered using the Disopred server (25) with a threshold of 5%.
Cloning of Mouse Full-length ADAM17, ADAM10, and ADAM9
Mutations of the PC sites were incorporated in the following constructs: mouse full-length ADAM17 WT cDNA (with a C-terminal Myc tag (EQKLISEEDL)) was cloned into pSECtag. Mutagenic primers were incorporated into the mouse full-length ADAM17 WT with restriction-free PCR cloning (26). Mutations in ADAM9 as well as in ADAM10 (with a C-terminal HA tag (YPYDVPDYA)) were incorporated by Transfer PCR as previously described (27). All sequences were confirmed by DNA sequencing.
Transfection of Mouse Embryonic Fibroblasts (MEFs) and Shedding Assays
Shedding experiments were performed in Adam17−/− MEFs or Adam10/17−/− MEFs generated from knock-out mice, as previously described (28, 39). Briefly, MEFs were generated from either wild type (WT) or Adam17−/− embryos at E13.5. Double knock out Adam10/17−/− MEFs were derived from E9.5 embryos. Plasmids encoding AP-tagged substrates were constructed by inserting partial cDNAs for human transforming growth factor (TGF) α, betacellulin (BTC), or EphB4 into the 3′ end of human placental AP cDNA on a pRc/CMV-based expression vector pAlPh. AP-TGFα was co-transfected with ADAM17 WT or mutants into Adam17−/− MEFs. AP-BTC was co-transfected with ADAM10 WT or mutants into Adam10/17−/− MEFs. AP-EphB4 was co-transfected with ADAM9 WT or mutants into Adam10/17−/− MEFs. All cells were transfected with LipofectamineTM (Invitrogen) according to manufacturer's instructions. Fresh Opti-MEM (Invitrogen) was added after 12 h, incubated for 1 h, and then replaced with Opti-MEM containing either 20 ng/ml of phorbol 12-myristate 13-acetate or 2.5 μm ionomycin, which was collected after 1 h. Evaluation of AP activity by colorimetric assays was performed as described previously (29). No AP activity was present in conditioned media of non-transfected cells.
Western Blot Analysis of the Pro- and Mature Forms of Overexpressed ADAM17, ADAM10, or ADAM9 in HEK293 Cells
HEK293 cells were grown in Nunc six-well plates and cultured in DMEM containing penicillin-streptomycin and 10% FBS, 37 °C, and 5% CO2 to ∼80% confluence. Mouse full-length ADAM17 WT and mutants, mouse full-length ADAM10 WT and mutants, and mouse full-length ADAM9 WT and mutants were transfected with jetPEI® reagent (PolyPlus) according to the manufacturer's instructions. Cells were harvested after overnight incubation and cell surface biotinylation was performed with a Cell Surface Protein Isolation Kit (Pierce) according to the manufacturer's instructions with minor adjustments. For cell lysis, MMP inhibitors (1 μm marimastat and 10 mm 1,10-phenanthroline) and EDTA-free complete Protease Inhibitor Mixture (Roche) were added to the lysis buffer to prevent autodegradation of the mature form of ADAM17 (30, 31). Samples from the total lysate were collected immediately after lysis and analyzed by Western blot. Equal protein concentrations were loaded for each sample of the WT or mutant forms of each ADAM and separated by SDS-PAGE (8% polyacrylamide) and transferred to a nitrocellulose membrane. The blots were probed with polyclonal anti-rabbit sera against the cytoplasmic domain or pro-domain of ADAM17. ADAM10 samples were probed with rabbit anti-HA (Santa Cruz) and ADAM9 samples were probed with rabbit anti-ADAM9 antibodies (32). A horseradish peroxidase-conjugated goat anti-rabbit antibody (Santa Cruz) was used as a secondary antibody. Signal was detected with ECL (Pierce). The cell surface fraction blots were quantified using ImageJ to calculate the ratio between the pro- and mature forms.
Cloning, Expression, and Purification of WT or Mutant ADAM17 Proenzymes in Escherichia coli
The codon usage of WT human ADAM17 proenzyme DNA (pro-domain and catalytic domain Δ-signal sequence- Asp23–Val477) was optimized for expression in E. coli by GeneArt. The ADAM17 proenzyme was cloned into pET28 with an N-terminal His tag followed by the tobacco etch virus cleavage site SAGENLYFQGT. Mutations in the upstream site mutant (US), the boundary site mutant (BS), the double US/BS mutant (2M), and the inactivating EA mutant in the catalytic site together with the BS mutant (EA/BS) were introduced by PCR mutagenesis with the pET28 WT ADAM17 proenzyme as a template. Sequences were confirmed by DNA sequencing. ADAM17 proenzyme WT and mutants were expressed and purified according to the following procedure. Electrocompetent bacteria (E. coli BL21(DE3)) were transformed with the corresponding ADAM17 proenzyme plasmid and plated on LB plates containing 30 μg/ml of kanamycin. After overnight incubation at 37 °C, single colonies were selected and cultured in 10 ml of LB medium with 30 μg/ml of kanamycin overnight at 37 °C. These cultures were added to 1 liter of LB medium containing 30 μg/ml of kanamycin, and protein expression was induced with 200 μm isopropyl β-d-thiogalactopyranoside when the optical density of the culture reached 0.6. Immediately after induction, cells continued to grow at 15 °C overnight. Cells were harvested by centrifugation for 20 min at 5000 rpm. The pellets were resuspended with 100 ml of 50 mm Tris, 300 mm NaCl, 20 mm imidazole, 0.1 mg/ml of lysozyme, 1 μg/ml of DNase, and one tablet of protease inhibitor mixture (Roche Diagnostics), pH 8. The resuspended pellets were sonicated and centrifuged at 15,000 rpm for 45 min at 4 °C. The supernatant was applied to a 5-ml Ni2+ column (GE Healthcare). The column was washed with 10 times bed volume of buffer containing 50 mm Tris, 300 mm NaCl, and 20 mm imidazole, pH 8. The protein was eluted with buffer containing 50 mm Tris, 300 mm NaCl, and 250 mm imidazole, pH 8. The eluted protein was dialyzed against 50 mm Tris, pH 8, at 4 °C overnight. Following dialysis, the protein was applied to a HiTrap Q HP column (GE Healthcare). The column was washed with 10 times bed volume of 50 mm Tris, pH 8, and eluted with a salt gradient starting from 0 m NaCl, 50 mm Tris, pH 8, to 1 m NaCl, 50 mm Tris, pH 8, in 5 ml/min for 10 min. Fractions were collected and analyzed by 15% SDS-PAGE. The fractions containing the ADAM17 proenzyme WT or mutants were collected and concentrated using a Vivaspin concentrator with a molecular mass cutoff of 10 kDa to 1 mg/ml concentration. The protein concentration was determined using the extinction coefficient of 51760 cm−1 m−1.
Circular Dichroism (CD)
CD measurements were performed using an Aviv 202 spectropolarimeter with a 1-mm path length quartz cuvette at 25 °C. Wavelength scans were done in the 190–260-nm far-ultraviolet range. The CD spectra of ADAM17 proenzyme WT and mutants were recorded at a concentration of 0.1 mg/ml in 150 mm NaCl, 50 mm Tris buffer, pH 8, at 2 s/nm. Data sets were averaged and then normalized to the baseline at 255 nm.
Activation of Recombinant ADAM17 Proenzyme by Furin and ADAM17 Activity Assay
Purified ADAM17 proenzymes including WT, US, BS, 2M, and EA/BS mutants (0.2 mg/ml final concentration) were incubated with Furin (2 enzyme units, New England Biolab) in Furin assay buffer (100 mm HEPES, pH 7.5), 0.5% Triton X-100, 1 mm CaCl2, 1 mm 2-mercaptoethanol) at 37 °C for 3 h. 10 μl of each reaction was separated on a 12% SDS-PAGE and stained with 12% basic native gel stain Coomassie Blue solution. In addition, the catalytic activity of ADAM17 (10 nm) was measured by hydrolytic processing of the fluorogenic substrate Mca-PLAGAV-Dpa-RSSSR by monitoring the increasing fluorescence intensity at λex = 340 nm and λem = 380 nm for 40 min at 37 °C with a SynergyTM HT plate reader (BioTek). All reactions were performed in a fluorogenic buffer containing 50 mm Tris, pH 8, 150 mm NaCl, and 0.05% Brij-35.
Preparation of Furin-digested ADAM17 Proenzyme WT and Mutants for N-terminal Sequencing
Purified ADAM17 proenzyme WT and mutants were treated with Furin as described above and products were separated by SDS-PAGE and transferred to a PVDF membrane. The different bands were excised from the membrane and N-terminal sequence analysis was performed using a Procise 492 Protein Sequencer (Applied Biosystems, CA) according to the manufacturer's instructions.
Statistical Analysis
Data were assumed to meet normal distribution. The variance between groups that were statistically compared was similar. All data were expressed as mean ± S.D. p values were generated using two-tailed Student's t test (*, p < 0.05; **, p < 0.01; ***, p < 0.001). All statistical analyses were performed with Microsoft Excel software.
RESULTS
Activation Mechanism of ADAM Proenzymes: Prediction of a Novel Upstream PC Processing Motif
Many enzymes, including ADAMs, are synthesized as inactive proenzymes that undergo post-translational processing to become biologically active. In mammals, the PC family and specifically Furin has been shown to be responsible for the maturation of members of the ADAM family by processing at a PC consensus site at the boundary between the catalytic domain and the autoinhibitory pro-domain. To identify additional potential cleavage sites for PCs in the pro-domains of ADAMs, we subjected the sequences of different ADAMs to a proprotein convertase cleavage site prediction program (ProP server (24)), which predicts PC cleavage sites based on artificial neural networks. In addition, the sequences were analyzed with disorder prediction by the Disopred server (25) to provide information regarding the sequence properties of the PC cleavage sites. This analysis predicted two distinct PC sites in the pro-domains of ADAM8, -9, -10, -11, and -17 (Fig. 1, A–E). The first predicted site was the classical putative boundary PC site between the pro- and the catalytic domain (Fig. 1F): ADAM8-RETR196/Y197, ADAM9-RRRR205/A206, ADAM10-RKKR214/T215, ADAM11-RRKR229/Q230, and ADAM17-RVKR214/R215. Although ADAM pro-domains share low sequence homology, the second predicted site was located in the same region upstream to the boundary site (Fig. 1F): ADAM8-RVRR42/A43, ADAM9-RERR56/E57, ADAM10-RAKR51/A52, ADAM11-RLVR55/Q56, and ADAM17-RKR58/D59. In addition, the putative upstream PC cleavage site appeared to be located in a disordered region, which may indicate an increased accessibility to proteases.
FIGURE 1.

ADAM pro-domain sequence alignment of the potential PC sites. Disordered probability predicted by Disopred server (orange graph), with a threshold of 5% (orange dashed line), and propeptide cleavage potential as predicted by ProP server (blue column), with a threshold of 0.5 (blue dashed line) in the pro-domain sequence of ADAM8 (A), ADAM9 (B), ADAM10 (C), ADAM11 (D), and ADAM17 (E). Sequence alignment of the pro-domain of ADAM8, -9, -10, -11, and -17 in the predicted PC sites (F). Schematic presentation of the ADAM domains structure; SP, signal peptide; PRO, pro-domain; CAT, catalytic domain; DIS, disintegrin domain; CYT, cytoplasmic tail, and below the detailed sequence where the blue underline marks the predicted upstream PC site and red underline marks the boundary site (G). The residues marked in blue are the mutations introduced in the upstream PC sites and in red are the mutations in the boundary sites of ADAM9, -10, or -17 in the US, BS, and 2M mutants.
To evaluate the contribution of both the boundary site and the predicted upstream PC site to the physiological activation and maturation of different ADAMs, we mutated the respective sites in the full-length mouse ADAM17, ADAM10, and ADAM9 (Fig. 1G). To eliminate the PC consensus sites in both the boundary site and upstream PC site, we mutated the basic arginine residues in P1 and/or P4 in each of the ADAM sequences. The following mutants were generated for (a) the upstream PC site (US): ADAM17-R58A, ADAM10-R48A/R51G, and ADAM9-R48A/R51G, (b) the boundary site (BS): ADAM17-R211A/R214G, ADAM10-R210A/R213A, and ADAM9-R202A/R205G, and (c) a combination of both mutations designated as 2M double mutations (Fig. 1G).
A Mutation in the Upstream PC Site Motif of ADAMs9, -10, or -17 Interferes with Pro-domain Removal and Activation
To explore the role of the predicted upstream PC site in the maturation and shedding activity of ADAMs9, -10, and -17, we performed cell-based ectodomain shedding assays with mouse embryonic fibroblasts isolated from ADAM knock-out mice (Adam17−/− or Adam10/17−/− MEFs), which eliminate the endogenous activity of one or both of these major sheddases (28, 34, 39). These cells were transfected with either of the following full-length mouse ADAM mutants: WT, US, BS, 2M (Fig. 1), or the Glu > Ala mutant in the catalytic site as an inactive negative control. To monitor shedding, we co-transfected alkaline phosphatase (AP)-tagged cell surface substrates; TGFα, an EGFR-ligand to monitor the activity of ADAM17 (28, 34), BTC to study ADAM10 (28, 35), or Ephrin receptor B4 (EphB4), which is a substrate for overexpressed ADAM9 (36). The cells were stimulated with either phorbol 12-myristate 13-acetate (PMA) to stimulate ADAM17, or ionomycin to enhance the activity of ADAM10 (37–39), or left unstimulated in experiments with overexpressed ADAM9. In addition, the WT or mutant forms of each of these ADAM were overexpressed in HEK293 cells and analyzed by Western blot (see “Experimental Procedures”).
ADAM17, one of the best characterized ADAMs, has been implicated in the shedding of TGFα in mouse embryonic fibroblasts and primary keratinocytes (2, 28, 40). As expected, Adam17−/− MEFs transfected with the catalytically inactive Glu > Ala mutant showed very low levels of TGFα shedding activity with or without PMA stimulation (1.5 ± 0.9 and 1.25 ± 0.5%, respectively). Expression of WT ADAM17 significantly increased TGFα shedding, with a 5-fold increase in activity observed in the absence of PMA (5.6 ± 1.2%) and 20-fold activity after PMA treatment compared with the inactive Glu > Ala mutant (23.6 ± 5.6%) (Fig. 2A). The BS mutant had similar shedding activity as the WT 5.2 ± 0.8% without PMA and 20.1 ± 5.1% in the PMA-stimulated cells. These results suggest that the presence of a PC consensus site in the boundary is not necessary for the shedding activity of ADAM17. However, Adam17−/− MEFs transfected with either the US or 2M mutants showed very low levels of TGFα shedding activity (US, without PMA 1.4 ± 0.3% and with PMA 1.8 ± 0.5%; 2M without PMA 1.7 ± 0.3% and with PMA 1.8 ± 0.5%). Both mutants had a similarly low activity as the inactive Glu > Ala mutant, indicating that these mutations produced a catalytically inactive form of ADAM17. Interestingly, a single mutation in the predicted novel upstream PC site (Fig. 2A, US) completely prevented ADAM17-dependent shedding of TGFα, even though the canonical boundary site was still processed (Fig. 2B). Therefore, we conclude that processing at the newly predicted upstream PC site plays a crucial role in maturation or activation of ADAM17, whereas processing at the boundary site is not sufficient to allow production of active ADAM17.
FIGURE 2.

Cell surface shedding activity and Western blot analysis of ADAM17, ADAM10, and ADAM9 WT and PC sites mutants. A, shedding activity of Adam17−/− MEFs co-transfected with Glu > Ala, WT, US, BS, 2M mutants, and AP-TGFα. ADAM17-dependent shedding was stimulated by PMA and calculated by determining the ratio of AP activity in the supernatants and the AP activity in cell lysates (mean ± S.D., n = 3). B, Western blot of the total lysate (upper panel) or the immunoprecipitated membrane fractions of cell-surface biotinylated WT or mutant forms of ADAM17 (second panel) from HEK293 cells overexpressing ADAM17 WT and mutants using antibodies against the cytoplasmic portion of ADAM17. The third panel shows a Western blot probed with an antibody against the pro-domain of ADAM17 of the membrane fraction correlated to the respective constructs, the left lanes are the molecular markers as indicated. C, proenzyme to mature enzyme ratio quantified by the band densitometry in the membrane fraction blot of ADAM17. The dashed line marks the 1:1 ratio between the pro-form and the mature form where data above indicates a predominant pro-form and below indicates a predominant mature form (representative of 3 experiments). D, shedding activity of Adam10/17−/− MEFs co-transfected with the ADAM10 mutants Glu > Ala, WT, US, BS, or 2M together with AP-BTC. ADAM10 shedding was determined by calculating the ratio of AP activity in the supernatants and the AP activity in cell lysates (mean ± S.D., n = 3). E, Western blot of the total lysate (upper panel) or the membrane fraction (immunoprecipitation of cell-surface biotinylated samples; second panel) from HEK293 cells overexpressing ADAM10 WT, US, BS, 2M mutants using anti-HA antibody, the left lanes are the molecular markers as indicated. F, pro/mature ratio quantified by the band densitometry in the cell surface fraction blot of ADAM10 (representative of 3 experiments). G, shedding of EphB4 in Adam10/17−/− MEFs co-transfected with ADAM9 Glu > Ala, WT, US, BS, 2M mutants, and AP-EphB4 (mean ± S.D., n = 6). H, Western blot of the total lysates (upper panel) or the membrane fractions (immunoprecipitation of cell-surface biotinylated samples, second panel) from HEK293 cells overexpressing ADAM9 WT, US, BS, 2M mutants using antibodies against ADAM9, the left lanes are the molecular markers as indicated. I, pro/mature ratio quantified by band densitometry in the cell surface fraction blot of ADAM9 (representative of 6 experiments).
ADAMs are usually present in two forms: a pro-form including the pro-domain, and a mature form, where the pro-domain has been processed. In the lysates of cells expressing the individual ADAM17 constructs, a band of around 120 kDa was detected, which corresponds to the pro-form of ADAM17 (30) (Fig. 2B, upper panel). Following cell surface biotinylation and immunoprecipitation, we found that the pro-form of WT ADAM17 and the BS mutant were labeled, presenting a similar pattern as in the total lysate fraction. There was an increase of expression of the pro-form in both the BS and US mutants (Fig. 2B, second panel). However, in the US mutant a faster migrating dominant band appeared at ∼110 kDa, which thus migrated close to the mature form of ADAM17 (Fig. 2B, second panel), with a ratio between the pro- and mature form of 0.28 on the Western blot in this panel (Fig. 2C). Previous reports demonstrated that the mature form of ADAM17 can only be detected when an MMP inhibitor is present during cell lysis because of a post-lysis autodegradation of the mature ADAM17 (30, 31). As a control, we added untransfected cell lysates with or without MMP inhibitors to detect both forms (Fig. 2B, second panel, A17+ and A17−). This indicates that the catalytic activity of the US mutant ADAM17, which migrated slightly slower than the mature WT ADAM17, was inhibited, because it was unable to promote shedding of the cell surface substrate TGFα (Fig. 2A). Thus, preventing cleavage of the upstream PC site in the pro-domain also prevented the proper functional activation and maturation of ADAM17. In addition, another slower migrating band around 130 kDa appeared in lanes loaded with the US and 2M mutant samples. This band is most likely a sialylated pro-form of ADAM17 that is generated by modification of O-linked carbohydrates in the trans-Golgi network. This slower migrating pro-form is usually not seen in the endogenous ADAM17, presumably because it is efficiently processed before the sialylation occurs.
Interestingly, when we used an antibody against the pro-domain of ADAM17 in the membrane fraction, the intact pro-domain of ∼25 kDa was only detected in the US mutant (Fig. 2B, third panel). These results indicate that in the absence of cleavage at the upstream PC site, the pro-domain remains intact and bound to ADAM17, presenting as a pseudo proenzyme lacking proteolytic activity.
Cleavage at the Upstream PC Site Is Necessary for Full Activation of Both ADAM10 and ADAM9
Next, we tested the involvement of the newly identified upstream PC site in the maturation of other ADAMs. The shedding activity of ADAM10 was assessed by measuring BTC shedding, and ADAM9 WT activity was monitored by measuring EphB4 shedding in Adam10/17−/− MEFs. The inactive Glu > Ala mutant of ADAM10 did not exhibit detectable activity, even after stimulation with ionomycin (0.25 ± 0.1%; Fig. 2D). The shedding of BTC was strongly increased after co-transfection of Adam10/17−/− MEFs with WT ADAM10 (23.6 ± 5.8% ionomycin stimulated, 2.2 ± 0.85% constitutive). Similar to the ADAM17 BS mutant, the ADAM10 BS mutant did not have an effect on its maturation or activation (ionomycin-induced shedding, 17 ± 7.5%; without ionomycin, 2.9 ± 0.6% (Fig. 2D)). This suggests that for ADAM10, the PC consensus site in the boundary between the catalytic domain and the pro-domain was also not required for the sheddase activity of the protein. However, when the upstream PC site mutation was incorporated to generate US and 2M mutants, the ionomycin-stimulated shedding activity of ADAM10 was reduced to 10.4 ± 3 and 5 ± 2.3%, respectively, and constitutive shedding was 1.5 ± 0.5 and 1.4 ± 0.3%, respectively. These results show that a mutation in the upstream PC site cleavage impairs the function of ADAM10 as a sheddase, and are consistent with our identification of a regulatory role of the upstream PC site in ADAM17.
Similar to ADAM17, ADAM10 exists in two forms: as a ∼100 kDa pro-form and a ∼70 kDa mature form (12). Western blots of overexpressed ADAM10 in HEK293 cells confirmed the importance of the upstream PC site. In the total lysates, the predominant band was the pro-form ∼100 kDa for all transfected constructs, but in contrast to ADAM17, the mature form of the US mutant was also detected (Fig. 2E, upper panel). In the cell surface fraction, the mature form of ∼70 kDa could be detected for all variants (Fig. 2E, second panel). However, the mature form was enriched in the US sample, although the shedding activity was impaired (Fig. 2, D and E, the ratio between the pro- and mature form is 0.66, Fig. 2F). This result resembled the ADAM17 US mutant experiment where we could detect large amounts of mature and processed ADAM17 that was not active, presumably because of a stabilizing inhibitory interaction between the pro-domain and the catalytic domain.
The involvement of the upstream PC site in ADAM maturation was also apparent in ADAM9. ADAM9-mediated shedding of EphB4 from Adam10/17−/− MEFs transfected with the corresponding WT or mutant forms of ADAM9 provided evidence for a similar importance of the upstream PC site for the function of ADAM9 (Fig. 2G). In this experiment, the baseline-shedding activity was defined as the shedding from Adam10/17−/− MEFs transfected with the inactive Glu > Ala mutant as a negative control (9 ± 1.6%). The background levels of EphB4 shedding under these conditions could be due to low levels of endogenous ADAM9 or other proteases, except for ADAM10 and ADAM17, which are lacking in these cells. Shedding of EphB4 was significantly increased when the WT or BS mutant of ADAM9 were overexpressed, reaching 27.4 ± 5.6 and 25.2 ± 4.7% AP ratio, respectively. A mutation in the upstream PC site (US mutant) reduced the shedding activity to 19 ± 4.6%, which was a significant reduction, but not of the same magnitude compared with the effect of the US mutation in ADAM17 and ADAM10. The 2M mutant possessed lower shedding activity (13.8 ± 3.1%) than the US mutant, indicating that the presence of a PC boundary site could contribute to the functional maturation of ADAM9. Although, the effect of the ADAM9 US and 2M mutants are less striking than in the case of ADAM17 or ADAM10, they nevertheless, support the notion that the upstream PC site plays a role in the maturation and function of ADAM9.
ADAM9 exists in a pro-form of ∼110 kDa and a mature form of ∼80 kDa where the pro-domain is removed (14). Overexpression of the ADAM9 WT and mutants resulted in the appearance of the pro- and mature forms in the total lysate (Fig. 2H, upper panel). However, stronger bands were detected in the US mutant, whereas the pro-form was dominant in the 2M mutant. We found no significant differences between the WT and BS mutants, with the pro- and mature form present in both cases (Fig. 2H, second panel). The activation pattern of the US mutant resembled that observed in the ADAM17 and ADAM10 US mutants, where the mature from was predominant (the ratio between pro- and mature form is 0.19 (Fig. 2I)), even though the shedding activity was abolished or strongly reduced. There were almost no differences in the band pattern of the 2M mutant when comparing the total lysate fraction and the cell surface fraction, with the pro-form dominant due to the improper cleavage of PC in both sites. Taken together, these results demonstrate that the upstream PC site is a key cleavage site for the functional activation of several members of the ADAM family of metalloproteinases.
Purification and Activation of Recombinant Soluble ADAM17 Proenzyme Mutants
To analyze the molecular events occurring during the pro-domain removal by PCs in vitro, we recombinantly expressed and purified the ADAM17 proenzyme (pro-domain and catalytic domain Asp23-Val477) in E. coli to >95% purity (Fig. 3A). To assess the importance of PCs in ADAM17 proenzyme activation, both PC recognition sites were mutated similar to the previous section in the ADAM17 pro-domain sequence (Fig. 3B). The mutations in neither of the PC sites changed the overall secondary structures of the protein significantly as demonstrated in the CD measurements (Fig. 3C), indicating that the WT and mutant ADAM17 proenzymes are folded in a similar manner.
FIGURE 3.
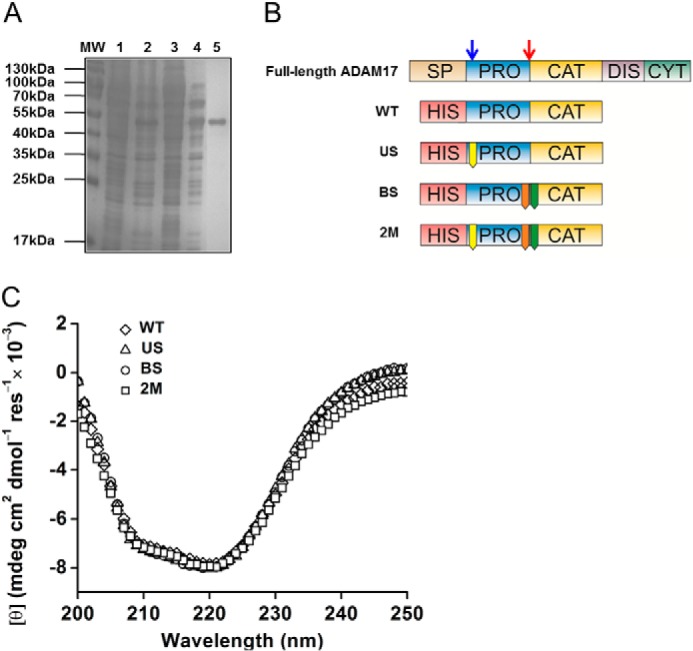
Purification and circular dichroism of recombinant ADAM17 proenzyme WT and mutants. A, representative SDS-PAGE of purification of ADAM17 proenzyme with a nickel-nitrilotriacetic acid column followed by anion exchange column: molecular mass (MW), nickel-nitrilotriacetic acid lysate load (1), nickel-nitrilotriacetic acid flow-through (2), anion exchange load (3), anion exchange flow-through (4), and anion exchange first peak (5). B, schematic presentation of ADAM17 domain structure; SP, signal peptide; PRO, pro-domain; CAT, catalytic domain; DIS, disintegrin domain; CYT, cytoplasmic tail. The blue arrow indicates the upstream PC site and the red arrow indicates the boundary site. Below are the recombinant ADAM17 proenzyme constructs with an N-terminal His tag: WT, pro-catalytic domains; US-R58A mutation in the Upstream PC site; BS, R211A/R214G mutation in the boundary site; and 2M, R58A/R211A/R214G containing both PC sites mutations. C, CD measurements of WT ADAM17 proenzyme and mutants.
The purified ADAM17 proenzyme WT and mutants were incubated with the PC Furin (see “Experimental Procedures” for details). The degradation patterns were analyzed using SDS-PAGE, and ADAM17 activity was measured by the proteolysis of a fluorogenic peptide mimicking the cleavage site of TNFα (41). The WT ADAM17 proenzyme was cleaved by Furin in both PC sites resulting in two protein fragments: a 35-kDa (Arg214-Val477) fragment corresponding to the catalytic domain (N-terminal sequence, 215RADPD), and a 15-kDa pro-domain fragment (Asp59-Arg213) (N-terminal sequence, 59DLQTS) (Fig. 4A, WT+). Importantly, the resulting product was able to cleave the fluorogenic peptide (Fig. 4B, WT+). This confirmed that the upstream PC site is indeed a relevant PC site. In agreement with our cell-based shedding experiments, full activation was also observed in the BS mutant after incubation with Furin. Similarly to the WT ADAM17 proenzyme, the BS mutant was cleaved at both the upstream PC site and at the boundary site, resulting in two protein fragments with the same N-terminal amino acid sequences as the WT (Fig. 4A, BS+) and an activated ADAM17 (Fig. 4B, BS+). These results demonstrate that the presence of the Furin cleavage consensus in the boundary site is not necessary for full activation of ADAM proenzyme.
FIGURE 4.
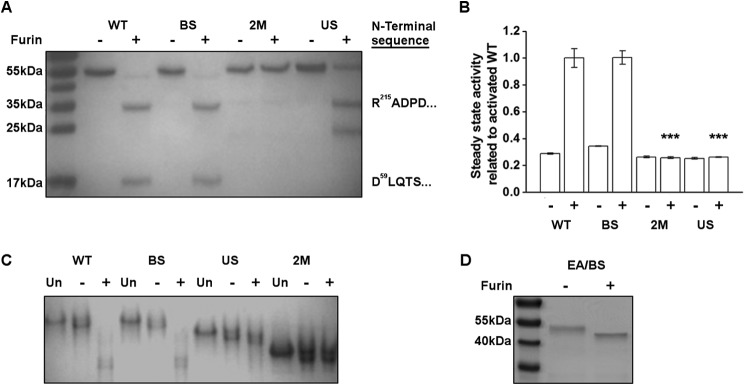
Cleavage of the upstream PC site is essential for dissociation of ADAM17 pro-domain and proenzyme activation. A, degradation pattern of the ADAM17 proenzyme by Furin. 5 μg of ADAM17 proenzyme WT or mutants were preincubated with 2 enzyme units of Furin for 3 h and analyzed by 12% SDS-PAGE: WT, BS, US, and 2M without Furin (−) and in the presence of Furin (+). N-terminal sequencing of the 35-kDa band of WT+, BS+, and US+ yielded in the same N terminus of the ADAM17 catalytic domain beginning at residue 215: RADPD. The lower 15-kDa band in WT+ and BS+ had the N-terminal sequence of DLQTS, corresponding to residue 59. B, fluorogenic peptide hydrolysis assays of the Furin-treated ADAM17 proenzyme. Purified ADAM17 proenzyme WT and mutants were preincubated with Furin as mentioned above and 10 nm protein samples were tested in the presence of 10 μm fluorogenic substrate for hydrolysis activity. The data were collected during steady state activity and normalized to the activity of the activated WT (mean ± S.D., n = 3). C, native gel analysis of ADAM17 proenzyme: WT, BS, US, and 2M untreated (UN), without Furin (−) and in the presence of Furin (+). D, EA/BS ADAM17 proenzyme mutants (R211A/R214G/E406A) in which the boundary site was deleted and ADAM17 was inactivated by the Glu > Ala mutation was processed by Furin and analyzed by SDS-PAGE as described above, without (−) or with (+) Furin.
As expected, the 2M mutant could not be cleaved by Furin and the protein remained full-length and inactive (Fig. 4, A and B, 2M+). Furin cleaved the US mutant only at the boundary site, resulting in two protein fragments: a 35-kDa catalytic domain (N-terminal sequence, 215RADPD) and a 20-kDa intact pro-domain, whereas some of the full-length protein (55 kDa) remained uncleaved (Fig. 4A, US+). Interestingly, although Furin cleaved the US mutant, the resulting product could not process the fluorogenic peptide (Fig. 4B, US+). This suggests that the unprocessed 20-kDa pro-domain fragment remains bound to the catalytic domain of the US mutant ADAM17 and inhibits its enzymatic activity, thus creating a pseudo proenzyme form.
To determine whether the pro-domain and the catalytic domain form a complex in the US mutant after Furin treatment, the resulting products were subjected to native gel electrophoresis. As expected, the degradation products of both WT ADAM17 proenzyme and the BS mutant resulted in a similar band migration patterns after incubation with Furin (Fig. 4C, WT+, BS+), indicating that the pro-domain is processed similarly in both cases. The US mutant showed no significant difference in its migration pattern with or without Furin treatment (Fig. 4C, US+) although Furin cleaved the boundary site, as shown in Fig. 4A, US+. This implies that an intact complex of the pro-domain and the catalytic domain co-migrates in a native gel even after cleavage at the boundary site RVKR214/R215. Accordingly, the 2M mutant, which was not cleaved by Furin, exhibited a similar behavior as the US mutant where the pro-domain remained in complex with the catalytic domain before and after treatment with Furin. The differences in the rates of band migration between these two mutants may be explained by the differences in electrostatic protein surface potentials resulting from the point mutations. Thus, full dissociation of the pro-domain is achieved only after a cleavage at the non-canonical upstream PC site RKR58/D59.
Secondary Cleavage at the Boundary Site Is Furin Independent
The data above shows that cleavage at the boundary site occurs after the primary cleavage at the upstream PC site even in the absence of the PC consensus site (Fig. 4B, BS mutant). In this in vitro system, the second cleavage can either be performed by an unspecific activity of Furin or by ADAM17 itself. To address this question, we generated a mutant ADAM17 proenzyme protein carrying the inactivating EA mutation and a BS mutation in the boundary site (EA/BS). This mutant allowed us to examine whether ADAM17 itself was responsible for cleavage at the boundary site after the initial cleavage by Furin at the upstream PC site. After treatment with Furin, the EA/BS mutant showed a slight reduction in molecular weight compared with the non-treated protein (Fig. 4D). This resulted in cleavage by Furin at the upstream PC site RKR58/D59 generating a ∼50 kDa fragment corresponding to the ADAM17 zymogen where the N-terminal His tag and Asp23-Arg58 residues were removed. The second cleavage at the boundary site RVKR214/R215 did not occur in this mutant, suggesting that neither Furin, due to the mutation in the boundary site, nor ADAM17, due to the inactive mutant, could cleave the boundary site. From these results, we conclude that the second cleavage in the boundary site can be processed by other proteases besides Furin and could be autocatalytic. However, the second cleavage event at the BS was dependent on the primary cleavage at the non-canonical upstream PC site RKR58/D59.
DISCUSSION
Two mechanisms have been proposed for the activation of ADAM proenzymes. One is the removal of the pro-domain by autocatalysis, which was demonstrated for ADAM8 (22) and ADAM28 (21). The second is the processing at a canonical PC cleavage site between the pro- and metalloprotease domain, which has been proposed and demonstrated as a pre-requisite for activation of several ADAMs, including ADAM9 (14), ADAM12 (18), ADAM15 (20), ADAM17 (30), and ADAM19 (19). Here we identify a crucial novel upstream PC cleavage site that has an important role in the activation of several ADAMs. Our results suggest that processing at this novel site needs to occur to allow the pro-domain to dissociate from the catalytic domain, at least in the case of ADAM17, and that processing at the previously identified boundary site between the pro- and metalloprotease domain is not sufficient to activate ADAM17.
Based on our findings, we propose that processing at the upstream PC cleavage site is a general novel activation mechanism that is shared by several ADAMs (Fig. 5). After ADAMs are translated and translocated across the ER membrane as inactive proenzymes, they are delivered to the Golgi compartment with the intact pro-domain. This is presumably meant to keep these ADAMs inactive and prevent inappropriate processing of growth factors and substrates, for example, to prevent intracrine signaling via prematurely released growth factors (42). After trafficking of the proenzyme to the later compartments of the secretory pathway, it encounters PCs such as Furin in the trans-Golgi network (Fig. 5A) (17, 23, 43). The pro-domain processing then takes place in the two distinct sites in the pro-domain, namely the upstream PC site discovered here, and the boundary site, which contains a PC consensus sequence, but can also be processed autocatalytically. The exact order of these processing events under physiological conditions remains to be established, and two scenarios are shown in Fig. 5. In one model, processing at the upstream site (Fig. 5B) is a pre-requisite for activating the ADAM, which then processes the BS (Fig. 5C), although PCs or other enzymes could also have a role in processing at the boundary site. Together, these two processing events result in the release of the inhibitory pro-domain, which is a pre-requisite for the activation of the ADAM (Fig. 5, D and E). In the second model, processing at the boundary generates an intermediate product, in which the upstream site is exposed to other proteases (Fig. 5, F and G). However, our results suggest that removal of the residual pro-domain requires processing at the upstream site, at least for ADAM17. If the upstream site is not processed, then the ADAM is presented as a pseudo-proenzyme on the cell surface, in which the pro-domain has been processed at the boundary site, but cannot dissociate without processing at the upstream PC site (Fig. 5H). The enzyme therefore remains in its latent form and is unable to process its substrates on the cell surface (Fig. 5I).
FIGURE 5.
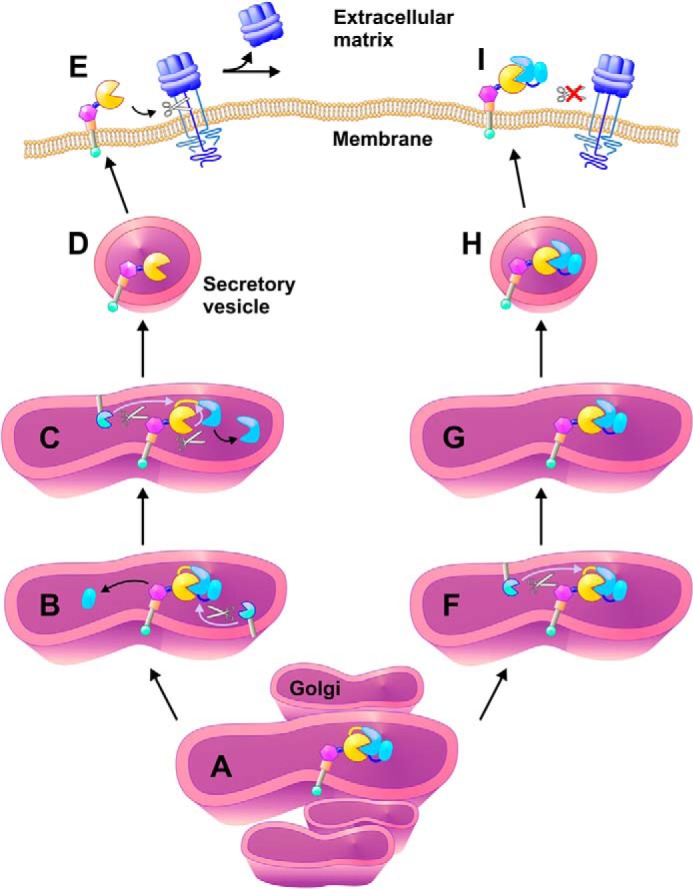
Schematic presentation of ADAM proenzyme activation by pro-domain removal. ADAMs are generated as an inactive proenzyme with an autoinhibitory pro-domain (catalytic domain of ADAM is represented by a yellow circle, the pro-domain is represented by two connected parts (blue) where the upstream PC site is located in the middle (dark blue line) and the boundary site is connected to the catalytic domain (yellow line)). The removal of the pro-domain occurs in the trans Golgi compartment (A) and the first cleavage event is performed by Furin or PC at the upstream PC site (B). This leads to conformational changes, which expose the boundary site for rapid cleavage by either Furin or other proteases (C). The activated ADAM is trafficking to the membrane (D) to shed cell surface substrates (E). However, when only the boundary site is cleaved as in the US mutant (F) the pro-domain remains attached to the catalytic domain, thus forming an inactive tightly bound binary complex (G). This pseudo-proenzyme form is delivered to the membrane (H) but is unable to shed cell surface substrates (I).
According to our results, only PC proteases are involved in the processing of the upstream PC site. This is consistent with the observation that Furin-deficient cells fail to activate ADAM17 (44). Moreover, a report by Schwarz et al. (45) demonstrated that a mutation in the boundary site did not affect the shedding activity of TNFα in cell-based assays, similar to our results with the BS mutant. Interestingly, in both studies, very little, if any of the mature form of ADAM17 was detected by Western blot. This is consistent with previous observations that overexpressed WT ADAM17 can usually only be detected as a pro-form, although the reason for this remains to be determined (38, 46). Nevertheless, the overexpressed ADAM17 is able to rescue defects in TGFα or TNFα shedding in Adam17−/− MEFs, demonstrating that it is catalytically active.
In contrast to previous studies showing that the activation of ADAM10 requires processing at the boundary site (12), our functional studies of the shedding activity of ADAM10 mutants lead us to a different conclusion. Expression of ADAM10 with a mutation in the boundary site (BS mutant) leads to a high level of BTC shedding, similar to expression of WT ADAM10. Thus the functional maturation of ADAM10 was not impaired by this mutation. The reason for this discrepancy remains to be established. In the case of ADAM9, it has been shown that deletion of the boundary site RRRR205/A206 did not hinder the maturation of ADAM9. Instead, the BS mutant appears to be processed at a different site, adjacent to the boundary site (14). Indeed, we have found that the ADAM9 BS mutant could promote shedding of EphB4 at a similar level as the WT protein, demonstrating that mutation in the boundary site did not prevent the functional maturation of ADAM9. By analogy to our results with the related ADAM17, it is possible that upon cleavage of the upstream PC site RERR56/E57 in ADAM9, the secondary cleavage occurs at a site adjacent to the boundary PC site by another protease, leading to proper activation.
It should be noted that the ectodomain shedding activities of ADAM10 and ADAM9 were not completely abolished by introduction of the US mutation, whereas the activity of ADAM17 was strongly affected by the US mutant (Fig. 2). However, introduction of the US and BS mutations into ADAM10 and ADAM9 further reduced their shedding activity. There could be several explanations for this phenomenon: 1) in contrast to ADAM17, the PC cleavage regulation of ADAM10 and ADAM9 may depend on both PC sites, 2) PCs are not the only proteases responsible for removal of the pro-domain of ADAM10 and ADAM9, therefore, other proteases could contribute to the removal of the pro-domain but with lower efficiency, 3) or the affinities of the pro-domains of ADAM10 and ADAM9 to their relative catalytic domains are not as high as ADAM17 pro-domain to ADAM17 catalytic domain. Nevertheless, these findings point to a general strategy for ADAM activation, where the newly discovered upstream PC site plays an important role in the maturation of the ADAM proenzymes.
Based on our predictions, ADAM8 also should possess an upstream PC site in its pro-domain (RVRR44/A45) in addition to its boundary site (RETR200/Y201), which has been suggested to be a suboptimal consensus cleavage sequence for Furin (22). The processing of ADAM8 at the boundary site between the pro- and metalloprotease domain depends on autocatalytic activity (21, 22). However, these previous studies did not address the possible requirement for processing at the upstream site by PC. In this scenario the autocatalytic cleavage could only occur after the initial cleavage by a PC at the upstream site. Interestingly, Hall et al. (47) had demonstrated that one of the fragments produced during the purification of ADAM8 begins with the N-terminal Ala45, providing validation to the processing of ADAM8 in the upstream PC site. Therefore, it would be interesting to revisit ADAM8 for similar studies of the contribution of the upstream site to PC-dependent activation.
Multiple cleavage sites within the pro-domain region have also been reported for other members of the MMP family (48, 49). It has been proposed that a primary cleavage occurs in a “bait” region that disrupts the pro-domain structure, creating an unstable intermediate at the boundary between the pro- and catalytic domains, making it accessible to PC activity for removal of the pro-domain (50). Comparably, the upstream PC site in the case of ADAMs is the bait region located in an approachable and accessible loop in which the cleavage leads to conformational changes creating an unstable intermediate and exposing the boundary site for proteolytic digestion. Moreover, previous reports showed that the pro-domain of ADAM17 is constituted by two independent subunits, one including Pro18–Ser54 and the second including Val55-Arg214 (33). The newly characterized upstream PC site RKR58/D59 is located after the N-terminal subunit, suggesting the existence of an N-terminal unit in ADAM17 pro-domain divided by the upstream PC site. Taking into account both the fact that the cysteine switch is not necessary for ADAM17 inhibition (13) and our results showing that cleavage at the upstream PC site leads to removal of the pro-domain, it is reasonable to assume that the N-terminal pro-domain plays a major role in the autoinhibitory activity.
In summary, by analyzing the molecular mechanism driving the activation of ADAMs, we have characterized a new key regulatory PC cleavage site in the pro-domain sequence. Our results demonstrate that Furin can cleave at two distinct sites located in the pro-domain: the putative boundary site and the newly characterized upstream PC site. Processing at the upstream site is particularly important for activation of the ADAM proenzyme, whereas PC-dependent processing at the boundary site does not appear to be essential. In the absence of the upstream PC site cleavage, the ADAM may be present on the cell surface as a “proenzyme-like” form. Our results provide new insights into the understanding of how the maturation of ADAMs is controlled by precisely orchestrated sequential processing of their pro-domains, which is essential for proper activation of these important regulators of cell-cell signaling.
Acknowledgment
We thank Dr. Benjamin Philip Born for help with figure preparation.
This work was supported by SaveMe FP7, ISF and by National Institutes of Health Grant GM64750 to C. P. B.
- ADAM
- A Disintegrin And Metalloprotease
- PC
- proprotein convertase
- BTC
- betacellulin
- MEF
- mouse embryonic fibroblast
- US
- upstream site
- BS
- boundary site
- 2M
- double US/BS
- AP
- alkaline phosphatase
- EphB4
- ephrin receptor B4
- PMA
- phorbol 12-myristate 13-acetate.
REFERENCES
- 1. Rooke J., Pan D., Xu T., Rubin G. M. (1996) KUZ, a conserved metalloprotease-disintegrin protein with two roles in Drosophila neurogenesis. Science 273, 1227–1231 [DOI] [PubMed] [Google Scholar]
- 2. Peschon J. J., Slack J. L., Reddy P., Stocking K. L., Sunnarborg S. W., Lee D. C., Russell W. E., Castner B. J., Johnson R. S., Fitzner J. N., Boyce R. W., Nelson N., Kozlosky C. J., Wolfson M. F., Rauch C. T., Cerretti D. P., Paxton R. J., March C. J., Black R. A. (1998) An essential role for ectodomain shedding in mammalian development. Science 282, 1281–1284 [DOI] [PubMed] [Google Scholar]
- 3. Qi H., Rand M. D., Wu X., Sestan N., Wang W., Rakic P., Xu T., Artavanis-Tsakonas S. (1999) Processing of the notch ligand δ by the metalloprotease Kuzbanian. Science 283, 91–94 [DOI] [PubMed] [Google Scholar]
- 4. Kuno K., Kanada N., Nakashima E., Fujiki F., Ichimura F., Matsushima K. (1997) Molecular cloning of a gene encoding a new type of metalloproteinase-disintegrin family protein with thrombospondin motifs as an inflammation associated gene. J. Biol. Chem. 272, 556–562 [DOI] [PubMed] [Google Scholar]
- 5. Sandy J. D., Verscharen C. (2001) Analysis of aggrecan in human knee cartilage and synovial fluid indicates that aggrecanase (ADAMTS) activity is responsible for the catabolic turnover and loss of whole aggrecan whereas other protease activity is required for C-terminal processing in vivo. Biochem. J. 358, 615–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Iba K., Albrechtsen R., Gilpin B. J., Loechel F., Wewer U. M. (1999) Cysteine-rich domain of human ADAM 12 (meltrin α) supports tumor cell adhesion. Am. J. Pathol. 154, 1489–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Buxbaum J. D., Liu K. N., Luo Y., Slack J. L., Stocking K. L., Peschon J. J., Johnson R. S., Castner B. J., Cerretti D. P., Black R. A. (1998) Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated α-secretase cleavage of the Alzheimer amyloid protein precursor. J. Biol. Chem. 273, 27765–27767 [DOI] [PubMed] [Google Scholar]
- 8. Koike H., Tomioka S., Sorimachi H., Saido T. C., Maruyama K., Okuyama A., Fujisawa-Sehara A., Ohno S., Suzuki K., Ishiura S. (1999) Membrane-anchored metalloprotease MDC9 has an α-secretase activity responsible for processing the amyloid precursor protein. Biochem. J. 343, 371–375 [PMC free article] [PubMed] [Google Scholar]
- 9. Colciaghi F., Borroni B., Pastorino L., Marcello E., Zimmermann M., Cattabeni F., Padovani A., Di Luca M. (2002) [alpha]-Secretase ADAM10 as well as αAPPs is reduced in platelets and CSF of Alzheimer disease patients. Mol. Med. 8, 67–74 [PMC free article] [PubMed] [Google Scholar]
- 10. Hooper N. M., Turner A. J. (2002) The search for alpha-secretase and its potential as a therapeutic approach to Alzheimer disease. Curr. Med. Chem. 9, 1107–1119 [DOI] [PubMed] [Google Scholar]
- 11. Van Wart H. E., Birkedal-Hansen H. (1990) The cysteine switch: a principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc. Natl. Acad. Sci. U.S.A. 87, 5578–5582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Anders A., Gilbert S., Garten W., Postina R., Fahrenholz F. (2001) Regulation of the α-secretase ADAM10 by its prodomain and proprotein convertases. FASEB J. 15, 1837–1839 [DOI] [PubMed] [Google Scholar]
- 13. Gonzales P. E., Solomon A., Miller A. B., Leesnitzer M. A., Sagi I., Milla M. E. (2004) Inhibition of the tumor necrosis factor-α-converting enzyme by its prodomain. J. Biol. Chem. 279, 31638–31645 [DOI] [PubMed] [Google Scholar]
- 14. Roghani M., Becherer J. D., Moss M. L., Atherton R. E., Erdjument-Bromage H., Arribas J., Blackburn R. K., Weskamp G., Tempst P., Blobel C. P. (1999) Metalloprotease-disintegrin MDC9: intracellular maturation and catalytic activity. J. Biol. Chem. 274, 3531–3540 [DOI] [PubMed] [Google Scholar]
- 15. Milla M. E., Leesnitzer M. A., Moss M. L., Clay W. C., Carter H. L., Miller A. B., Su J. L., Lambert M. H., Willard D. H., Sheeley D. M., Kost T. A., Burkhart W., Moyer M., Blackburn R. K., Pahel G. L., Mitchell J. L., Hoffman C. R., Becherer J. D. (1999) Specific sequence elements are required for the expression of functional tumor necrosis factor-α-converting enzyme (TACE). J. Biol. Chem. 274, 30563–30570 [DOI] [PubMed] [Google Scholar]
- 16. Molloy S. S., Bresnahan P. A., Leppla S. H., Klimpel K. R., Thomas G. (1992) Human furin is a calcium-dependent serine endoprotease that recognizes the sequence Arg-X-X-Arg and efficiently cleaves anthrax toxin protective antigen. J. Biol. Chem. 267, 16396–16402 [PubMed] [Google Scholar]
- 17. Nakayama K. (1997) Furin: a mammalian subtilisin/Kex2p-like endoprotease involved in processing of a wide variety of precursor proteins. Biochem. J. 327, 625–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Loechel F., Gilpin B. J., Engvall E., Albrechtsen R., Wewer U. M. (1998) Human ADAM 12 (meltrin α) is an active metalloprotease. J. Biol. Chem. 273, 16993–16997 [DOI] [PubMed] [Google Scholar]
- 19. Kang T., Zhao Y. G., Pei D., Sucic J. F., Sang Q. X. (2002) Intracellular activation of human adamalysin 19/disintegrin and metalloproteinase 19 by furin occurs via one of the two consecutive recognition sites. J. Biol. Chem. 277, 25583–25591 [DOI] [PubMed] [Google Scholar]
- 20. Lum L., Reid M. S., Blobel C. P. (1998) Intracellular maturation of the mouse metalloprotease disintegrin MDC15. J. Biol. Chem. 273, 26236–26247 [DOI] [PubMed] [Google Scholar]
- 21. Howard L., Maciewicz R. A., Blobel C. P. (2000) Cloning and characterization of ADAM28: evidence for autocatalytic pro-domain removal and for cell surface localization of mature ADAM28. Biochem. J. 348, 21–27 [PMC free article] [PubMed] [Google Scholar]
- 22. Schlomann U., Wildeboer D., Webster A., Antropova O., Zeuschner D., Knight C. G., Docherty A. J., Lambert M., Skelton L., Jockusch H., Bartsch J. W. (2002) The metalloprotease disintegrin ADAM8: processing by autocatalysis is required for proteolytic activity and cell adhesion. J. Biol. Chem. 277, 48210–48219 [DOI] [PubMed] [Google Scholar]
- 23. Steiner D. F. (1998) The proprotein convertases. Curr. Opin. Chem. Biol. 2, 31–39 [DOI] [PubMed] [Google Scholar]
- 24. Duckert P., Brunak S., Blom N. (2004) Prediction of proprotein convertase cleavage sites. Protein Eng. Des. Sel. 17, 107–112 [DOI] [PubMed] [Google Scholar]
- 25. Ward J. J., Sodhi J. S., McGuffin L. J., Buxton B. F., Jones D. T. (2004) Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 337, 635–645 [DOI] [PubMed] [Google Scholar]
- 26. van den Ent F., Löwe J. (2006) RF cloning: a restriction-free method for inserting target genes into plasmids. J. Biochem. Biophys. Methods 67, 67–74 [DOI] [PubMed] [Google Scholar]
- 27. Erijman A., Dantes A., Bernheim R., Shifman J. M., Peleg Y. (2011) Transfer-PCR (TPCR): a highway for DNA cloning and protein engineering. J. Struct. Biol. 175, 171–177 [DOI] [PubMed] [Google Scholar]
- 28. Sahin U., Weskamp G., Kelly K., Zhou H. M., Higashiyama S., Peschon J., Hartmann D., Saftig P., Blobel C. P. (2004) Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J. Cell Biol. 164, 769–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zheng Y., Schlondorff J., Blobel C. P. (2002) Evidence for regulation of the tumor necrosis factor α-convertase (TACE) by protein-tyrosine phosphatase PTPH1. J. Biol. Chem. 277, 42463–42470 [DOI] [PubMed] [Google Scholar]
- 30. Schlöndorff J., Becherer J. D., Blobel C. P. (2000) Intracellular maturation and localization of the tumour necrosis factor α convertase (TACE). Biochem. J. 347, 131–138 [PMC free article] [PubMed] [Google Scholar]
- 31. McIlwain D. R., Lang P. A., Maretzky T., Hamada K., Ohishi K., Maney S. K., Berger T., Murthy A., Duncan G., Xu H. C., Lang K. S., Häussinger D., Wakeham A., Itie-Youten A., Khokha R., Ohashi P. S., Blobel C. P., Mak T. W. (2012) iRhom2 regulation of TACE controls TNF-mediated protection against Listeria and responses to LPS. Science 335, 229–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Weskamp G., Krätzschmar J., Reid M. S., Blobel C. P. (1996) MDC9, a widely expressed cellular disintegrin containing cytoplasmic SH3 ligand domains. J. Cell Biol. 132, 717–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Buckley C. A., Rouhani F. N., Kaler M., Adamik B., Hawari F. I., Levine S. J. (2005) Amino-terminal TACE prodomain attenuates TNFR2 cleavage independently of the cysteine switch. Am. J. Physiol. Lung Cell Mol. Physiol. 288, L1132–1138 [DOI] [PubMed] [Google Scholar]
- 34. Zheng Y., Saftig P., Hartmann D., Blobel C. (2004) Evaluation of the contribution of different ADAMs to tumor necrosis factor alpha (TNFα) shedding and of the function of the TNFα ectodomain in ensuring selective stimulated shedding by the TNFα convertase (TACE/ADAM17). J. Biol. Chem. 279, 42898–42906 [DOI] [PubMed] [Google Scholar]
- 35. Sanderson M. P., Erickson S. N., Gough P. J., Garton K. J., Wille P. T., Raines E. W., Dunbar A. J., Dempsey P. J. (2005) ADAM10 mediates ectodomain shedding of the betacellulin precursor activated by p-aminophenylmercuric acetate and extracellular calcium influx. J. Biol. Chem. 280, 1826–1837 [DOI] [PubMed] [Google Scholar]
- 36. Guaiquil V., Swendeman S., Yoshida T., Chavala S., Campochiaro P. A., Blobel C. P. (2009) ADAM9 is involved in pathological retinal neovascularization. Mol. Cell Biol. 29, 2694–2703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Reddy P., Slack J. L., Davis R., Cerretti D. P., Kozlosky C. J., Blanton R. A., Shows D., Peschon J. J., Black R. A. (2000) Functional analysis of the domain structure of tumor necrosis factor-α converting enzyme. J. Biol. Chem. 275, 14608–14614 [DOI] [PubMed] [Google Scholar]
- 38. Horiuchi K., Le Gall S., Schulte M., Yamaguchi T., Reiss K., Murphy G., Toyama Y., Hartmann D., Saftig P., Blobel C. P. (2007) Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phorbol esters and calcium influx. Mol. Biol. Cell 18, 176–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Le Gall S. M., Bobé P., Reiss K., Horiuchi K., Niu X. D., Lundell D., Gibb D. R., Conrad D., Saftig P., Blobel C. P. (2009) ADAMs 10 and 17 represent differentially regulated components of a general shedding machinery for membrane proteins such as transforming growth factor α, L-selectin, and tumor necrosis factor α. Mol. Biol. Cell 20, 1785–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Merlos-Suárez A., Ruiz-Paz S., Baselga J., Arribas J. (2001) Metalloprotease-dependent protransforming growth factor-α ectodomain shedding in the absence of tumor necrosis factor-α-converting enzyme. J. Biol. Chem. 276, 48510–48517 [DOI] [PubMed] [Google Scholar]
- 41. Solomon A., Akabayov B., Frenkel A., Milla M. E., Sagi I. (2007) Key feature of the catalytic cycle of TNF-α converting enzyme involves communication between distal protein sites and the enzyme catalytic core. Proc. Natl. Acad. Sci. U.S.A. 104, 4931–4936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wiley H. S., Woolf M. F., Opresko L. K., Burke P. M., Will B., Morgan J. R., Lauffenburger D. A. (1998) Removal of the membrane-anchoring domain of epidermal growth factor leads to intracrine signaling and disruption of mammary epithelial cell organization. J. Cell Biol. 143, 1317–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Molloy S. S., Thomas L., VanSlyke J. K., Stenberg P. E., Thomas G. (1994) Intracellular trafficking and activation of the furin proprotein convertase: localization to the TGN and recycling from the cell surface. EMBO J. 13, 18–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Srour N., Lebel A., McMahon S., Fournier I., Fugère M., Day R., Dubois C. M. (2003) TACE/ADAM-17 maturation and activation of sheddase activity require proprotein convertase activity. FEBS Lett. 554, 275–283 [DOI] [PubMed] [Google Scholar]
- 45. Schwarz J., Broder C., Helmstetter A., Schmidt S., Yan I., Müller M., Schmidt-Arras D., Becker-Pauly C., Koch-Nolte F., Mittrücker H. W., Rabe B., Rose-John S., Chalaris A. (2013) Short-term TNFα shedding is independent of cytoplasmic phosphorylation or furin cleavage of ADAM17. Biochim. Biophys. Acta 1833, 3355–3367 [DOI] [PubMed] [Google Scholar]
- 46. Yoda M., Kimura T., Tohmonda T., Morioka H., Matsumoto M., Okada Y., Toyama Y., Horiuchi K. (2013) Systemic overexpression of TNF α-converting enzyme does not lead to enhanced shedding activity in vivo. Plos One 8, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hall T., Leone J. W., Wiese J. F., Griggs D. W., Pegg L. E., Pauley A. M., Tomasselli A. G., Zack M. D. (2009) Autoactivation of human ADAM8: a novel pre-processing step is required for catalytic activity. Biosci. Rep. 29, 217–228 [DOI] [PubMed] [Google Scholar]
- 48. Rosenblum G., Meroueh S., Toth M., Fisher J. F., Fridman R., Mobashery S., Sagi I. (2007) Molecular structures and dynamics of the stepwise activation mechanism of a matrix metalloproteinase zymogen: challenging the cysteine switch dogma. J. Am. Chem. Soc. 129, 13566–13574 [DOI] [PubMed] [Google Scholar]
- 49. Golubkov V. S., Cieplak P., Chekanov A. V., Ratnikov B. I., Aleshin A. E., Golubkova N. V., Postnova T. I., Radichev I. A., Rozanov D. V., Zhu W., Motamedchaboki K., Strongin A. Y. (2010) Internal cleavages of the autoinhibitory prodomain are required for membrane type 1 matrix metalloproteinase activation, although furin cleavage alone generates inactive proteinase. J. Biol. Chem. 285, 27726–27736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Golubkov V. S., Chekanov A. V., Shiryaev S. A., Aleshin A. E., Ratnikov B. I., Gawlik K., Radichev I., Motamedchaboki K., Smith J. W., Strongin A. Y. (2007) Proteolysis of the membrane type-1 matrix metalloproteinase prodomain: implications for a two-step proteolytic processing and activation. J. Biol. Chem. 282, 36283–36291 [DOI] [PubMed] [Google Scholar]