

Background: Endosome-based protein trafficking in the heart is poorly understood.
Results: Functional targeting and expression of the T-type Ca2+ channel (TTCC) in the atria requires the endosomal protein, EHD3.
Conclusion: Impaired endosomal transport leads to cardiac rhythm and conduction disorders.
Significance: Understanding endosome-based protein trafficking in the heart may provide new therapeutic targets for cardiovascular disease.
Keywords: cytoskeleton, endosome, heart, ion channel, membrane trafficking, ankyrin, arrhythmia
Abstract
Proper trafficking of membrane-bound ion channels and transporters is requisite for normal cardiac function. Endosome-based protein trafficking of membrane-bound ion channels and transporters in the heart is poorly understood, particularly in vivo. In fact, for select cardiac cell types such as atrial myocytes, virtually nothing is known regarding endosomal transport. We previously linked the C-terminal Eps15 homology domain-containing protein 3 (EHD3) with endosome-based protein trafficking in ventricular cardiomyocytes. Here we sought to define the roles and membrane protein targets for EHD3 in atria. We identify the voltage-gated T-type Ca2+ channels (CaV3.1, CaV3.2) as substrates for EHD3-dependent trafficking in atria. Mice selectively lacking EHD3 in heart display reduced expression and targeting of both Cav3.1 and CaV3.2 in the atria. Furthermore, functional experiments identify a significant loss of T-type-mediated Ca2+ current in EHD3-deficient atrial myocytes. Moreover, EHD3 associates with both CaV3.1 and CaV3.2 in co-immunoprecipitation experiments. T-type Ca2+ channel function is critical for proper electrical conduction through the atria. Consistent with these roles, EHD3-deficient mice demonstrate heart rate variability, sinus pause, and atrioventricular conduction block. In summary, our findings identify CaV3.1 and CaV3.2 as substrates for EHD3-dependent protein trafficking in heart, provide in vivo data on endosome-based trafficking pathways in atria, and implicate EHD3 as a key player in the regulation of atrial myocyte excitability and cardiac conduction.
Introduction
Endosome-based protein trafficking systems mediate a wide range of cellular processes. Through a complex network of vesicular transport systems, endosomes facilitate neuronal plasticity, internalization of membrane proteins targeted for recycling or degradation, nutrient endocytosis, anterograde trafficking of newly synthesized proteins from the Golgi apparatus to the plasma membrane, as well as retrograde protein trafficking from the membrane (1–5). Because of the development of new in vivo models to study endosome-based protein trafficking, we are now beginning to understand the molecular underpinnings of endosome-based protein trafficking in the context of the intact physiological environment. Furthermore, we have now identified that defects in endosome-based processes may result in disease (4, 6, 7).
Cardiac membrane excitability depends on the proper organization and expression of specific ion channels, pumps, exchangers, and transporters within the plasma membrane. The localization of these membrane-bound proteins requires transport through endosomal protein trafficking pathways. C-terminal Eps15 homology domain-containing (EHD)2 proteins are endocytic regulatory proteins. Four EHD gene products (EHD1–4) are differentially expressed across all tissue types (8). Years of research in surrogate cells systems and model organisms has established the general role of EHD proteins as regulators of endosomal trafficking. EHDs are known to mediate protein trafficking from the Golgi to the plasma membrane along with internalization, recycling, and degradation pathways (9–13). EHDs primarily mediate clathrin-independent pathways. However, EHD1 mediates the endosome-based recycling of select membrane proteins initially internalized by clathrin-dependent pathways (14–16). Disruption of their function results in impaired intracellular protein trafficking and devastating consequences for the cell. These proteins are highly conserved across species, emphasizing their fundamental role in cell biology. Indeed, the human and mouse isoforms of EHD1 share 99.6% sequence homology (17).
Despite their putative roles, remarkably little is known about what specific membrane protein substrates require EHD-based endosomal systems for their proper trafficking and function, particularly in complex vertebrate cells including cardiomyocytes. Recent data indicates that EHD proteins are critical mediators of protein trafficking in the ventricle (4, 6, 7). For example, we identified EHD3 as a key regulator of anterograde trafficking of the Na+/Ca2+ exchanger (NCX), and the voltage-gated L-type Ca2+ channel (LTCC) in the ventricle (4, 7). Each of these proteins plays an important role in maintaining cardiac membrane excitability and facilitating excitation-contraction coupling in the heart (18–20). However, the role of EHD3 beyond the ventricle is unknown and unstudied.
Here, using an unbiased approach, we identify the cardiac voltage-gated T-type Ca2+ channels (TTCC), CaV3.1 and CaV3.2, as novel substrates for EHD3-dependent trafficking. CaV3.1 and CaV3.2 are critical mediators of action potential initiation and conduction from the sinoatrial node through the atria and into the atrioventricular node (AVN) in heart (21, 22). Using a cardiac-selective model of EHD3 deficiency, we demonstrate defects in CaV3.1 and CaV3.2 expression, localization, and function in EHD3−/− atrial myocytes. Furthermore, EHD3 co-immunoprecipitates with CaV3.1 and CaV3.2. In line with known roles for CaV3.1 and CaV3.2 in cardiac conduction system, loss of EHD3 in heart results severe cardiac rhythm and conduction defects. These new data are the first to implicate EHD3-dependent endosomal trafficking as a key mediator of cell excitability and electrical conduction in the atria.
EXPERIMENTAL PROCEDURES
Generation of Cardiac-selective Mouse Model of EHD3 Deficiency
To test the in vivo cardiac intrinsic roles of EHD3, we utilized a conditional null mutant allele where the 5′ UTR and exon 1 of the mouse EHD3 gene (Ehd3) was flanked by LoxP sites (Ehd3f/f) and therefore deleted in the presence of Cre recombinase (23) (Fig. 1A). We selectively eliminated EHD3 in cardiomyocytes by crossing these animals with mice expressing αMHC-Cre (24); homozygous conditional knock-out mice are referred to as EHD3 cKO throughout the report. Mice were born at expected Mendelian ratios, were healthy and fertile with body weights comparable with WT littermates, and displayed lack of EHD3 by PCR and immunoblot. Mice were genotyped by PCR. Three primers in a single duplex PCR amplified the WT allele (377 bp) and mutant allele (488 bp). Primers were as follows: 5′ CAA CAA GAG TGT CAG GAA ACC TGA ACT A-3′; 5′-CTG GGA AAC TGC AGA ACA TCA GGG AAC A-3′; 5′-ATG AGG GAC TCA AGG GGC AAG TCC TGG A-3′ (Fig. 1B). Mice utilized in this study were between 8 and 12 weeks of age.
FIGURE 1.

Cacna1h transcript levels are reduced in EHD3−/− heart. A, representative heat map of microarray data generated by analyzing relative transcript levels (∼25,000 mRNA targets) in WT versus EHD3−/− hearts (n = 3 each). B, select genes of interest with known roles in endosomal transport or excitation contraction coupling that were significantly different in expression between WT and EHD3−/− hearts. C, quantitative PCR results of Cacna1h levels between WT and EHD3−/− hearts. Both actin and GAPDH are included as controls (right).
ECG Experiments
ECG recordings of ambulatory mice were obtained using implanted radiotelemeters (DSI, St. Paul, MN) (25). Recordings were obtained from mice that were conscious, post-exercise, post-isoprenaline injection, and post-exercise plus isoprenaline injection. For baseline heart rate analysis, continuous ECG data were collected from WT and EHD3 cKO mice for 1 h on seven separate days. For stress tests, mice were run on a treadmill for a maximum of 45 min or until exhaustion, then immediately injected with isoprenaline (0.5 mg/kg), or injected with isoprenaline in the absence of exercise. Non-sustained and sustained arrhythmias were identified using standard guidelines (26).
Isolation of Atrial Myocytes
Mouse hearts were excised and perfused on a Langendorff setup as described (7). Following enzymatic digestion, the heart was transferred to a dissecting microscope and atria were physically separated from the ventricles and placed into modified Tyrode solution (in mm: NaCl (137); KCl (5.4); MgCl2 (4.5); NaH2PO4 (0.16), NaHCO3 (3); CaCl2 (10); HEPES-NaOH (20); glucose (10); taurine (1.3 mg/ml), pH 7.4) supplemented with collagenase II (Worthington, Type II) and protease (P-5147, Sigma). The atria were diced into small sections using microscissors. The solution was maintained in a 37 °C water bath, and the tissue was manually agitated every 1 min for 5 min total. After 5 min, the suspension was centrifuged at 1000 rpm for 2 min. The preparation was resuspended in 500 μl of modified KB solution (in mm: l-glutamic acid potassium salt (100); potassium aspartate (10); KCl (25); KH2PO4 (10); MgSO4 (2); taurine (20); creatine (5); EGTA (0.5); glucose (20); HEPES-KOH (5); BSA (1 mg/ml), pH 7.2, with KOH). Tissue was manually agitated by pipette for 4–5 min to dissociate single myocytes. This process yielded ∼5,000–10,000 viable atrial myocytes/preparation. As the atria were physically dissociated from the ventricle, contamination by ventricular myocytes was minimal. However, atrial myocytes were further validated using two additional criteria. First, atrial myocytes are morphologically dissimilar from ventricular myocytes (Fig. 2, E–G). Atrial myocytes are visibly smaller, thinner, and spindle shaped compared with ventricular myocytes. Second, prior to all electrophysiology experiments, whole cell membrane capacitance was measured. On average, the capacitance of ventricular myocytes ranges from 130 to 220 pF, whereas atrial myocytes range from 30 to 65 pF (27, 28). Myocytes with a capacitance >85 pF were excluded from experiments. The average capacitance was 56.92 ± 21.29 and 57.25 ± 17.81 pF (±S.D.; p = N.S.) for WT and EHD3 cKO myocytes, respectively. Myocytes were stored in modified KB solution and were viable for >5 h.
FIGURE 2.

Generation of cardiac-specific EHD3-deficient mouse (EHD cKO). A, gene targeting map. LoxP site were inserted and flanked exon1 of the Ehd3 gene. Crossing this mouse with one expressing Cre recombinase under control of the αMHC promoter selectively eliminates EHD3 expression from cardiac myocytes. B, top, representative genotype PCR for EHD+/− (377 bp + 488 bp product), EHD3−/− (488 bp product), and WT (377 bp product). C, PCR indicating the EHD3−/− mouse above also expressed Cre recombinase (300 bp product) resulting in an EHD3 cKO mouse. D, immunoblot of EHD3 in WT versus EHD3 cKO heart lysate. E, light field image of freshly isolated myocytes displaying an atrial myocyte (black arrowhead) alongside a ventricular myocyte (white arrowhead, scale bar = 10 μm). Light field image of a freshly isolated atrial (F) myocyte and ventricular (G) myocyte (scale bars = 5 μm).
Immunoblots and Immunostaining
Immunoblots of atrial lysates were performed as described (29, 30). Briefly, atria were harvested from the hearts of adult, age-matched littermates, and immediately placed into ice-cold homogenization buffer (in mm: 50 Tris-HCl, 10 NaCl, 320 sucrose, 5 EDTA, 2.5 EGTA; supplemented with 1:1000 protease inhibitor mixture and 1:1000 PMSF (Sigma)). Following quantification, tissue lysates were analyzed on Mini-PROTEAN tetra cell (Bio-Rad) on a 4–15% precast TGX gel (Bio-Rad). Gels were transferred to a nitrocellulose membrane using the Mini-PROTEAN tetra cell (Bio-Rad). Membranes were blocked for 1 h at room temperature using a 3% BSA solution or 5% milk solution and incubated with primary antibody overnight at 4 °C. Densitometry analysis was done using Image Lab software (Bio-Rad). For all experiments, protein values were normalized against an internal loading control (GAPDH).
Antibodies
The following antibodies were used in this study: affinity-purified mouse monoclonal antibody directed against mouse CaV3.1 (NeuroMab, 75–206) and CaV3.2 (NeuroMab, 75–095), mouse monoclonal anti-NCX1 (Swant, Bellinzona, Switzerland), rabbit polyclonal anti-ankyrin-B (31), mouse monoclonal anti-GAPDH (Fitzgerald Industries, Co., Wicklow, Ireland), affinity-purified rabbit polyclonal antibody directed at EHD3 (NLKRMQDQLQAQ).
Electrophysiology
Membrane currents were assessed by use of an Axopatch-200B amplifier and a CV-203BU head stage (Axon Instruments). Experimental control, data acquisition, and data analysis were accomplished with the use of software package PClamp 10 with the Digidata 1440A acquisition system (Axon Instruments). Patch pipettes were pulled from thin-walled glass capillary tubes (Sutter Instruments). The electrode resistance ranged from 2 to 4 mΩ. L- and T-type calcium currents were recorded at room temperature (20–22 °C) with pipette resistances <3.0 MΩ. The external solution contained the following (in mmol/liter): TEA-Cl (137); CsCl (5.4); MgCl2 (1); CaCl (1); 4-AP (4); HEPES (10); glucose (11) adjusted to pH 7.4 with CsOH. The internal solution contained the following (in mmol/liter): CsCl (120); TEA-Cl: (20) MgCl-6H2O; Mg-ATP (5.2); HEPES (10); EGTA (10) adjusted to pH 7.2 with CsOH. For L-type calcium current measurement, myocytes were held at −50 mV followed by 300-ms depolarizing steps to voltages ranging from −40 to +50 mV in 5-mV increments. For total calcium current, myocytes were clamped at −90 mV and subjected to the same step protocol as above. T-type current was determined by subtracting the former from the latter. Sodium current (INa) was recorded at room temperature (20–22 °C) with pipette resistances <2.0 MΩ. The external solution contained the following (in mmol/liter). The extracellular bathing solution contained (in mm): NaCl (5), MgCl2 (1), CaCl2 (1.8), CdCl2 (0.1), glucose (11), CsCl (132.5), and HEPES (20); pH was adjusted to 7.4 with CsOH. The internal solution contained the following (in mmol/liter): NaCl (5), CsF (135), EGTA (10), MgATP (5), HEPES (5), pH 7.2. To assess the INa density, myocytes were held at −160 mV and stepped to various test potentials from −100 to 30 mV in 5-mV increments, with 200-ms duration pulses and 2800-ms interpulse intervals. Voltage dependence of inactivation was assessed by holding the cells at −160 mV utilizing a 300-ms test pulse from −140 to −40 mV to prime inactivation followed by a step to −40 mV; interpulse duration was 2700 ms. Appropriate whole cell capacitance and series resistance compensation (≥60%) was applied along with leak subtraction.
Microarray
Total RNA was isolated from the hearts of 3–4-month-old WT and EHD3−/− mice using the Qiagen RNeasy Mini Kit per the manufacturer's instructions (Qiagen Inc., Valencia, CA). RNA was purified and cRNA was generated as previously described (32). Relative expression levels of cRNA were assessed using Affymetrix GeneChip Mouse Gene 2.0 ST Array (Affymetrix, Santa Clara, CA). Signal intensities were analyzed by Affymetrix Expression Console software. Background correction and quantile normalization was performed to adjust technical bias, and gene expression levels were summarized by robust multiarray method (33). A filtering method based on percentage of arrays above noise cutoff was applied to filter out low expression genes. Linear model was employed to detect differentially expressed genes. To improve the estimates of variability and statistical tests for differential expression, a variance smoothing method with fully moderated t-statistic was employed for this study (34). The significance level was adjusted by controlling the mean number of false positives (35).
Real-time PCR
Total RNA was isolated from the hearts of 3–4-month-old WT and EHD3−/− mice using the Qiagen RNeasy Mini Kit per the manufacturer instructions (Qiagen Inc., Valencia, CA). RNA was subsequently treated with TURBO DNase (Invitrogen). First strand cDNA was synthesized from 200 ng of RNA as a template using the SuperScript III First Strand Synthesis System (Invitrogen). Real-time PCR was conducted on fast optical 96-well reaction plates using the StepOne Plus real-time PCR system (Invitrogen) in a 10-μl final volume, using 0.5 μl of TaqMan primer (FAM-labeled), 2 μl of PCR-grade H2O, and 5.0 μl of TaqMan Master Mix per well. 2.5 μl of a 1:5 cDNA dilution was added to each well. The oligonucleotide primer for Cacna1h (Mm00445382_m1) was from Applied Biosystems. Gene expression was calculated relative to GAPDH (Mm99999915_g1) and β-actin (Mm00607939_s1). Data are reported as fold-change (2(−ΔΔCT)). CT values are normalized to control samples.
Immunofluorescence
Isolated atrial myocytes from WT and EHD3 cKO hearts were fixed and permeabilized in 100% ethanol at −20 °C. Cells were blocked in 2% gelatin from cold water fish skin (Sigma, G7041) and 0.075 Triton X-100 in PBS. Cells were stained with primary antibodies overnight at 4 °C in blocking solution. Cells were washed three times in ice-cold blocking solution. After washes were complete, secondary antibodies were applied in blocking solution for 2 h at room temperature. Secondary antibodies included Alexa-conjugated donkey anti-mouse 488 and 568, and donkey anti-rabbit 488 and 568. Cells were imaged on an LSM 780 confocal microscope (Carl Zeiss). Myocytes were images using identical confocal settings between genotypes. At least 20 myocytes were examined for each staining protocol.
Co-immunoprecipitation
Protein A-conjugated agarose beads (AffiGel; Bio-Rad) were incubated with either control mouse Ig or anti-EHD3 Ig in co-immunoprecipitation binding buffer (PBS with 0.1% Triton X-100 and protease inhibitor mixture (Sigma)) for 12 h at 4 °C. Beads were centrifuged and washed three times in ice-cold PBS. WT atrial tissue lysates were added to the washed beads, along with protease inhibitor mixture and co-immunoprecipitation binding buffer, and incubated for 12 h at 4 °C. The reactions were washed three times in ice-cold co-immunoprecipitation buffer. The samples were eluted and the proteins were separated by SDS-PAGE prior to immunoblots with antibodies specific for CaV3.1, CaV3.2, or EHD3. Due to low copy number of CaV3.2 in adult atria, the input lysates were scaled.
Statistics
All values are presented as mean ± S.E. Categorical variables were assessed by χ square test and Fisher's exact test. Continues variables were analyzed using a two-tailed Student's t test. Statistical analyses were conducted using GraphPad Prism version 4 (GraphPad Software, Inc., La Jolla, CA). p values <0.05 were considered significant.
RESULTS
Unbiased Screen Reveals Reduced Cacna1h in EHD3 Null Heart
EHD-based protein pathways are poorly defined in heart. As an initial and unbiased first step to define EHD3-dependent pathways in the mammalian heart, we identified differentially expressed mRNAs between WT and EHD3−/− mice (mice globally lacking EHD3). Approximately 25,000 unique transcripts were evaluated. Surprisingly, fewer than 300 transcripts showed significant differences in mRNA expression between WT and EHD3−/− hearts (Fig. 1). In fact, only a small subset of transcripts met the strict criteria of having both a 1.5-fold change in expression and a p value of <0.05 as assessed by analysis of variance. These data suggest that the EHD3-dependent pathways have a relatively selective impact on the regulation of the cardiac transcriptional program.
From this transcript profile, we focused on gene candidates with direct roles in either cardiac excitation-contraction coupling or endosomal protein trafficking, including Slc8a1 (Na/Ca exchanger, NCX), Sln (sarcolipin), or Strn (striatin) (19, 36–38). These genes (33 in all) are summarized in Table 1 and Fig. 1B. A representative heat map reflecting this data are show in Fig. 1A. Notably, the gene encoding one isoform of the cardiac TTCC. Specifically, Cacna1h mRNA was reduced by 67% in the EHD3−/− hearts (Fig. 1B, p < 0.01). Although the depolarization phase of the atrial action potential is primarily mediated by the voltage-gated sodium channel, NaV1.5, the T-type calcium current may also facilitate this process (21, 39). Furthermore, T-type calcium current is dysregulated in cardiac disease and linked with the maladaptive hypertrophic response associated with heart failure (40–42). Based on the severity of the expression change and role in vertebrate physiology, we focused on our studies on the putative link between EHD3 and TTCC in heart.
TABLE 1.
Select genes identified in EHD3−/− screen
Select down-regulated genes.
| Gene name | Protein | Function | -Fold increase/decrease (vs WT) | p value |
|---|---|---|---|---|
| Ehd3 | EHD3 (negative control) | Regulator of endosomal transport | −3.6359 | 0.0001 |
| Sln | Sarcolipin | SERCA regulating protein. Dominantly expressed in atria, with smaller ventricular expression | −1.9642 | 0.016 |
| Slc8a1 | Na/Ca exchanger | Promotes proper Ca homeostasis and EC coupling | −1.6789 | 0.0031 |
| Cacna1h | T-type Ca channel, α1H subunit | (Also known as CaV3.2) Intimately involved in pacemaker activity of SAN cells, small distribution in ventricle | −1.6789 | 0.047 |
| Strn | Striatin | Involved in vesicular trafficking and signal transduction | −1.483 | 0.0009 |
| Vit | Vitrin | Part of the Willebrand A domain, involved in cell adhesion | −1.4811 | 0.0051 |
| Stard10 | StarD | StarD family is involved in protein-related lipid transfer and lipid transport systems | −1.4192 | 0.0335 |
| Trdn | Triadin | Regulating RyR2 response to SR luminal Ca | −1.4098 | 0.086 |
| Scn4b | β1 subunit of voltage-gated NaV1.4 | Excitability of myocyte; slows inactivation and hastens recovery. Mutations linked to LQTS 3 | −1.3139 | 0.067 |
| Scn10a | Voltage-gated NaV1.8 | Regulation of myocyte excitability. Loss of function mutants associated with heart block and ventricular arrhythmia | −1.2786 | 0.0213 |
| Foxi1 | Forkhead box I1 | Regulation of H+-ATPase (proton pump) | −1.2813 | 0.011 |
| Taar7b | Trace amine-associated receptor, 7B | G-protein coupled chemosensory receptor, little known regarding function in heart | −1.2705 | 0.0125 |
| Rcan1 | Rcan | Regulator of calcineurin | −1.2634 | 0.0409 |
| Snx10 | Sorting nexin 10 | Involved in endosomal sorting complexes, localized to early endosomes | −1.2581 | 0.0034 |
| Atp4b | H+/K+ pump | Acidification of early endosomes after internalization | −1.2243 | 0.0040 |
| Ak4 | Adenylate kinase 4 | Governs metabolic response, AMP-dependent signaling, localized to mitochondrial matrix | −1.2158 | 0.0071 |
| Rep15 | RAB15 effector protein | Involved in protein recycling out of the endosomal recycling compartment | −1.1973 | 0.0227 |
| Myom1 | Myomesin 1 | Sarcomeric protein; known to be upregulated in dilated myopathies | −1.197 | 0.0015 |
| Rab39 | RAB39 | Member of the Ras oncogene family, linked to endosomal trafficking | −1.195 | 0.05 |
| Slc26a3 | Chloride/sulfate transporter | Ion homeostasis | 1.7762 | 0.0011 |
| Ide | Insulin degrading enzyme | Metabolic response, response to natriuretic peptide-mediated signaling, etc. | 1.509 | 0.0002 |
| Kcne1 | Voltage-gated K+ channel β subunit, Isk-related subfamily | Beta subunit of the slow, voltage gated K channel. Mutations linked to torsade de pointes | 1.397 | 0.0241 |
| Pcdhb2 | Protocadherin β2 | Cadherin family protein, structural & cell adhesion | 1.3568 | 0.024 |
| Cldn10 | Claudin 10 | Component of tight junctions | 1.3396 | 0.0165 |
| Kcnj8 | Inward-rectifying K channel | K(ATP) channel, membrane excitability | 1.2272 | 0.0253 |
| Adrb2 | β2-AR | Mediates response to catecholamines | 1.2092 | 0.044 |
| Snx39 | Sorting nexin 29 | Involved in endosomal sorting complexes | 1.2066 | 0.0279 |
| Atp2a3 | SR/ER Ca ATPase3 (SERCA3) | Regulated SR Ca uptake in heart, minor component | 1.1804 | 0.0341 |
| Calb1 | Calbindin 1 | Ca binding protein, mediates Ca absorption, heart function unclear | 1.1482 | 0.0178 |
| Rab11fip2 | RAB11 interacting protein 2 | Known to interact with EHD3 in early endosomal transport | 1.1312 | 0.01 |
| Scnn1b | Beta unit of the nonvoltage-gated Na channel | Maintains proper epithelial tension, epithelial distribution | 1.1065 | 0.0136 |
As a first step to identify the potential link between EHD3 and TTCC, we performed quantitative PCR measurements of mRNA transcripts levels in WT and EHD3−/− heart, probing specifically for Cacna1h to validate our microarray analyses (Fig. 1C). Using both actin and GAPDH probes as controls, Cacna1h mRNA expression was decreased by >50% in EHD3−/− hearts compared with WT (p < 0.05; Fig. 1C).
EHD3 cKO Atria Display Reduced CaV3.1 and CaV3.2 Protein Expression
Cardiac activity may be influenced by non-cardiac extrinsic factors (i.e. sympathetic and parasympathetic inputs). To avoid confounding effects on the heart due to loss of EHD3 in non-cardiac tissues we generated mice selectively lacking EHD3 in cardiac myocytes (see “Experimental Procedures,” EHD3 cKO). Consistent with the design of the targeting construct (Fig. 2, A and B), myocytes from EHD3 cKO hearts displayed nearly complete elimination of EHD3 protein expression (Fig. 1C). As this animal line represents a more selective model to study myocyte-specific effects of EHD3 on cardiac function, the remaining experiments in this study utilized the EHD3 cKO mouse.
Although not enriched in ventricle, the vertebrate atria expresses two TTCC gene products CaV3.1 (Cacna1g) and CaV3.2 (Cacna1h) (21). The dominant isoform in the adult atria is CaV3.1, whereas CaV3.2 is linked with embryonic and postnatal cardiac development (43). Therefore, we analyzed the protein expression of both CaV3.1 and CaV3.2 in WT and EHD3 cKO atria. Notably, expression of CaV3.1 and CaV3.2 is reduced by ∼40 and 60% in atria isolated from EHD3 cKO mice, respectively (Fig. 3, A and B, p < 0.01). As expected, CaV3.1 and CaV3.2 levels were minimal in ventricle compared with mouse atria. Notably, expression of Cacna1g (Cav3.1) was not significantly reduced in microarray experiments. This suggests that the loss of this major TTCC subtype from EHD3 cKO atria is likely due to a post-translational mechanism.
FIGURE 3.
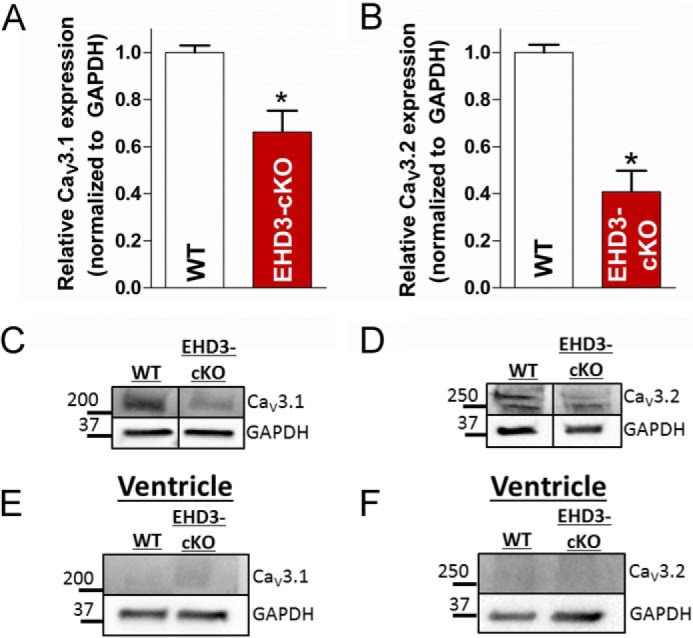
CaV3.1 and CaV3.2 protein levels are reduced in EHD3 cKO atria. A, CaV3.1 expression was decreased by ∼35% in EHD3 cKO atrial lysates assessed by immunoblot (n = 4 WT, n = 6 EHD3 cKO, p < 0.05). B, CaV3.2 expression was decreased by ∼60% in EHD3 cKO atrial lysates assessed by immunoblot (n = 4 WT, n = 6 EHD3 cKO, p < 0.001). B and C, representative blots of data in A and B. E and F, CaV3.1 and CaV3.2 were undetectable in ventricular lysates.
EHD3 Is Required for CaV3.1 and CaV3.2 Targeting in Atrial Myocytes
Based on our immunoblot data and known roles for EHDs, we tested if the membrane targeting of CaV3.1 and CaV3.2 was disrupted in EHD3 cKO atrial myocytes (Fig. 4). In WT myocytes, both CaV3.1 and CaV3.2 were expressed both on the plasma membrane and over the atrial Z-line (mouse atrial myocytes have a less well developed T-tubule network, but maintain well organized Z-line structure) (44). In line with previously published data, ankyrin-B expression and localization were disrupted in the EHD3-deficient atrial myocytes (4). In contrast, whereas markers of the Z-line are clearly present in EHD3 cKO atrial myocytes, we observed loss of both CaV3.1 and CaV3.2 localization. In fact, both proteins show diffuse punctate staining in EHD3 cKO myocytes, consistent with reduced CaV3.1 and CaV3.2 expression observed by immunoblot (Fig. 3). In summary, whereas these immunofluorescence data do not explicitly assign a role for EHD3 in the insertion of these ion channels into the surface membrane, the results clearly a role for EHD3 in the targeting of CaV3.1 and CaV3.2 in atrial myocytes.
FIGURE 4.

EHD3 is required for proper CaV3.1 and CaV3.2 targeting in atrial myocytes. A and B, compared with WT atrial myocytes, CaV3.1 and CaV3.2 are mis-localized in EHD3 cKO atrial myocytes. Ankyrin-B is utilized as a control for cell quality. (Scale bar = 10 μm.)
EHD3 Co-immunoprecipitates with Cav3.1 and Cav3.2
In non-cardiac tissue, EHD proteins are linked with a host of membrane ion channels, transporters, and receptors (4, 45–48). However, loss of CaV3.1 and CaV3.2 expression and targeting in EHD3 cKO atria may represent an indirect response to myocyte remodeling in the face of altered cell trafficking pathways. Therefore, as a first step to establish the relationship between EHD3 and CaV3.1/CaV3.2 we performed co-immunoprecipitation experiments from detergent-soluble lysates from mouse atria, immobilizing EHD3 Ig on beads. Notably, even in high salt conditions, we observed co-immunoprecipitation of both CaV3.1 and CaV3.2 with EHD3 (Fig. 5, A and B). In contrast, NaV1.5 did not co-immunoprecipitate with EHD3 supporting that NaV1.5 trafficking is likely independent of EHD3 (Fig. 5E). These data support that EHD3 is localized within the same in vivo protein complexes with CaV3.1 and CaV3.2.
FIGURE 5.

EHD3 associates with CaV3.1 and CaV3.2. EHD3 Ig immobilized Protein A-conjugated agarose beads immunoprecipitates CaV3.1 (A) and CaV3.2 (B). C and D, EHD3 was included as a positive control. E, NaV1.5 does not associate with EHD3 in co-immunoprecipitation experiments.
EHD3 Is Required for Atrial T-type Ca2+ Current
To test the functional relevance of impaired EHD3-dependent targeting of CaV3.1 and CaV3.2, we assessed ICa,T in atrial myocytes isolated from WT and EHD3 cKO hearts (Fig. 6). ICa,T was significantly decreased across all physiologically relevant voltages (Fig. 6, A and B), and peak ICa,T was reduced by ∼50% (Fig. 6C) in EHD3 cKO atrial myocytes compared with WT (p < 0.05). Notably, we also observed loss of L-type calcium channel (ICa,L; LTCC) in EHD3 cKO atrial myocytes, consistent with our prior report linking EHD3 with Cav1.2 targeting in ventricular myocytes (4)(Fig. 6D). Notably, loss of EHD3 was selective for ICa versus other atrial myocyte currents, as we observed no difference in INa properties in EHD3 cKO atrial myocytes compared with WT (Fig. 6, E and F). The data are consistent with results from co-immunoprecipitation experiments showing inability of EHD3 to associate with Nav1.5 (Fig. 5E). Atrial myocyte size was comparable between WT and EHD3 cKO myocytes as measured by cell capacitance (56.9 ± 21.3 versus 57.2 ± 17.8 pF, respectively, p > 0.99). Together, our data demonstrate that EHD3 is requisite for normal atrial ICa,T and ICa,L.
FIGURE 6.

Voltage-gated Ca2+ currents are decreased in EHD3 cKO atrial myocytes. A, current-voltage relationship of the ICa,T in WT and EHD3 cKO atrial myocytes. ICa,T is significantly different across all relevant physiologically voltages (n = 11 WT, n = 14 atria). B, peak ICa,T is significantly decreased in EHD3 cKO myocytes (−0.558 versus −0.292 pA/pF, WT versus EHD3 cKO p < 0.05). C, current-voltage relationship of the ICa,L in WT and EHD3 cKO atrial myocytes. ICa,L is significantly different across all relevant physiologically voltages (n = 11 WT, n = 14 EHD3 cKO atria). D, peak ICa,L is significantly decreased in EHD3 cKO myocytes (−1.71 versus −1.09 pA/pF, WT versus EHD3 cKO, p < 0.01). E and F, INa and INa inactivation are both unchanged by loss of EHD3 in atrial myocytes. G, representative patch clamp protocol applied to an atrial myocyte to assess ICa,T and ICa,L at a test potential of −20 mV. H, representative raw current traces of both T-type and L-type current in WT and EHD3 cKO atrial myocytes.
EHD3 cKO Mice Display Defects in Atrial Function
Both TTCC and LTCC play critical roles in atrial myocyte automaticity, atrial conduction, and atrioventricular conduction (49). Therefore, we hypothesized that disruption of both TTCC and LTCC function in EHD3 cKO mice would result in defects in atrial physiology. We implanted WT and EHD3 cKO mice with radiotelemeters and assessed cardiac ECGs in conscious mice at both baseline and following acute stress (injection of β-adrenergic agonist, isoprenaline). Consistent with our hypothesis, compared with age-matched WT littermates, EHD3 cKO mice displayed severe rhythm and conduction abnormalities (Fig. 7). For example, at baseline, EHD3 cKO mice displayed significant heart rate variability compared with WT littermates (Fig. 7, A, B, and F). In resting EHD3 cKO mice, we regularly observed heart rate fluctuations from 400 to 650 bpm within the span of 10 s. This variation was regularly sustained for more than 5 min with interspersed durations of normal sinus rhythm. This phenotype was not observed in WT mice (100 versus 0%, EHD3 cKO versus WT; p < 0.01). In response to sympathetic stimulation (0.2 mg/kg of Iso), EHD3 cKO mice displayed a high frequency of atrioventricular block, with multiple episodes of block observed in all mice. Unlike EHD3 cKO mice, atrioventricular block was only observed in one WT mouse after Iso treatment, and this occurred in only one instance in this mouse (100 versus 17%, p < 0.05, Fig. 7F). EHD3 cKO conduction phenotypes are clearly shown in Fig. 7, C and D, by the presence of non-propagating P waves. These traces indicate that whereas the heart displayed normal sinus rhythm, the cardiac action potential did not properly conduct into the ventricle (lack of QRS complex). Last, EHD3 cKO mice showed a high incidence of sinus pause following adrenergic stimulation (Fig. 7E). This phenotype was only regularly observed in EHD3 cKO mice (but observed only once in one WT mouse after Iso). In summary, at baseline and following sympathetic stimulation, EHD3 cKO mice display aberrant atrial phenotypes.
FIGURE 7.

EHD3 deficiency results in severe conduction and rhythm abnormalities. A and B, representative ECG data from conscious WT (A) and EHD3 cKO (B) mice, note the high heart rate variability in the EHD3 cKO mouse. C, representative ECG data from WT mouse post-Iso injection. D, representative ECG data from EHD3 cKO mouse post-Iso injection demonstrating repeated incidences of atrioventricular conduction block (indicated by arrowheads). E, representative ECG data of an EHD3 cKO mouse post-Iso injection demonstrating repeated incidences of sinoatrial node pause. Black arrows indicate R to R interval present during normal sinus rhythm. F, statistical analysis of observed incidence of heart rate variability, atrioventricular, and sinoatrial node pause block shown as relative percentage (p < 0.01).
DISCUSSION
The in vivo roles of endosome-based pathways for the targeting/transport of membrane proteins in heart are not well studied. Here, we define a key role for EHD3 in atrial function, as EHD deficiency in mice results in defects in cardiac automaticity and conduction. At the cellular level, we identify that EHD3 is critical for the selective expression, targeting, and function of both T-type and L-type calcium channels. Observed phenotypes in EHD3 cKO mice are consistent with atrial phenotypes found in animals deficient in either LTCC or TTCC (50–52). In summary, these findings identify the TTCC as a novel target for EHD3-dependent endosomal trafficking and demonstrate that loss of a single endosome-based protein is sufficient to disrupt normal cardiac function.
At baseline, the atrial action potential is primarily initiated by the INa, with both the LTCC and TTCC facilitating membrane depolarization (53). Furthermore, LTCC and TTCC play unique roles related to Ca2+ homeostasis and contractility (54). Intriguingly, data from the Molkentin lab suggest that CaV3.1-mediated Ca2+ entry may have anti-hypertrophic effects (55). During heart failure, TTCC expression and function is increased and plays a more prominent role in the atria (40–42, 56). For these reasons, targeting of the TTCC as a potential therapeutic against heart failure has recently gained interest, but its effectiveness remains unclear (41, 57, 58). EHD3 expression is significantly increased during heart failure (6). Our current study implicates EHD3 in the regulation of CaV3.1 and CaV3.2 in the atria. It is intriguing, then, to speculate that the increased expression and function of the TTCC during heart failure relies on EHD3-dependent mechanisms. Furthermore, the TTCC is well established to facilitate both sinoatrial and atrioventricular pacemaker activity (59, 60). We previously demonstrated EHD3 to be expressed within the sinoatrial node (4). Thus, the observation of rhythm and conduction disorders in EHD3 cKO mice was consistent with this prior work. It is important to note that whereas whole animal atrial phenotypes are consistent with loss of TTCC and LTCC function (50, 52), we cannot rule out secondary molecular defects in EHD3 cKO hearts that may contribute to observed phenotypes. Furthermore, beyond membrane protein function and electrical activity, EHDs have been linked with the biogenesis/organization of key membrane domains in skeletal muscle (61). As atrial dysfunction is linked to both dysfunction in electrical function as well as defects in atrial substrate, future experiments will be important to define any potential new roles of EHD3 in atrial function, both at baseline and in disease. Finally, our microarray experiments were utilized as an unbiased method to predict new pathways regulated by EHD3 in heart. These data noted a decrease in the levels of Cacna1h that encodes the minor component of T-type calcium current in the atria, Cav3.2. In contrast, we did not observe a significant difference in Cacna1g that encodes Cav3.1, the dominant component of atrial T-type current in the adult mouse. Our subsequent findings in EHD3 cKO adult atria revealed reduced Cav3.1 protein expression, aberrant Cav3.1 immunostaining, and reduced T-type current (despite no difference in Cacna1g mRNA levels). We therefore surmise that observed Cav3.1 defects in EHD3 cKO atria are most likely related to a post-translational trafficking defect. The roles of EHD3 in the transcriptional regulation of Cacna1h are not currently known but an important area of future investigation.
A surprising, yet consistent observation of this work (as well as past studies) is the apparent selectivity of EHD-based endosome pathways. For example, the unbiased mRNA screen identified fewer than 300 of ∼25,000 transcripts differentially expressed between WT and EHD3−/− hearts (Fig. 1; Table 1). Furthermore, although also found at the plasma membrane of atrial and ventricular myocytes, we observed no difference in NaV1.5 in EHD3 cKO hearts and NaV1.5 did not co-immunoprecipitate with EHD3 (Figs. 5 and 6) (4). These data suggest that EHD3-dependent protein trafficking in the heart is independent of “bulk” protein trafficking mechanisms, and more likely have evolved to mediate the transport of a specific subset of membrane bound proteins. The selectivity of EHD3-dependent transport mechanisms may be advantageous in developing new approaches to therapies that target the endosome. It is of particular interest that, thus far, EHD3-mediated protein trafficking in the heart is responsible for the proper trafficking, targeting, and expression of several proteins that are intimately involved in excitation-contraction coupling. Currently our knowledge of EHD3-dependent mechanism indicates that the NCX, LTCC, and TTCC all require EHD3 for their proper function. Notably, these proteins are differentially regulated during electrical remodeling. In particular, the increased function and expression NCX and TTCC in heart failure are considered to be substrates for arrhythmia (62). We hypothesize that by targeting EHD3-dependent trafficking (e.g. decreasing EHD3-mediated endosomal transport), a new approach to anti-arrhythmic therapy may be developed. This approach may offer a way to specifically tune ion channel trafficking to tune cardiac excitability in disease. Indeed, this potential has sparked the active development of EHD inhibitors (63, 64). We have previously demonstrated that in vitro EHD3 overexpression is sufficient to tune cell excitability (7), suggesting that targeting endosomal transport may be a feasible approach. However, further characterization of the EHD-dependent trafficking system in the heart is needed.
Although it will be important to define additional membrane protein substrates of the EHD3 pathway, it will also be critical to define the molecular machinery of EHD-linked endosomes. There are four EHD gene products expressed in the heart. Given the number of proteins that are trafficked through endosomes, it is unlikely that these four proteins alone are sufficient to confer selectivity for cardiac membrane protein trafficking. We predict that the Rab family of GTPases may play important roles in EHD-dependent pathways in heart. In fact, our microarray data indicate that several Rab GTPases or Rab effector proteins are altered by the loss of EHD3 (Table 1). Rab GTPases and Rab effectors are tightly linked in all components of the endosomal trafficking system, and have long been known to coordinate vesicular trafficking (65, 66). Furthermore, both Rab and Rab effectors are known binding partners of EHD proteins (46, 67–69). Currently, there are >60 Rab GTPases with an equal number of Rab effectors. The combinatorics of these proteins is believed to confer target specificity of endosomal transport. An important future goal will be to decode how Rab/Rab effector interactions with EHDs may dictate substrate specificity. In fact, the use of in vivo models for EHD tissue- and cell-specific deletion may offer important insight into defining the endosome machinery in heart, as well as how defects in these pathways may cause defects in cardiac physiology.
This work was supported, in whole or in part, by National Institutes of Health Grants HL114252 (to J. C.), HL084583, HL083422, and HL114893 (to P. J. M.), and CA105489, CA87986, CA99163, and CA116552 (to H. B.).
- EHD
- C-terminal Eps15 homology domain-containing protein
- EHD3
- C-terminal Eps15 homology domain-containing protein 3
- EHD3 cKO
- cardiac-specific EHD3 knockout mouse
- CaV3.1
- voltage-gated Ca2+ channel 3.1
- CaV3.2
- voltage-gated Ca2+ channel 3.2
- LTCC
- L-type Ca2+ channel
- NCX
- Na+/Ca2+ exchanger
- TTCC
- T-type Ca2+ channel
- Iso
- isoproterenol.
REFERENCES
- 1. Fei H., Grygoruk A., Brooks E. S., Chen A., Krantz D. E. (2008) Trafficking of vesicular neurotransmitter transporters. Traffic 9, 1425–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wolfe B. L., Trejo J. (2007) Clathrin-dependent mechanisms of G protein-coupled receptor endocytosis. Traffic 8, 462–470 [DOI] [PubMed] [Google Scholar]
- 3. De Matteis M. A., Luini A. (2008) Exiting the Golgi complex. Nat. Rev. Mol. Cell Biol. 9, 273–284 [DOI] [PubMed] [Google Scholar]
- 4. Curran J., Makara M. A., Little S. C., Musa H., Liu B., Wu X., Polina I., Alecusan J. S., Wright P., Li J., Billman G. E., Boyden P. A., Gyorke S., Band H., Hund T. J., Mohler P. J. (2014) EHD3-dependent endosome pathway regulates cardiac membrane excitability and physiology. Circ. Res. 115, 68–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang Z., Edwards J. G., Riley N., Provance D. W., Jr., Karcher R., Li X. D., Davison I. G., Ikebe M., Mercer J. A., Kauer J. A., Ehlers M. D. (2008) Myosin Vb mobilizes recycling endosomes and AMPA receptors for postsynaptic plasticity. Cell 135, 535–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gudmundsson H., Curran J., Kashef F., Snyder J. S., Smith S. A., Vargas-Pinto P., Bonilla I. M., Weiss R. M., Anderson M. E., Binkley P., Felder R. B., Carnes C. A., Band H., Hund T. J., Mohler P. J. (2012) Differential regulation of EHD3 in human and mammalian heart failure. J. Mol. Cell Cardiol. 52, 1183–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gudmundsson H., Hund T. J., Wright P. J., Kline C. F., Snyder J. S., Qian L., Koval O. M., Cunha S. R., George M., Rainey M. A., Kashef F. E., Dun W., Boyden P. A., Anderson M. E., Band H., Mohler P. J. (2010) EH domain proteins regulate cardiac membrane protein targeting. Circ. Res. 107, 84–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pohl U., Smith J. S., Tachibana I., Ueki K., Lee H. K., Ramaswamy S., Wu Q., Mohrenweiser H. W., Jenkins R. B., Louis D. N. (2000) EHD2, EHD3, and EHD4 encode novel members of a highly conserved family of EH domain-containing proteins. Genomics 63, 255–262 [DOI] [PubMed] [Google Scholar]
- 9. Chukkapalli S., Amessou M., Dekhil H., Dilly A. K., Liu Q., Bandyopadhyay S., Thomas R. D., Bejna A., Batist G., Kandouz M. (2014) Ehd3, a regulator of vesicular trafficking, is silenced in gliomas and functions as a tumor suppressor by controlling cell cycle arrest and apoptosis. Carcinogenesis 35, 877–885 [DOI] [PubMed] [Google Scholar]
- 10. Daumke O., Lundmark R., Vallis Y., Martens S., Butler P. J., McMahon H. T. (2007) Architectural and mechanistic insights into an EHD ATPase involved in membrane remodelling. Nature 449, 923–927 [DOI] [PubMed] [Google Scholar]
- 11. Galperin E., Benjamin S., Rapaport D., Rotem-Yehudar R., Tolchinsky S., Horowitz M. (2002) EHD3: a protein that resides in recycling tubular and vesicular membrane structures and interacts with EHD1. Traffic 3, 575–589 [DOI] [PubMed] [Google Scholar]
- 12. Miliaras N. B., Wendland B. (2004) EH proteins: multivalent regulators of endocytosis (and other pathways). Cell Biochem. Biophys. 41, 295–318 [DOI] [PubMed] [Google Scholar]
- 13. Wei S., Xu Y., Shi H., Wong S. H., Han W., Talbot K., Hong W., Ong W. Y. (2010) EHD1 is a synaptic protein that modulates exocytosis through binding to snapin. Mol. Cell. Neurosci. 45, 418–429 [DOI] [PubMed] [Google Scholar]
- 14. Lin S. X., Grant B., Hirsh D., Maxfield F. R. (2001) Rme-1 regulates the distribution and function of the endocytic recycling compartment in mammalian cells. Nat. Cell. Biol. 3, 567–572 [DOI] [PubMed] [Google Scholar]
- 15. Guilherme A., Soriano N. A., Furcinitti P. S., Czech M. P. (2004) Role of EHD1 and EHBP1 in perinuclear sorting and insulin-regulated GLUT4 recycling in 3T3-L1 adipocytes. J. Biol. Chem. 279, 40062–40075 [DOI] [PubMed] [Google Scholar]
- 16. Picciano J. A., Ameen N., Grant B. D., Bradbury N. A. (2003) Rme-1 regulates the recycling of the cystic fibrosis transmembrane conductance regulator. Am. J. Physiol. Cell Physiol. 285, C1009–C1018 [DOI] [PubMed] [Google Scholar]
- 17. Naslavsky N., Caplan S. (2005) C-terminal EH-domain-containing proteins: consensus for a role in endocytic trafficking, EH? J. Cell Sci. 118, 4093–4101 [DOI] [PubMed] [Google Scholar]
- 18. Mohler P. J., Schott J. J., Gramolini A. O., Dilly K. W., Guatimosim S., duBell W. H., Song L. S., Haurogné K., Kyndt F., Ali M. E., Rogers T. B., Lederer W. J., Escande D., Le Marec H., Bennett V. (2003) Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature 421, 634–639 [DOI] [PubMed] [Google Scholar]
- 19. Pott C., Philipson K. D., Goldhaber J. I. (2005) Excitation-contraction coupling in Na+-Ca2+ exchanger knockout mice: reduced transsarcolemmal Ca2+ flux. Circ. Res. 97, 1288–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bers D. M. (2002) Cardiac excitation-contraction coupling. Nature 415, 198–205 [DOI] [PubMed] [Google Scholar]
- 21. Ono K., Iijima T. (2010) Cardiac T-type Ca2+ channels in the heart. J. Mol. Cell. Cardiol. 48, 65–70 [DOI] [PubMed] [Google Scholar]
- 22. Vaidyanathan R., O'Connell R. P., Deo M., Milstein M. L., Furspan P., Herron T. J., Pandit S. V., Musa H., Berenfeld O., Jalife J., Anumonwo J. M. (2013) The ionic bases of the action potential in isolated mouse cardiac Purkinje cell. Heart Rhythm 10, 80–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. George M., Rainey M. A., Naramura M., Foster K. W., Holzapfel M. S., Willoughby L. L., Ying G., Goswami R. M., Gurumurthy C. B., Band V., Satchell S. C., Band H. (2011) Renal thrombotic microangiopathy in mice with combined deletion of endocytic recycling regulators EHD3 and EHD4. PLoS ONE 6, e17838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Agah R., Frenkel P. A., French B. A., Michael L. H., Overbeek P. A., Schneider M. D. (1997) Gene recombination in postmitotic cells: targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J. Clin. Invest. 100, 169–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. DeGrande S., Nixon D., Koval O., Curran J. W., Wright P., Wang Q., Kashef F., Chiang D., Li N., Wehrens X. H., Anderson M. E., Hund T. J., Mohler P. J. (2012) CaMKII inhibition rescues proarrhythmic phenotypes in the model of human ankyrin-B syndrome. Heart Rhythm 9, 2034–2041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mitchell G. F., Jeron A., Koren G. (1998) Measurement of heart rate and Q-T interval in the conscious mouse. Am. J. Physiol. 274, H747–H751 [DOI] [PubMed] [Google Scholar]
- 27. Roca T. P., Pigott J. D., Clarkson C. W., Crumb W. J., Jr. (1996) L-type calcium current in pediatric and adult human atrial myocytes: evidence for developmental changes in channel inactivation. Pediatr. Res. 40, 462–468 [DOI] [PubMed] [Google Scholar]
- 28. Heubach J. F., Trebess I., Wettwer E., Himmel H. M., Michel M. C., Kaumann A. J., Koch W. J., Harding S. E., Ravens U. (1999) L-type calcium current and contractility in ventricular myocytes from mice overexpressing the cardiac β2-adrenoceptor. Cardiovasc. Res. 42, 173–182 [DOI] [PubMed] [Google Scholar]
- 29. Cunha S. R., Mohler P. J. (2008) Obscurin targets ankyrin-B and protein phosphatase 2A to the cardiac M-line. J. Biol. Chem. 283, 31968–31980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kline C. F., Kurata H. T., Hund T. J., Cunha S. R., Koval O. M., Wright P. J., Christensen M., Anderson M. E., Nichols C. G., Mohler P. J. (2009) Dual role of K ATP channel C-terminal motif in membrane targeting and metabolic regulation. Proc. Natl. Acad. Sci. U.S.A. 106, 16669–16674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Abdi K. M., Mohler P. J., Davis J. Q., Bennett V. (2006) Isoform specificity of ankyrin-B: a site in the divergent C-terminal domain is required for intramolecular association. J. Biol. Chem. 281, 5741–5749 [DOI] [PubMed] [Google Scholar]
- 32. Irizarry R. A., Bolstad B. M., Collin F., Cope L. M., Hobbs B., Speed T. P. (2003) Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 31, e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Irizarry R. A., Hobbs B., Collin F., Beazer-Barclay Y. D., Antonellis K. J., Scherf U., Speed T. P. (2003) Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264 [DOI] [PubMed] [Google Scholar]
- 34. Yu L., Gulati P., Fernandez S., Pennell M., Kirschner L., Jarjoura D. (2011) Fully moderated T-statistic for small sample size gene expression arrays. Stat. Appl. Genet. Mol. Biol. 10, pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gordon A., Glazko G., Qiu X., Yakovlev A. (2007) Control of the mean number of false discoveries, Bonferroni and stability of multiple testing. Ann. Appl. Stat. 1, 179–190 [Google Scholar]
- 36. Periasamy M., Bhupathy P., Babu G. J. (2008) Regulation of sarcoplasmic reticulum Ca2+-ATPase pump expression and its relevance to cardiac muscle physiology and pathology. Cardiovasc. Res. 77, 265–273 [DOI] [PubMed] [Google Scholar]
- 37. Tsai C. T., Lai L. P., Hwang J. J., Lin J. L., Chiang F. T. (2008) Molecular genetics of atrial fibrillation. J. Am. Coll. Cardiol 52, 241–250 [DOI] [PubMed] [Google Scholar]
- 38. Benoist M., Gaillard S., Castets F. (2006) The striatin family: a new signaling platform in dendritic spines. J. Physiol. Paris 99, 146–153 [DOI] [PubMed] [Google Scholar]
- 39. Ono K., Iijima T. (2005) Pathophysiological significance of T-type Ca2+ channels: properties and functional roles of T-type Ca2+ channels in cardiac pacemaking. J. Pharmacol. Sci. 99, 197–204 [DOI] [PubMed] [Google Scholar]
- 40. Izumi T., Kihara Y., Sarai N., Yoneda T., Iwanaga Y., Inagaki K., Onozawa Y., Takenaka H., Kita T., Noma A. (2003) Reinduction of T-type calcium channels by endothelin-1 in failing hearts in vivo and in adult rat ventricular myocytes in vitro. Circulation 108, 2530–2535 [DOI] [PubMed] [Google Scholar]
- 41. Kinoshita H., Kuwahara K., Takano M., Arai Y., Kuwabara Y., Yasuno S., Nakagawa Y., Nakanishi M., Harada M., Fujiwara M., Murakami M., Ueshima K., Nakao K. (2009) T-type Ca2+ channel blockade prevents sudden death in mice with heart failure. Circulation 120, 743–752 [DOI] [PubMed] [Google Scholar]
- 42. Yasui K., Niwa N., Takemura H., Opthof T., Muto T., Horiba M., Shimizu A., Lee J. K., Honjo H., Kamiya K., Kodama I. (2005) Pathophysiological significance of T-type Ca2+ channels: expression of T-type Ca2+ channels in fetal and diseased heart. J. Pharmacol. Sci. 99, 205–210 [DOI] [PubMed] [Google Scholar]
- 43. Niwa N., Yasui K., Opthof T., Takemura H., Shimizu A., Horiba M., Lee J. K., Honjo H., Kamiya K., Kodama I. (2004) Cav3.2 subunit underlies the functional T-type Ca2+ channel in murine hearts during the embryonic period. Am. J. Physiol. Heart Circ. Physiol. 286, H2257–H2263 [DOI] [PubMed] [Google Scholar]
- 44. Dobrev D., Teos L. Y., Lederer W. J. (2009) Unique atrial myocyte Ca2+ signaling. J. Mol. Cell. Cardiol. 46, 448–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jović M., Naslavsky N., Rapaport D., Horowitz M., Caplan S. (2007) EHD1 regulates β1 integrin endosomal transport: effects on focal adhesions, cell spreading and migration. J. Cell Sci. 120, 802–814 [DOI] [PubMed] [Google Scholar]
- 46. Naslavsky N., Boehm M., Backlund P. S., Jr., Caplan S. (2004) Rabenosyn-5 and EHD1 interact and sequentially regulate protein recycling to the plasma membrane. Mol. Biol. Cell 15, 2410–2422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bar M., Schuster S., Leibman M., Ezer R., Avni A. (2014) The function of EHD2 in endocytosis and defense signaling is affected by SUMO. Plant Mol. Biol. 84, 509–518 [DOI] [PubMed] [Google Scholar]
- 48. Naslavsky N., McKenzie J., Altan-Bonnet N., Sheff D., Caplan S. (2009) EHD3 regulates early-endosome-to-Golgi transport and preserves Golgi morphology. J. Cell Sci. 122, 389–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lakatta E. G., Maltsev V. A., Vinogradova T. M. (2010) A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart's pacemaker. Circ. Res. 106, 659–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mangoni M. E., Traboulsie A., Leoni A. L., Couette B., Marger L., Le Quang K., Kupfer E., Cohen-Solal A., Vilar J., Shin H. S., Escande D., Charpentier F., Nargeot J., Lory P. (2006) Bradycardia and slowing of the atrioventricular conduction in mice lacking CaV3.1/α1G T-type calcium channels. Circ. Res. 98, 1422–1430 [DOI] [PubMed] [Google Scholar]
- 51. Le Quang K., Naud P., Qi X. Y., Duval F., Shi Y. F., Gillis M. A., Comtois P., Tardif J. C., Li D., Levesque P. C., Dobrev D., Charpentier F., Nattel S. (2011) Role of T-type calcium channel subunits in post-myocardial infarction remodelling probed with genetically engineered mice. Cardiovasc. Res. 91, 420–428 [DOI] [PubMed] [Google Scholar]
- 52. Goonasekera S. A., Hammer K., Auger-Messier M., Bodi I., Chen X., Zhang H., Reiken S., Elrod J. W., Correll R. N., York A. J., Sargent M. A., Hofmann F., Moosmang S., Marks A. R., Houser S. R., Bers D. M., Molkentin J. D. (2012) Decreased cardiac L-type Ca2+ channel activity induces hypertrophy and heart failure in mice. J. Clin. Investig. 122, 280–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Alvarez J. L., Rubio L. S., Vassort G. (1996) Facilitation of T-type calcium current in bullfrog atrial cells: voltage-dependent relief of a G protein inhibitory tone. J. Physiol. 491, 321–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jaleel N., Nakayama H., Chen X., Kubo H., MacDonnell S., Zhang H., Berretta R., Robbins J., Cribbs L., Molkentin J. D., Houser S. R. (2008) Ca2+ influx through T- and L-type Ca2+ channels have different effects on myocyte contractility and induce unique cardiac phenotypes. Circ. Res. 103, 1109–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nakayama H., Bodi I., Correll R. N., Chen X., Lorenz J., Houser S. R., Robbins J., Schwartz A., Molkentin J. D. (2009) α1G-dependent T-type Ca2+ current antagonizes cardiac hypertrophy through a NOS3-dependent mechanism in mice. J. Clin. Investig. 119, 3787–3796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chiang C. S., Huang C. H., Chieng H., Chang Y. T., Chang D., Chen J. J., Chen Y. C., Chen Y. H., Shin H. S., Campbell K. P., Chen C. C. (2009) The Cav3.2 T-type Ca2+ channel is required for pressure overload-induced cardiac hypertrophy in mice. Circ. Res. 104, 522–530 [DOI] [PubMed] [Google Scholar]
- 57. Giordanetto F., Knerr L., Wållberg A. (2011) T-type calcium channels inhibitors: a patent review. Expert Opin. Ther. Pat. 21, 85–101 [DOI] [PubMed] [Google Scholar]
- 58. Sandmann S., Bohle R. M., Dreyer T., Unger T. (2000) The T-type calcium channel blocker mibefradil reduced interstitial and perivascular fibrosis and improved hemodynamic parameters in myocardial infarction-induced cardiac failure in rats. Virchows Arch. 436, 147–157 [DOI] [PubMed] [Google Scholar]
- 59. Mesirca P., Torrente A. G., Mangoni M. E. (2015) Functional role of voltage gated Ca2+ channels in heart automaticity. Front. Physiol. 6, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mesirca P., Torrente A. G., Mangoni M. E. (2014) T-type channels in the sino-atrial and atrioventricular pacemaker mechanism. Pflugers Arch. 466, 791–799 [DOI] [PubMed] [Google Scholar]
- 61. Posey A. D., Jr., Swanson K. E., Alvarez M. G., Krishnan S., Earley J. U., Band H., Pytel P., McNally E. M., Demonbreun A. R. (2014) EHD1 mediates vesicle trafficking required for normal muscle growth and transverse tubule development. Dev. Biol. 387, 179–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bers D. M., Pogwizd S. M., Schlotthauer K. (2002) Upregulated Na/Ca exchange is involved in both contractile dysfunction and arrhythmogenesis in heart failure. Basic Res. Cardiol. 97, I36–42 [DOI] [PubMed] [Google Scholar]
- 63. Kamens A. J., Eisert R. J., Corlin T., Baleja J. D., Kritzer J. A. (2014) Structured cyclic peptides that bind the EH domain of EHD1. Biochemistry 53, 4758–4760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Han W., Chartier D., Li D., Nattel S. (2001) Ionic remodeling of cardiac Purkinje cells by congestive heart failure. Circulation 104, 2095–2100 [DOI] [PubMed] [Google Scholar]
- 65. Stenmark H. (2009) Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 10, 513–525 [DOI] [PubMed] [Google Scholar]
- 66. Stenmark H., Olkkonen V. M. (2001) The Rab GTPase family. Genome Biol. 2, REVIEWS3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kobayashi H., Fukuda M. (2013) Rab35 establishes the EHD1-association site by coordinating two distinct effectors during neurite outgrowth. J. Cell Sci. 126, 2424–2435 [DOI] [PubMed] [Google Scholar]
- 68. Naslavsky N., Rahajeng J., Sharma M., Jovic M., Caplan S. (2006) Interactions between EHD proteins and Rab11-FIP2: a role for EHD3 in early endosomal transport. Mol. Biol. Cell 17, 163–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sharma M., Giridharan S. S., Rahajeng J., Caplan S., Naslavsky N. (2010) MICAL-L1: an unusual Rab effector that links EHD1 to tubular recycling endosomes. Commun. Integr. Biol. 3, 181–183 [DOI] [PMC free article] [PubMed] [Google Scholar]