

Background: Chk1 and the MCM complex play important roles in DNA replication and replication checkpoint.
Results: Chk1 phosphorylates MCM3.
Conclusion: Phosphorylation of MCM3 by Chk1 regulates normal DNA replication and checkpoint.
Significance: These results provide insights into our understanding of DNA replication control and replication checkpoint activation.
Keywords: Cell Cycle, DNA Damage, DNA Replication, Phosphorylation, Signal Transduction
Abstract
Mechanisms controlling DNA replication and replication checkpoint are critical for the maintenance of genome stability and the prevention or treatment of human cancers. Checkpoint kinase 1 (Chk1) is a key effector protein kinase that regulates the DNA damage response and replication checkpoint. The heterohexameric minichromosome maintenance (MCM) complex is the core component of mammalian DNA helicase and has been implicated in replication checkpoint activation. Here we report that Chk1 phosphorylates the MCM3 subunit of the MCM complex at Ser-205 under normal growth conditions. Mutating the Ser-205 of MCM3 to Ala increased the length of DNA replication track and shortened the S phase duration, indicating that Ser-205 phosphorylation negatively controls normal DNA replication. Upon replicative stress treatment, the inhibitory phosphorylation of MCM3 at Ser-205 was reduced, and this reduction was accompanied with the generation of single strand DNA, the key platform for ataxia telangiectasia mutated and Rad3-related (ATR) activation. As a result, the replication checkpoint is activated. Together, these data provide significant insights into the regulation of both normal DNA replication and replication checkpoint activation through the novel phosphorylation of MCM3 by Chk1.
Introduction
The DNA damage response is essential for the maintenance of genome stability in eukaryotic cells and functions as a barrier to tumorigenesis (1, 2). A critical effector of the DNA damage response signal is the protein kinase, Chk12 (3). Chk1 responds largely, if not entirely, to DNA damage that affects DNA replication (3, 4). Consistently, loss of CHK1 causes early embryonic lethality in mice (5, 6).
In addition to its roles in the DNA damage response and checkpoint activation, Chk1 also regulates DNA replication under normal growth conditions. Chk1 interacts with proteins involved in DNA replication in the absence of DNA damage, such as proliferating cell nuclear antigen, Tim/Tipin, and Claspin (7–9). Inhibiting the activity of human Chk1 results in increases in both DNA replication initiation and DNA breakage (10). Furthermore, in the absence of Chk1, stalled replication forks are prone to collapse or aberrantly refire, leading to either irreversible S phase arrest or premature chromosome condensation (11). However, how exactly Chk1 regulates DNA replication is unclear.
The MCM complex (containing MCM2–7), Cdc45, and the GINS (from the Japanese Go-Ichi-Ni-San meaning 5-1-2-3, after the four related subunits of the complex Sld5, Psf1, Psf2, and Psf3) complex are collectively termed the CMG complex, which forms the core of DNA helicase for DNA replication (12–14). The MCM complex has recently been identified as an important component of the DNA damage response through regulating the replication checkpoint (15–17). MCM proteins undergo substantial phosphorylation both under normal growth conditions and during DNA damage by a variety of protein kinases, including cyclin-dependent kinases, the cyclin-dependent kinase-like Cdc7, ataxia telangiectasia mutated (ATM), and ATR (15, 18–24). However, the functional significance of such phosphorylation seems to be context-dependent and largely remains unknown.
We previously demonstrated that Chk1 interacts with the MCM complex (25). Here we report that human Chk1 phosphorylates MCM3 mainly at Ser-205, and this phosphorylation keeps normal DNA replication under control through negatively controlling the replication fork progression. Replicative stress induces Chk1 phosphorylation followed by its release from chromatin where Ser-205 phosphorylation of MCM3 mainly occurs. As a result, the level of Ser-205 phosphorylated MCM3 is reduced by replicative stress. The reduction in Ser-205 phosphorylation of MCM3 is accompanied with the generation of long stretches of single strand DNA (ssDNA), the critical platform for maximal activation of ATR. This seems to be important for subsequent Chk1 phosphorylation and likely the amplification and maintenance of replication checkpoint.
EXPERIMENTAL PROCEDURES
Cell Cultures, Transfection, and Chk1 Inhibitor Treatment
HEK293T, HeLa, U2-OS, and A549 cells were cultured in DMEM with 10% fetal bovine serum (FBS). HEK293T cells were transfected with calcium phosphate, whereas other cell lines were transfected with Lipofectamine 2000 (Invitrogen) or X-tremeGENE (Roche Applied Science) according to the manufacturers' protocols. Cell fractionation was conducted as described previously (26). To inhibit the catalytic activity of Chk1 in vitro, His-Chk1 proteins purified from baculovirus were preincubated with 500 nm Chk1 inhibitor (PF-003946901, Pfizer) for 120 min on ice before the in vitro kinase assay. For cell culture experiments, cells were pretreated with 1 or 2 μm Chk1 inhibitor for 6 h followed by procedures to detect MCM3 phosphorylation.
Immunoblotting and Antibodies
Immunoblotting was carried out as described previously (27). Anti-Chk1 (DCS-1310 and G4), anti-CyclinA (H-432), and anti-ATR (N-19) antibodies were from Santa Cruz Biotechnology. Anti-phospho-Ser-345-Chk1, anti-phospho-RRX(S/T) (PKA substrate), anti-phospho-Ser/Thr, anti-phospho-Tyr/Thr-Cdc2, and anti-phospho-Ser-216-Cdc25C were from Cell Signaling Technology. Anti-MCM7, anti-CyclinB, and mouse α-BrdU/IdU (B44 clone) were from BD Pharmingen. Rat α-BrdU/CldU was from AbD Serotec. Anti-MCM2, anti-FLAG, anti-HA, anti-GFP, and anti-Myc were described previously (26, 28).
Plasmid Construction
Myc-, FLAG-, or HA-tagged MCMs were generated using standard polymerase chain reactions (PCRs) with lentiviral vectors for each MCM (28) as the template. MCM3 full length (FL) and fragments were cloned in pcDNA3.1 with an HA tag. The forward primers used are: 5′-CCGCTCGAGATGTATCCTTACGACGTTCCAGACTATGC-3′ (for both FL and fragment 1), 5′-CTGCTCGAGATGTATCCTTACGACGTTCCAGACTATGCCATGAGCAAGGATGCTCAG-3′ (fragment 2), 5′-CCGCTCGAGATGTATCCTTACGACGTTCCAGACTATGCCGGGGACATCAATATTCTT-3′ (fragment 3), and 5′-CTGCTCGAGATGTATCCTTACGACGTTCCAGACTATGCCCCTGAGCAGGATCGGGAG-3′ (fragment 4). The reverse primers used are: 5′-CCGGAATTCTCAGATGAGGAAGATGATGCCCTCAG-3′ (for both FL and fragment 4), 5′-CCGGAATTCTCACTGCTTAACATTACAGGC-3′ (fragment 1), 5′-GGTGAATTCTCAACGGATGTGGCTGCCATT-3′ (fragment 2), and 5′-GGCGAATTCTCAATCCATCTGATCCAGCAT-3′ (fragment 3). Amino acid mutation was carried out using the QuikChange mutagenesis kit according to the manufacturer's protocol (Stratagene). The mutagenesis primers used are: 5′-CCATAGAGCGACGTTATGCTGATCTCACCACCCTG-3′ (S205A forward); 5′-CAGGGTGGTGAGATCAGCATAACGTCGCTCTATGG-3′ (S205A reverse); 5′-ACTGGCCGGGGCTCCGCTGGAGTGGGTCTGACG-3′ (S419A forward); 5′-CGTCAGACCCACTCCAGCGGAGCCCCGGCCAGT-3′ (S419A reverse); 5′-CCATAGAGCGACGTGATTCTGATCTCGCCGCCCTGGTGGCCTTTCCC-3′ (ASDLAA forward); and 5′-GGGAAAGGCCACCAGGGCGGCGAGATCAGAATCACGTCGCTCTATGG-3′ (ASDLAA reverse).
In Vitro Chk1 Kinase Assay
HEK293T cells were transfected with plasmids expressing differently tagged MCM constructs for 48 h, lysed in lysis buffer (50 mm Tris-HCl, pH 7.6, 150 mm NaCl, 10 mm NaF, 1 mm Na3VO4, 1 mm PMSF, 1 mm DTT, 10 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin A, and 0.2% Nonidet P-40), and sonicated. The supernatants were immunoprecipitated (IPed) with anti-HA antibodies overnight and collected with Protein A/G beads (Thermo Scientific). The beads were incubated with calf intestine alkaline phosphatase (CIAP; New England Biolabs) at 37 °C for 1 h and then used as the substrate to incubate with His-Chk1 purified from baculovirus sf9 cells. The in vitro kinase reaction was assembled in reaction buffer (20 mm HEPES, pH 7.4, 10 mm MgCl2, 5 mm EGTA, 25 mm β-glycerol phosphatase, 1 mm Na3VO4, and 1 mm DTT) with or without 10 mm ATP for 30 min at 37 °C. The reaction was stopped by addition of an equal volume of 2× SDS sample buffer. Samples were boiled for 5 min and separated by SDS-PAGE followed by immunoblotting with the anti-Ser(P)/Thr(P) mixture overnight at 4 °C. The same membrane was stripped and reblotted with anti-HA antibodies. For radiolabeling experiments, 1 μCi of 32P-labeled γ-ATP was added into the reaction, and the reaction was monitored by radiography.
DNA Fiber Assay
DNA fiber analyses were carried out as reported previously with modifications (29, 30). U2-OS cells were split into a 60-mm dish and transfected with pCas9 vectors targeting 3′- and 5′-UTRs of human MCM3 (31). After 24 h, cells were transfected again with pCas9 vectors plus HA-MCM3 wild type (WT) or S205A for another 48 h. The cells were pulsed with 250 μm CldU for 20 min, washed, and incubated with 25 μm IdU for another 20 min. The cells were collected and diluted to 250–400 cells/μl. The fiber assay was then followed as reported (29), and the image was taken under a fluorescence microscope. A scale bar was taken under the same condition (100× oil lens). We focused on the second track that is stained with the green color as it represents ongoing DNA replication forks. The length of the DNA replication track was calibrated by using NIH ImageJ software and adjusted by a scale bar of 10 μm. The experiments were performed two times in duplicate. A minimum of 100 tracks was measured in each duplicate. Data represent median and S.D.
Mass Spectrometry for MCM3 Phosphorylation Identification
HA-MCM3 WT or mutants underwent in vitro phosphorylation by His-Chk1, were separated by SDS-PAGE, and were Coomassie-stained. The bands were excised and in-gel trypsin-digested, and peptides were extracted with 50% acetonitrile in 5% formic acid and then resuspended in 0.1% formic acid. The analysis of the resulting peptides was performed with an Orbitrap Elite hybrid mass spectrometer (Thermo Electron, San Jose, CA) equipped with a Waters nanoACQUITY UPLC system (Waters, Taunton, MA). Spectra were recorded by data-dependent methods with an alternative full scan followed by 20 MS/MS scans. Data were analyzed using Mascot Daemon (Matrix Science, Boston, MA) with a setting of 10 ppm for parent ions and 0.8 Da for product ions. Carbamidomethylation of Cys residues was set as a fixed modification, and oxidation of Met and phosphorylation on Ser, Thr, and Tyr residues were set as variable modifications. Phosphorylation sites were further verified by manual examination of each tandem mass spectrum of phosphopeptides.
RESULTS
Chk1 Phosphorylates MCM3 Both in Vitro and in Vivo
We recently reported that Chk1 interacts with the MCM complex in cells (25). Because Chk1 is a Ser/Thr protein kinase, which preferentially phosphorylates proteins at RXX(S/T) sites similarly to PKA (3, 32), we asked whether Chk1 could phosphorylate any member of the MCM complex.
To this end, we overexpressed tagged MCM proteins in HEK293T cells, IPed with antibodies against the corresponding tag, and treated the IP beads with CIAP to remove pre-existing phosphorylation events. We used the dephosphorylated beads as the substrate and His-Chk1 purified from baculovirus as the kinase to perform the in vitro kinase assay. A fragment of Cdc25C containing a known Chk1 phosphorylation site, Ser-216, fused to GST was used as the positive control. To detect any potential phosphorylation signal of MCMs, we used a mixture of phospho-Ser/Thr antibodies (containing an equal ratio of anti-Ser(P)-216-Cdc25C, the known Chk1 target; anti-RRX(pS/pT) (where pS is phosphoserine and pT is phosphothreonine), the substrate for PKA; pan-anti-Ser(P)/Thr(P); and anti-Tyr(P)/Thr(P)-Cdc2). This antibody mixture readily detected phosphorylation of Cdc25C (Fig. 1A, lane 14). Remarkably, we observed that MCM3 and to a much lesser extent MCM4, -5, and -7 were phosphorylated (Fig. 1A, lane 10). We also detected phosphorylation of non-MCM proteins (e.g. the band marked with an asterisk in Fig. 1, A and C), likely coming from IP, by Chk1 or associating kinases.
FIGURE 1.

Phosphorylation of MCM3 by Chk1. A, HEK293T cells were transfected with tagged MCM proteins for 48 h, lysed, IPed with antibodies against the tag, and collected on Protein A/G-agarose beads. The beads were treated with 30 units of CIAP at 37 °C for 1 h, and the in vitro kinase assay was performed in the presence or absence of 10 mm ATP with His-Chk1 as the protein kinase. A GST-tagged Cdc25 fragment (200–250) was used as the positive control. The membrane was probed with an antibody mixture at an equal ratio that can recognize potential Chk1 phosphorylation sites. The same membrane was stripped, cut into small sections based on the protein marker, and reblotted with antibodies against Chk1, the tags for MCMs, or GST. Protein markers are shown on the left. Arrows indicate MCM proteins that were phosphorylated. The asterisk indicates a non-MCM protein that was also phosphorylated. B, HEK293T cells were transfected with HA-MCM3, IPed with anti-HA antibodies, treated or not with CIAP, and blotted with the anti-Ser(P)/Thr(P) mixture. The same membrane was stripped and reblotted with anti-HA antibodies. C, an in vitro kinase assay was performed as in A except that the His-Chk1 protein was pretreated with 500 nm Chk1specific inhibitor (Chk1i) for 2 h on ice. Short and long exposures of the ant-Ser(P)/Thr(P) antibody mixture are illustrated to show the difference in the phospho-MCM3 level between cell cultures and in vitro kinase reaction. The arrow denotes HA-MCM3 protein, and the arrowhead in the anti-Chk1 blot indicates autophosphorylated Chk1 proteins. The asterisk indicates the same non-MCM protein as in A.
We noticed that MCM3 proteins were already phosphorylated in cells (Fig. 1B, without CIAP treatment). However, the level of phospho-MCM3 in cells was much lower than that from the in vitro kinase reaction (Fig. 1C, lanes 1 and 5, between short and long exposure, indicated by the arrow), suggesting that Chk1 robustly phosphorylates MCM3 in vitro.
To determine whether the observed MCM3 phosphorylation is dependent on Chk1, we pretreated His-Chk1 proteins with a specific Chk1 inhibitor, PF-003946901 (33), prior to the in vitro kinase reaction. The results showed that phosphorylation of MCM3 was significantly reduced by the Chk1 inhibitor (Fig. 1C, lanes 5 and 6, short exposure, band indicated by the arrow). The inhibitor did inhibit the catalytic activity of Chk1 as indicated by the disappearance of the slow migrating band of His-Chk1 arising from its own autophosphorylation (Fig. 1C, lanes 5 and 6, the anti-Chk1 blot, band denoted by the arrowhead). To further confirm that MCM3 phosphorylation is dependent on the catalytic activity of Chk1, we carried out in vitro kinase assays using either WT or kinase-dead D148A Chk1 proteins as the kinase. The results showed that, unlike the WT, the kinase-dead Chk1 failed to phosphorylate MCM3 (Fig. 2A), suggesting that MCM3 phosphorylation is dependent on the catalytic activity of Chk1.
FIGURE 2.

MCM3 phosphorylation dependent on Chk1. A, the same in vitro kinase was performed as in Fig. 1A using either Chk1 WT or a kinase-dead (KD) mutant as the kinase. B, HEK293T cells were transfected with HA-MCM3. After 48 h, cells were treated with 1 or 2 μm Chk1 inhibitor for 6 h, lysed, IPed with anti-HA antibodies, and immunoblotted with the anti-Ser(P)/Thr(P) mixture. The same membrane was stripped and reblotted with anti-HA antibodies. Protein expression in whole cell extracts (WCE) was assessed. C, HEK293T cells were treated or not with 1 μm Chk1 inhibitor (Chk1i) for 12 h, lysed, IPed with anti-MCM3 antibodies, and immunoblotted with the indicated antibodies. D, cells treated with 1 μm Chk1 inhibitor for 6 and 12 h were analyzed for the cell cycle profile.
To confirm these results in vivo, we treated HEK293T cells expressing HA-MCM3 WT with the Chk1 inhibitor and examined MCM3 phosphorylation by immunoblotting. The results showed that the Chk1 inhibitor reduced phosphorylation of HA-MCM3 in a dose-dependent manner (Fig. 2B). Such reduction was not due to inhibition of expression of Chk1 or HA-MCM3 proteins (Fig. 2B). Furthermore, the level of phosphorylated endogenous MCM3 protein was also reduced by the Chk1 inhibitor (Fig. 2C). To confirm the effect of the Chk1 inhibitor in cell culture, we monitored the protein level of a known downstream cell cycle target of Chk1, Cdc25A. Cdc25A undergoes Chk1-dependent phosphorylation followed by proteasome-dependent degradation (34). Thus, if Chk1 is inhibited, we expected to observe an increase in the protein level of Cdc25A. Our data showed that this is indeed the case (Figs. 2, B and C). To preclude the possibility that such changes were due to altered cell cycle distribution by the Chk1 inhibitor, we monitored the cell cycle profile in the presence of the Chk1 inhibitor. The results did not reveal an obvious cell cycle change even after 12 h of Chk1 inhibitor treatment (Fig. 2D). We also noticed that the Chk1 inhibitor did not completely block MCM3 phosphorylation, suggesting that other kinases might also phosphorylate MCM3 albeit to a much lesser degree. Together, these data suggest that MCM3 phosphorylation is mainly dependent on Chk1.
Ser-205 of MCM3 Is the Major Site Phosphorylated by Chk1
We next sought to determine the phosphorylation site(s) of MCM3 by Chk1. We first asked which region of MCM3 contains the phosphorylation signal. We generated four fragments targeting different regions of human MCM3 (Fig. 3A; note that new analysis identified an additional 45 residues at the N terminus of human MCM3 with GenBankTM accession number NM_002388.4; thus the FL MCM3 is 853 amino acids, not the previously reported 808 amino acids). We overexpressed HA-tagged MCM3 FL or the four fragments into HEK293T cells, IPed with anti-HA antibodies, and examined protein phosphorylation using the anti-Ser(P)/Thr(P) mixture. We repeatedly observed that fragment 1, but not the other three fragments, was phosphorylated to an extent similar to the FL (Fig. 3B, lanes 1 and 4, and data not shown). Protein sequence analysis revealed that human MCM3 contains a potential Chk1 phosphorylation site at Ser-205 in fragment 1 (also see Fig. 4A). Thus, we mutated Ser-205 to Ala in HA-MCM3 FL or fragment 1. As a control, we mutated Ser-419 of MCM3 to Ala because Ser-419 also matches to the consensus phosphorylation site of Chk1. We then measured phosphorylation of HA-MCM3 FL (WT, S205A, or S419A) and fragment 1 (WT or S205A). The results showed that mutating Ser-205 to Ala completely abolished phosphorylation of both the FL and the fragment 1 of MCM3 (Fig. 3B, lanes 1 and 2 and lanes 4 and 5). In contrast, the S419A mutation had almost no effect (Fig. 3B, lanes 1 and 3). To determine whether Ser-205 is the major phosphorylation site of MCM3 by Chk1, we performed an in vitro kinase assay using HA-MCM3 WT or the S205A mutant as the substrate in the presence of [γ-32P]ATP. The results showed that the level of phosphorylated S205A mutant was significantly lower than that of the WT (Fig. 3C).
FIGURE 3.

Identification of MCM3 phosphorylation sites. A, schematic diagram of MCM3 FL and four fragments (1–4). AAA+, ATPase domain; NTD, nucleic acid-binding domain. B, HEK293T cells were transfected with HA-MCM3 FL (WT, S205A, or S419A) or fragment 1 (WT or S205A) for 48 h, IPed with anti-HA antibodies, and blotted with the anti-Ser(P)/Thr(P) mixture (left). The same membrane was stripped and reblotted with anti-HA antibodies (right). C, HA-MCM3 WT or S205A mutant was collected from transfected HEK293T cells, treated with CIAP, and used as the substrate for in vitro kinase assay in the presence of [γ-32P]ATP. The SDS-polyacrylamide gel was stained with Coomassie Blue (lower), dried, and exposed to x-ray radiography (upper).
FIGURE 4.
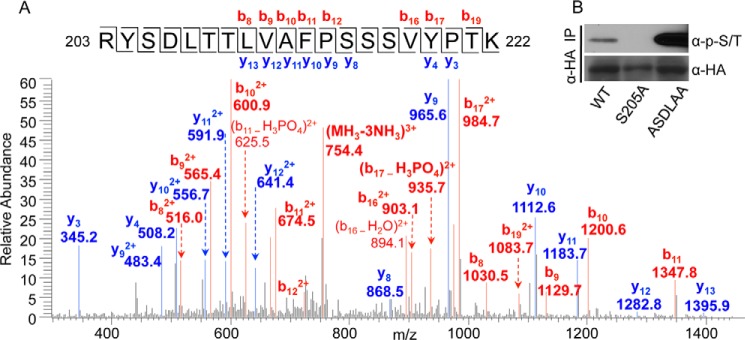
Ser-205 is the major site of MCM3 phosphorylated by Chk1. A, HA-MCM3 WT proteins were collected by IP from HEK293T cells, treated with CIAP, used in the in vitro kinase reaction, and run on SDS-PAGE, and the corresponding band was excised and used for mass spectrometry analyses. Phosphorylation was detected for the peptide 203–222 (one of the following four residues: Tyr-204, Ser-205, Thr-208, and Thr-209). B, HEK293T cells were transfected with HA-MCM3 FL (WT, S205A, or ASDLAA) for 48 h, IPed with anti-HA antibodies, and blotted with the anti-Ser(P)/Thr(P) mixture. The same membrane was stripped and reblotted with anti-HA antibodies.
To further confirm these results, we isolated HA-MCM3 WT proteins after in vitro kinase reaction and identified its phosphorylation sites by mass spectrometry. Even without peptide enrichment, we detected a number of phosphopeptides of MCM3 (such as Thr-243, Thr-428, Thr-719, Ser-337, Ser-717, Ser-756, and Ser-773), indicating that 1) the phosphosignal of these peptides was abundant and 2) Chk1 and likely other kinases from the co-IP phosphorylated MCM3. Among them, a peptide (203–222) contains the above identified Chk1 consensus site, Ser-205 (Fig. 4A). However, the mass spectrometric results can only demonstrate that one of four sites in this peptide (Tyr-204, Ser-205, Thr-208, or Thr-209) is phosphorylated (Fig. 4A). To determine which one of the four sites is phosphorylated, we mutated the YSDLTT motif to ASDLAA in MCM3. The assumption was that if phosphorylation occurred on one of these three non-Chk1 sites then we would not be able to detect the phosphosignal by mass spectrometry or immunoblotting. Despite repeated attempts, we failed to detect the same peptide (203–222) from the ASDLAA MCM3 mutant by mass spectrometry (data not shown), indicating that mutating these three residues might have affected ionization and detection of this peptide by mass spectrometry. Then we asked whether the phosphosignal was lost for the ASDLAA mutant of MCM3. In contrast, the ASDLAA mutant was still phosphorylated in cells (Fig. 4B), whereas the S205A mutant of HA-MCM3 completely lost the phosphosignal (Fig. 4B). These results suggest that it is Ser-205 in this peptide that is phosphorylated. The phosphosignal for the ASDLAA mutant was actually stronger than that of the WT (Fig. 4B), indicating that mutating these three residues (Tyr-204, Thr-208, and Thr-209) might have provided a better conformation of MCM3 to be phosphorylated by Chk1 at Ser-205.
We noticed that after phosphopeptide enrichment additional phosphorylation sites of MCM3 were identified. Again, the majority of them do not match to the Chk1 consensus site, indicating that MCM3 was phosphorylated by other co-IPed kinases. To our surprise, we detected phosphopeptide for Ser-419 despite the fact that Ser-419 was not phosphorylated in cells (Fig. 3B). We consider phosphorylation of Ser-419 as a minor event based on the following reasons. First, it was only detected after phosphopeptide enrichment, suggesting that its abundance is much lower than Ser(P)-205. Second, it was only detected after in vitro kinase reaction, suggesting that Ser-419 is a weak target at best in cells. Together, these lines of evidence suggest that Ser-205 is the major site of MCM3 phosphorylated by Chk1.
Ser-205 Phosphorylation of MCM3 Negatively Regulates Normal Cell Cycle Progression
We then asked what is the biological consequence of Ser-205 phosphorylation of MCM3? Because Ser-205 is already phosphorylated in unperturbed cells, the non-phosphorylated S205A, but not a phosphomimic mutant, should provide insights into the function of Ser-205 phosphorylation in cells. We first observed that Ser-205 phosphorylation did not affect the interaction between MCM3 and Chk1 or other MCM components (Fig. 5, A and B), indicating that it is unlikely involved in protein complex assembly.
FIGURE 5.

Ser-205 phosphorylation of MCM3 negatively regulates DNA replication and cell cycle progression. A, HEK293T cells were transfected with Myc-Chk1 and HA-MCM3 WT or S205A, IPed with anti-Myc antibodies, and blotted with anti-HA antibodies. The same membrane was stripped and reblotted with anti-Myc antibodies. B, HEK293T cells were transfected with FLAG-MCM2 and HA-MCM3 WT or S205A, IPed with anti-FLAG antibodies, and blotted with anti-HA antibodies followed by stripping and reprobing with anti-FLAG antibodies. C, U2-OS cells were transfected twice in consecutive days with the CRISPR/Cas9 system that targets the 3′- and 5′-UTRs of human MCM3. 48 h after the second transfection (total 72 h of knockdown), cells were collected and immunoblotted with the indicated antibodies. The Cas9 is expressed as a FLAG fusion protein. D, cell cycle profile of U2-OS cells after 72 h of transfection with the control pCas9 or pCas9-MCM3 knockdown (KD) vector. E, DNA fiber assay. U2-OS cells were transfected twice in consecutive days with the CRISPR/Cas9 vectors targeting both the 3′- and 5′-UTRs of MCM3. During the second transfection, HA-MCM3 WT or S205A vectors were also transfected. 48 h after the second transfection, DNA replication in cells was analyzed by the DNA fiber assay as detailed under “Experimental Procedures.” Red and green represent initial and subsequent replication tracks, respectively, taken under a fluorescence microscope (100× oil lens). F, a DNA fiber assay was carried out in U2-OS cells expressing HA-MCM3 WT or S205A mutant under the background of depletion of endogenous MCM3 by the CRISPR/Cas system. The length of actively replicating DNA tracks was analyzed using NIH ImageJ software and adjusted to a standard scale bar of 10 μm taken under the same condition. A minimum of 100 tracks was measured from duplicated experiments, and the experiment was repeated twice. Data represent median and S.D. (error bars). G, U2-OS cells grown on glass coverslips were co-transfected with the CRISPR/Cas MCM3 knockdown plasmid and HA-MCM3 WT or S205A for 72 h, blocked at the G2/M phase by treatment with 200 ng/ml nocodazole for 20 h, and released into the cell cycle. Cells were pulsed with 40 μm BrdU for 15 min at 4, 8, 12, 16, 20, and 24 h after nocodazole release; fixed; immunostained with antibodies against HA and BrdU; and visualized under a fluorescence microscope. The percentage of BrdU-positive cells in HA-negative or HA-positive populations was determined. Data represent median and S.D. (error bars) from duplicate experiments repeated twice. More than 50 cells were counted in each duplicate. WCE, whole cell extracts.
Because the MCM complex is an important factor for DNA replication in cells (12), we assessed the effect of Ser-205 phosphorylation on DNA replication and S phase progression. We utilized the CRISPR/Cas system (31) targeting the 3′- and 5′-UTRs of human MCM3 to reduce the expression of endogenous proteins in U2-OS cells (Fig. 5C). Results showed that depletion of MCM3 did not affect cell viability or the cell cycle profile (Fig. 5D), similar to a previous report (35), indicating that cells can tolerate significant depletion of MCM3 for growth or that a residual amount of MCM3 is sufficient for cell proliferation. Such depletion allowed us to assess the function of HA-MCM3 WT or S205A by overexpression in cells and monitoring DNA replication using the DNA fiber assay (29). We measured the length of actively replicating DNA tracks, and the results showed that cells expressing the HA-MCM3 WT had DNA replication track length of ∼6.3 μm, similar to that of parental cells (Fig. 5F). However, the track length from S205A cells was ∼9.6 μm (Fig. 5F), a ∼50% increase compared with the WT.
Given the increased replication track length (Fig. 5F), we expect that cells expressing the S205A mutant might progress through the S phase faster than cells expressing the MCM3 WT. To this end, we knocked down endogenous MCM3 and overexpressed HA-MCM3 WT or S205A mutant in these cells. We then blocked the cells at G2/M phase by nocodazole treatment, released them into the cell cycle, collected cells at different time points with BrdU pulse labeling, and stained them with anti-BrdU and anti-HA antibodies. The BrdU-positive population represents S phase cells. This method allowed us to specifically monitor the S phase progression of MCM3 WT- or S205A mutant-expressing cells. The results showed that the duration of the S phase (indicated by the percentage of BrdU-positive cells) was shorter in cells expressing the S205A mutant than in cells expressing the WT or the control (Fig. 5G). Flow cytometry-based BrdU incorporation analysis also revealed shortened S phase in S205A cells (data not shown). These results suggest that the S205A mutant accelerated S phase progression. In turn, they indicate that phosphorylation of MCM3 at Ser-205 plays a negative role in keeping DNA replication under control in unperturbed cells.
Ser-205 Phosphorylation Is Involved in Replication Checkpoint Activation
We recently reported that the MCM complex plays an important role in recruiting Chk1 onto chromatin, a critical step for DNA damage-induced Chk1 phosphorylation (25). Consistent with this idea, we found that phosphorylated MCM3 proteins were mainly located on chromatin-enriched cellular compartments (Fig. 6A). Chk1 undergoes DNA damage-dependent phosphorylation and release from chromatin (Fig. 6B, lower panel) (36, 37), leading to a reduction in its interaction with the MCM complex (25). These findings led us to predict that DNA damage reduces Ser-205 phosphorylation of MCM3 because Chk1, the major kinase that phosphorylates MCM3, will be released from chromatin after DNA damage. To test this idea, we examined the effect of a replicative stress agent, hydroxyurea, on MCM3 phosphorylation. The results showed that hydroxyurea (or other DNA-damaging agents like the topoisomerase 1 inhibitors) treatment indeed reduced the levels of both total cellular and chromatin-bound phospho-MCM3 proteins (Fig. 6B, IP results). However, the reduction reached the lowest level (∼50% of that before treatment) at 4–8 h post-treatment. This agrees with the fact that a fraction of Chk1 remains on chromatin even after DNA damage (Fig. 6B, lower panel) (36, 37). Another possibility is that other kinases might also contribute to MCM3 phosphorylation.
FIGURE 6.

Roles of MCM3 phosphorylation in replication checkpoint activation. A, HEK293T cells were transfected with HA-MCM3 WT, fractionated into chromatin and non-chromatin fractions, IPed with anti-HA antibodies, and blotted with anti-Ser(P)/Thr(P) antibodies followed by stripping and reprobing with anti-HA antibodies. B, HEK293T cells were transfected with HA-MCM3 for 48 h and treated with 2 mm hydroxyurea (HU) for 0, 1, 4 and 8 h. Whole cell extracts (WCE) or chromatin-enriched fraction were IPed with anti-HA antibodies and blotted with the anti-Ser(P)/Thr(P) mixture. The same membrane was stripped and reblotted with anti-HA antibodies. Phosphorylated Chk1 (pChk1) and total Chk1 in the extracts were also monitored. The band intensity of both the Ser(P)/Thr(P) and the HA blots was analyzed using NIH ImageJ software, and the relative intensity of the Ser(P)/Thr(P) blot after adjusting against the anti-HA blot intensity is shown from three replicates. C, U2-OS cells grown on glass coverslips were co-transfected with the CRISPR/Cas MCM3 knockdown (KD) plasmid and HA-MCM3 WT or S205A for 72 h, treated with 1.5 μg/ml aphidicolin for 1 h, fixed, and immunostained with anti-HA and anti-RPA antibodies. Data represent the number of RPA foci per cell from 30–50 cells in duplicate. D, representative images of RPA foci from C. Scale bar, 10 μm. E, quantitation results of RPA focus size by NIH ImageJ software from cells in D. Data represent median and S.D. (error bars) from 30–50 cells in duplicate. *, p < 0.05. F, U2-OS cells were co-transfected with the CRISPR/Cas MCM3 knockdown plasmid and HA-MCM3 WT or S205A for 72 h and treated with 1.5 μg/ml aphidicolin for 1 h, and protein expression was analyzed. The phospho-Chk1 blot was stripped and reprobed with anti-Chk1 antibodies. The protein band intensity of Chk1 and phospho-Chk1 in lanes 2 and 4 was quantitated using NIH ImageJ software. The relative level of phospho-Chk1 was normalized to that of total Chk1, which shows a ∼40% increase averaged from three blots. Cyto + Sol Nu, cytosol and soluble nuclear proteins.
To determine the functional significance of the reduction in Ser-205 phosphorylation of MCM3, we examined replication checkpoint activation in cells expressing MCM3 WT or the S205A mutant. To this end, we overexpressed HA-MCM3 WT or the S205A mutant in U2-OS cells whose endogenous MCM3 was reduced; treated cells with aphidicolin, a DNA polymerase inhibitor; and measured focus formation of RPA, the key player in initiating and amplifying the replication checkpoint signal through activating ATR (4). The number of RPA foci was slightly higher in the S205A mutant cells than in the WT (Fig. 6C). Most interestingly, we noticed that the average size of RPA foci appears to be larger in S205A cells than in WT cells (Fig. 6D). Summaries from two independent experiments (Fig. 6E) suggest that such difference in the size of RPA foci did not originate from the variability of the immunofluorescence signals in different slides.
The presence of long stretches of ssDNA is important for the maintenance of ATR-dependent replication checkpoint (3, 4). Therefore, we examined ATR-dependent phosphorylation of Chk1 at Ser-345, the downstream event that depends on ssDNA and is also a gold standard of checkpoint activation (3, 4). We found that cells expressing the S205A mutant consistently displayed a ∼40% increase in Chk1 phosphorylation compared with cells expressing the WT by aphidicolin (Fig. 6F, lanes 2 and 4). In our opinion, this is a significant increase as it represents extra increase in the checkpoint signal. In addition, the amount of other checkpoint proteins (e.g. ATR) that are available might have limited the extent by which the checkpoint signal can be further increased by the S205A mutant. Nevertheless, these data are consistent with the results showing larger RPA foci, indicating enhanced replication checkpoint activation in S205A-expressing cells, although we cannot preclude that the increased Chk1 phosphorylation is just the result of aberrant DNA replication caused by the S205A mutant. A phosphomimic S205E mutant showed a level of Chk1 phosphorylation similar to that of the WT (data not shown), indicating that this might not be a perfect phosphomimic. Together, these data suggest that a replicative stress-induced reduction in Ser-205 phosphorylation of MCM3 is important for replication checkpoint activation.
DISCUSSION
Here we have illustrated a novel mechanism by which Chk1 regulates DNA replication and replication checkpoint through inducing an inhibitory phosphorylation of MCM3. These findings shed significant light on our understanding of both normal DNA replication and replication checkpoint activation. Under normal growth conditions, Chk1 phosphorylates MCM3 mainly at Ser-205, which in turn keeps normal DNA replication and cell cycle progression under control. In response to replicative stress, phosphorylation of MCM3 at Ser-205 is reduced likely due to the release of Chk1 from chromatin. A reduction in this inhibitory phosphorylation of MCM3 seems to facilitate the activation of replication checkpoint through promoting the generation of long stretches of ssDNA, the critical platform for ATR activation. A recent report showed that Chk1 associates with the TopBP1-binding protein Treslin, another component of the DNA replication machinery (38). Similarly, Chk1 constitutively phosphorylates Treslin and negatively inhibits normal DNA replication (38). Together with our results, these lines of evidence suggest that Chk1-dependent phosphorylation of replication machinery proteins (such as MCM3 and Treslin) may be a general mechanism by which Chk1 negatively regulates DNA replication under normal conditions. They are also consistent with previous studies demonstrating that inhibiting Chk1 leads to increased DNA replication (10).
Our data showed that although Chk1 is the major player that phosphorylates MCM3 at Ser-205 in cells other kinases might also contribute to MCM3 phosphorylation. MCM3 was reported to be phosphorylated by the death-associated protein kinase at the same Ser-205 site (previously Ser-160 due to the lack of the N-terminal 45 amino acids) (39). Similarly, such phosphorylation did not affect the MCM complex assembly (39). However, the biological significance of death-associated protein kinase-dependent MCM3 phosphorylation was unclear.
An interesting question that warrants further analysis in the future is how exactly MCM3 phosphorylation negatively regulates DNA replication. It is tempting to speculate that such phosphorylation might inhibit the helicase activity of the MCM complex, leading to the uncoupling between the helicase and the DNA polymerase (4). The generation of a long stretch of ssDNA is in agreement with this idea. In addition, recent studies have illustrated important roles of MCM3 in both the assembly of the MCM complex and the helicase activity. The distal C terminus and phosphorylation at Ser-112 of MCM3 are important for the MCM complex assembly (22, 40). Furthermore, a recent report showed that phosphorylation of fly MCM3 by checkpoint kinase 2 negatively regulates the helicase activity of the MCM complex (41). Together with our data, they suggest that the inhibitory phosphorylation of MCM3 by Chk1/2 may be evolutionally conserved. Our attempts to test the impact of MCM3 Ser-205 phosphorylation on the helicase activity of the CMG complex have been unsuccessful so far largely due to technical issues in isolating the entire CMG complex containing either the MCM3 WT or the S205A mutant with the correct stoichiometry. This limited us in drawing a definite conclusion on the effect of MCM3 Ser-205 phosphorylation on the helicase activity at this time. Nevertheless, these new lines of evidence reveal a previously unappreciated role of the MCM3 component in the function of the MCM complex in both DNA replication and replication checkpoint activation.
Acknowledgments
We thank Randal Tibbetts and Toshiya Tsuji for reagents and Jin Sil Chung for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants R00 CA126173, a career development award from the NCI, and R01 CA163214 from the NCI (to Y. Z.). This work was also supported by American Cancer Society Pilot Grant ACS IRG-91-022-15 (to Y. Z.).
- Chk1
- checkpoint kinase 1
- ATR
- ataxia telangiectasia mutated and Rad3-related
- CIAP
- calf intestine alkaline phosphatase
- MCM
- minichromosome maintenance
- ssDNA
- single strand DNA
- CMG
- Cdc45/MCM2–7/GINS
- FL
- full length
- IP
- immunoprecipitation
- IPed
- immunoprecipitated
- CldU
- 5-chloro-2′-deoxyuridine
- BrdU
- 5-bromodeoxyuridine
- IdU
- iododeoxyuridine
- CRISPR
- clustered regularly interspaced short palindromic repeat
- RPA
- replication protein A.
REFERENCES
- 1. Harper J. W., Elledge S. J. (2007) The DNA damage response: ten years after. Mol. Cell 28, 739–745 [DOI] [PubMed] [Google Scholar]
- 2. Jackson S. P., Bartek J. (2009) The DNA-damage response in human biology and disease. Nature 461, 1071–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang Y., Hunter T. (2014) Roles of Chk1 in cell biology and cancer therapy. Int. J. Cancer 134, 1013–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cimprich K. A., Cortez D. (2008) ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 9, 616–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu Q., Guntuku S., Cui X. S., Matsuoka S., Cortez D., Tamai K., Luo G., Carattini-Rivera S., DeMayo F., Bradley A., Donehower L. A., Elledge S. J. (2000) Chk1 is an essential kinase that is regulated by Atr and required for the G2/M DNA damage checkpoint. Genes Dev. 14, 1448–1459 [PMC free article] [PubMed] [Google Scholar]
- 6. Takai H., Tominaga K., Motoyama N., Minamishima Y. A., Nagahama H., Tsukiyama T., Ikeda K., Nakayama K., Nakanishi M. (2000) Aberrant cell cycle checkpoint function and early embryonic death in Chk1(−/−) mice. Genes Dev. 14, 1439–1447 [PMC free article] [PubMed] [Google Scholar]
- 7. Unsal-Kaçmaz K., Mullen T. E., Kaufmann W. K., Sancar A. (2005) Coupling of human circadian and cell cycles by the timeless protein. Mol. Cell. Biol. 25, 3109–3116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Scorah J., Dong M. Q., Yates J. R., 3rd, Scott M., Gillespie D., McGowan C. H. (2008) A conserved proliferating cell nuclear antigen-interacting protein sequence in Chk1 is required for checkpoint function. J. Biol. Chem. 283, 17250–17259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Taricani L., Shanahan F., Parry D. (2009) Replication stress activates DNA polymerase α-associated Chk1. Cell Cycle 8, 482–489 [DOI] [PubMed] [Google Scholar]
- 10. Syljuåsen R. G., Sørensen C. S., Hansen L. T., Fugger K., Lundin C., Johansson F., Helleday T., Sehested M., Lukas J., Bartek J. (2005) Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol. Cell. Biol. 25, 3553–3562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zachos G., Rainey M. D., Gillespie D. A. (2005) Chk1-dependent S-M checkpoint delay in vertebrate cells is linked to maintenance of viable replication structures. Mol. Cell. Biol. 25, 563–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bochman M. L., Schwacha A. (2009) The Mcm complex: unwinding the mechanism of a replicative helicase. Microbiol. Mol. Biol. Rev. 73, 652–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tye B. K., Sawyer S. (2000) The hexameric eukaryotic MCM helicase: building symmetry from nonidentical parts. J. Biol. Chem. 275, 34833–34836 [DOI] [PubMed] [Google Scholar]
- 14. Bell S. P., Dutta A. (2002) DNA replication in eukaryotic cells. Annu. Rev. Biochem. 71, 333–374 [DOI] [PubMed] [Google Scholar]
- 15. Cortez D., Glick G., Elledge S. J. (2004) Minichromosome maintenance proteins are direct targets of the ATM and ATR checkpoint kinases. Proc. Natl. Acad. Sci. U.S.A. 101, 10078–10083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tercero J. A., Longhese M. P., Diffley J. F. (2003) A central role for DNA replication forks in checkpoint activation and response. Mol. Cell 11, 1323–1336 [DOI] [PubMed] [Google Scholar]
- 17. Tsao C. C., Geisen C., Abraham R. T. (2004) Interaction between human MCM7 and Rad17 proteins is required for replication checkpoint signaling. EMBO J. 23, 4660–4669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pereverzeva I., Whitmire E., Khan B., Coué M. (2000) Distinct phosphoisoforms of the Xenopus Mcm4 protein regulate the function of the Mcm complex. Mol. Cell. Biol. 20, 3667–3676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Masai H., Taniyama C., Ogino K., Matsui E., Kakusho N., Matsumoto S., Kim J. M., Ishii A., Tanaka T., Kobayashi T., Tamai K., Ohtani K., Arai K. (2006) Phosphorylation of MCM4 by Cdc7 kinase facilitates its interaction with Cdc45 on the chromatin. J. Biol. Chem. 281, 39249–39261 [DOI] [PubMed] [Google Scholar]
- 20. Sheu Y. J., Stillman B. (2010) The Dbf4-Cdc7 kinase promotes S phase by alleviating an inhibitory activity in Mcm4. Nature 463, 113–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Montagnoli A., Valsasina B., Brotherton D., Troiani S., Rainoldi S., Tenca P., Molinari A., Santocanale C. (2006) Identification of Mcm2 phosphorylation sites by S-phase-regulating kinases. J. Biol. Chem. 281, 10281–10290 [DOI] [PubMed] [Google Scholar]
- 22. Lin D. I., Aggarwal P., Diehl J. A. (2008) Phosphorylation of MCM3 on Ser-112 regulates its incorporation into the MCM2–7 complex. Proc. Natl. Acad. Sci. U.S.A. 105, 8079–8084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shi Y., Dodson G. E., Mukhopadhyay P. S., Shanware N. P., Trinh A. T., Tibbetts R. S. (2007) Identification of carboxyl-terminal MCM3 phosphorylation sites using polyreactive phosphospecific antibodies. J. Biol. Chem. 282, 9236–9243 [DOI] [PubMed] [Google Scholar]
- 24. Yoo H. Y., Shevchenko A., Shevchenko A., Dunphy W. G. (2004) Mcm2 is a direct substrate of ATM and ATR during DNA damage and DNA replication checkpoint responses. J. Biol. Chem. 279, 53353–53364 [DOI] [PubMed] [Google Scholar]
- 25. Han X., Aslanian A., Fu K., Tsuji T., Zhang Y. (2014) The interaction between checkpoint kinase 1 (Chk1) and the minichromosome maintenance (MCM) complex is required for DNA damage-induced Chk1 phosphorylation. J. Biol. Chem. 289, 24716–24723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang J., Han X., Feng X., Wang Z., Zhang Y. (2012) Coupling cellular localization and function of checkpoint kinase 1 (chk1) in checkpoints and cell viability. J. Biol. Chem. 287, 25501–25509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang J., Engle S., Zhang Y. (2011) A new in vitro system for activating the cell cycle checkpoint. Cell Cycle 10, 500–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tsuji T., Ficarro S. B., Jiang W. (2006) Essential role of phosphorylation of MCM2 by Cdc7/Dbf4 in the initiation of DNA replication in mammalian cells. Mol. Biol. Cell 17, 4459–4472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Merrick C. J., Jackson D., Diffley J. F. (2004) Visualization of altered replication dynamics after DNA damage in human cells. J. Biol. Chem. 279, 20067–20075 [DOI] [PubMed] [Google Scholar]
- 30. Petermann E., Woodcock M., Helleday T. (2010) Chk1 promotes replication fork progression by controlling replication initiation. Proc. Natl. Acad. Sci. U.S.A. 107, 16090–16095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., Zhang F. (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen P., Luo C., Deng Y., Ryan K., Register J., Margosiak S., Tempczyk-Russell A., Nguyen B., Myers P., Lundgren K., Kan C. C., O'Connor P. M. (2000) The 1.7 Å crystal structure of human cell cycle checkpoint kinase Chk1: implications for Chk1 regulation. Cell 100, 681–692 [DOI] [PubMed] [Google Scholar]
- 33. Zhang Y. W., Brognard J., Coughlin C., You Z., Dolled-Filhart M., Aslanian A., Manning G., Abraham R. T., Hunter T. (2009) The F box protein Fbx6 regulates Chk1 stability and cellular sensitivity to replication stress. Mol. Cell 35, 442–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sørensen C. S., Syljuåsen R. G., Falck J., Schroeder T., Rönnstrand L., Khanna K. K., Zhou B. B., Bartek J., Lukas J. (2003) Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell 3, 247–258 [DOI] [PubMed] [Google Scholar]
- 35. Ibarra A., Schwob E., Méndez J. (2008) Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc. Natl. Acad. Sci. U.S.A. 105, 8956–8961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang Y. W., Otterness D. M., Chiang G. G., Xie W., Liu Y. C., Mercurio F., Abraham R. T. (2005) Genotoxic stress targets human Chk1 for degradation by the ubiquitin-proteasome pathway. Mol. Cell 19, 607–618 [DOI] [PubMed] [Google Scholar]
- 37. Smits V. A., Reaper P. M., Jackson S. P. (2006) Rapid PIKK-dependent release of Chk1 from chromatin promotes the DNA-damage checkpoint response. Curr. Biol. 16, 150–159 [DOI] [PubMed] [Google Scholar]
- 38. Guo C., Kumagai A., Schlacher K., Shevchenko A., Shevchenko A., Dunphy W. G. (2015) Interaction of chk1 with treslin negatively regulates the initiation of chromosomal DNA replication. Mol. Cell 57, 492–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bialik S., Berissi H., Kimchi A. (2008) A high throughput proteomics screen identifies novel substrates of death-associated protein kinase. Mol. Cell. Proteomics 7, 1089–1098 [DOI] [PubMed] [Google Scholar]
- 40. Frigola J., Remus D., Mehanna A., Diffley J. F. (2013) ATPase-dependent quality control of DNA replication origin licensing. Nature 495, 339–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ilves I., Tamberg N., Botchan M. R. (2012) Checkpoint kinase 2 (Chk2) inhibits the activity of the Cdc45/MCM2–7/GINS (CMG) replicative helicase complex. Proc. Natl. Acad. Sci. U.S.A. 109, 13163–13170 [DOI] [PMC free article] [PubMed] [Google Scholar]