

Abstract
OBJECTIVES:
Prucalopride is effective at alleviating symptoms of chronic constipation in women. The aim of this study was to assess the efficacy of 12 weeks of prucalopride treatment compared with placebo in men with chronic constipation.
METHODS:
This was a multicenter, stratified, randomized, parallel-group, double-blind, placebo-controlled, phase 3 study (ClinicalTrials.gov identifier: NCT01147926). The primary end point was the proportion of patients with a mean of three or more spontaneous complete bowel movements (SCBMs) per week across the treatment period. Efficacy end points were assessed using daily electronic diaries, global assessment of the severity of constipation and efficacy of treatment, and Patient Assessment of Constipation—Symptoms (PAC-SYM) and Patient Assessment of Constipation—Quality of Life (PAC-QOL) questionnaires.
RESULTS:
In total, 374 patients were enrolled in the study. Significantly more patients achieved a mean of three or more SCBMs per week in the prucalopride group (37.9%) than in the placebo group (17.7%, P<0.0001). The proportion of patients rating their constipation treatment as “quite a bit” to “extremely” effective at the final on-treatment visit was 46.7 and 30.4% in the prucalopride and placebo groups, respectively. The difference between treatment groups was statistically significant (P<0.0001). The proportion of patients with an improvement of at least 1 point in PAC-QOL satisfaction subscale score was 52.7 and 38.8% in the prucalopride and placebo groups, respectively (P=0.0035). Prucalopride had a good safety profile and was well tolerated.
CONCLUSIONS:
Prucalopride is effective, has a good safety profile, and is well tolerated for the treatment of men with chronic constipation.
INTRODUCTION
Constipation is a common, often chronic, gastrointestinal motility disorder characterized by a diversity of symptoms including bloating, straining, abdominal pain, lumpy or hard stools, sensation of incomplete evacuation, and infrequent defecation (fewer than three bowel movements per week) (1, 2, 3). The estimated global prevalence of chronic constipation is 14% ((ref. 4) and it is more common in women and the elderly (5). Individual symptoms are frequently severe and can significantly impair patients' health-related quality of life (HRQoL) (6) and decrease work productivity (1). Poor HRQoL in patients with constipation has been shown to be a predictor of increased health-care utilization (7).
A large proportion of patients are not satisfied with traditional treatment options for constipation (e.g., prescription and over-the-counter laxatives, and fiber), mainly owing to lack of efficacy (1, 8). Prucalopride is a 5-hydroxytryptamine receptor-4 (5-HT4) agonist that stimulates intestinal motility. Four phase 3, double-blind clinical trials with similar study designs have shown prucalopride to be significantly more effective than placebo at alleviating symptoms of constipation and improving HRQoL after 12 weeks of treatment (9, 10, 11, 12, 13, 14). Prucalopride is currently licensed in Europe for the treatment of chronic constipation in women in whom laxatives fail to provide adequate relief. A recent systematic review demonstrated the efficacy of prucalopride and other 5-HT4 agonists on outcomes that are important to the patient, and highlighted its favorable safety profile (15).
Although men were included in all of the pivotal phase 3 trials, they accounted for ~10% of patients enrolled. This reflects the smaller number of men who seek medical care for chronic constipation. Therefore, the primary objective of this study was to assess the efficacy of prucalopride compared with placebo over 12 weeks of treatment in men with chronic constipation. Consistent with previous trials, the primary end point was defined as the proportion of patients with a mean of three or more spontaneous complete bowel movements (SCBMs) per week evaluated over the entire treatment period. Secondary objectives were to evaluate the effect on symptoms, HRQoL, global assessment of illness severity, safety, and tolerability of prucalopride compared with placebo in this population.
METHODS
Patients
Men aged 18 years and older with chronic constipation were eligible for inclusion in the study. Chronic constipation was defined, according to the Rome III criteria (3), as two or fewer SCBMs (bowel movements that are not preceded within 24 h by the use of laxatives or enema and that result in a feeling of complete evacuation) per week, as well as one or more of the following for at least 6 months: at least 25% of stools being very hard or hard; sensation of incomplete evacuation following at least 25% of defecations; and straining at defecation for at least 25% of the time.
Patients with drug-induced constipation, or constipation secondary to causes such as endocrine disorders, metabolic disorders, neurological disorders, organic disorders of the large bowel, or surgery, were excluded from the trial. In addition, patients were excluded if they had a history of clinically significant (as evaluated by the investigator) cancer, cardiac, vascular, liver, pulmonary, endocrine, neurological, or psychiatric disorders, or metabolic disturbances. Individuals were also excluded if they had previously used prucalopride.
The study was carried out in accordance with the International Conference on Harmonisation of Good Clinical Practice guidelines (16), the principles of the Declaration of Helsinki (17), and local ethical and legal requirements. The study protocol was approved by local ethics committees before study initiation, and all patients provided written informed consent before inclusion in the trial.
Trial design
This was a multicenter, stratified, randomized, parallel-group, double-blind, placebo-controlled, phase 3 study (ClinicalTrials.gov identifier: NCT01147926). It was carried out at 66 sites across Europe (Belgium, Bulgaria, Czech Republic, Denmark, France, Germany, the Netherlands, Poland, Romania, and the United Kingdom) from September 2010 to October 2013.
The trial design is shown in Figure 1. After an initial screening visit, there was a 2-week run-in phase; patients who required a colonoscopy after screening had a 4-week run-in and those who had been using agents that influence bowel habits, which were stopped at screening, had a 3-week run-in. At baseline, patients were randomized (using a central interactive web-based response system or an interactive voice response system) 1:1 to receive prucalopride or placebo once daily. Randomization was stratified on the basis of number of complete bowel movements (CBMs) per week during the run-in period (0 or >0) and by country. They were instructed to take their treatment before breakfast. Patients younger than 65 years took prucalopride 2 mg once daily, whereas those aged 65 years or older started on a daily dose of prucalopride 1 mg. The dose was increased to 2 mg in the case of insufficient response (mean of <3 SCBMs per week in the previous 2 weeks), evaluated at week 2 or 4. No further dose adjustments were allowed. Patients returned to the treatment center at weeks 2, 4, 8, and 12 after the baseline visit (week 0).
Figure 1.
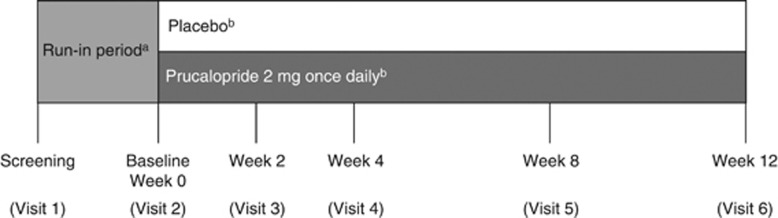
Study design. aTwo to four weeks. bElderly patients (≥65 years old) started on prucalopride 1 mg once daily (or matching placebo); this could be increased to 2 mg once daily (or matching placebo) at the week 2 or week 4 visit if efficacy was insufficient.
The use of agents that influence bowel habits, such as prokinetics, anticholinesterases, or anticholinergic drugs, was disallowed during the trial. Laxative use was also disallowed during the trial, except for bisacodyl that was allowed as a rescue medication if patients had not had a bowel movement for three or more consecutive days. The use of rescue medication was not permitted in the 24 h before or 48 h after the baseline visit.
Assessments
Patients were asked to keep daily electronic diaries (e-diaries) for the duration of the study to record bowel movement frequency (the date and time of each bowel movement) and consistency (using the Bristol Stool Scale) (18), degree of straining (no straining, mild straining, moderate straining, severe straining, or very severe straining), feeling of complete evacuation after a bowel movement (yes or no), and use of rescue medication or enema, if any. The primary efficacy end point was the proportion of patients with an average weekly frequency of three or more SCBMs per week (responders) over 12 weeks of treatment. The proportion of responders during each week and each 4 weekly period was also assessed. Secondary efficacy variables derived from the e-diary data were evaluated over the 12-week treatment period.
At baseline, week 4, and week 12, patients completed a global assessment of the severity of their constipation over the past 2 weeks, which they were asked to assess on a 5-point scale (0, absent; 4, very severe). At baseline and weeks 4 and 12, participants were asked to complete the Patient Assessment of Constipation—Symptoms (PAC-SYM) questionnaire (19). This validated psychometric instrument comprises 12 items that assess the severity of constipation symptoms over the past 2 weeks on three subscales (stool, abdominal, and rectal symptoms) using a 5-point Likert-type scale (0, absent; 4, very severe). The overall score is calculated as the mean rating across all items. The clinically significant cutoff was defined as an improvement in total PAC-SYM score of at least 1 point from baseline.
The Patient Assessment of Constipation—Quality of Life (PAC-QOL) questionnaire was administered at baseline and weeks 4 and 12. The PAC-QOL questionnaire assesses constipation-related HRQoL parameters on four subscales (physical discomfort, psychosocial discomfort, worries and concerns, and satisfaction) that are scored on a 5-point Likert-type scale (0, none/not at all; 4, extremely/all the time) (20, 21).
Adverse events and safety
The occurrence of treatment-emergent adverse events (TEAEs) was recorded from signing of informed consent at screening to the final visit. All adverse events (AEs) starting at or after first dosing and occurring up to 5 days after the final dose of investigational product were considered to be TEAEs. In addition, all AEs that started before the first dosing but that worsened in severity at or after the first dose were considered to be TEAEs. Serious TEAEs were recorded until 30 days after the final dose of the investigational product. Vital signs were monitored, electrocardiography was carried out, and samples for blood and urine tests were collected at screening, at baseline, and at the 4- and 12-week visits or on early discontinuation. Physical examinations were carried out at screening and at week 12 or early discontinuation.
Statistical methods
Previous pivotal phase 3 studies of prucalopride showed that, during treatment, the percentage of men with three or more SCBMs per week was in the range 11–21% for placebo and 20–29% for prucalopride, on the basis of a sample of 20–30 men per treatment group per study (~10% of the study population). For this study, a sample size of 174 patients per treatment arm was used, which was considered sufficient to detect a 14% difference in response rate between placebo (14%) and prucalopride (28%) at a power of 90%.
The safety population included all randomized patients who took at least one dose of the trial medication. The modified intent-to-treat (mITT) population included all randomized patients who took at least one dose of the trial medication, but excluded 12 patients from one site where a serious breach in good clinical practice was identified before unblinding. All efficacy analyses were carried out using data from the mITT population. Sensitivity analysis of efficacy parameters was carried out using data from two subsets of the mITT population: the per protocol population (which excluded patients who discontinued investigational product before day 28 of the treatment duration or had another important protocol violation that potentially could have influenced efficacy) and the completers population (which excluded patients who discontinued the study, or whose treatment duration in the study was <81 days).
Data on demographics, baseline characteristics, and AEs were presented descriptively. For e-diary end points, patients with fewer than 14 days of diary data were assumed to be non-responders. For patients with 7 or more days of data after the first week of treatment but fewer than 84 days of e-diary data, the information from the last 7 days was repeatedly copied for all missing days until the end of the study. A Cochran–Mantel–Haenszel test, stratified for the randomization stratification factors (0 or >0 CBMs per week during the run-in period and country), was used to compare the primary end point for prucalopride vs. placebo. Sensitivity analyses of the primary end point were performed using the following: a generalized linear mixed model for repeated measures (with treatment, week, treatment × week, country, and number of CBMs/week during the run-in period (0 or >0) as factors and baseline SCBM as a covariate, fitted by the direct likelihood technique); multiple imputation to impute missing weekly frequencies of SCBMs (on the basis of the treatment group to which the participant with missing data belonged); and the Cochran–Mantel–Haenszel test (as for the primary efficacy analysis). These analyses were conducted on the completers' population and on the per protocol population). Subgroup analyses were carried out on the primary end point comparing results by country and by number of CBMs at baseline (0 or >0).
RESULTS
Patient characteristics and baseline demographics
In total, 374 patients were randomized, of whom four did not receive treatment and were excluded from the safety population (Supplementary Figure 1 online). The primary and secondary analyses were carried out on the mITT population, which included 181 patients taking placebo and 177 patients taking prucalopride, and excluded 12 patients owing to a serious breach of good clinical practice at one site. Sensitivity analyses of the primary end point were carried out on two subpopulations of the mITT population: the per protocol population (prucalopride, 146; placebo, 145) and the completers population (prucalopride, 146; placebo, 147). The majority of randomized patients completed the study (318 (85.0%)). The main reasons for discontinuation were withdrawal of consent (19 (5.1%)), TEAE(s) (13 (3.5%)), and non-compliance (9 (2.4%)). The discontinuation rates were similar in the two treatment groups (prucalopride, 29 patients (15.5%); placebo, 27 patients (14.4%)).
Baseline demographics of the safety population are presented in Table 1.The majority of patients were white (96.8%) with a mean age of 58.5 years (s.d., 16.91 years). Patients had an average duration of constipation of 9.2 years (s.d., 11.63 years). At baseline, 9.7% of patients had no SBMs per week, and 28.1% had an average of between zero and one SBM per week. The most common constipation-related symptoms at baseline were a feeling of not completely emptying the bowels (24.4%), straining (22.2%), and infrequent defecation (19.5%). There was a statistically significant difference between groups in the proportion feeling that they do not completely empty their bowels at baseline (P=0.009), reported by more subjects in the prucalopride treatment group (30.4%) than the placebo group (18.3%).
Table 1. Baseline demographics of the safety population.
| Placebo (N=186) | Prucalopride (N=184) | Total (N=370) | |
|---|---|---|---|
| Age, years | |||
| Mean (s.d.) | 58.5 (16.28) | 58.4 (17.57) | 58.5 (16.91) |
| <65 Years, n (%) | 115 (61.8) | 104 (56.5) | 219 (59.2) |
| Race, n (%) | |||
| White | 179 (96.2) | 179 (97.3) | 358 (96.8) |
| Black | 3 (1.6) | 5 (2.7) | 8 (2.2) |
| Asian/other | 4 (2.2) | 0 (0.0) | 4 (1.1) |
| Mean (s.d.) duration of constipation, years | 9.2 (11.35) | 9.2 (11.94) | 9.2 (11.63) |
| Number of SBMs per week,a n (%) | |||
| 0 | 14 (7.5) | 22 (12.0) | 36 (9.7) |
| >0 To ≤1 | 48 (25.8) | 56 (30.4) | 104 (28.1) |
| >1 To ≤2 | 102 (54.8) | 90 (48.9) | 192 (51.9) |
| >2 To ≤3 | 10 (5.4) | 8 (4.3) | 18 (4.9) |
| >3 | 12 (6.5) | 8 (4.3) | 20 (5.4) |
| Main symptom,a n (%) | |||
| Feeling of not completely emptying the bowels | 34 (18.3) | 56 (30.4) | 90 (24.4) |
| Straining | 44 (23.7) | 38 (20.8) | 82 (22.2) |
| Infrequent defecation | 42 (22.6) | 30 (16.4) | 72 (19.5) |
| Hard stools | 23 (12.4) | 25 (13.7) | 48 (13.0) |
| Abdominal bloating | 22 (11.8) | 16 (8.7) | 38 (10.3) |
| Abdominal pain | 21 (11.3) | 18 (9.8) | 39 (10.5) |
SBM, spontaneous bowel movement.
Average over past 6 months, as estimated by patients at screening.
However, there was not a significant difference between groups in the number of CBMs (0 or >0) during the run-in period (P=0.2869).
Efficacy
Primary end point
Over 12 weeks of treatment, statistically significantly more patients achieved a mean of three or more SCBMs per week in the prucalopride group (37.9%) than in the placebo group (17.7% P<0.0001; Figure 2). The number needed to treat was 5 (95% confidence interval (CI), 4, 10). The results of sensitivity analyses using the per protocol and completers populations, a generalized linear mixed model for repeated measures, multiple imputation, and logistic regression, were consistent with the primary analysis (data not shown).
Figure 2.

Proportion of patients with a mean frequency of three or more SCBMs per week. *P<0.005, **P≤0.0001. SCBM, spontaneous complete bowel movement.
The proportion of patients achieving three or more SCBMs per week was statistically significantly (P<0.005) greater in the prucalopride group than in the placebo group over weeks 1–4, 5–8, and 9–12 (prucalopride, 29.9%, 41.2%, and 39.0%, respectively; placebo, 14.9%, 23.2%, and 23.8%, respectively; Figure 2). In the subgroup analysis by country, the proportion of responders was higher in the prucalopride group (range, 20.0–66.7%) than in the placebo group (range, 0–55.6%) for all countries. In the subgroup analysis by baseline severity, there was a significantly higher proportion of responders in the prucalopride group than in the placebo group for those patients with zero CBMs during run-in (31.8 and 10.0%, P=0.0003) and for those patients with a mean of more than zero CBMs per week during run-in (43.5 and 23.8%, P=0.0081).
Secondary end points
Supplementary Table 1 presents the secondary end point variables. Statistically significantly more patients treated with prucalopride than those treated with placebo achieved a mean of three or more SCBMs per week in addition to an increase of one or more SCBM per week for at least 75% of the whole treatment period and at least 75% of the last third of the treatment period (prucalopride, 27.7% placebo, 12.2% P=0.0002). The proportion of patients with three or more SBMs, CBMs, and bowel movements per week, respectively, over the 12-week study period was statistically significantly higher in the prucalopride group (77.4%, 39.5%, and 80.8%, respectively) than in the placebo treatment group (60.8%, 21.5%, and 68.0% P=0.0011, P=0.0001, and P=0.0064, respectively).
At baseline, the mean percentage of SBMs with hard/very hard consistency was 51.2% (s.d., 39.57%) in the placebo group and 56.2% (s.d., 39.79%) in the prucalopride group. During the treatment period, this average percentage was 31.9% (s.d., 29.86%) in the placebo group and 26.9% (s.d., 28.27%) in the prucalopride group. From the intake of investigational product on day 1, the median time to the first SCBM was 218.9 h (CI, 143.93, 291.43) in the placebo group and 110.3 h (CI, 70.80, 172.77) in the prucalopride group, P=0.009.
The mean (s.d.) number of bisacodyl tablets used per week during the treatment period in the placebo and prucalopride treatment groups was 1.0 (1.76) and 0.6 (1.56), which corresponds to a mean (s.d.) decrease vs. run-in of 0.7 (1.84) and 1.0 (1.96), respectively. Overall, 43.1% of patients in the placebo group and 58.8% of those in the prucalopride treatment group used no rescue medication during the treatment period.
At baseline, 56.2% of patients in the placebo group and 67.6% of those in the prucalopride group rated their constipation as severe or very severe. At the final on-treatment assessment, constipation was rated as severe or very severe by 30.4% of patients who received placebo and by 21.9% of those who received prucalopride. The distribution of constipation severity across the five possible responses (absent, mild, moderate, severe, and very severe) was statistically significantly different (P<0.05) between the treatment groups at all time points, with more individuals in the prucalopride treatment group rating their constipation as severe or very severe at baseline and more in the placebo treatment group, rating their constipation as severe or very severe at all time points on therapy. The proportion of patients rating their constipation treatment as “quite a bit” or “extremely” effective at the final on-treatment assessment was 30.4% and 46.7% in the placebo and prucalopride treatment groups, respectively. The difference between the treatment groups was statistically significant (P<0.0001).
The mean PAC-SYM overall score was 1.75 (s.d., 0.67) at baseline for patients in the placebo group and 1.84 (s.d., 0.66) in the prucalopride group. The mean change from baseline (improvement) in the PAC-SYM overall score was numerically greater in the prucalopride group (–0.76; s.d., 0.77) than in the placebo group (–0.59; s.d., 0.76) at the final on-treatment assessment; however, this change was not statistically significant (P=0.0623).
There was also no statistically significant difference (P=0.3152) in the proportion of patients with a clinically significant improvement in the total PAC-SYM score of at least 1 point from baseline between the placebo (30.4%) and prucalopride (34.9%) treatment groups. In the overall population, the PAC-SYM stool-symptom subscale score was highest at baseline, with 63% of patients scoring their stool symptoms as severe to very severe (average score ≥3). For abdominal and rectal symptoms, these proportions were 27% and 11%, respectively. Significantly more patients had stool symptom improvements of at least 1 point in the prucalopride group (53.3% (n=90)) than in the placebo group (36.3% (n=62); P=0.0005) at the final on-treatment assessment. There was no statistically significant between-group difference at the final on-treatment assessment for the abdominal symptom (placebo, 35.1% (n=60); prucalopride, 39.1% (n=66); P=0.4874) and rectal symptom subscales (placebo, 29.2% (n=50); prucalopride, 34.9% (n=59); P=0.2759).
The mean change from baseline in the PAC-QOL score at the final on-treatment assessment was statistically significantly greater in the prucalopride group (–0.79 (s.d., 0.84)) than in the placebo group (–0.59 (s.d., 0.82); P=0.0158); a reduction in PAC-QOL score indicates an improvement. The proportion of patients with at least a 1 point improvement in the overall PAC-QOL score at the final on-treatment assessment was 32.7% for placebo and 40.2% for prucalopride (P=0.0755). For the PAC-QOL patient satisfaction subscale, 52.7% of patients taking prucalopride showed an improvement of at least 1 point compared with 38.8% of patients in the placebo group (P=0.0035). For the other PAC-QOL subscales, the differences between the proportions of patients in the prucalopride group and in the placebo group with an improvement of at least 1 point at the final on-treatment assessment were: 50.3% vs. 39.2% (P=0.0249) for the physical discomfort subscale, 30.2% vs. 24.6% (P=0.1234) for the psychosocial discomfort subscale, and 37.3% vs. 29.2% (P=0.0637) for the worries and concerns subscale.
Safety
An overview of the most common TEAEs in the safety population is shown in Table 2, and the complete list of TEAEs is provided as Supplementary Material (Supplementary Table 2). A total of 78 patients (42.4%) in the prucalopride group and 64 patients (34.4%) in the placebo group experienced at least one TEAE. These were considered by the investigator to be related to the investigational product in 42 patients (22.8%) and 25 patients (13.4%), respectively. The relative risk of experiencing a TEAE in the prucalopride group compared with the placebo group was 1.23 (CI, 0.95, 1.60); there was no statistically significant difference in the rate of TEAEs between the prucalopride and placebo groups. The most common TEAEs associated with the use of prucalopride were gastrointestinal disorders (diarrhea, nausea, and abdominal pain) and headache. Gastrointestinal disorders were experienced by 37 patients (20.1%) in the prucalopride group compared with 26 (14.0%) in the placebo group. Headache was experienced by 17 patients (9.2%) who received prucalopride and 7 patients (3.8%) taking placebo.
Table 2. Summary of TEAEs in the safety population.
| Placebo (N=186), n (%) | Prucalopride (N=184), n (%) | |
|---|---|---|
| ≥1 TEAE | 64 (34.4) | 78 (42.4) |
| ≥1 TEAE considered treatment-related | 25 (13.4) | 42 (22.8) |
| ≥1 Mild TEAE as worst severity | 37 (19.9) | 51 (27.7) |
| ≥1 Moderate TEAE as worst severity | 24 (12.9) | 24 (13.0) |
| ≥1 Severe TEAE as worst severity | 3 (1.6) | 3 (1.6) |
| ≥1 Serious TEAE | 4 (2.2) | 1 (0.5) |
| ≥1 Serious TEAE considered treatment-related | 0 (0.0) | 0 (0.0) |
| Deaths | 0 (0.0) | 0 (0.0) |
| ≥1 TEAE leading to permanent discontinuation | 7 (3.8) | 6 (3.3) |
| Types of TEAE | ||
| Gastrointestinal disorders | 26 (14.0) | 37 (20.1) |
| Diarrhea | 3 (1.6) | 12 (6.5) |
| Nausea | 4 (2.2) | 11 (6.0) |
| Abdominal pain | 11 (5.9) | 8 (4.3) |
| Nervous system disorders | 11 (5.9) | 22 (12.0) |
| Headache | 7 (3.8) | 17 (9.2) |
| Dizziness | 3 (1.6) | 4 (2.2) |
| Infections and infestations | 16 (8.6) | 10 (5.4) |
| Influenza | 4 (2.2) | 1 (0.5) |
| Nasopharyngitis | 5 (2.7) | 1 (0.5) |
TEAE, treatment-emergent adverse event.
TEAEs include the most common types (affecting >2% of the treatment group).
The majority of patients with at least one TEAE experienced TEAEs that were mild to moderate in severity (prucalopride, 75 (96.2%); placebo, 61 (95.3%)). Serious TEAEs were experienced by one patient taking prucalopride (0.5% atrial fibrillation) and four patients in the placebo group (2.2% one case each of myocardial ischemia, lower limb fracture, glottis carcinoma, and atelectasis), none of which were considered by the investigator to be related to the investigational product. The case of atrial fibrillation occurred in a patient with a previous medical history of the condition; the investigator considered this episode to be of moderate severity and unrelated to treatment. The arrhythmia resolved with medical management the following day and the patient completed the study. The overall incidence of cardiovascular AEs was low, reported by two subjects in the placebo group (one case of angina pectoris and one case of myocardia ischemia) and one subject in the prucalopride treatment group (one case of coronary artery occlusion). There were no fatalities in the study. Six patients (3.3%) in the prucalopride group and seven patients (3.8%) in the placebo group experienced TEAEs that led to permanent discontinuation of the investigational product, all of which were mild to moderate in severity. The most common TEAEs leading to permanent discontinuation in the prucalopride treatment group were diarrhea, nausea, headache, and dizziness, which were reported for 3 (1.6%), 2 (1.1%), 3 (1.6%), and 2 (1.1%) patients, respectively (patients could have more than one TEAE leading to discontinuation). The mean changes from baseline in clinical laboratory and electrocardiogram parameters were generally small and no changes were considered clinically relevant. There was one event of electrocardiogram QT prolonged in the prucalopride group, in a subject who reported no other concomitant AEs, that was noted at week 4, before returning to normal at week 12. This event was considered to be non-serious and did not lead to treatment discontinuation.
DISCUSSION
In this study, the efficacy of prucalopride once daily compared with placebo was assessed over 12 weeks of treatment in men with chronic constipation. Prucalopride was significantly more effective than placebo at increasing the number of bowel movements and improving the mean HRQoL score in this population.
These findings were confirmed across all sensitivity analyses and are consistent with the overall results from the four previously published phase 3 trials, which enrolled predominantly female patients (9, 10, 11, 12, 13, 14). The design of the present study is comparable to that of the pivotal phase 3 trials, and the same primary end point (the proportion of patients with a mean of three or more SCBMs per week) was investigated in all studies. In this study, the difference in response to prucalopride compared with placebo (therapeutic gain) was 20.2%, which is consistent with that seen in the four pivotal phase 3 trials of prucalopride (9.9–20.0% (refs 9, 10, 11, 14). This demonstrates that the response rate to prucalopride is similar in men and women.
Analysis of the secondary end points generally supports the findings of the primary end point. Fewer patients used rescue medication in the prucalopride patient group (41.2%) than in the placebo group (56.9%). The improvement in HRQoL in patients receiving prucalopride compared with those receiving placebo is also consistent with previous studies (14, 15). In addition, at the final on-treatment visit, patients treated with prucalopride were less likely to rate their symptoms as severe, and more likely to rate the treatment as effective, than those in the placebo group.
Whereas there was a significant improvement in PAC-SYM subscale scores for stool symptoms in favor of prucalopride, there was no statistically significant difference in overall PAC-SYM response or abdominal and rectal symptom subscales between prucalopride and placebo. These findings conflict with those of the previous phase 3 trials, which demonstrated significantly greater improvements in the overall PAC-SYM score in patients receiving prucalopride than in those taking placebo (9, 10, 13, 14). It is likely that the smaller sample size in the present study than in the previous trials may have contributed to the inability to achieve statistical significance for this end point. A separate integrated analysis of the subgroup of male patients from six clinical studies demonstrated a significantly greater improvement in the PAC-SYM total score in prucalopride-treated than in placebo-treated patients, providing support for the requirement for a large sample size to be able to detect this difference statistically (Shire, data on file).
Prucalopride had a good safety profile and was well tolerated in this trial; this is also consistent with previous studies (15, 22, 23, 24). No fatal TEAEs occurred and the incidence of other serious TEAEs was low across both treatment groups. The most common TEAEs associated with the use of prucalopride were gastrointestinal disorders (diarrhea, nausea, and abdominal pain) and headache. The types and frequencies of TEAEs, clinical laboratory abnormalities, and other safety parameters in the prucalopride treatment group were comparable to those in the placebo group. No new safety signals were identified. In particular, this study demonstrated a good cardiovascular safety profile for prucalopride. Unlike the previous, less selective 5-HT4 agonists that were withdrawn from the market because of the association with fatal arrhythmias related to QT prolongation and other cardiovascular AEs, prucalopride is highly selective. The safety of prucalopride has been confirmed in a number of previous large-scale studies, including a thorough QT study and a trial specifically in elderly patients (22, 23, 24).
A potential limitation of this study is that only European study sites were included and that the trial was restricted to 12 weeks. However, as this is the first prucalopride investigation to focus solely on efficacy in men, a larger sample size of male patients was achieved in this trial compared with the previous studies.
On the basis of these findings, it can be concluded that prucalopride once daily, taken for 12 weeks, is effective in the treatment of chronic constipation in men. Both the efficacy and safety results of this study were in line with observations in the overall prucalopride population studied to date, which has included predominantly female patients. Prucalopride was generally well tolerated and no new safety signals were identified.
Study Highlights

Acknowledgments
Writing support was provided by Dr Rosalind Morley and Dr Vivienne Johnson of PharmaGenesis London, and this study was funded by Shire.
Guarantor of the article: Yan Yiannakou, MD.
Specific author contributions: R.K. contributed to the concept and design of the study. A.L., H.P., K.E., R.K., and Y.Y. were involved in data acquisition and analysis. All authors contributed to data interpretation and critically reviewed the manuscript. All authors approved the final version of the manuscript.
Financial support: This study was funded in full by Shire-Movetis NV.
Potential competing interests: H.P. has received lecture fees and travel grants from Shire. Y.Y. has received lecture fees, a travel and educational grants from Shire. K.E. has received a travel grant from Shire. A.L. is an employee and shareholder of Shire. R.K. was an employee of Shire at time of the study. D.S. was an employee and shareholder of Shire at the time of the study. The remaining authors declare no conflict of interest.
Footnotes
SUPPLEMENTARY MATERIAL is linked to the online version of the paper at http://www.nature.com/ajg
Supplementary Material
References
- Johanson JF, Kralstein J. Chronic constipation: a survey of the patient perspective. Aliment Pharmacol Ther. 2007;25:599–608. doi: 10.1111/j.1365-2036.2006.03238.x. [DOI] [PubMed] [Google Scholar]
- Herz MJ, Kahan E, Zalevski S, et al. Constipation: a different entity for patients and doctors. Fam Pract. 1996;13:156–9. doi: 10.1093/fampra/13.2.156. [DOI] [PubMed] [Google Scholar]
- Longstreth GF, Thompson WG, Chey WD, et al. Functional bowel disorders. Gastroenterology. 2006;130:1480–91. doi: 10.1053/j.gastro.2005.11.061. [DOI] [PubMed] [Google Scholar]
- Suares NC, Ford AC. Prevalence of, and risk factors for, chronic idiopathic constipation in the community: systematic review and meta-analysis. Am J Gastroenterol. 2011;106:1582–91. doi: 10.1038/ajg.2011.164. [DOI] [PubMed] [Google Scholar]
- Wald A, Scarpignato C, Mueller-Lissner S, et al. A multinational survey of prevalence and patterns of laxative use among adults with self-defined constipation. Aliment Pharmacol Ther. 2008;28:917–30. doi: 10.1111/j.1365-2036.2008.03806.x. [DOI] [PubMed] [Google Scholar]
- Belsey J, Greenfield S, Candy D, et al. Systematic review: impact of constipation on quality of life in adults and children. Aliment Pharmacol Ther. 2010;31:938–49. doi: 10.1111/j.1365-2036.2010.04273.x. [DOI] [PubMed] [Google Scholar]
- Irvine EJ, Ferrazzi S, Pare P, et al. Health-related quality of life in functional GI disorders: focus on constipation and resource utilization. Am J Gastroenterol. 2002;97:1986–93. doi: 10.1111/j.1572-0241.2002.05843.x. [DOI] [PubMed] [Google Scholar]
- Muller-Lissner S, Tack J, Feng Y, et al. Levels of satisfaction with current chronic constipation treatment options in Europe—an internet survey. Aliment Pharmacol Ther. 2013;37:137–45. doi: 10.1111/apt.12124. [DOI] [PubMed] [Google Scholar]
- Tack J, van Outryve M, Beyens G, et al. Prucalopride (Resolor) in the treatment of severe chronic constipation in patients dissatisfied with laxatives. Gut. 2009;58:357–65. doi: 10.1136/gut.2008.162404. [DOI] [PubMed] [Google Scholar]
- Camilleri M, Kerstens R, Rykx A, et al. A placebo-controlled trial of prucalopride for severe chronic constipation. N Engl J Med. 2008;358:2344–54. doi: 10.1056/NEJMoa0800670. [DOI] [PubMed] [Google Scholar]
- Quigley EM, Vandeplassche L, Kerstens R, et al. Clinical trial: the efficacy, impact on quality of life, and safety and tolerability of prucalopride in severe chronic constipation—a 12-week, randomized, double-blind, placebo-controlled study. Aliment Pharmacol Ther. 2009;29:315–28. doi: 10.1111/j.1365-2036.2008.03884.x. [DOI] [PubMed] [Google Scholar]
- Tack J, Quigley E, Camilleri M, et al. Efficacy and safety of oral prucalopride in women with chronic constipation in whom laxatives have failed: an integrated analysis. United European Gastroenterol J. 2013;1:48–59. doi: 10.1177/2050640612474651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tack J, Stanghellini V, Dubois D, et al. Effect of prucalopride on symptoms of chronic constipation. Neurogastroenterol Motil. 2014;26:21–7. doi: 10.1111/nmo.12217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke M, Zou D, Yuan Y, et al. Prucalopride in the treatment of chronic constipation in patients from the Asia-Pacific region: a randomized, double-blind, placebo-controlled study. Neurogastroenterol Motil. 2012;24:999–e541. doi: 10.1111/j.1365-2982.2012.01983.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin A, Camilleri M, Kolar G, et al. Systematic review with meta-analysis: highly selective 5-HT4 agonists (prucalopride, velusetrag or naronapride) in chronic constipation. Aliment Pharmacol Ther. 2014;39:239–53. doi: 10.1111/apt.12571. [DOI] [PubMed] [Google Scholar]
- ICH harmonised tripartite guidelines for good clinical practice 1996 ICH Harmonised Tripartite Guidelines for Good Clinical Practice, 1996; ; http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R1_Guideline.pdf Accessed 11 June 2012.
- World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. http://www.wma.net/e/policy/b3.htm [Cited 12 August 2009].
- Lewis SJ, Heaton KW. Stool form scale as a useful guide to intestinal transit time. Scand J Gastroenterol. 1997;32:920–4. doi: 10.3109/00365529709011203. [DOI] [PubMed] [Google Scholar]
- Frank L, Kleinman L, Farup C, et al. Psychometric validation of a constipation symptom assessment questionnaire. Scand J Gastroenterol. 1999;34:870–7. doi: 10.1080/003655299750025327. [DOI] [PubMed] [Google Scholar]
- Dubois D, Gilet H, Viala-Danten M, et al. Psychometric performance and clinical meaningfulness of the Patient Assessment of Constipation-Quality of Life questionnaire in prucalopride (RESOLOR) trials for chronic constipation. Neurogastroenterol Motil. 2010;22:e54–63. doi: 10.1111/j.1365-2982.2009.01408.x. [DOI] [PubMed] [Google Scholar]
- Marquis P, De La Loge C, Dubois D, et al. Development and validation of the Patient Assessment of Constipation Quality of Life questionnaire. Scand J Gastroenterol. 2005;40:540–51. doi: 10.1080/00365520510012208. [DOI] [PubMed] [Google Scholar]
- Camilleri M, Beyens G, Kerstens R, et al. Safety assessment of prucalopride in elderly patients with constipation: a double-blind, placebo-controlled study. Neurogastroenterol Motil. 2009;21:1256–e117. doi: 10.1111/j.1365-2982.2009.01398.x. [DOI] [PubMed] [Google Scholar]
- Mendzelevski B, Ausma J, Chanter DO, et al. Assessment of the cardiac safety of prucalopride in healthy volunteers: a randomized, double-blind, placebo- and positive-controlled thorough QT study. Br J Clin Pharmacol. 2012;73:203–9. doi: 10.1111/j.1365-2125.2011.04088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman H, Pasternack M. The action of the novel gastrointestinal prokinetic prucalopride on the HERG K+ channel and the common T897 polymorph. Eur J Pharmacol. 2007;554:98–105. doi: 10.1016/j.ejphar.2006.10.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.