

Background: Proper regulation of DSB resection is key to genome maintenance.
Results: 14-3-3s bind to Exo1 and restrain its damage association and resection activity by counteracting the function of PCNA.
Conclusion: Exo1 activity is controlled by both positive and negative regulators to ensure a proper level of DNA end resection.
Significance: Our data reveal a key mechanism that controls the DNA end resection process.
Keywords: 14-3-3 protein, checkpoint control, DNA damage response, DNA repair, proliferating cell nuclear antigen (PCNA), DNA resection, Exo1
Abstract
The DNA end resection process dictates the cellular response to DNA double strand break damage and is essential for genome maintenance. Although insufficient DNA resection hinders homology-directed repair and ATR (ataxia telangiectasia and Rad3 related)-dependent checkpoint activation, overresection produces excessive single-stranded DNA that could lead to genomic instability. However, the mechanisms controlling DNA end resection are poorly understood. Here we show that the major resection nuclease Exo1 is regulated both positively and negatively by protein-protein interactions to ensure a proper level of DNA resection. We have shown previously that the sliding DNA clamp proliferating cell nuclear antigen (PCNA) associates with the C-terminal domain of Exo1 and promotes Exo1 damage association and DNA resection. In this report, we show that 14-3-3 proteins interact with a central region of Exo1 and negatively regulate Exo1 damage recruitment and subsequent resection. 14-3-3s limit Exo1 damage association, at least in part, by suppressing its association with PCNA. Disruption of the Exo1 interaction with 14-3-3 proteins results in elevated sensitivity of cells to DNA damage. Unlike Exo1, the Dna2 resection pathway is apparently not regulated by PCNA and 14-3-3s. Our results provide critical insights into the mechanism and regulation of the DNA end resection process and may have implications for cancer treatment.
Introduction
The integrity and stability of the cellular genome is safeguarded by sophisticated DNA damage surveillance and repair systems. DNA double strand breaks (DSBs)2 are one of the most dangerous types of DNA damage, which, if left unrepaired or misrepaired, could lead to genomic instability, immunodeficiency, cancer, and premature aging (1, 2). In response to DSBs, cells activate a DNA damage response system with two major components, DNA repair and cell cycle checkpoints, which are both controlled by the process of 5′-to-3′ DNA resection to produce a single-stranded DNA (ssDNA) structure at the ends (3–5). A DSB can be repaired by nonhomologous end joining or by homologous recombination (HR). Although DNA end resection promotes HR, it averts nonhomologous end joining (6, 7). The checkpoint response induced by DSBs coordinates DNA repair with other cellular processes (e.g. the cell cycle) or promotes cell death or senescence when the damage is too severe (8, 9). Induction of the checkpoint response is dependent on the activation of two related protein kinases, ATM and ATR. When activated, these kinases initiate downstream responses to DNA damage by signaling through protein phosphorylation cascades (10, 11). Although DNA end resection promotes ATR activation, it attenuates ATM activation (12–14). Therefore, DSB resection plays a pivotal role in determining the mode of the overall DNA damage response.
A two-step model has recently been proposed for DSB resection, consisting of an initial endonucleolytic cleavage to generate short 3′ overhangs followed by extended resection to enable repair by HR and ATR checkpoint activation. DNA resection is believed to be initiated by MRX/MRN and Sae2/CtIP, which mediate the endonucleolytic cleavage of the 5′ strand at the DNA break. Although this endocleavage step is not absolutely required to resect DSBs with “clean” ends, it is essential for the resection of DSBs with 5′ ends that are blocked by covalently linked proteins or chemical adducts. Extended, long-range resection is carried out by two nucleases, Exo1 and Dna2 (15–18). Although insufficient resection hinders HR and ATR checkpoint activation, excessive resection by these nucleases can have deleterious consequences because ssDNA is more prone to degradation and breakage of the 3′ strand DNA could cause loss of genetic information (19). Furthermore, persistent checkpoint signaling induced by excessive ssDNA could lead to cell death (20). Therefore, mechanisms to prevent overresection must exist to prevent genomic instability, but little is known about how the extent of DNA resection is controlled properly.
To address this issue, we investigated the regulation of the Exo1-mediated resection pathway. Exo1 is a member of the RAD2 family of nucleases that plays a crucial role in DNA replication, recombination, repair, and checkpoint activation. Its function has been implicated in a wide range of biological processes, including genome maintenance, meiosis, and telomere regulation as well as class switch recombination and somatic hypermutation in lymphocytes (21, 22). Although Exo1 deficiency causes defects in DNA repair and meiosis and an elevated susceptibility to cancer (23, 24), inappropriately higher levels of Exo1 activity could also have detrimental effects. Consistent with this notion, deletion of Exo1 in yeast or mice reverses the phenotypes caused by loss of function of Cdc13 or telomerase (which results in uncapped or dysfunctional telomeres), including cell survival and life span (25–27). Deletion of Exo1 in yeast also rescues the replication fork instability and DNA damage sensitivity caused by functional disruption of Rad53 (28–30). These observations suggest that Exo1 activity is normally restrained to prevent overresection of DNA breaks. However, the mechanisms for regulating Exo1 activity are unclear. In this study, we characterized Exo1 damage recruitment and resection activity using cultured human cells and Xenopus egg extracts. Our results indicate that a central region of Exo1 negatively regulates its damage recruitment and subsequent DNA end resection. The function of the central region is mediated by 14-3-3 proteins, which directly bind to this region. The interaction between Exo1 and 14-3-3s has been reported recently. However, the functional consequences of this interaction for DNA end resection have not been measured (31, 32). We show that 14-3-3 interaction limits Exo1 damage association and resection activity, in part by suppressing the positive regulator PCNA from binding to the C terminus of Exo1 (33). We also show that negative regulation of Exo1 by 14-3-3s is important for cell survival after DNA damage.
EXPERIMENTAL PROCEDURES
Plasmids, Antibodies, and Chemicals
GFP-tagged or FLAG-tagged wild-type or mutant Exo1 expression constructs in the pEGFP-C1 or pCAG07 vector were generated through PCR and site-directed mutagenesis. To generate human Exo1 baculoviral expression constructs, DNA sequences encoding C-terminally His-tagged Exo1(WT), Exo1(ΔCR), Exo1(D173A), and Exo1(ΔCR-D173A) were inserted into the Gateway donor vector pDONR221 and then into pDEST8 through BP and LR recombination reactions, respectively, according to the protocols of the manufacturer (Life Technologies). To generate mCherry-Difopein(WT) and mCherry-Difopein(MUT), DNA fragments encoding wild-type Difopein (SADGA PHCVP RDLSW LDLEA NMCLP GAAGL DSADG APHCV PRDLS WLDLE ANMCL PGAAG LE) or its mutant form (SADGA PHCVP RDLSW LKLKA NMCLP GAAGL DSADG APHCV PRDLS WLKLK ANMCL PGAAG LE) were chemically synthesized (Genewiz) and inserted into pmCherry-C1. The same sequences were inserted in pGEX4T2 to generate GST-Difopein(WT) and GST-Difopein(MUT) expression constructs. His-xDna2(1–712) was generated through PCR cloning. His-PCNA in pET28a(+), shExo1-1, and shExo1-2 in pLKO.1 have been described before (33, 34). All constructs were verified by sequencing. Primer sequences are available upon request. Xenopus Dna2 antibodies were raised in rabbits against a bacterially expressed His-tagged fusion protein containing the N-terminal 712 amino acids of xDna2. Xenopus Exo1 and PCNA antibodies have been described previously (33). Human Exo1 antibodies were generated in rabbits, in collaboration with Millipore, against residues 533–548 (KENNLHESEYGDQEGK). Anti-FLAG (Sigma, catalog no. F1804), anti-His (LifeTein, catalog no. LT0426), anti-14-3-3 (Cell Signaling Technology, catalog no. 8312), anti-GFP (Clontech, catalog no. 632569) and anti-mCherry (BioVison, catalog no. 5993-100), anti-RPA (Bethyl, catalog no. A300-244A), and anti-pS4/S8-RPA (Bethyl, catalog no. A300-245A) antibodies were purchased from the respective vendors. Camptothecin (catalog no. C9911) was purchased from Sigma.
Cell Culture, Transfection, Laser Microirradiation and Live-cell Imaging, and Clonogenic Assay
Human U2OS and HEK293T cells were cultured in DMEM with 10% FBS at 37 °C with 5% CO2. Plasmid DNA was transfected into U2OS and HEK293T cells using TransIT-LT1 (Mirus) transfection reagent according to the protocols of the manufacturer. A customized laser microirradiation and live-cell imaging system has been described before (33, 35). A lower laser power than that used in Ref. 33 was used in this study to better reveal the differences between various Exo1 protein fragments. To determine DNA damage sensitivity using the clonogenic assay, 1000 cells were plated on 10-cm dishes. Twenty-four hours after plating, cells were treated with dimethyl sulfoxide or camptothecin for 3 h. After 2 weeks of culturing, cell colonies were stained with crystal violet (0.2% in 50% methanol, 30 min), destained with H2O, and counted.
Xenopus Nuclear Extract, Immunodepletion, Immunoprecipitation, Immunoblotting, and DNA Binding Assay
Xenopus nucleoplasmic protein extract (NPE) was prepared from synthetic nuclei assembled in crude egg extract as described previously (36). To deplete xExo1 from the Xenopus NPE, 10 μl protein A-agarose beads coupled with 40 μl of xExo1 antiserum, xDna2 antiserum, or both xExo1 and xDna2 antiserum were incubated with 50 μl of extract for 45 min at 4 °C. Beads were then removed from the extract by low-speed centrifugation (5000 rpm, 1 min) in a desktop microcentrifuge. The extract supernatant was then subjected to two additional rounds of depletion under the same conditions. Coimmunoprecipitation experiments in human HEK293T cells for assessing protein-protein interactions were performed essentially as described previously (33). Briefly, cells expressing the various proteins indicated in Figs. 2, A and B, and 3, A and B, were washed once with ice-cold PBS and lysed in cell lysis buffer (20 mm Tris-HCl (pH 8.0), 1 mm EDTA, 150 mm NaCl, 0.5% Nonidet P-40, and 1 mm PMSF) for 1 h at 4 °C. The cell lysate was then centrifuged at 12,000 rpm for 15 min, and the supernatant was incubated with protein A-Sepharose beads prebound by anti-GFP antibodies or with anti-FLAG M2-conjugated Sepharose (Sigma) for 2 h at 4 °C. Beads were washed three times with the same lysis buffer, and the immunoprecipitates were eluted with protein sample buffer followed by SDS-PAGE. For coimmunoprecipitation of endogenous 14-3-3s and xExo1 in the Xenopus extract shown in Fig. 6, 10 μl of protein A-Sepharose beads prebound with 2 μl anti-xExo1 antiserum or control rabbit antiserum were incubated with 50 μl of nuclear extract at 4 °C for 2 h. Beads were then washed three times with the cell lysis buffer described above, and the proteins bound were eluted with protein sample buffer followed by SDS-PAGE. Immunoblotting was performed using DyLight 800- and DyLight 680-conjugated secondary antibodies (Pierce) and an Odyssey infrared imaging system (LI-COR Biosciences), as described previously (33).
FIGURE 2.

14-3-3 proteins bind to the central domain of Exo1 and suppress its damage association. A, coimmunoprecipitation of 14-3-3s with FLAG-tagged Exo1(WT), Exo1(ΔCR), and Exo1(CR) expressed in human 293T cells. The immunoprecipitates (IP) and 5% of the input proteins are shown. IB, immunoblot. B, coimmunoprecipitation of 14-3-3s with GFP-Exo1(WT) or GFP-Exo1(ΔCR) in 293T cells expressing mCherry, mCherry-Difopein(WT), or mCherry-Difopein(MUT). C, left panels, representative images for the damage association of GFP-Exo1(WT) (top panel) or GFP-Exo1(ΔCR) (bottom panel) in cells overexpressing mCherry, mCherry-Difopein(WT), or mCherry-Difopein(MUT). Right panels, quantified results for the damage association of GFP-Exo1(WT) (top panel) or GFP-Exo1(ΔCR) (bottom panel) during the first 20 min after laser irradiation. Each data point is the average of independent measurements from five cells. Error bars represent mean ± S.D.
FIGURE 3.
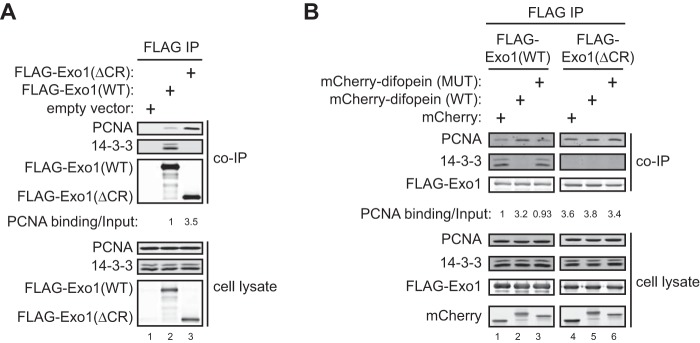
The interaction of Exo1 with PCNA is suppressed by binding of 14-3-3 proteins to the central region of Exo1. A, coimmunoprecipitation (IP) of PCNA and 14-3-3s with FLAG-Exo1(WT) or FLAG-Exo1(ΔCR) shows that deletion of the central region enhances binding to PCNA while interfering with binding to 14-3-3s. B, coimmunoprecipitation of PCNA and 14-3-3s with FLAG-Exo1(WT) or FLAG-Exo1(ΔCR) in 293T cells overexpressing mCherry, mCherry-Difopein(WT), or mCherry-Difopein(MUT).
FIGURE 6.

14-3-3 proteins repress Exo1-mediated DNA end resection in a Xenopus egg extract. A, purified recombinant GST, GST-Difopein(WT), and GST-Difopein(MUT) were expressed in and purified from BL21(DE3) E. coli cells. MW, molecular weight. B, effects of GST, GST-Difopein(WT), or GST-Difopein(MUT) on the association of xExo1 with 14-3-3 proteins in the Xenopus NPE. The asterisk denotes a nonspecific band detected by the anti-14-3-3 antibody. IP, immunoprecipitation. C, effects of GST, GST-Difopein(WT) or GST-Difopein(MUT) on DNA end resection in the extract. Vertical lines were added for sample lane identification. D, effects of GST-Difopein(WT) or GST-Difopein(MUT) on DNA end resection in the mock-depleted (depl) and xExo1-depleted or xDna2-depleted extracts. Vertical lines were added for sample lane identification. E, effects of GST-Difopein(WT) or GST-Difopein(MUT) on DNA end resection mediated by added Exo1(WT)-His or Exo1(ΔCR)-His in the extract depleted of both xExo1 and xDna2. Vertical lines were added for sample lane identification.
To compare the damage association of Exo1(WT)-His and Exo1(ΔCR)-His in Xenopus extracts, a one-end 5′ biotinylated, 2-kb dsDNA fragment was generated by PCR with a 5′ biotinylated primer. The DNA fragment immobilized on 3-μl streptavidin magnetic beads (1 μg of DNA/μl beads) was incubated with 10 μl of NPE that was depleted of endogenous xExo1 and supplemented with 1.25 μm Exo1(WT)-His or Exo1(ΔCR)-His recombinant proteins. After 5 min of incubation at room temperature, beads were washed six times each with 200 μl of egg lysis buffer (250 mm sucrose, 2.5 mm MgCl2, 50 mm KCl, 10 mm HEPES (pH 7.7)) supplemented with 1% Triton X-100, and DNA-associated proteins were eluted with protein sample buffer, followed by SDS-PAGE and Western blotting.
Recombinant Protein Expression and Purification
Recombinant, C-terminally His-tagged Exo1(WT), Exo1(D173A), Exo1(ΔCR), and Exo1(ΔCR-D173A) were expressed in Sf9 insect cells using a Bac-to-Bac baculovirus expression system with Gateway Technology (Life Technologies) according to the protocols of the manufacturer. Briefly, His-tagged Exo1 and its mutants were cloned into the Gateway donor vector pDONR221 through PCR and BP recombination and then transferred into pDEST8 through LR recombination. Subsequently, the destination vectors expressing Exo1 and its mutants were used to transform DH10Bac Escherichia coli cells that contain a baculovirus shuttle vector and a helper plasmid to generate recombinant bacmid DNA. After verifying the inserts via a PCR-based approach, the bacmids were transfected into Sf9 cells using Cellfectin II transfection reagent (Life Technologies) to produce recombinant baculoviruses. After two rounds of amplification in Sf9 cells, the medium containing baculoviruses were used to infect Sf9 cells for 72 h to produce recombinant proteins. His-tagged PCNA, and GST-tagged Difopein(WT) and Difopein(MUT) were expressed in the E. coli strain BL21(DE3). All His-tagged and GST-tagged proteins were affinity-purified using HisPur cobalt resin (Pierce) and glutathione-agarose beads (Pierce), respectively, according to the protocols of the manufacturer. Purified proteins were dialyzed in PBS containing 10% glycerol, frozen in liquid nitrogen, and stored at −80 °C.
DNA Substrate Labeling and DNA End Resection in Xenopus Extracts and with Purified Proteins
The 3′ 32P-labeled, 6-kb DNA substrate for resection was prepared as described previously (33). A typical resection reaction in the Xenopus extract contained 6 μl of treated or untreated NPE (immunodepletion and/or addition of recombinant proteins) supplemented with an ATP regenerating system (2 mm ATP, 20 mm phosphocreatine, and 5 μg/ml creatine phosphokinase) and 1.5 μl of radiolabeled DNA substrate (2 ng/μl). In a typical in vitro resection reaction, 30 nm Exo1-His (or as indicated otherwise) and 2 ng/μl radiolabeled DNA substrate were incubated in the reaction buffer (20 mm Hepes (pH 7.5), 50 mm KCl, 0.5 mm DTT, 5 mm MgCl2, and 5% glycerol) at room temperature. Following incubation, 1.5-μl reactions were stopped at the indicated times by incubation with 10 μl of stop buffer (8 mm EDTA, 0.13% phosphoric acid, 10% Ficoll, 0.2% bromphenol blue, 0.5% SDS, and 80 mm Tris-HCl (pH 8.0)) supplemented with proteinase K (2 μg/μl) for 2 h at 37 °C. Samples were then resolved on 0.8% TAE-agarose gels overnight, followed by gel drying and autoradiography as described previously (33).
RESULTS
The Central Domain of Exo1 Negatively Regulates Association with Sites of DNA Damage
In response to DSBs, Exo1 is recruited to damage sites to resect the DNA ends and generate ssDNA that promotes the activation of HR and the ATR checkpoint (5). To determine how Exo1 damage recruitment is controlled in cells, we transfected human U2OS cells with wild-type or truncated Exo1 fused to GFP. A laser microirradiation method was used to introduce DNA damage in transfected cells, followed by live cell imaging of the recruitment of GFP-Exo1 fusion proteins to the sites of DNA damage. Using this approach, we previously identified a PCNA-interacting protein box in the C terminus of Exo1 (amino acids 750–846) that promotes the stable association of Exo1 with damaged DNA (33). Correspondingly, the GFP-Exo1(1–750) protein lacking the PCNA-interacting protein box exhibited a very low level of damage association compared with the GFP-Exo1 full-length protein (Fig. 1) (33). Interestingly, further deletion of the central region spanning Exo1 residues 508–750 partially restored the damage association activity. As shown in Fig. 1, the truncated GFP-Exo1(1–507) mutant was rapidly and efficiently recruited to damage, with a peak signal even stronger than that of GFP-Exo1(WT). However, association of GFP-Exo1(1–507) with damage was very transient in comparison with GFP-Exo1(WT). An Exo1 fragment containing both the central region and the C-terminal PCNA-interacting domain (Exo1(508–846)) also exhibited much reduced damage association compared with the C-terminal domain alone (Exo1(751–846)) (Fig. 1). Although the central region (CR) itself did not localize to damage, a deletion of this region from the full-length Exo1 (Exo1(ΔCR)) protein dramatically increased the extent and duration of the association of Exo1 with DNA damage (compare the damage association of GFP-Exo1(WT) and GFP-Exo1(ΔCR) in Fig. 1). Taken together, these results indicate that the central region of Exo1 is a functionally independent domain that negatively regulates the association of Exo1 with DNA damage and that the central region limits Exo1 damage association, apparently by suppressing the function of both the N-terminal and C-terminal domains. In addition to damage recruitment, deletion of the central region in Exo1 dramatically increases its nucleolar localization (compare GFP-Exo1(WT) with GFP-Exo1(ΔCR) in Fig. 1), although the functional significance of this observation remains to be determined.
FIGURE 1.

The central domain of Exo1 limits its association with DNA damage. A, left panel, wild-type Exo1 and the truncation mutants used in this study. Right panel, representative images showing the association of GFP-tagged Exo1 and deletion mutants with microirradiation-induced damage. Red lines indicate the sites of laser irradiation in cells. B, the time course for damage association by GFP-Exo1 and truncation mutants is shown during the first 20 min after laser irradiation. Each data point is the average of independent measurements from five cells. Error bars represent mean ± S.D.
14-3-3 Proteins Bind to the Central Region of Exo1
The central domain of Exo1 might, in turn, be regulated by protein factors that bind to this domain and control Exo1 interactions with damaged DNA. The 14-3-3 family of dimeric proteins are attractive candidates that regulate a diverse array of cellular processes, including DNA damage response (37, 38). It has been reported recently that EXO1 in budding yeast interacts with two 14-3-3 proteins and that these 14-3-3 proteins suppress the function of EXO1 in processing replication intermediates at stalled forks (32). In the absence of 14-3-3s, extensive ssDNA structures accumulate behind replication forks, consistent with Exo1 activity, although this remains to be confirmed (32). Human Exo1 interacts with at least six of the seven 14-3-3 human paralogs in a yeast two-hybrid assay. However, the functional significance of this interaction in DSB resection has not been determined (31, 32).
To determine whether the central domain of Exo1 interacts with 14-3-3 proteins in human cells, we performed coimmunoprecipitation experiments in cells expressing FLAG-tagged Exo1(WT), Exo1(ΔCR), or Exo1(CR) in HEK293T cells using an anti-FLAG antibody. As shown in Fig. 2A, wild-type Exo1 associated with multiple isoforms of endogenous 14-3-3 proteins (detected with a pan-14-3-3 antibody) (31, 32). The central region of Exo1 expressed in human cells was also associated with 14-3-3s, whereas deletion of the central region completely abrogated the interaction of Exo1 with 14-3-3s (Fig. 2A). Together, these data indicate that the central domain of Exo1 is both necessary and sufficient for the physical interaction of Exo1 with 14-3-3s.
Association with 14-3-3 Proteins Limits Exo1 Damage Recruitment
We next asked whether the interaction with 14-3-3 proteins restrains Exo1 damage association. Directly assessing the role of 14-3-3s in Exo1 damage association using knockdown or knockout approaches is very challenging because Exo1 interacts with at least six of the seven 14-3-3 proteins encoded by different genes in human cells, many of which are functionally redundant (Fig. 2A) (31, 32, 39). To circumvent this problem, we used Difopein, a potent peptide antagonist that specifically blocks the binding of all 14-3-3 isoforms to their target proteins (40, 41). This antagonist contains two tandem Arg-18 peptide repeats that are connected by a flexible linker. The 20-mer Arg-18 was isolated as a non-phosphorylated peptide that binds and inhibits 14-3-3s (42). Difopein is believed to function as an inhibitor by blocking two independent protein binding sites in the 14-3-3 dimer (40). As a control, we generated a mutant form of Difopein with two key residues in each of the Arg-18 repeat (Asp-12 and Glu-14) mutated into lysine, which abolishes the interaction between Arg-18 and 14-3-3s (40). To determine whether Difopein can indeed block the interaction of Exo1 with 14-3-3 proteins, we transfected mCherry, mCherry-Difopein(WT), or mCherry-Difopein(MUT) into human cells expressing GFP-Exo1(WT), followed by coimmunoprecipitation using a GFP antibody. As shown in Fig. 2B, overexpression of mCherry-Difopein(WT), but not mCherry-Difopein(MUT) or mCherry alone, completely abrogated the association of GFP-Exo1(WT) with 14-3-3s.
To test whether the association of 14-3-3s with Exo1 regulates its recruitment to damage, we examined the effects of overexpression of mCherry-Difopein(WT), mCherry-Difopein(MUT), or mCherry on the damage association of GFP-Exo1(WT) in human cells after laser irradiation. Overexpression of mCherry-Difopein(WT) indeed drastically enhanced the damage association of GFP-Exo1(WT) compared with mCherry-Difopein(MUT) and mCherry alone (Fig. 2C, top panel). In contrast, mCherry-Difopein(WT) did not affect the damage association of GFP-Exo1(ΔCR), which lacks the ability to bind to 14-3-3s (Fig. 2, A and B and C, bottom panel). These data strongly suggest that 14-3-3 proteins limit Exo1 damage association by binding to the central region of Exo1.
14-3-3s Suppress Exo1 Binding to PCNA
Because the Exo1 central domain suppresses the recruitment of its C-terminal domain to sites of damage (Fig. 1), it is possible that 14-3-3s interfere with Exo1 binding to PCNA as the primary mechanism of regulating damage association. In support of this idea, we found that FLAG-Exo1(ΔCR) exhibited a much higher level of PCNA-binding activity compared with FLAG-Exo1(WT) (Fig. 3A). Moreover, overexpression of mCherry-Difopein(WT), but not mCherry-Difopein(MUT) or mCherry alone, substantially increased the PCNA binding of FLAG-Exo1(WT) (Fig. 3B). In contrast, mCherry-Difopein(WT) overexpression did not significantly affect the PCNA binding activity of FLAG-Exo1(ΔCR), which lacks the 14-3-3 interaction site (Fig. 3B). These results demonstrate that 14-3-3s restrain the damage recruitment of Exo1 by suppressing its interaction with PCNA. The central domain of Exo1 also suppresses the damage recruitment of Exo1(1–507) (Fig. 1), suggesting that 14-3-3s may also counteract the damage localization function of the Exo1 N-terminal domain by another mechanism.
The Central Region of Exo1 Restrains DSB Resection Activity
The suppression of Exo1 damage localization by its central domain (Fig. 1 and 2) could down-regulate DSB resection activity. To test this, we used a cell-free NPE system derived from Xenopus eggs that faithfully recapitulates the DNA damage response in cells (43, 44). A 3′ 32P-labeled, 6-kb dsDNA fragment was used as a substrate for DNA end resection. As shown previously, DNA end resection occurs on this dsDNA fragment in Xenopus NPE in the 5′-to-3′ direction under proper control and can be visualized on agarose gels (33, 45). As in cells, bulk DNA end resection in Xenopus NPE is carried out by the Exo1 (xExo1) and Dna2 (xDna2) nucleases acting in two parallel pathways (16, 18, 46, 47). Correspondingly, immunodepletion of either xExo1 or xDna2 from NPE partially abolishes DNA end resection (33, 47).
To investigate whether the central domain of Exo1 regulates resection activity in vitro, we first generated recombinant Exo1(WT)-His or Exo1(ΔCR)-His proteins using a baculovirus expression system (Fig. 4A). These proteins exhibited the same level of exonuclease activity in vitro, which could be stimulated by PCNA to a similar extent, suggesting that the central domain does not regulate the intrinsic nuclease activity of Exo1 (Fig. 4, B and C). The corresponding catalytically inactive mutants of these proteins, Exo1(D173A)-His and Exo1(ΔCR-D173A)-His, purified under the same conditions, did not exhibit nuclease activity, authenticating the source of the nuclease activity in reactions containing Exo1(WT)-His or Exo1(ΔCR)-His (Fig. 4D).
FIGURE 4.

Interaction of 14-3-3s with the central domain of Exo1 does not affect the exonuclease activity of Exo1 in vitro. A, recombinant Exo1(WT)-His, Exo1(D173A)-His, Exo1(ΔCR)-His, and Exo1(ΔCR-D173A)-His proteins were purified from overexpressing Sf9 cells. MW, molecular weight. B, comparison of the exonuclease activities of purified recombinant Exo1(WT)-His and Exo1(ΔCR)-His in vitro toward a 3′ 32P-labeled, 6-kb dsDNA fragment. The reactions included 30 nm of Exo1(WT)-His or Exo1(ΔCR)-His incubated with 2 ng/μl of the DNA substrate. C, PCNA stimulated the resection activity of Exo1(WT)-His and Exo1(ΔCR)-His to a similar extent in vitro using the same reaction conditions as in B. D, in vitro DNA resection activities of Exo1(WT)-His and the mutant proteins shown under the reaction conditions described in B.
Next, we codepleted xExo1 and xDna2 from NPE and then added an equimolar amount of recombinant Exo1(WT)-His or Exo1(ΔCR)-His proteins to the depleted extract (Fig. 5A). Consistent with the results obtained in human cells, more of the Exo1(ΔCR)-His protein was associated with a bead-immobilized DNA fragment compared with Exo1(WT)-His (Fig. 5B). Correspondingly, a much higher level of DNA resection was observed for Exo1(ΔCR)-His compared with Exo1(WT)-His in reactions using a radiolabeled DNA substrate added to the double-depleted extracts (Fig. 5C). These results directly demonstrate that the central domain of Exo1 limits DSB resection activity by restraining the association of Exo1 with damaged DNA.
FIGURE 5.

The central region of Exo1 limits DNA end resection activity in a Xenopus egg extract. A, immunodepletion (depl) of the Xenopus Dna2 protein (xDna2) or of both xDna2 and xExo1 from the Xenopus NPE. B, physical association of Exo1(WT)-His and Exo1(ΔCR)-His with a bead-immobilized, 2-kb DNA fragment in xExo1-depleted NPE. C, DNA end resection activity toward a 3′ 32P-labeled, 6-kb dsDNA fragment in the Exo1 and Dna2 codepleted NPE depicted in A that was supplemented with Exo1(WT)-His or Exo1(ΔCR)-His. Vertical lines were added for sample lane identification.
14-3-3 Proteins Limit Exo1-mediated DNA End Resection
We next asked whether 14-3-3s mediate the inhibitory effect of the Exo1 central domain on DNA resection activity. To address this question, we first examined DNA end resection in the Xenopus NPE supplemented with purified recombinant GST-Difopein(WT), GST-Difopein(MUT), or GST (Fig. 6A). Consistent with the results obtained in human cells (Fig. 2B), GST-Difopein(WT), but not GST-Difopein(MUT) or GST alone, added to the Xenopus extract blocked the interaction between xExo1 and 14-3-3s (Fig. 6B). Consequently, GST-Difopein(WT) increased DNA end resection activity in NPE, especially at the 20-min time point compared with reactions supplemented with GST-Difopein(MUT) or GST alone. These results indicate that 14-3-3s indeed negatively regulate DNA end resection in this cell-free system (Fig. 6C).
Next we determined whether the Exo1 pathway underlies the observed effects of the 14-3-3 inhibitor Difopein on DNA end resection in the Xenopus extract. xExo1 was immunodepleted from the Xenopus NPE, and GST-Difopein(WT) or GST-Difopein(MUT) was added before measuring DNA end resection activity. In the mock-depleted extract, GST-Difopein(WT) enhanced DNA resection compared with GST-Difopein(MUT), whereas GST-Difopein(WT) had no effect on resection activity in the xExo1-depleted extract (Fig. 6D). In contrast, depletion of Dna2 from the extract did not interfere with GST-Difopein(WT) stimulation of DNA end resection (Fig. 6D). These results indicate that 14-3-3s selectively regulate Exo1-mediated DNA resection without affecting the Dna2 resection pathway. In further support of this idea, Dna2 was not detectably bound by 14-3-3s (data not shown). Taken together, these data strongly suggest that Exo1 resection activity is selectively repressed by 14-3-3s.
To further examine the mechanism by which 14-3-3 proteins negatively regulate Exo1-mediated DNA resection, we determined whether a physical interaction between 14-3-3s and the central domain of Exo1 is required for this regulation. To this end, we immunodepleted both xExo1 and xDna2 from the Xenopus extract and then added recombinant Exo1(WT)-His or Exo1(ΔCR)-His to the depleted extract. Subsequently, GST-Difopein(WT) or GST-Difopein(MUT) was added to the extracts, and the resection of a radiolabeled DNA fragment was monitored. As before, the addition of GST-Difopein(WT), but not GST-Difopein(MUT), dramatically increased DNA resection in the extract containing Exo1(WT)-His (Fig. 6E). Resection activity was unaffected by GST-Difopein(WT) in the extract containing Exo1(ΔCR)-His (Fig. 6E). These results show that interaction of 14-3-3s with the central domain of Exo1 functions to restrain the extent of Exo1-mediated DNA resection.
Regulation of Exo1 by 14-3-3s Is Important for Cell Survival after DNA Damage
Uncontrolled, excessive resection of DSBs may decrease cell viability. Therefore, we determined whether disruption of the repressive interaction between Exo1 and 14-3-3s would sensitize cells to DNA damage. To this end, we overexpressed mCherry-Difopein(WT) or mCherry-Difopein(MUT) stably in control knockdown or Exo1 knockdown cells (Fig. 7A). Cells were then treated with camptothecin, a topoisomerase I inhibitor that induces DSBs mainly in S phase (48). As shown in Fig. 7B, cells overexpressing mCherry-Difopein(WT) exhibited increased levels of RPA phosphorylation at Ser-4/Ser-8 (a marker of DNA resection) after camptothecin treatment compared with cells overexpressing mCherry-Difopein(MUT). This result is consistent with the idea that a disruption of 14-3-3 function by Difopein increases DNA resection (Figs. 6 and 7B) (49, 50). Depletion of Exo1 abolished the hyperphosphorylation of RPA caused by Difopein expression (Fig. 7B). To determine whether functional disruption of 14-3-3s affects cell viability after DNA damage, we next performed a clonogenic assay to measure cell survival after DNA damage. Indeed, cells overexpressing mCherry-Difopein(WT) were more sensitive to camptothecin than cells expressing mCherry-Difopein(MUT) (compare a and d in Fig. 7C). Importantly, this hypersensitivity was largely abrogated when Exo1 was depleted from cells (compare e and f with d in Fig. 7C). These results collectively suggest that negative regulation of Exo1 resection activity by 14-3-3s is important for cell survival after DNA damage.
FIGURE 7.

The interaction of Exo1 with 14-3-3s promotes cell survival after DNA damage. A, protein levels of Exo1, mCherry-Difopein, and PCNA in control knockdown or Exo1 knockdown U2OS cells expressing mCherry-Difopein(WT) or mCherry-Difopein(MUT). B, RPA phosphorylation at Ser-4/Ser-8 in control knockdown or Exo1 knockdown cells expressing either mCherry-Difopein(WT) or mCherry-Difopein(MUT) after treatment with dimethyl sulfoxide (DMSO), 0.2 μm camptothecin (CPT), or 1 μm CPT for 3 h, followed by a 3-h recovery period. C, results of a clonogenic survival assay for control knockdown and Exo1 knockdown cells expressing either mCherry-Difopein(WT) or mCherry-Difopein(MUT). Cells were treated with 1 or 0.2 μm camptothecin for 3 h, followed by a 3-h recovery period. Data represent the mean ± S.D. of three independent experiments. *, p < 0.05; **, p < 0.01 (paired Student's t test). D, a model for the regulation of Exo1 in DNA end resection. The association of Exo1 with DNA damage is controlled by Exo1 protein-protein interactions with PCNA and 14-3-3 proteins. PCNA associates with the PCNA-interacting protein box in the C-terminal domain of Exo1 to promote retention at sites of DNA damage and processive resection of DNA breaks. 14-3-3 proteins interact with the central domain of Exo1 and suppress the binding of PCNA to Exo1, thereby limiting the association of Exo1 with DNA damage. The coordinate regulation of Exo1 by the opposing activities of PCNA and 14-3-3s ensures a proper level of DSB resection by Exo1.
DISCUSSION
This study has uncovered key elements of a mechanism for controlling Exo1-mediated DNA resection, an activity that is required for both DNA repair and checkpoint activation after DSB damage (Fig. 7D). Our data indicate that Exo1 function in DNA resection is regulated both positively and negatively through protein-protein interactions. Although association with PCNA promotes Exo1 damage retention and its processivity in resection, interaction with 14-3-3 proteins represses the association of Exo1 with damaged DNA and resection activity (Figs. 1, 2, and 4–6) (33). The mechanism by which 14-3-3s restrain Exo1-mediated DNA resection is achieved, at least in part, by interfering with Exo1 binding to PCNA (Fig. 3). This coordinated regulation ensures efficient, but limited, DNA end resection that is crucial for genome maintenance and cell survival after DNA damage (Fig. 7).
We have shown previously that, after DSB damage, the ring-shaped DNA sliding clamp PCNA loads onto DNA ends and binds directly to the PCNA-interacting protein box in the C terminus of Exo1, tethering Exo1 to damaged DNA and, thereby, increasing its processivity during resection (33). This positive regulation ensures the efficient resection that is required for the timely activation of HR and the ATR checkpoint. Although insufficient resection hinders HR and ATR activation, overresection can have several undesirable consequences. Excessive ssDNA generated by hyperresection may exhaust the pool of RPA protein. Exposure of the unprotected 3′ ssDNA may result in further damage, leading to genomic instability with a possible loss of genetic information (19, 51). The combined activities of Exo1 and Dna2 have the potential to cause extensive resection of DNA that must be effectively controlled to promote efficient repair with minimal DNA sequence loss. The results described in this study revealed that 14-3-3 proteins play a key role in limiting DNA resection by Exo1. Through direct binding to the central domain of Exo1, 14-3-3s restrain Exo1 damage association and subsequent DNA resection (Figs. 1, 2, 5, and 6). Mechanistically, we found that 14-3-3s limit Exo1 damage recruitment and resection by suppressing Exo1 binding to PCNA (Fig. 3). Reduction in PCNA binding may be achieved directly through conformation changes in Exo1 or indirectly through Exo1 modification or interaction with other protein factors upon 14-3-3 binding. Because 14-3-3 proteins do not localize to sites of DNA damage (data not shown) and because 14-3-3η (which interacts with Exo1 (31)) did not affect the nuclease activity of recombinant Exo1 in vitro (data not shown), we suggest that 14-3-3s do not modulate the nuclease activity of Exo1 at the sites of DNA damage but, instead, decrease the recruitment and retention of Exo1 at sites of DNA damage.
Our finding of the negative regulation of Exo1 resection by 14-3-3s is consistent with a recent genetic study by Engels et al. (32) in budding yeast, which shows that Exo1-mediated processing of stalled replication forks is suppressed by 14-3-3 proteins in this organism. Deletion of the two 14-3-3 genes in yeast results in an Exo1-dependent accumulation of ssDNA gaps behind replication forks, suggesting that Exo1 resection activity is also kept in check by 14-3-3s during replication (32). Although the general role of Exo1 in DNA resection is conserved in yeast and vertebrates, there are significant organismal differences in Exo1 regulation. For example, the 14-3-3-binding region of human Exo1 is not conserved in the budding yeast Exo1 protein. Moreover, although PCNA directly binds Exo1 and promotes Exo1 damage association and resection in vertebrates, no PCNA-binding motifs have been identified in yeast Exo1 (33, 52). Interestingly, yeast and human Exo1 both are regulated by another DNA clamp, the 9-1-1 complex, by an unknown mechanism (52, 53). Many other candidate regulators of Exo1 function in DSB resection have been identified, including MRN, CtIP, Ku, RPA, SOSS1, BLM, SWR1, ATM, ATR, Rad53, and CDK, in human cells or in yeast, either directly or indirectly (28–32, 54–67). The identification in this study of the key functional domains in Exo1 and their direct binding partners has provided a mechanistic framework for understanding the coordination of these Exo1 regulators in controlling DNA resection. In contrast to Exo1, we obtained no evidence that the Dna2 pathway is regulated by 14-3-3s (Fig. 6). Recent studies indicate that Dna2 resection activity is instead suppressed by FANCD2 in human cells, RAD9 in budding yeast, and Pxd1 in fission yeast, either directly or indirectly (68–70). It remains to be determined how these two pathways are coordinately regulated to ensure the proper level of DNA resection during DSB repair.
The strict regulation of cellular nucleases is crucial for accurate DNA repair and cell survival (71). Our results demonstrate that regulation of Exo1 resection by 14-3-3s is important for cell survival after DNA damage (Fig. 7C). The decreased cell viability in the absence of 14-3-3-mediated regulation of Exo1 may result from toxic aberrant repair products following uncontrolled DNA end resection or from decoupling of homology-mediated repair from DNA resection (Figs. 6 and 7). Previous studies in yeast and mice indicate that deletion of Exo1 can rescue various phenotypes that result from functional disruption of the checkpoint kinase Rad53 or telomerase, such as DNA damage hypersensitivity, chromosomal instability, and aging (25–30). It will be important to determine whether uncontrolled Exo1 resection contributes to these phenotypes and whether 14-3-3 regulation plays a role in these contexts. Our results also suggest that the DNA end resection process can be exploited for cancer treatment. Given the replication stress and genomic instability in many cancers, it can be envisioned that small molecule inhibitors that specifically target the interaction of 14-3-3 proteins with Exo1 might be effective as chemosensitizers for cancer therapy.
Acknowledgments
We thank Hong Yan, Sheila Stewart, and Andrey Shaw for Xenopus Dna2, human Dna2, and human 14-3-3 expression constructs, respectively.
This work was supported, in whole or in part, by National Institutes of Health Grants R01GM098535 (to Z. Y.) and P01CA92584 (to T. E.). This work was also supported by American Cancer Society Research Scholar Grant RSG-13-212-01-DMC (to Z. Y.) and a Washington University Siteman Career Award in Breast Cancer Research (to Z. Y.).
- DSB
- double strand break
- ssDNA
- single-stranded DNA
- PCNA
- proliferating cell nuclear antigen
- HR
- homologous recombination
- NPE
- nucleoplasmic protein extract
- CR
- central region
- ATM
- ataxia telangiectasia mutated
- ATR
- ataxia telangiectasia and Rad3 related
- MRN
- Mre11-Rad50-NBS1
- MRX
- Mre11-Rad50-Xrs2
- RPA
- replication factor A
- BLM
- Bloom syndrome, RecQ helicase-like
- CDK
- cyclin-dependent kinase.
REFERENCES
- 1. Hoeijmakers J. H. (2009) DNA damage, aging, and cancer. N. Engl. J. Med. 361, 1475–1485 [DOI] [PubMed] [Google Scholar]
- 2. Lord C. J., Ashworth A. (2012) The DNA damage response and cancer therapy. Nature 481, 287–294 [DOI] [PubMed] [Google Scholar]
- 3. Ciccia A., Elledge S. J. (2010) The DNA damage response: making it safe to play with knives. Mol. Cell 40, 179–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jackson S. P., Bartek J. (2009) The DNA-damage response in human biology and disease. Nature 461, 1071–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Symington L. S., Gautier J. (2011) Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 45, 247–271 [DOI] [PubMed] [Google Scholar]
- 6. Lieber M. R. (2010) The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 79, 181–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Moynahan M. E., Jasin M. (2010) Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 11, 196–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Halazonetis T. D., Gorgoulis V. G., Bartek J. (2008) An oncogene-induced DNA damage model for cancer development. Science 319, 1352–1355 [DOI] [PubMed] [Google Scholar]
- 9. Stracker T. H., Roig I., Knobel P. A., Marjanović M. (2013) The ATM signaling network in development and disease. Front. Genet. 4, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cimprich K. A., Cortez D. (2008) ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 9, 616–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shiloh Y., Ziv Y. (2013) The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 14, 197–210 [PubMed] [Google Scholar]
- 12. Shiotani B., Zou L. (2009) Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol. Cell 33, 547–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. You Z., Shi L. Z., Zhu Q., Wu P., Zhang Y. W., Basilio A., Tonnu N., Verma I. M., Berns M. W., Hunter T. (2009) CtIP links DNA double-strand break sensing to resection. Mol. Cell 36, 954–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zou L., Elledge S. J. (2003) Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300, 1542–1548 [DOI] [PubMed] [Google Scholar]
- 15. Huertas P. (2010) DNA resection in eukaryotes: deciding how to fix the break. Nat. Struct. Mol. Biol. 17, 11–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mimitou E. P., Symington L. S. (2008) Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455, 770–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mimitou E. P., Symington L. S. (2009) DNA end resection: many nucleases make light work. DNA Repair 8, 983–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhu Z., Chung W. H., Shim E. Y., Lee S. E., Ira G. (2008) Sgs1 helicase and two nucleases dna2 and exo1 resect DNA double-strand break ends. Cell 134, 981–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen H., Lisby M., Symington L. S. (2013) RPA coordinates DNA end resection and prevents formation of DNA hairpins. Mol. Cell 50, 589–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rossiello F., Herbig U., Longhese M. P., Fumagalli M., d'Adda di Fagagna F. (2014) Irreparable telomeric DNA damage and persistent DDR signalling as a shared causative mechanism of cellular senescence and ageing. Curr. Opin. Genet. Dev. 26, 89–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bardwell P. D., Woo C. J., Wei K., Li Z., Martin A., Sack S. Z., Parris T., Edelmann W., Scharff M. D. (2004) Altered somatic hypermutation and reduced class-switch recombination in exonuclease 1-mutant mice. Nat. Immunol. 5, 224–229 [DOI] [PubMed] [Google Scholar]
- 22. Tran P. T., Erdeniz N., Symington L. S., Liskay R. M. (2004) EXO1-A multi-tasking eukaryotic nuclease. DNA Repair 3, 1549–1559 [DOI] [PubMed] [Google Scholar]
- 23. Schaetzlein S., Chahwan R., Avdievich E., Roa S., Wei K., Eoff R. L., Sellers R. S., Clark A. B., Kunkel T. A., Scharff M. D., Edelmann W. (2013) Mammalian Exo1 encodes both structural and catalytic functions that play distinct roles in essential biological processes. Proc. Natl. Acad. Sci. U.S.A. 110, E2470–E2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wei K., Clark A. B., Wong E., Kane M. F., Mazur D. J., Parris T., Kolas N. K., Russell R., Hou H., Jr., Kneitz B., Yang G., Kunkel T. A., Kolodner R. D., Cohen P. E., Edelmann W. (2003) Inactivation of Exonuclease 1 in mice results in DNA mismatch repair defects, increased cancer susceptibility, and male and female sterility. Genes Dev. 17, 603–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bertuch A. A., Lundblad V. (2004) EXO1 contributes to telomere maintenance in both telomerase-proficient and telomerase-deficient Saccharomyces cerevisiae. Genetics 166, 1651–1659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maringele L., Lydall D. (2004) Telomerase- and recombination-independent immortalization of budding yeast. Genes Dev. 18, 2663–2675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schaetzlein S., Kodandaramireddy N. R., Ju Z., Lechel A., Stepczynska A., Lilli D. R., Clark A. B., Rudolph C., Kuhnel F., Wei K., Schlegelberger B., Schirmacher P., Kunkel T. A., Greenberg R. A., Edelmann W., Rudolph K. L. (2007) Exonuclease-1 deletion impairs DNA damage signaling and prolongs lifespan of telomere-dysfunctional mice. Cell 130, 863–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cotta-Ramusino C., Fachinetti D., Lucca C., Doksani Y., Lopes M., Sogo J., Foiani M. (2005) Exo1 processes stalled replication forks and counteracts fork reversal in checkpoint-defective cells. Mol. Cell 17, 153–159 [DOI] [PubMed] [Google Scholar]
- 29. Morin I., Ngo H. P., Greenall A., Zubko M. K., Morrice N., Lydall D. (2008) Checkpoint-dependent phosphorylation of Exo1 modulates the DNA damage response. EMBO J. 27, 2400–2410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Segurado M., Diffley J. F. (2008) Separate roles for the DNA damage checkpoint protein kinases in stabilizing DNA replication forks. Genes Dev. 22, 1816–1827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Andersen S. D., Keijzers G., Rampakakis E., Engels K., Luhn P., El-Shemerly M., Nielsen F. C., Du Y., May A., Bohr V. A., Ferrari S., Zannis-Hadjopoulos M., Fu H., Rasmussen L. J. (2012) 14-3-3 checkpoint regulatory proteins interact specifically with DNA repair protein human exonuclease 1 (hEXO1) via a semi-conserved motif. DNA Repair 11, 267–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Engels K., Giannattasio M., Muzi-Falconi M., Lopes M., Ferrari S. (2011) 14-3-3 Proteins regulate exonuclease 1-dependent processing of stalled replication forks. PLoS Genet. 7, e1001367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen X., Paudyal S. C., Chin R. I., You Z. (2013) PCNA promotes processive DNA end resection by Exo1. Nucleic Acids Res. 41, 9325–9338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim I. K., Kiefer J. R., Ho C. M., Stegeman R. A., Classen S., Tainer J. A., Ellenberger T. (2012) Structure of mammalian poly(ADP-ribose) glycohydrolase reveals a flexible tyrosine clasp as a substrate-binding element. Nat. Struct. Mol. Biol. 19, 653–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pei H., Zhang L., Luo K., Qin Y., Chesi M., Fei F., Bergsagel P. L., Wang L., You Z., Lou Z. (2011) MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature 470, 124–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Walter J., Sun L., Newport J. (1998) Regulated chromosomal DNA replication in the absence of a nucleus. Mol. Cell 1, 519–529 [DOI] [PubMed] [Google Scholar]
- 37. Peng C. Y., Graves P. R., Thoma R. S., Wu Z., Shaw A. S., Piwnica-Worms H. (1997) Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science 277, 1501–1505 [DOI] [PubMed] [Google Scholar]
- 38. Reinhardt H. C., Yaffe M. B. (2013) Phospho-Ser/Thr-binding domains: navigating the cell cycle and DNA damage response. Nat. Rev. Mol. Cell Biol. 14, 563–580 [DOI] [PubMed] [Google Scholar]
- 39. Morrison D. K. (2009) The 14-3-3 proteins: integrators of diverse signaling cues that impact cell fate and cancer development. TiCB 19, 16–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Masters S. C., Fu H. (2001) 14-3-3 proteins mediate an essential anti-apoptotic signal. J. Biol. Chem. 276, 45193–45200 [DOI] [PubMed] [Google Scholar]
- 41. Xu Z., Fulop Z., Wu G., Pone E. J., Zhang J., Mai T., Thomas L. M., Al-Qahtani A., White C. A., Park S. R., Steinacker P., Li Z., Yates J., 3rd, Herron B., Otto M., Zan H., Fu H., Casali P. (2010) 14-3-3 adaptor proteins recruit AID to 5′-AGCT-3′-rich switch regions for class switch recombination. Nat. Struct. Mol. Biol. 17, 1124–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang B., Yang H., Liu Y. C., Jelinek T., Zhang L., Ruoslahti E., Fu H. (1999) Isolation of high-affinity peptide antagonists of 14-3-3 proteins by phage display. Biochemistry 38, 12499–12504 [DOI] [PubMed] [Google Scholar]
- 43. Costanzo V., Gautier J. (2004) Xenopus cell-free extracts to study DNA damage checkpoints. Methods Mol. Biol. 241, 255–267 [DOI] [PubMed] [Google Scholar]
- 44. Knipscheer P., Räschle M., Schärer O. D., Walter J. C. (2012) Replication-coupled DNA interstrand cross-link repair in Xenopus egg extracts. Methods Mol. Biol. 920, 221–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liao S., Toczylowski T., Yan H. (2008) Identification of the Xenopus DNA2 protein as a major nuclease for the 5′->3′ strand-specific processing of DNA ends. Nucleic Acids Res. 36, 6091–6100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gravel S., Chapman J. R., Magill C., Jackson S. P. (2008) DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 22, 2767–2772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liao S., Toczylowski T., Yan H. (2011) Mechanistic analysis of Xenopus EXO1's function in 5′-strand resection at DNA double-strand breaks. Nucleic Acids Res. 39, 5967–5977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pommier Y. (2006) Topoisomerase I inhibitors: camptothecins and beyond. Nat. Rev. Cancer 6, 789–802 [DOI] [PubMed] [Google Scholar]
- 49. Sartori A. A., Lukas C., Coates J., Mistrik M., Fu S., Bartek J., Baer R., Lukas J., Jackson S. P. (2007) Human CtIP promotes DNA end resection. Nature 450, 509–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shiotani B., Nguyen H. D., Håkansson P., Maréchal A., Tse A., Tahara H., Zou L. (2013) Two distinct modes of ATR activation orchestrated by Rad17 and Nbs1. Cell Rep. 3, 1651–1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Toledo L. I., Altmeyer M., Rask M. B., Lukas C., Larsen D. H., Povlsen L. K., Bekker-Jensen S., Mailand N., Bartek J., Lukas J. (2013) ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155, 1088–1103 [DOI] [PubMed] [Google Scholar]
- 52. Ngo G. H., Balakrishnan L., Dubarry M., Campbell J. L., Lydall D. (2014) The 9-1-1 checkpoint clamp stimulates DNA resection by Dna2-Sgs1 and Exo1. Nucleic Acids Res. 42, 10516–10528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Karras G. I., Fumasoni M., Sienski G., Vanoli F., Branzei D., Jentsch S. (2013) Noncanonical role of the 9-1-1 clamp in the error-free DNA damage tolerance pathway. Mol. Cell 49, 536–546 [DOI] [PubMed] [Google Scholar]
- 54. Adkins N. L., Niu H., Sung P., Peterson C. L. (2013) Nucleosome dynamics regulates DNA processing. Nat. Struct. Mol. Biol. 20, 836–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bolderson E., Tomimatsu N., Richard D. J., Boucher D., Kumar R., Pandita T. K., Burma S., Khanna K. K. (2010) Phosphorylation of Exo1 modulates homologous recombination repair of DNA double-strand breaks. Nucleic Acids Res. 38, 1821–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cannavo E., Cejka P., Kowalczykowski S. C. (2013) Relationship of DNA degradation by Saccharomyces cerevisiae Exonuclease 1 and its stimulation by RPA and Mre11-Rad50-Xrs2 to DNA end resection. Proc. Natl. Acad. Sci. U.S.A. 110, E1661–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Eid W., Steger M., El-Shemerly M., Ferretti L. P., Peña-Diaz J., König C., Valtorta E., Sartori A. A., Ferrari S. (2010) DNA end resection by CtIP and exonuclease 1 prevents genomic instability. EMBO Rep. 11, 962–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. El-Shemerly M., Hess D., Pyakurel A. K., Moselhy S., Ferrari S. (2008) ATR-dependent pathways control hEXO1 stability in response to stalled forks. Nucleic Acids Res. 36, 511–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Langerak P., Mejia-Ramirez E., Limbo O., Russell P. (2011) Release of Ku and MRN from DNA ends by Mre11 nuclease activity and Ctp1 is required for homologous recombination repair of double-strand breaks. PLoS Genet. 7, e1002271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mimitou E. P., Symington L. S. (2010) Ku prevents Exo1 and Sgs1-dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO J. 29, 3358–3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nicolette M. L., Lee K., Guo Z., Rani M., Chow J. M., Lee S. E., Paull T. T. (2010) Mre11-Rad50-Xrs2 and Sae2 promote 5′ strand resection of DNA double-strand breaks. Nat. Struct. Mol. Biol. 17, 1478–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nimonkar A. V., Genschel J., Kinoshita E., Polaczek P., Campbell J. L., Wyman C., Modrich P., Kowalczykowski S. C. (2011) BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 25, 350–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nimonkar A. V., Ozsoy A. Z., Genschel J., Modrich P., Kowalczykowski S. C. (2008) Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc. Natl. Acad. Sci. U.S.A. 105, 16906–16911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shim E. Y., Chung W. H., Nicolette M. L., Zhang Y., Davis M., Zhu Z., Paull T. T., Ira G., Lee S. E. (2010) Saccharomyces cerevisiae Mre11/Rad50/Xrs2 and Ku proteins regulate association of Exo1 and Dna2 with DNA breaks. EMBO J. 29, 3370–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sun J., Lee K. J., Davis A. J., Chen D. J. (2012) Human Ku70/80 protein blocks exonuclease 1-mediated DNA resection in the presence of human Mre11 or Mre11/Rad50 protein complex. J. Biol. Chem. 287, 4936–4945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tomimatsu N., Mukherjee B., Catherine Hardebeck M., Ilcheva M., Vanessa Camacho C., Louise Harris J., Porteus M., Llorente B., Khanna K. K., Burma S. (2014) Phosphorylation of EXO1 by CDKs 1 and 2 regulates DNA end resection and repair pathway choice. Nat. Commun. 5, 3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yang S. H., Zhou R., Campbell J., Chen J., Ha T., Paull T. T. (2013) The SOSS1 single-stranded DNA binding complex promotes DNA end resection in concert with Exo1. EMBO J. 32, 126–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bonetti D., Villa M., Gobbini E., Cassani C., Tedeschi G., Longhese M. P. (2015) Escape of Sgs1 from Rad9 inhibition reduces the requirement for Sae2 and functional MRX in DNA end resection. EMBO Rep. 16, 351–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Karanja K. K., Lee E. H., Hendrickson E. A., Campbell J. L. (2014) Preventing over-resection by DNA2 helicase/nuclease suppresses repair defects in Fanconi anemia cells. Cell Cycle 13, 1540–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhang J. M., Liu X. M., Ding Y. H., Xiong L. Y., Ren J. Y., Zhou Z. X., Wang H. T., Zhang M. J., Yu Y., Dong M. Q., Du L. L. (2014) Fission yeast Pxd1 promotes proper DNA repair by activating Rad16XPF and inhibiting Dna2. PLoS Biol. 12, e1001946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tsutakawa S. E., Lafrance-Vanasse J., Tainer J. A. (2014) The cutting edges in DNA repair, licensing, and fidelity: DNA and RNA repair nucleases sculpt DNA to measure twice, cut once. DNA Repair 19, 95–107 [DOI] [PMC free article] [PubMed] [Google Scholar]