

Background: Modification of proteins with small ubiquitin-related modifier has many important functions in cell biology.
Results: Substrate-attached SUMO chains are processed by the yeast enzymes Ulp1 and Ulp2 via different mechanisms.
Conclusion: Ulp2 recognizes chains with three or more SUMO units and releases single SUMO molecules from their distal ends.
Significance: Ulp2 controls the dynamic length range of cellular SUMO chains.
Keywords: protease, protein degradation, Saccharomyces cerevisiae, small ubiquitin-like modifier (SUMO), sumoylation
Abstract
Sumoylation is a post-translational modification essential in most eukaryotes that regulates stability, localization, activity, or interaction of a multitude of proteins. It is a reversible process wherein counteracting ligases and proteases, respectively, mediate the conjugation and deconjugation of SUMO molecules to/from target proteins. Apart from attachment of single SUMO moieties to targets, formation of poly-SUMO chains occurs by the attachment of additional SUMO molecules to lysine residues in the N-terminal extensions of SUMO. In Saccharomyces cerevisiae there are apparently only two SUMO(Smt3)-specific proteases: Ulp1 and Ulp2. Ulp2 has been shown to be important for the control of poly-SUMO conjugates in cells and to dismantle SUMO chains in vitro, but the mechanism by which it acts remains to be elucidated. Applying an in vitro approach, we found that Ulp2 acts sequentially rather than stochastically, processing substrate-linked poly-SUMO chains from their distal ends down to two linked SUMO moieties. Furthermore, three linked SUMO units turned out to be the minimum length of a substrate-linked chain required for efficient binding to and processing by Ulp2. Our data suggest that Ulp2 disassembles SUMO chains by removing one SUMO moiety at a time from their ends (exo mechanism). Apparently, Ulp2 recognizes surfaces at or near the N terminus of the distal SUMO moiety, as attachments to this end significantly reduce cleavage efficiency. Our studies suggest that Ulp2 controls the dynamic range of SUMO chain lengths by trimming them from the distal ends.
Introduction
The family of small ubiquitin-related modifiers (SUMOs)3 is ubiquitously expressed and highly conserved throughout the eukaryotic kingdom (1). Sumoylation, the covalent attachment of a SUMO moiety to a substrate, typically modulates activity, localization, interactions, or stability of a modified target protein (2–5). So far hundreds of different proteins have been found to be sumoylated at some stage (6–16). This makes SUMO the most widely used ubiquitin-like protein, its interactome probably only being exceeded by ubiquitin itself. Although there are four SUMO paralogs in human cells, the yeast Saccharomyces cerevisiae only has one SUMO, which is also known as Smt3 (2, 5).
SUMO moieties are linked to their substrates via isopeptide bonds between the C-terminal carboxyl group of SUMO and the ϵ-amino groups of lysine residues exposed on the surface of the substrates. Often, but not always, these lysine residues reside in sumoylation consensus sequences (ΨKX(D/E), where Ψ is hydrophobic residue, and D/E is acidic residue) (17). Like ubiquitin, SUMO forms polymeric chains in vivo and in vitro (18–25). The chain members are linked via lysine residues located near the N terminus. Although human SUMO2 and SUMO3 mainly form chains via Lys-11 (26), which is embedded within a consensus sumoylation site, yeast Smt3 has three acceptor lysines (Lys-11, -15, and -19) that can serve as alternative attachment sites for additional SUMO molecules, with Lys-15 being the predominant lysine used during Smt3 chain formation (27). There is some cross-talk between the SUMO and the ubiquitin pathways. If recognized and targeted by SUMO-targeted ubiquitin ligases/ULS, substrates with poly-SUMO chains are further modified by the attachment of ubiquitin moieties (28–32). The latter modification can then lead to degradation by the proteasome (33).
Sumoylation is a cyclic process of conjugation and deconjugation. SUMO molecules are synthesized as inactive precursors, which require processing to expose a diglycine motif at the C terminus, thereby becoming conjugation-competent (34). Both precursor processing and deconjugation are carried out by specialized cysteine proteases. Although there are >20 different deubiquitylating enzymes known in S. cerevisiae (35), to date only two SUMO-specific proteases have been identified in this organism, namely the ubiquitin-like protein-specific proteases Ulp1 and Ulp2 (36–38). Another structurally distinct protein, the metalloprotease Wss1, has been suggested to remove ubiquitin from SUMO and to depolymerize SUMO chains; the latter activity, however, is controversial (39–41). Two classes of SUMO-specific cysteine proteases have been identified. The first one is the Ulp/SENP group. More recently, the mammalian desumoylating isopeptidase 1 (DeSI-1) protein has been identified as a SUMO-specific protease, the active site cysteine residue of which resides in a papain-like fold that is structurally distinct from the Ulp fold (42). SUMO-cleaving proteases of the Ulp/SENP group share a Ulp domain (UD) as a common feature. The UDs comprise ∼200 amino acids and provide the SUMO peptidase activity (43). ∼27% identity is found between the UDs of Ulp1 and Ulp2, whereas their non-catalytic domains exhibit no obvious similarity (37, 38). Human cells bear six different SUMO-specific proteases, SENP1, -2, -3, -5, -6, and -7 (44–49), which are grouped into two branches according to their domain structures. SENPs showing unconventional domain architecture (namely SENP6 and SENP7) are part of the Ulp2-containing branch; the others belong to the Ulp1 family (44, 45, 49, 50).
Ulp1 and Ulp2 have a distinct subset of substrates and non-redundant functions. Ulp1 is essential in S. cerevisiae and localizes to the nuclear pore (37, 38). One of its essential functions is Smt3 precursor processing, but it is also involved in deconjugation of Smt3 from substrates (36). By contrast, Ulp2 is found in the nucleoplasm, does not process Smt3 precursors efficiently, and is primarily active in poly-Smt3 chain deconjugation (19, 37, 38). Even though dispensable for vegetative growth, Ulp2 is involved in re-commencement of cell cycle progression after checkpoint arrest, chromosomal segregation, and meiosis (37, 38, 51). The N-terminal domain of Ulp2 has been found to be necessary and sufficient for nuclear localization of the protease, whereas the C-terminal non-catalytic domain is required for efficient depolymerization of large poly-Smt3 conjugates in vivo (52). Even though poly-SUMO chains appear to be of low abundance in wild-type cells under favorable conditions, dismantling these chains appears to be important for cell vitality. In the absence of Ulp2, high molecular weight conjugates accumulate in cells (19). Ulp2Δ mutants suffer from a severe growth defect, elevated levels of chromosome loss, and sensitivity to several different stresses (37, 38, 51, 53, 54). These severe phenotypes are efficiently suppressed by replacing Smt3 with mutated versions impaired in the formation of chains due to mutations or deletions of the relevant Lys residues in the N-terminal domain (19, 33). Together these data indicated that Ulp2 has an important role in Smt3 homeostasis by controlling the abundance of poly-sumoylated proteins.
Although a domain of Ulp1 has been crystallized and its enzymatic mechanism studied in some detail (43), very little is known about the Ulp2 enzyme. One of the main reasons for this is that Ulp2 does not lend itself well to analysis. Yield and purification efficiency of recombinant Ulp2 is poor, and in vitro it exhibits only weak activity (19, 37, 55). The non-catalytic domains of Ulp2 have been predicted to be intrinsically disordered to a large extent (52).
As mentioned above, SENP6 and SENP7 are the human SUMO-specific proteases closest to yeast Ulp2. Like Ulp2, SENP6 and SENP7 concentrate in the nucleus, although at least SENP6 is also found in the cytoplasm (56), and they are able to disassemble poly-SUMO chains, whereas they show little activity in SUMO precursor processing (56–58). Consistent with a role in chain depolymerization, SENP6 and SENP7 show a clear preference for SUMO2/3 over SUMO1 (56, 57), and SENP6 does not exhibit any activity toward Smt3 (59). SENP6 was found to be inactive for deconjugation of model substrates with single SUMO moieties and prefers substrates with three or more SUMO2/3 moieties (56).
In 2011 a study on the mechanism of SUMO chain disassembly by SENP6 was published and pointed toward a stochastic mechanism for that enzyme (60). The authors surmised that this might be a mechanism shared by all SENPs and possibly non-human desumoylation enzymes as well. This notion was in line with the previous observation that SENP2, SENP6, and SENP7 catalytic domains randomly cleave poly-SUMO chains (57). DeSI-1 exhibits a stochastic activity toward SUMO2/3 chains similar to the SENP UD (42).
To investigate how Ulp2 processes SUMO chains, we set up an in vitro system in which recombinant Ulp2 activity is analyzed with designed artificial substrates containing linear Smt3 chains as well as with substrates with authentic lysine-linked Smt3 chains. We found that Ulp2 does not employ a distributive endo mechanism but an ordered exo mechanism. We show that Ulp2 sequentially cleaves off single Smt3 moieties from poly-Smt3 chains starting at the most distal moiety, leaving the last two Smt3 molecules attached to the substrate. Our findings indicate that the protease needs to recognize surfaces at or near the N terminus of the distal Smt3 molecule of a chain plus two more Smt3 moieties to bind and process it. Proteins linked to chains of less than three linked Smt3 moieties are not recognized as substrates. Our data support a model in which Ulp2 acts as a safeguard against extensive poly-sumoylation by keeping Smt3 chain length within the borders of a certain dynamic range.
EXPERIMENTAL PROCEDURES
Cloning of Constructs Encoding Substrates with Linear Smt3 Chains
Plasmids driving synthesis of GFP-based substrates with N-terminal linear ubiquitin-capped Smt3 polymers in Escherichia coli were cloned stepwise building on one another. First, a sequence encoding ubiquitin was amplified by PCR using a forward primer that attached an EcoRI restriction site, a FLAG tag-encoding sequence and a SacI restriction site (in that order) to the gene, and a reversed primer with an overhang consisting of an NsiI restriction site followed by the first 16 bp of enhanced green fluorescent protein gene (eGFP). eGFP was amplified applying a corresponding forward primer and a reversed primer that added an HA tag followed by a KpnI restriction site to the 3′ end of the amplicon. The two DNA fragments were fused by overlap PCR. The resulting fusion product was digested with EcoRI and KpnI and ligated into pAM16, a pET11a derivative with an EcoRI site downstream of the promoter and a His6 tag encoding sequence preceded by a KpnI site. Utilizing the intrinsic NsiI site, n copies of ΔN17SMT3 flanked by the restriction sites NsiI and PstI were inserted into the initial construct between the ubiquitin and eGFP-encoding sequences, making use of the compatibility of NsiI and PstI overhangs. The same strategy was used to clone constructs featuring several linked ubiquitin units. To generate SMT3-led constructs, the SMT3 coding sequence was amplified and linked to a 5′ SacI and a 3′ NsiI restriction site. Via a SacI-NsiI digest of the respective construct, ubiquitin was replaced by SMT3. The same strategy was used to generate an MBP-capped construct. To obtain Smt3 chain constructs without N-terminal FLAG tag, an AgeI restriction site was inserted into the corresponding ORF after the SacI site upstream of ubiquitin. Then, SMT3 was amplified by PCR with a forward primer that added an AgeI restriction site followed by the sequence of a TEV protease recognition site (GAG AAC TTG TAC TTC CAG AGC) to the amplicon and a reversed primer generating a 3′ NsiI restriction site. The resulting product was inserted into the prepared vector via AgeI-NsiI digest and ligation. Analogously, eGFP was amplified with primers attaching a TEV protease recognition site after an AgeI restriction site to the 5′ end of the amplicon and an HA tag followed by a KpnI restriction site to the 3′ end and inserted into a AgeI/KpnI-digested vector. To obtain a maltose-binding protein (MBP)-capped Smt3 chain without a FLAG tag, an ORF-encoding ubiquitin was inserted in front of MBP in the corresponding MBP-4xSmt3-GFP construct.
To obtain a size standard for FLAG-Smt3, the SMT3 ORF was amplified with primers adding an AgeI restriction site to the 5′ end and a TEV protease recognition site followed by a KpnI site to the 3′ end of the amplicon. To obtain a size standard for ΔN17Smt3, the truncated SMT3 ORF was amplified with primers adding an AgeI restriction site followed by a TEV protease recognition site to the 5′ end and a TEV protease recognition site followed by a KpnI restriction site to the 3′ end of the amplicon. The resulting fragments were inserted into a pET11a-based vector. All constructs were verified by sequencing. A full list of primers used for this study is available upon request.
Expression of Substrate Constructs in E. coli
All constructs were expressed in the E. coli strain BL21-CodonPlus. Cells were cultivated at 37 °C in LB medium supplemented with ampicillin (100 μg/ml) and chloramphenicol (25 μg/ml) to an optical density at 600 nm (A600) of ∼0.5. The liquid culture was briefly cooled on ice, and plasmid expression was induced by the addition of isopropyl-β-d-thiogalactoside to a final concentration of 0.5 mm. Further fermentation was carried out at 30 °C for 3.5 h. In the case of the MBP-capped construct, the LB medium was supplemented with 1% glucose throughout all steps.
Purification of Ulp Substrates
All eGFP substrates fused to various types of chains were affinity-purified via a two-step protocol that made use of the N-terminal FLAG tag and the C-terminal hexahistidine tag present on all but one construct. In brief, bacteria pelleted from expression cultures were resuspended in 15 μl of lysis buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 10% glycerol, 4 mm MgCl2, 1× protease inhibitor mixture (cOmplete, EDTA-free; Roche Applied Science), 2.5 mg/ml lysozyme (Sigma), 0.8 mg/ml DNase I (Roche Applied Science), 1 mm PMSF) per absorbance units of cells. The mixture was incubated for 10 min at 4 °C then incubated on ice for 2 min. Per 1 ml of suspension, 500 μl of glass beads (∅ = 0.1–0.11 mm, Sartorius) were added. The lysate was subjected to 3 repeats of 90 s vigorous vortexing followed by 90 s of incubation on ice. The crude extract was cleared from cell debris by centrifugation at 20,000 × g for 30 min at 4 °C. The supernatant was recovered, and sodium chloride and imidazole were added to final concentrations of 500 and 10 mm, respectively. This solution was then diluted 1:3 with nickel purification buffer (50 mm Tris, pH 7.4, 500 mm NaCl, 10% glycerol, 1 mm MgCl2) containing 10 mm imidazole, and 400 μl of equilibrated nickel-SepharoseTM high performance (GE Healthcare) was added. After 1 h of incubation on a rotating wheel at 4 °C, the resin was transferred to a drop column and washed with 15 ml of nickel purification buffer supplemented with 20 mm imidazole. Proteins were eluted with 200 mm imidazole. Buffer was exchanged to FLAG purification buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 10% glycerol, 1 mm MgCl2), and 50 μl of equilibrated anti-FLAG M2 resin (Sigma) was added. FLAG affinity purification was performed according to the manufacturer's specifications. To produce the substrates without the N-terminal FLAG tag, elution was performed by incubation with 1 unit of AcTEV™ Protease (Invitrogen) in TEV protease buffer (50 mm Tris-HCl, pH 8.0, 1 mm DTT, 0.5 mm EDTA) for 8 h in the case of GFP and 5xSmt3-GFP, and by incubation with 10 nm Ubp41 for 8 h at 4 °C. All purified proteins were analyzed by anti-HA Western blotting. Removal of the FLAG tag was checked by anti-FLAG M5 Western blotting.
Purification of FLAG-Smt3 and ΔN17Smt3 Standards
FLAG-Smt3 and ΔN17Smt3 were purified as described for the Ulp2 substrates except for the elution step from anti-FLAG M2 resin. After binding and washing, elution was performed by incubation with 1 unit of AcTEV™ Protease (Invitrogen) in TEV protease buffer (50 mm Tris-HCl, pH 8.0, 0.5 mm EDTA, 1 mm DTT) for 16 h. Then the supernatants were collected and incubated with another unit of AcTEV™ Protease for eight more hours. Purified proteins were checked by anti-Smt3 Western blotting.
Expression and Purification of Ubc9 Carrying Authentic Smt3 Chains
To obtain MBP-tagged Ubc9, the ORF encoding Ubc9 was cloned into pMALc2x vector (New England Biolabs) via HindIII/PstI. Plasmids pACYC-Duet-Aos1p/Uba2p and pRSF-Duet-Ubc9/Smt3p driving expression of the machinery for the in vivo sumoylation system in E. coli were kind gifts from Dr. James Wohlschlegel (61). A plasmid (pRSF-Duet-Ubc9/Smt3p-K11R,K15R,K19R) expressing a mutant version of Smt3 impaired in chain formation was generated and kindly provided by Dr. Marion Schnellhardt. A BL21(DE3) strain carrying all three plasmids was grown in 100 ml of LB medium supplemented with ampicillin, chloramphenicol, kanamycin (50 μg/ml) and 1% glucose to an A600 of ∼0.5, and protein expression was induced by the addition of isopropyl-β-d-thiogalactoside to a final concentration of 0.3 mm. Further fermentation was carried out for 20 h at 20 °C.
To obtain Smt3-conjugated Ubc9, cells were lysed as described above for the artificial Ulp substrates. The resultant crude extract was mixed with 500 μl of amylose resin (New England Biolabs). Binding was allowed for 1 h at 4 °C while rotating. Unbound material was removed by subsequent washing with a total volume of 10 ml of amylose purification buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 17% glycerol). Bound proteins were eluted with 10 mm maltose in amylose purification buffer. Purified proteins were analyzed by anti-Smt3 Western blotting.
Ulp Constructs and Their Expression
ORFs encoding Ulp1 and Ulp2 were amplified from genomic yeast DNA using forward primers that attached a PstI restriction site followed by a TEV protease recognition site to the 5′ end of the amplicons and a FLAG tag plus a HindIII restriction site to their 3′ ends. The PCR products were inserted into pMALc2x vector via PstI-HindIII cloning. To obtain the active domain of Ulp2 only, the ULP2 sequence encoding amino acids 411–693 was amplified with primers adding a PstI restriction site followed by a TEV protease recognition site to the 5′ end of the amplicon and a FLAG tag plus a HindIII restriction site to its 3′ end. The truncated ULP2 ORF was then ligated into pMALc2x vector after PstI/HindIII digest. All constructs were verified by sequencing. E. coli BL21-CodonPlus cells were transformed with the respective plasmid and grown in LB medium supplemented with ampicillin (100 μg/ml), chloramphenicol (25 μg/ml), and 1% glucose at 37 °C to an A600 of 0.5. Expression was then induced by the addition of isopropyl-β-d-thiogalactoside to final concentrations of 1 or 0.4 mm for Ulp2 or Ulp1, respectively. Further fermentation was performed at 20 °C for 21 h with shaking. Expression cultures were harvested by centrifugation at 2800 × g for 10 min at 4 °C. Cell pellets were washed with ice-cold water and stored at −20 °C until further use.
Expression and Purification of Ubp41
Ubp41 (62) was expressed from the pET15b derivative pHUbp41 (kind gift from Dr. Rohan Baker) in the E. coli strain BL21-CodonPlus. Cells were grown in LB medium supplemented with ampicillin and chloramphenicol to late exponential phase (A600 ≈ 0.8) at 37 °C. Protein expression was induced by the addition of isopropyl-β-d-thiogalactoside to a final concentration of 0.4 mm and carried out at 30 °C for 6 h. Cells were harvested and resuspended in 15 μl of buffer U (50 mm Na2HPO4/NaH2PO4, pH 7.5, 300 mm NaCl, 12 mm imidazole, 10 mm DTT, 4 mm MgCl2, 30% glycerol) per absorbance unit of cells. The suspension was then frozen at −20 °C overnight. His6-tagged recombinant Ubp41 was purified in batch mode on nickel-SepharoseTM High Performance (GE Healthcare) as described above for the Ulp substrates with different buffers. In brief, 1× protease inhibitor mixture (cOmplete, EDTA-free), 2.5 mg/ml lysozyme (Sigma), 0.8 mg/ml DNase I, and 1 mm PMSF were added to the thawed cell suspension. After 10 min of incubation on ice, 500 μl/ml glass beads were added, and cells were broken by 3 repeats of 90 s of vigorous vortexing followed by 90 s incubation on ice. The crude extract was cleared from cell debris by centrifugation at 30,000 × g for 20 min at 4 °C. The supernatant was recovered and mixed with 500 μl of equilibrated nickel beads. The following procedure matches the purification protocol described above for the Ulp substrates with the exception that all buffers contained 30% glycerol and 1 mm DTT.
Preparation of Cell Lysates with Ulp Enzymes
The entire lysis was performed at 4 °C. Lysis buffer was composed as follows: 50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 10 mm DTT, 4 mm MgCl2, 1 mm EDTA, 2.5 mg/ml Lysozyme (Sigma), 1× protease inhibitor mixture (cOmplete, EDTA-free), 0.8 mg/ml DNase I, 1 mm PMSF. For each A600 unit of cells (corresponds to the amount of cells in 1 ml of a culture with A600 = 1), 15 μl of lysis buffer was added to the frozen pellet. Cells were resuspended by gentle manual shaking. The mixture was incubated on the bench (at 4 °C) for 5 min followed by a 5-min incubation on ice. For each 1 ml of suspension, 500 μl of glass beads (∅ = 0.1–0.11 mm, Sartorius) were added, and cells were subjected to 4 repeats of 1 min vigorous vortexing followed by 1 min of incubation on ice. The lysates were cleared from cell debris by centrifugation at 19,000 × g for 30 min. The supernatant was transferred to a fresh tube and immediately used in Ulp assays.
Ulp2 Purification
The entire purification was performed at 4 °C. To 5 ml of lysate of E. coli containing MBP-Ulp2-FLAG, 40 μl of anti-FLAG M2 resin (Sigma) was added. Binding was allowed for 1 h while rotating. Subsequently, resin beads were washed 3× with 1 ml of buffer M (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm DTT). Specifically bound proteins were eluted for 1 h with 100 μl of buffer M containing 100 μg/ml FLAG peptide (Sigma) while rotating.
Western Blot Detection
Western blots were prepared using standard techniques. Briefly, the entire volume of each sample obtained from activity assays was subjected to SDS-PAGE in 10% or 12% gels. Separated proteins were transferred to nitrocellulose membranes by semidry blotting. To detect low molecular weight species, the protocol was modified as follows. The SDS-PAGE running buffer of the outer tank was supplemented with 100 mm sodium acetate; the transfer buffer did not contain SDS; transfer was performed onto two layers of membranes of which only the upper one was then treated further; after transfer was completed, the membranes were incubated in a boiling water bath for 40 min and then dried on the bench before further processing. Membranes were blocked against unspecific antibody binding by incubation in 5% nonfat dry milk powder in phosphate-buffered saline (PBS; 1.8 mm KH2PO4, 10 mm Na2HPO4, 2.7 mm KCl, 137 mm NaCl, pH 7.4) for 2 h at room temperature with gentle shaking. Primary antibodies were diluted in 5% nonfat dry milk powder in PBS-T (PBS + 0.1% Tween 20). Antibody detection was allowed overnight at 4 °C with gentle rocking. HA-tag antibody (3F10, Sigma) was used at 1:5,000 dilution, FLAG tag antibody (anti-FLAG M5; Sigma) was used at a 1:2,000 dilution, polyclonal Smt3 antiserum was used at a 1:10,000 dilution, and ubiquitin antibody (P4D1; Santa Cruz Biotechnology) was used at 1:2000 dilution. Membranes were washed with PBS-T 3 times for 10 min. Secondary antibody, either horseradish peroxidase (HRP)-coupled goat anti-rat IgG (Abcam) or HRP-coupled polyclonal anti-mouse IgG (Sigma) or HRP-linked donkey anti-rabbit IgG (GE Healthcare), was diluted to 1:5000 in 5% nonfat dry milk powder in PBS-T and incubated with the blots for 50 min at room temperature with gentle agitation. Blots were washed as before, and luminescence was induced by brief incubation with 100 mm Tris-HCl, pH 8.5, 2.5 mm luminol, 400 μm p-coumaric acid, 0.02% hydrogen peroxide. Exposed films were developed in an AGFA Curix 60 machine.
In Vitro Desumoylation Assay
Desumoylation assays were carried out in 1.5-ml Protein LoBind Tubes (Eppendorf). Substrates and lysates were diluted in activity test buffer (10 mm Tris-HCl, pH 8.0, 150 mm NaCl, 10 mm DTT, 1 mm EDTA). The reaction mixture was composed as follows; 6 μl (diluted) lysate (or purified MBP-Ulp2-FLAG) was mixed with 2 μl (diluted) substrate and 12 μl activity test buffer. Cleavage was allowed to proceed at 30 °C for 2 h (unless indicated differently). By the end of assay time, reaction mixtures were subjected to centrifugation at 19,000 × g for 5 min at 4 °C. Seventeen microliters of supernatant were mixed with Laemmli SDS sample buffer (63 mm Tris-HCl, 10% glycerol, 2% SDS, 0.0025% bromphenol blue, pH 6.8) and boiled for 5 min. Cleavage products were analyzed by SDS-PAGE and Western blotting.
In Vitro Deubiquitylation Assay
Deubiquitylation assays were carried out as the desumoylation assays described above with the exception that the 20-μl reaction mix contained 2 μl of purified Ubp41 instead of Ulp2 (lysate).
Ulp2 Binding Assay
The entire assay was carried out at 4 °C. Crude extracts of E. coli cells expressing MBP-Ulp2(C624A)-FLAG were prepared as described above. The lysate was diluted 1:3 in buffer A (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 17% glycerol, 1 mm DTT). Per 1 ml of undiluted lysate, 50 μl of amylose resin (New England Biolabs) was added, and the mixture was incubated for 1.5 h with gentle agitation. Another aliquot of amylose resin was treated identically but without lysate (for controls). Afterward, beads were collected by 1 min of centrifugation at 500 × g, transferred to a fresh tube, and washed in batch: once with 10 column volumes of binding buffer then 4 times with 10 column volumes of buffer B (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 17% glycerol, 1 mm DTT, 0.1 mg/ml BSA). In the last washing step, the beads were transferred to fresh LoBindTM tubes (Eppendorf) and split into aliquots of 20 μl of resin/tube. Substrate was added to a final concentration of ∼100 nm to each aliquot of resin with or without immobilized Ulp2(C624A). Binding was allowed for 1.5 h while rotating. Unbound proteins were subsequently removed by washing once with 1 ml of buffer B and transfer to fresh tubes followed by washing twice with 1 ml of Buffer A. Bound proteins were eluted by boiling the resin in 40 μl of Laemmli buffer for 5 min. Binding was analyzed by SDS-PAGE and Western blotting.
To determine the binding capacity of Ulp2 to MBP-4xSmt3-GFP, MBP-Ulp2-FLAG was immobilized on anti-FLAG M2 resin (Sigma), and unbound material was removed as described above for the assay utilizing amylose resin. In the last washing step, the beads were transferred to fresh LoBindTM tubes (Eppendorf) and split into aliquots of 10 μl of coated resin/tube. Instead of substrates carrying a FLAG tag, their equivalents without a FLAG tag were used in these assays. Further steps were as described above for binding assays that used amylose resin for immobilization.
RESULTS
Ulp2 Processes poly-Smt3 Chains in a Sequential Manner
In a previous work that investigated in vitro activity of Ulp2, the recombinant enzyme was found to exhibit low activity toward test substrates carrying a single Smt3 unit (37). In that study Ulp2 was expressed as a GST fusion. This construct yielded low amounts of soluble material in our hands (data not shown). To increase expression of functional enzyme, we fused the ORF of Ulp2 to MBP, which was reported to have positive effects on solubility and folding of proteins to which it linked (63). Additionally, we added a FLAG tag to the C terminus of the fusion to enable purification if needed. For comparison, we constructed an analogous fusion of Ulp1.
To investigate substrate preference and the selection of cleavage sites by Ulp2, we designed a set of test substrates (Fig. 1). In all of these constructs, the Smt3 chains were linked to enhanced GFP. GFP is well characterized (64), reasonably stable, and can be tracked by fluorescence detection during purification. The Smt3 moieties in the chains attached to GFP were fused in a head-to-tail manner mimicking isopeptide-linked chains. As Smt3 chains are predominantly linked via one of the three acceptor lysines Lys-11, Lys-15, or Lys-19 in vivo, all acceptor Smt3 molecules within a chain were N-terminal-truncated lacking the first 17 amino acids (ΔN17Smt3) to imitate this linkage. In addition, the test substrates, contained three tags: an N-terminal FLAG tag that allowed tracking of the distal Smt3 moiety as well as purification and an HA tag at the C terminus of GFP, which was used to monitor the GFP-containing leaving groups of the cleavage reaction, followed by a C-terminal hexahistidine tag for purification purposes.
FIGURE 1.

Schematic representation of test substrate design. eGFP protein served as an anchor protein for Smt3/ubiquitin (Ubi) chains. Each construct was equipped with an HA tag and a hexahistidine (His6) tag at the C terminus. With one exception (B(I)), all constructs additionally had a FLAG tag replacing the N-terminal methionine residue. Smt3 units were fused in a head-to-tail fashion. To mimic natural Smt3 linkages, all but the most distal Smt3 unit were N-terminally truncated, lacking the first 17 amino acid residues (ΔN17Smt3 = amino acids 18–98)). The construct abbreviations used in the further course of this paper are given under each illustration. A, general constructs. n = 0–4; i = 1–3; I, constructs with Smt3 chains of different length; II, constructs with ubiquitin-capped Smt3 chains of different length. Ubiquitin moieties were fused head-to-tail. Each ubiquitin molecule was led by an extra glycine (Gly) residue, which served as a spacer. III, control molecule featuring 5 identical (ΔN17Smt3) chain members. B, constructs with N-terminal obstruction. Constructs were designed to evaluate the importance of an accessible distal end of a substrate-attached SUMO chain for Ulp2 activity. Red cross = uncleavable peptide bond (G98A).
To assay Ulp activity, a purified 5xSmt3-GFP substrate was incubated with various dilutions of lysates from E. coli cells expressing the Ulp constructs under conditions described in detail under “Experimental Procedures” (Fig. 2, A and B). To check whether any nonspecific cleavage reactions derived from unrelated components of the bacterial extract would interfere with the assay, we prepared a construct in which Ulp2 was rendered inactive by replacing the active cysteine (Cys-624) with alanine. Lysate from E. coli cells expressing ULP2-C624A was used as a negative control in desumoylation assays. Lysates containing inactive Ulp2 did not have any impact on the integrity of the test substrate used (Fig. 2A). Therefore, all cleavage activity observed with extracts expressing wild-type Ulp1 or Ulp2 can be assigned to the respective Ulps.
FIGURE 2.

Ulp2 removes single Smt3 moieties from the distal end of a substrate-attached Smt3 chain. 5xSmt3-GFP substrates were incubated with E. coli lysates containing Ulp1 or Ulp2 diluted in activity test buffer (1:1 = undiluted lysate). As a control, a lysate was used that contained the inactive C624A variant of Ulp2. Substrates were incubated with different dilutions of Ulp2-containing lysates for 2 h at 30 °C. Reaction products were then analyzed by SDS-PAGE and anti-HA Western blotting. The full-length substrate is indicated on the left-hand side of each blot, and cleavage products are indicated on the right. A, analysis of Ulp2 activity profile using either E. coli lysate containing MBP-Ulp2-FLAG or purified MBP-Ulp2-FLAG. Purified MBP-Ulp2-FLAG was diluted in activity test buffer (ATB). A control sample contained only activity test buffer instead of enzyme solution (right lane). 5xSmt3-GFP substrate was treated with dilutions of MBP-Ulp2-FLAG (1:1 1:2, 1:5, 1:10, 1:100) for 2 h at 30 °C. B, analysis of Ulp1 activity profile. C, schematic representation of mechanistic differences between Ulp2 and Ulp1. Ulp2 binds to three Smt3 units and works by cleaving single Smt3 units off the end of a chain (exo). Ulp1 requires only a single Smt3 molecule to bind and can cleave randomly after any Smt3 moiety inside the chain (endo).
Even though similar to what was previously observed by others, in our system activity levels of Ulp2 were far lower than those of Ulp1, and they were sufficient to assay processing activity and observe cleavage reactions to completion. Contrary to what was observed for SENP6 (60), Ulp2 mainly showed an ordered sequential processing activity in which first the most distal Smt3 moiety of the chain was cleaved off followed by the second (then most distal) one, etc. (Fig. 2A). This cleavage pattern was different from that of Ulp1, which produced a more distributive pattern of cleavage products, suggesting that the link between the most distal Smt3 units is cleaved with an efficiency similar to cleavage of other Smt3-Smt3 links within a chain (Fig. 2B). Thus, the results of these desumoylation assays suggest that Ulp2 operates by an exo mechanism, whereas Ulp1 employs a stochastic (endo) mode to act on a substrate-attached Smt3 chain (Fig. 2C).
To check whether the first 17 amino acid residues of Smt3, which are present in the most distal moiety (with the exception of the first methionine) but not in the other Smt3 units in a chain, confers some bias to the enzyme's preference, we prepared a construct in which not only the internal Smt3 copies are truncated but the distal moiety as well. As shown in Fig. 3, deletion of the first 17 residues on the distal Smt3 had no apparent effect on the cleavage pattern produced by Ulp2.
FIGURE 3.
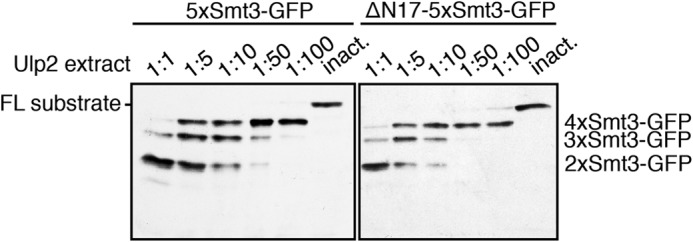
Ulp2 does not discriminate between full-length Smt3 and N-terminally truncated (ΔN17) Smt3. Comparison of band patterns derived from Ulp2-mediated processing of substrates harboring Smt3 chains led either by full-length (FL) Smt3 or by N-terminally truncated (ΔN17) Smt3. These 5xSmt3-GFP substrates were incubated with E. coli lysates containing Ulp1 or Ulp2 diluted in activity test buffer (1:1, undiluted lysate). As a control, a lysate was used that contained the inactive C624A variant of Ulp2. Substrates were incubated with different dilutions of Ulp2-containing lysates for 2 h at 30 °C. Reaction products were then analyzed by SDS-PAGE and anti-HA Western blotting. inact., inactive.
The substrates used in the assays described above are linear head-to-tail fusions. To test whether the cleavage of linear Smt3 chains by Ulp2 reflects its action on lysine-linked chains, we employed an in vivo sumoylation system reconstituted in E. coli to obtain a substrate with authentic Lys-linked poly-Smt3 chains (61). The SUMO-conjugating enzyme Ubc9 catalyzes its own sumoylation both in yeast cells (24) and in the E. coli-based system. This enabled the generation of poly-sumoylated Ubc9 (Fig. 4, A and B). Notably, if the system is supplied with Smt3, in which the major acceptor lysine residues (Lys-11, -15, and -19) used for chain formation (19) are exchanged to arginines, Smt3 chain formation is inhibited (Fig. 4A). This indicates that the isopeptide bonds between Smt3 moieties in the poly-sumoylated Ubc9 molecules are preferentially formed via one of the canonical linkage sites in Smt3. As shown in Fig. 4C, Ulp2 processes the Ubc9-attached chains starting from the most distal Smt3 moiety, similar to what was observed before for the linear chains fused to GFP. We conclude that the results on the Ulp2 mechanism we obtained with our linear Smt3 chains well reflect its activity on authentic K-linked Smt3 chains.
FIGURE 4.

Ulp2 dismantles authentic K-linked poly-Smt3 chains from their distal ends. A, MBP-Ubc9 conjugated to Smt3 was produced in an E. coli-based in vivo sumoylation system. Isopeptide bonds in larger Smt3 chains are formed mostly via the canonical acceptor lysines Lys-11,15,19. B, schematic representation of test substrate obtained from reconstituted sumoylation system in E. coli (in this example: MBP-Ubc9–4xSmt3). C, MBP-Ubc9 conjugated to poly-Smt3 chains of different lengths was incubated with E. coli lysate containing Ulp2 diluted in activity test buffer (1:1 = undiluted lysate). As a control, a lysate was used that contained the inactive C624A variant of Ulp2. Substrates were incubated with different dilutions of Ulp2-containing lysates for 2 h at 30 °C. Reaction products were then analyzed by SDS-PAGE and anti-Smt3 Western blotting. Lysate-derived signals are indicated by the asterisks (*).
In vitro, Ulp1 is highly active even in very low concentrations. Therefore, from a dilution series as shown in Fig. 2B, we could not rule out the possibility that Ulp1 actually acts as an exo enzyme, but due to a high processivity, its cleavage pattern would be reminiscent of an endo mechanism. To address this, we monitored Smt3 chain cleavage by Ulp1 over time, tracking the N-terminal FLAG tag of the substrate chain in the reaction mix. Notably, however, we were only able to detect released oligo-Smt3 chains when we reduced the speed of the cleavage reaction by performing the activity assay on ice (Fig. 5A). At 30 °C, apparently the reaction was too fast for these intermediates to be detected (Fig. 5B). Based upon these findings, we conclude that Ulp1 initially acts as an endo enzyme cleaving randomly within a poly-Smt3 chain. Subsequent to the initial cleavage, Ulp1 apparently acts in a highly progressive manner on the distal leaving group, rapidly dismantling it down to monomeric Smt3.
FIGURE 5.
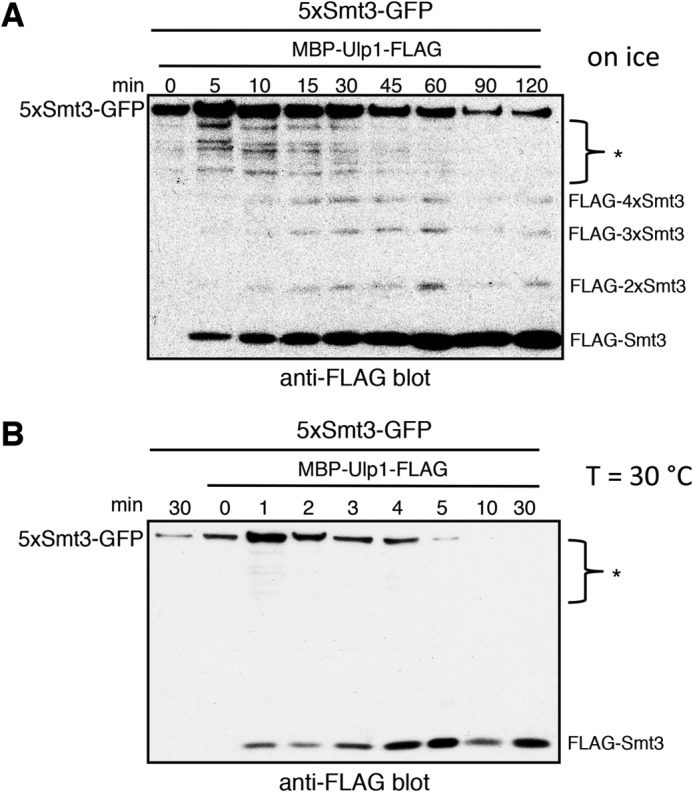
Ulp1 liberates poly-Smt3 from a substrate chain. Purified 5xSmt3-GFP was incubated with a 1:2000 dilution of lysate containing MBP-Ulp1-FLAG. The lysate was diluted in reaction buffer. At the indicated time points, samples were withdrawn, and the reaction was stopped by the addition of SDS Laemmli buffer and subsequent boiling. Reaction mixtures were then separated by SDS-PAGE and subjected to Western blot detection of the FLAG tag. Extract-derived or unspecific bands are indicated by asterisks (*). The full-length substrate is indicated on the left-hand side of each blot, and cleavage products are indicated on the right. A, assay was performed on ice. B, assay was performed at 30 °C.
By contrast, tracking of the substrate-containing leaving group of Smt3 chain processing by detection of the C-terminal HA tag (Fig. 2A) indicated that Ulp2 cleaves off one Smt3 moiety at a time. To verify this, we made use of the N-terminal FLAG tag attached to the most distal Smt3 moiety of the substrate-attached chain. Immunodetection of FLAG-tagged polypeptides confirmed that the first Smt3 is exclusively, or nearly so, cleaved off as a single moiety (mono-Smt3). This was verified for different enzyme dilutions (Fig. 6A) as well as by monitoring formation of FLAG-Smt3 over time (Fig. 6, B and C).
FIGURE 6.

Ulp2 almost exclusively liberates single moieties from Smt3 chains. E. coli lysates containing MBP-Ulp2-FLAG were diluted in activity test buffer, and an undiluted lysate containing the inactive variant Ulp2(C624A) was used as a control. Extract-derived or unspecific bands are indicated by asterisks (*). FL, full-length. A, purified 5xSmt3-GFP was incubated with the indicated dilutions of lysates containing MBP-Ulp2-FLAG for 2 h at 30 °C. Reaction products were then analyzed by SDS-PAGE and anti-FLAG Western blotting. B, purified 5xSmt3-GFP was incubated with a 1:10 dilution of lysate containing MBP-Ulp2-FLAG at 30 °C. At the indicated time points samples were withdrawn, and the reaction was stopped by the addition of SDS Laemmli buffer and subsequent boiling. Reaction mixtures were then separated by SDS-PAGE and subjected to Western blot detection of FLAG tag. The arrow indicates the size of FLAG-Smt3. inact., inactive. C, left panel, same as B, but with undiluted MBP-Ulp2-FLAG extract and Smt3 Western blot detection. Right panel, purified FLAG-Smt3 and ΔN17Smt3 were separated on a gel together with a 5xSmt3-GFP + Ulp2 reaction mix and controls and subjected to Western blot detection to determine product sizes. The FLAG-Smt3-His6 band derives from incomplete digestion by TEV protease during purification.
Ulp2 Needs at Least Three Smt3 Moieties to Process a Target
Ulp2 was previously found to dismantle poly-Smt3 chains (19). Based on those and our current findings that Ulp2 proceeds sequentially, we asked what the minimal requirement is for a substrate-attached Smt3 chain to serve as a Ulp2 target. To this end, we incubated substrates featuring 1–5 Smt3 moieties in a reaction with Ulp2. As shown in Fig. 7A, chains of less than three members are hardly processed by Ulp2. Two Smt3 moieties attached to eGFP represent the final species produced in the reaction, as can already be seen in Fig. 2. Therefore, Ulp2 seems to require a chain of at least three linked Smt3 molecules to efficiently process a target. In addition, we tested this with a variety of ubiquitin-capped substrates (Fig. 7B). This series of substrates verified the finding that Ulp2 does not recognize mono-Smt3 as a target. These experiments furthermore confirmed that cleavage only occurs if the number of Smt3 moieties in a chain is larger than two. Moreover, the results established that ubiquitin moieties replacing Smt3 in a chain are incompatible with recognition by Ulp2.
FIGURE 7.

Ulp2 preferentially acts on substrates with chains composed of three or more Smt3 units. The indicated substrates were incubated with E. coli lysates containing MBP-Ulp2-FLAG diluted in activity test buffer. An undiluted lysate containing the inactive variant Ulp2(C624A) was used as a control. Purified Smt3 chains consisting of 1–5 moieties linked to GFP-HA (A) or purified, ubiquitin-capped substrates consisting of 0–4 Smt3 moieties linked to GFP-HA (B) were subjected to digestion by MBP-Ulp2-FLAG in E. coli lysate for 2 h at 30 °C. Reaction products were analyzed by SDS-PAGE and anti-HA Western blotting. inact., inactive.
Ulp2 Needs to Access an N-terminal Region or Surrounding Surfaces of the Distal SUMO Moiety for Efficient Cleavage Activity
As Ulp2 clearly preferentially selects the distal Smt3 moiety of a chain for cleavage, we asked whether or in which way obstruction of the N terminus would affect substrate processing. To address this question, we tested substrates with different N termini (Fig. 1B). First, we compared our standard 5xSmt3-GFP substrate, which carries an N-terminal FLAG tag, to an otherwise identical substrate lacking the tag (ΔFLAG-5xSmt3-GFP). As shown in Fig. 8A, the N-terminal tag on the Smt3 chain had a very small, if any, effect on processing of the substrate by Ulp2. Next, we obstructed the distal end of the Smt3 chain by rendering the first or the first two Smt3 molecules uncleavable by exchanging the C-terminal glycine residue with alanine (G98A). As shown in Fig. 8B, Ulp2-mediated processing was strongly inhibited by uncleavable Smt3 at the distal end of the chain. These findings suggest that Ulp2 needs to recognize and access surfaces at or near the N terminus of the distal Smt3 moiety to efficiently depolymerize a poly-Smt3 chain. Of note, adding pure unanchored and uncleavable 3xSmt3 chains to a reaction did not have an inhibitory effect (data not shown), suggesting that the reduced processivity in the assay is not due to the substrate with uncleavable Smt3 units at the distal ends acting as an intrinsic inhibitor. In another variant we exchanged the full-length Smt3 copy with maltose-binding protein (MBP-4xSmt3-GFP). The MBP at the distal end of the chain also caused a strong inhibition of processing by Ulp2. Two uncleavable Smt3 units, however, caused a higher obstruction effect than MBP, which is unproportional to the size. Two Smt3 moieties have a combined molecular mass of ∼23 kDa, whereas MBP has a molecular mass of ∼42.5 kDa. Taken together, these findings demonstrated that cleavage by Ulp2 has to start at the most distal Smt3 unit. Obstruction of the distal unit of a chain severely impairs its processing by Ulp2. In contrast, processing by Ulp1 was not impaired by obstruction of the N terminus (Fig. 8C).
FIGURE 8.

Blocking access to the N terminus at the distal end of a Smt3 chain interferes with cleavage by Ulp2. The indicated substrates were incubated with E. coli lysates containing MBP-Ulp2-FLAG diluted in activity test buffer for 2 h at 30 °C. An undiluted lysate containing the inactive variant (inact.) Ulp2(C624A) was used as a control. Reaction products were analyzed by SDS-PAGE and anti-HA Western blotting. A, comparison of FLAG-tagged (5xSmt3-GFP) and untagged (ΔFLAG-5xSmt3-GFP) substrate variants in a Ulp2 cleavage assay. B, assessing the impact of uncleavable extensions at the distal ends of Smt3 chains on processing by Ulp2. The first two substrates harbor the uncleavable Smt3(G98A) variant in the first or the first two positions. The third substrate carries MBP at the distal end of the chain. C, as in B but using E. coli lysates containing Ulp1 instead of Ulp2. none = no lysate in reaction mix, instead more activity test buffer was used.
In contrast to the effects of uncleavable Smt3 or MBP at the distal end of a Smt3 chain, attaching ubiquitin to it, surprisingly, had only a mild inhibitory effect, if any, on processing by Ulp2 (Fig. 9A). Even increasing the number of capping ubiquitin moieties did not hamper their Ulp2-mediated depolymerization. Apparently, Ulp2 tolerates a sufficiently flexible appendix, such as the ones represented by ubiquitin or ubiquitin chains, on the distal moiety of an Smt3 chain. Of note, Ulp2 did not exhibit any activity toward the ubiquitin linkages, confirming previous findings (37) (Fig. 9B). By contrast, these linkages were readily cleaved by the human ubiquitin-specific protease Ubp41 in a control experiment (Fig. 9C).
FIGURE 9.

Multiple units of ubiquitin at the distal end of an Smt3 chain do not interfere with processing of an Smt3 chain by Ulp2. A, the indicated substrates were incubated with E. coli lysates containing MBP-Ulp2-FLAG diluted in activity test buffer for 2 h at 30 °C. An undiluted lysate containing the inactive variant Ulp2(C624A) was used as a control. Reaction products were analyzed by SDS-PAGE and anti-HA Western blotting. B, the indicated substrates were incubated with undiluted E. coli lysates containing MBP-Ulp2-FLAG diluted in activity test buffer for 2 h at 30 °C. Reaction products were analyzed by SDS-PAGE and anti-ubiquitin (Ubi) Western blotting. C, same as B but using purified Ubp41 instead of Ulp2 lysate.
Ulp2 Binds to Smt3 Chains of Three or More Units
Observing that Ulp2 processes Smt3 chains of three or more members, we next asked whether this was due to an inability of the enzyme to bind to shorter chains. To address this question, we set up a binding assay in which an inactive variant of Ulp2 (Ulp2(C624A)) was immobilized via its MBP tag on amylose resin. We then incubated Smt3 chains of different lengths linked to GFP with the enzyme-coated resin. None of the substrates exhibited any binding to amylose resin (Fig. 10); therefore, all the observed signals in this binding assay were specific. We found that Ulp2 binds to Smt3 chains of three and more members (Fig. 10). Notably, affinity does not increase substantially for substrates with chains longer than three units, suggesting that Ulp2 has three Smt3 binding sites that need to be occupied simultaneously to achieve full cooperative binding. Substrates equipped with mono- or di-Smt3, in contrast, were hardly bound if at all. Of note, rendering Smt3 moieties in a chain uncleavable does not interfere with the ability of Ulp2 to bind to them (Fig. 10B). To determine whether this also holds true for the MBP-capped substrate chain, we immobilized Ulp2 via its C-terminal FLAG tag and employed substrate chains lacking their FLAG tags. Apparently, Ulp2 is still able to capture MBP-4xSmt3-GFP, although less efficiently than an uncapped Smt3 chain (Fig. 10C).
FIGURE 10.

Ulp2 binds to SUMO chains of three and more members. Input, volumes of each substrate preparation equal to the volumes used in the binding assays were analyzed by SDS-PAGE and anti-HA Western blotting. A, MBP-Ulp2(C624A)-FLAG was immobilized on amylose resin and subsequently incubated with ∼100 nm concentrations of substrates with chains of different lengths (+). As a control, the same amount of each substrate was incubated with pure amylose resin (−). After 1.5 h of incubation, unbound proteins were washed off, and proteins were eluted by boiling in Laemmli buffer. Three-fourths of each elution volume was analyzed by SDS-PAGE and anti-HA Western blotting. B, assessing the ability of Ulp2 to bind to Smt3 chains capped by one or two units of Smt3(G98A). The same procedure was used as for A. C, assessing the ability of Ulp2 to bind to Smt3 chains capped by MBP. MBP-Ulp2(yA)-FLAG was immobilized on anti-FLAG M2 resin and subsequently incubated with ∼100 nm concentrations of substrates with chains of different lengths (+) from which the N-terminal FLAG tag had been enzymatically removed after purification. As a control, the same amount of each substrate was incubated with pure anti-FLAG M2 resin (−). After 1.5 h of incubation, unbound proteins were washed off, and proteins were eluted by boiling in Laemmli buffer. Three-fourths of each elution volume was analyzed by SDS-PAGE and anti-HA Western blotting. Arrows on right-hand side indicate bands of 5xSmt3-GFP and MBP-4xSmt3-GFP.
DISCUSSION
In this study we have demonstrated that Ulp2 dismantles Smt3 chains from their distal ends. This finding is in contrast to what was reported for SENP6 and SENP7, which are grouped together with Ulp2 in one subfamily of the Ulp/SENP-type proteases (57, 60). Data on SENP6, which is regarded as the human SENP most closely related to S. cerevisiae Ulp2, pointed toward a “stochastic mechanism” in which the enzyme cleaves different SUMO2/3-SUMO2/3 linkages within a chain with similar efficiencies (60). Based on these observations, it was proposed that this stochastic mode of SUMO chain processing might be universal throughout the SENP family and possibly for non-human SUMO proteases as well (60). Apparently, however, this does not apply to Ulp2, which clearly prefers to start with the distal Smt3 moiety of a substrate chain indicating that it operates from the end of a chain (exo mechanism). Similar modes of chain processing have been observed for deubiquitylating enzymes (65). A deubiquitylating enzyme activity (Usp14) that trims ubiquitin chains from their distal end is found associated with the 19S regulatory particle of the 26S proteasome (66). It was proposed that this editing function of Usp14 protects proteins with short ubiquitin chains from untimely degradation by the proteasome. We envision a similar function of Ulp2 in the control of the length of substrate-linked SUMO chains, thereby preventing substrates from being subjected untimely to SUMO-targeted ubiquitin ligase/ULS-mediated proteolytic targeting.
For Ulp1, similar to what was reported for SENP6 (60), we saw when detecting the GFP-containing leaving group that it produced a rather arbitrary product pattern, suggesting that it initiates Smt3 chain cleavage at internal linkages in a chain (endo mechanism) (Fig. 2B). However, when we followed the released distal Smt3 by its FLAG tag, we mainly detected monomeric FLAG-Smt3 (Fig. 5B). Only when the enzyme was slowed down by lowering the incubation temperature of the assay, small amounts of released distal leaving groups representing oligomeric unanchored Smt3 chains carrying the distal FLAG-Smt3 were detected. These findings suggest that Ulp1 acts on Smt3 chains by first stochastically cleaving internal Smt3-Smt3 linkages and then by rapidly dismantling the distal leaving group down to monomeric Smt3. Although further investigations are required to elucidate the mechanistic details of the Ulp1 mode of action, it is clear that Ulp1 operates by a mechanism distinct from the exo activity displayed by Ulp2.
The main categorization of SUMO-specific proteases within the Ulp/SENP family is based upon one criterion: all SENPs showing unconventional domain architecture are part of the Ulp2-containing branch; the others belong to the Ulp1 subfamily (44). As previously shown, Ulp2, SENP6, and SENP7 are all inactive or very inefficient in SUMO precursor processing (37, 38, 56). Similar to SENP6 and SENP7, we saw that Ulp2 has a preference for longer poly-SUMO chains over mono- or di-SUMO. Ulp2 exo activity, however, is clearly distinct from what was reported for SENP6 (60). Considering the crude classification criteria mentioned above, it would not be entirely surprising that there would be major differences between the SUMO proteases within a particular branch. It is worth noting that the aforementioned mechanistic study on SENP6 indicating that it acts by an endo mechanism were conducted using its active domain (UD) only. We found for Ulp2 that even its UD by itself dismantles an Smt3 chain from its distal end (Fig. 11). These findings indicated that there are already features within the UD of Ulp2 that determine its exo mode of action that are apparently absent from the SENP6 UD. It remains a possibility that the UD of the latter mammalian enzyme by itself exhibits no directionality in processing but that the non-catalytic domains enjoin the protease to a sequential mode. The UD of Ulp2 (residues 411–693) does not contain a recognizable canonical SIM, indicating that it binds to the substrate using non-canonical SUMO binding surfaces.
FIGURE 11.
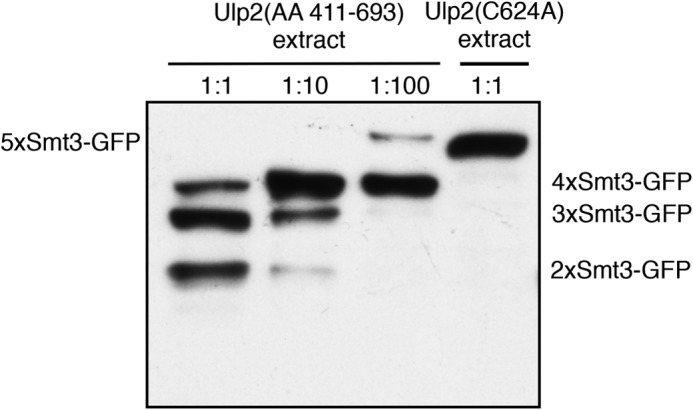
The Ulp2 catalytic domain (UD) by itself displays an exo activity on Smt3 chains. 5xSmt3-GFP substrates were incubated with E. coli lysates containing the Ulp2 catalytic domain (amino acids (AA) 411–693; UD)] diluted in activity test buffer. As a control, a lysate was used that contained the full-length, inactive variant of Ulp2(C624A). Substrates were incubated with different dilutions of UD-containing lysates for 2 h at 30 °C. Reaction products were then analyzed by SDS-PAGE and anti-HA Western blotting. The full-length substrate is indicated on the left-hand side of the blot, and cleavage products are indicated on the right.
Tracking the FLAG tag of the distal Smt3 moiety in a substrate-attached chain, we could show that Ulp2 mainly cleaves off single Smt3 moieties. Staining against Smt3 confirmed this finding (Fig. 6C). Notably, however, we saw that over time minor amounts of larger species are formed. Their relative amounts, however, are negligible and, therefore, not decisive when asking about the principle mechanism. In this context it is important for judging the relative amounts of products analyzed by anti-Smt3 Western blotting that the signal for equimolar amounts of di-Smt3 is twice as strong as for mono-Smt3. What is more, when subjecting authentic poly-Smt3 chains to processing by Ulp2, we detected only single Smt3 molecules being liberated (Fig. 4C). As all Smt3 molecules are of the same size in these substrates, the signal was much stronger than for the artificial Smt3 chains in which the product signal is divided into FLAG-Smt3 and ΔN17Smt3. In this context it is necessary to take into account that our Smt3 antibody apparently does not detect ΔN17Smt3 as efficiently as full-length Smt3, which leads to a limited detection of this species on the respective blot (see Fig. 6C).
Clearly, it would be desirable to work exclusively with purified recombinant Ulp2 instead of crude extracts. However, purification was poor in yield and linked to a significant loss of activity (data not shown). Therefore, and because there were no interfering proteases in the E. coli extract causing unspecific cleavage of the substrates in the reaction mix, we used the latter for most of the experiments. In addition, we showed in a representative assay that purified Ulp2 produced a cleavage pattern indistinguishable from Ulp2 in lysates (Fig. 2A).
Some of the rare cleavage events or effects observed in our in vitro assays are probably very unlikely to occur in vivo. For instance, based upon the low reactivity of Ulp2 toward mono-SUMO or two-membered chains in comparison to substrates with three or more linked SUMO moieties, cleavage of mono- or di-SUMO by Ulp2 are highly unlikely events in vivo. Likewise, blocking the N terminus of a substrate-linked SUMO chain might not be physiologically relevant. However, it showed us that Ulp2 requires access to the distal end of a SUMO chain to exhibit maximal activity toward that substrate. When we observed that substrates with uncleavable SUMO at the distal end of substrate-attached chains (Smt3(G98A)-4xSmt3-GFP and 2xSmt3(G98A)-3xSmt3-GFP) were inefficiently processed, we considered the possibility that the reduced cleavage of substrates is due to the fact that the uncleavable chains act as intrinsic inhibitors by binding and trapping Ulp2. However, this appears not to be the case, as the addition of 3xSmt3(G98A) chains did not interfere with the cleavage of 5xSmt3-GFP (data not shown). Taken together, our results thus point to the existence of a site or a domain near the N terminus or its surrounding surfaces of Smt3 that is recognized by Ulp2. This site seems to be less accessible in internal units of a chain.
In a binding test we found that Ulp2 binds Smt3 chains of three and more units. Interestingly, the affinity was similar for substrates with chains of three, four, or five Smt3 units, suggesting that Ulp2 has multiple Smt3 binding sites that have to be occupied simultaneously to enable high affinity binding. Based on our data, we, therefore, propose that Ulp2 has to bind to three consecutive Smt3 molecules in a chain to cleave off the distal Smt3 molecule. On this distal unit, Ulp2 recognizes a patch around the N terminus. The latter SUMO interaction site on Ulp2 may position the chain such that the enzyme can cleave the distal Smt3 unit in front of the two additional bound Smt3 moieties. Data obtained with the isolated Ulp2 UD suggest that such a binding site is contained in this domain and does not resemble a canonical SIM.
In vivo, SUMO chains can be further modified with ubiquitin by SUMO-targeted ubiquitin ligase/ULS enzymes (32), which may lead to proteasomal degradation of the tagged substrate. Based upon the observations made either with uncleavable Smt3 or MBP at the distal end of an Smt3 chain, it would have been a sensible model that ubiquitylation of Smt3 chains renders them inaccessible for desumoylation by Ulp2 by obstructing their distal ends. Surprisingly, that does not seem to be the case, as at least linear attachment of one or several ubiquitin moieties is insufficient to do so (Fig. 10). It remains a possibility that longer ubiquitin chains or ubiquitin chains with a different topology, e.g. Arg-48-linked chains, interfere with reactivity of Ulp2 toward an Smt3 chain and thereby efficiently direct a so-marked substrate to degradation by the proteasome.
In this work, we gained insight into the general mechanism by which Ulp2 processes Smt3 chains. The ulp2 knock-out strain accumulates high molecular weight poly-Smt3 aggregates (19, 33, 37, 38). This suggests that Ulp2 is the main, if not only player controlling Smt3 chain length by desumoylation. We saw that Ulp2 stops processing substrate-linked poly-SUMO chains when they are shorter than three members. Based on our data, we propose that Ulp2 controls the dynamic range of SUMO chains by trimming them from their distal ends.
Large parts of Ulp2 are predicted to lack a stable tertiary fold (52). This and its reluctant behavior in recombinant expression make it hard to gain structural insight into its interaction with substrate chains. The results of our biochemical approach, however, allowed us to obtain a rough picture, but many questions such as the following remain. Which parts or residues of Smt3 are necessary and sufficient for the interaction? Which are the relevant areas in Ulp2 necessary for binding to substrates and for the cleavage reaction? We will address these details in future experiments.
Acknowledgments
We thank Drs. Rohan Baker, Marion Schnellhardt, and James Wohlschlegel for providing plasmids.
Footnotes
- SUMO
- small ubiquitin-related modifier
- eGFP
- enhanced GFP
- Smt3
- suppressor of mif2 3
- Ulp
- ubiquitin-like modifier protease
- UD
- Ulp domain
- SENP
- sentrin protease
- ULS
- ubiquitin ligase for SUMO conjugates
- TEV
- tobacco etch virus
- MBP
- maltose-binding protein.
REFERENCES
- 1. Hochstrasser M. (2009) Origin and function of ubiquitin-like proteins. Nature 458, 422–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Johnson E. S. (2004) Protein modification by SUMO. Annu. Rev. Biochem. 73, 355–382 [DOI] [PubMed] [Google Scholar]
- 3. Wilkinson K. A., Henley J. M. (2010) Mechanisms, regulation, and consequences of protein SUMOylation. Biochem. J. 428, 133–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hay R. T. (2013) Decoding the SUMO signal. Biochem. Soc. Trans. 41, 463–473 [DOI] [PubMed] [Google Scholar]
- 5. Flotho A., Melchior F. (2013) Sumoylation: a regulatory protein modification in health and disease. Annu. Rev. Biochem. 82, 357–385 [DOI] [PubMed] [Google Scholar]
- 6. Wohlschlegel J. A., Johnson E. S., Reed S. I., Yates J. R., 3rd (2004) Global analysis of protein sumoylation in Saccharomyces cerevisiae. J. Biol. Chem. 279, 45662–45668 [DOI] [PubMed] [Google Scholar]
- 7. Zhou W., Ryan J. J., Zhou H. (2004) Global analyses of sumoylated proteins in Saccharomyces cerevisiae. Induction of protein sumoylation by cellular stresses. J. Biol. Chem. 279, 32262–32268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Denison C., Rudner A. D., Gerber S. A., Bakalarski C. E., Moazed D., Gygi S. P. (2005) A proteomic strategy for gaining insights into protein sumoylation in yeast. Mol. Cell Proteomics 4, 246–254 [DOI] [PubMed] [Google Scholar]
- 9. Hannich J. T., Lewis A., Kroetz M. B., Li S. J., Heide H., Emili A., Hochstrasser M. (2005) Defining the SUMO-modified proteome by multiple approaches in Saccharomyces cerevisiae. J. Biol. Chem. 280, 4102–4110 [DOI] [PubMed] [Google Scholar]
- 10. Panse V. G., Hardeland U., Werner T., Kuster B., Hurt E. (2004) A proteome-wide approach identifies sumoylated substrate proteins in yeast. J. Biol. Chem. 279, 41346–41351 [DOI] [PubMed] [Google Scholar]
- 11. Wykoff D. D., O'Shea E. K. (2005) Identification of sumoylated proteins by systematic immunoprecipitation of the budding yeast proteome. Mol. Cell Proteomics 4, 73–83 [DOI] [PubMed] [Google Scholar]
- 12. Vertegaal A. C., Andersen J. S., Ogg S. C., Hay R. T., Mann M., Lamond A. I. (2006) Distinct and overlapping sets of SUMO-1 and SUMO-2 target proteins revealed by quantitative proteomics. Mol. Cell Proteomics 5, 2298–2310 [DOI] [PubMed] [Google Scholar]
- 13. Golebiowski F., Matic I., Tatham M. H., Cole C., Yin Y., Nakamura A., Cox J., Barton G. J., Mann M., Hay R. T. (2009) System-wide changes to SUMO modifications in response to heat shock. Sci. Signal. 2, ra24. [DOI] [PubMed] [Google Scholar]
- 14. Schou J., Kelstrup C. D., Hayward D. G., Olsen J. V., Nilsson J. (2014) Comprehensive identification of SUMO2/3 targets and their dynamics during mitosis. PLoS ONE 9, e100692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schimmel J., Eifler K., Sigurðsson J. O., Cuijpers S. A., Hendriks I. A., Verlaan-de Vries M., Kelstrup C. D., Francavilla C., Medema R. H., Olsen J. V., Vertegaal A. C. (2014) Uncovering SUMOylation dynamics during cell-cycle progression reveals FoxM1 as a key mitotic SUMO target protein. Mol. Cell 53, 1053–1066 [DOI] [PubMed] [Google Scholar]
- 16. Hendriks I. A., D'Souza R. C., Yang B., Verlaan-de Vries M., Mann M., Vertegaal A. C. (2014) Uncovering global SUMOylation signaling networks in a site-specific manner. Nat. Struct. Mol. Biol. 21, 927–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Da Silva-Ferrada E., Lopitz-Otsoa F., Lang V., Rodríguez M. S., Matthiesen R. (2012) Strategies to Identify Recognition Signals and Targets of SUMOylation. Biochem. Res. Int. 2012, 875148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tatham M. H., Jaffray E., Vaughan O. A., Desterro J. M., Botting C. H., Naismith J. H., Hay R. T. (2001) Polymeric chains of SUMO-2 and SUMO-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J. Biol. Chem. 276, 35368–35374 [DOI] [PubMed] [Google Scholar]
- 19. Bylebyl G. R., Belichenko I., Johnson E. S. (2003) The SUMO isopeptidase Ulp2 prevents accumulation of SUMO chains in yeast. J. Biol. Chem. 278, 44113–44120 [DOI] [PubMed] [Google Scholar]
- 20. Ulrich H. D. (2008) The fast-growing business of SUMO chains. Mol. Cell 32, 301–305 [DOI] [PubMed] [Google Scholar]
- 21. Bruderer R., Tatham M. H., Plechanovova A., Matic I., Garg A. K., Hay R. T. (2011) Purification and identification of endogenous polySUMO conjugates. EMBO Rep. 12, 142–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vertegaal A. C. (2010) SUMO chains: polymeric signals. Biochem. Soc. Trans. 38, 46–49 [DOI] [PubMed] [Google Scholar]
- 23. Da Silva-Ferrada E., Xolalpa W., Lang V., Aillet F., Martin-Ruiz I., de la Cruz-Herrera C. F., Lopitz-Otsoa F., Carracedo A., Goldenberg S. J., Rivas C., England P., Rodríguez M. S. (2013) Analysis of SUMOylated proteins using SUMO-traps. Sci. Rep. 3, 1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Klug H., Xaver M., Chaugule V. K., Koidl S., Mittler G., Klein F., Pichler A. (2013) Ubc9 sumoylation controls SUMO chain formation and meiotic synapsis in Saccharomyces cerevisiae. Mol. Cell 50, 625–636 [DOI] [PubMed] [Google Scholar]
- 25. Srikumar T., Lewicki M. C., Costanzo M., Tkach J. M., van Bakel H., Tsui K., Johnson E. S., Brown G. W., Andrews B. J., Boone C., Giaever G., Nislow C., Raught B. (2013) Global analysis of SUMO chain function reveals multiple roles in chromatin regulation. J. Cell Biol. 201, 145–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Matic I., van Hagen M., Schimmel J., Macek B., Ogg S. C., Tatham M. H., Hay R. T., Lamond A. I., Mann M., Vertegaal A. C. (2008) In vivo identification of human small ubiquitin-like modifier polymerization sites by high accuracy mass spectrometry and an in vitro to in vivo strategy. Mol. Cell Proteomics 7, 132–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bencsath K. P., Podgorski M. S., Pagala V. R., Slaughter C. A., Schulman B. A. (2002) Identification of a multifunctional binding site on Ubc9p required for Smt3p conjugation. J. Biol. Chem. 277, 47938–47945 [DOI] [PubMed] [Google Scholar]
- 28. Perry J. J., Tainer J. A., Boddy M. N. (2008) A SIM-ultaneous role for SUMO and ubiquitin. Trends Biochem. Sci. 33, 201–208 [DOI] [PubMed] [Google Scholar]
- 29. Hunter T., Sun H. (2008) Crosstalk between the SUMO and ubiquitin pathways. Ernst. Schering Found Symp. Proc. 1–16 [DOI] [PubMed] [Google Scholar]
- 30. Geoffroy M. C., Hay R. T. (2009) An additional role for SUMO in ubiquitin-mediated proteolysis. Nat. Rev. Mol. Cell Biol. 10, 564–568 [DOI] [PubMed] [Google Scholar]
- 31. Praefcke G. J., Hofmann K., Dohmen R. J. (2012) SUMO playing tag with ubiquitin. Trends Biochem. Sci. 37, 23–31 [DOI] [PubMed] [Google Scholar]
- 32. Sriramachandran A. M., Dohmen R. J. (2014) SUMO-targeted ubiquitin ligases. Biochim. Biophys. Acta 1843, 75–85 [DOI] [PubMed] [Google Scholar]
- 33. Uzunova K., Göttsche K., Miteva M., Weisshaar S. R., Glanemann C., Schnellhardt M., Niessen M., Scheel H., Hofmann K., Johnson E. S., Praefcke G. J., Dohmen R. J. (2007) Ubiquitin-dependent proteolytic control of SUMO conjugates. J. Biol. Chem. 282, 34167–34175 [DOI] [PubMed] [Google Scholar]
- 34. Johnson E. S., Schwienhorst I., Dohmen R. J., Blobel G. (1997) The ubiquitin-like protein Smt3p is activated for conjugation to other proteins by an Aos1p/Uba2p heterodimer. EMBO J. 16, 5509–5519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hutchins A. P., Liu S., Diez D., Miranda-Saavedra D. (2013) The repertoires of ubiquitinating and deubiquitinating enzymes in eukaryotic genomes. Mol. Biol. Evol. 30, 1172–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li S. J., Hochstrasser M. (1999) A new protease required for cell-cycle progression in yeast. Nature 398, 246–251 [DOI] [PubMed] [Google Scholar]
- 37. Li S. J., Hochstrasser M. (2000) The yeast ULP2 (SMT4) gene encodes a novel protease specific for the ubiquitin-like Smt3 protein. Mol. Cell. Biol. 20, 2367–2377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schwienhorst I., Johnson E. S., Dohmen R. J. (2000) SUMO conjugation and deconjugation. Mol. Gen. Genet. 263, 771–786 [DOI] [PubMed] [Google Scholar]
- 39. Mullen J. R., Chen C. F., Brill S. J. (2010) Wss1 is a SUMO-dependent isopeptidase that interacts genetically with the Slx5-Slx8 SUMO-targeted ubiquitin ligase. Mol. Cell. Biol. 30, 3737–3748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Su D., Hochstrasser M. (2010) A WLM protein with SUMO-directed protease activity. Mol. Cell. Biol. 30, 3734–3736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stingele J., Schwarz M. S., Bloemeke N., Wolf P. G., Jentsch S. (2014) A DNA-dependent protease involved in DNA-protein crosslink repair. Cell 158, 327–338 [DOI] [PubMed] [Google Scholar]
- 42. Shin E. J., Shin H. M., Nam E., Kim W. S., Kim J. H., Oh B. H., Yun Y. (2012) DeSUMOylating isopeptidase: a second class of SUMO protease. EMBO Rep. 13, 339–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mossessova E., Lima C. D. (2000) Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Mol. Cell 5, 865–876 [DOI] [PubMed] [Google Scholar]
- 44. Mukhopadhyay D., Dasso M. (2007) Modification in reverse: the SUMO proteases. Trends Biochem. Sci. 32, 286–295 [DOI] [PubMed] [Google Scholar]
- 45. Hay R. T. (2007) SUMO-specific proteases: a twist in the tail. Trends Cell Biol. 17, 370–376 [DOI] [PubMed] [Google Scholar]
- 46. Yeh E. T. (2009) SUMOylation and De-SUMOylation: wrestling with life's processes. J. Biol. Chem. 284, 8223–8227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kim J. H., Baek S. H. (2009) Emerging roles of desumoylating enzymes. Biochim. Biophys. Acta 1792, 155–162 [DOI] [PubMed] [Google Scholar]
- 48. Kolli N., Mikolajczyk J., Drag M., Mukhopadhyay D., Moffatt N., Dasso M., Salvesen G., Wilkinson K. D. (2010) Distribution and paralogue specificity of mammalian deSUMOylating enzymes. Biochem. J. 430, 335–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nayak A., Müller S. (2014) SUMO-specific proteases/isopeptidases: SENPs and beyond. Genome Biol. 15, 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hickey C. M., Wilson N. R., Hochstrasser M. (2012) Function and regulation of SUMO proteases. Nat. Rev. Mol. Cell Biol. 13, 755–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schwartz D. C., Felberbaum R., Hochstrasser M. (2007) The Ulp2 SUMO protease is required for cell division following termination of the DNA damage checkpoint. Mol. Cell. Biol. 27, 6948–6961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kroetz M. B., Su D., Hochstrasser M. (2009) Essential role of nuclear localization for yeast Ulp2 SUMO protease function. Mol. Biol. Cell 20, 2196–2206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Strunnikov A. V., Aravind L., Koonin E. V. (2001) Saccharomyces cerevisiae SMT4 encodes an evolutionarily conserved protease with a role in chromosome condensation regulation. Genetics 158, 95–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bachant J., Alcasabas A., Blat Y., Kleckner N., Elledge S. J. (2002) The SUMO-1 isopeptidase Smt4 is linked to centromeric cohesion through SUMO-1 modification of DNA topoisomerase II. Mol. Cell 9, 1169–1182 [DOI] [PubMed] [Google Scholar]
- 55. Drag M., Salvesen G. S. (2008) DeSUMOylating enzymes: SENPs. IUBMB Life 60, 734–742 [DOI] [PubMed] [Google Scholar]
- 56. Mukhopadhyay D., Ayaydin F., Kolli N., Tan S. H., Anan T., Kametaka A., Azuma Y., Wilkinson K. D., Dasso M. (2006) SUSP1 antagonizes formation of highly SUMO2/3-conjugated species. J. Cell Biol. 174, 939–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lima C. D., Reverter D. (2008) Structure of the human SENP7 catalytic domain and poly-SUMO deconjugation activities for SENP6 and SENP7. J. Biol. Chem. 283, 32045–32055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shen L. N., Geoffroy M. C., Jaffray E. G., Hay R. T. (2009) Characterization of SENP7, a SUMO-2/3-specific isopeptidase. Biochem. J. 421, 223–230 [DOI] [PubMed] [Google Scholar]
- 59. Kim K. I., Baek S. H., Jeon Y. J., Nishimori S., Suzuki T., Uchida S., Shimbara N., Saitoh H., Tanaka K., Chung C. H. (2000) A new SUMO-1-specific protease, SUSP1, that is highly expressed in reproductive organs. J. Biol. Chem. 275, 14102–14106 [DOI] [PubMed] [Google Scholar]
- 60. Békés M., Prudden J., Srikumar T., Raught B., Boddy M. N., Salvesen G. S. (2011) The dynamics and mechanism of SUMO chain deconjugation by SUMO-specific proteases. J. Biol. Chem. 286, 10238–10247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wohlschlegel J. A., Johnson E. S., Reed S. I., Yates J. R., 3rd. (2006) Improved identification of SUMO attachment sites using C-terminal SUMO mutants and tailored protease digestion strategies. J. Proteome. Res. 5, 761–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Baek S. H., Choi K. S., Yoo Y. J., Cho J. M., Baker R. T., Tanaka K., Chung C. H. (1997) Molecular cloning of a novel ubiquitin-specific protease, UBP41, with isopeptidase activity in chick skeletal muscle. J. Biol. Chem. 272, 25560–25565 [DOI] [PubMed] [Google Scholar]
- 63. Fox J. D., Waugh D. S. (2003) Maltose-binding protein as a solubility enhancer. Methods Mol. Biol. 205, 99–117 [DOI] [PubMed] [Google Scholar]
- 64. Tsien R. Y. (1998) The green fluorescent protein. Annu. Rev. Biochem. 67, 509–544 [DOI] [PubMed] [Google Scholar]
- 65. Komander D., Clague M. J., Urbé S. (2009) Breaking the chains: structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 10, 550–563 [DOI] [PubMed] [Google Scholar]
- 66. Lam Y. A., Xu W., DeMartino G. N., Cohen R. E. (1997) Editing of ubiquitin conjugates by an isopeptidase in the 26S proteasome. Nature 385, 737–740 [DOI] [PubMed] [Google Scholar]